
Abstract
The energy level alignments at donor/acceptor interfaces in organic photovoltaics (OPVs) play a decisive role in device performance. However, little is known about the interfacial energetics in polymer OPVs due to technical issues of the solution process. Here, the frontier ortbial line-ups at the donor/acceptor interface in high performance polymer OPVs, PTB7/PC71BM, were investigated using in situ UPS, XPS and IPES. The evolution of energy levels during PTB7/PC71BM interface formation was investigated using vacuum electrospray deposition, and was compared with that of P3HT/PC61BM. At the PTB7/PC71BM interface, the interface dipole and the band bending were absent due to their identical charge neutrality levels. In contrast, a large interfacial dipole was observed at the P3HT/PC61BM interface. The measured photovoltaic energy gap (EPVG) was 1.10 eV for PTB7/PC71BM and 0.90 eV for P3HT/PC61BM. This difference in the EPVG leads to a larger open-circuit voltage of PTB7/PC71BM than that of P3HT/PC61BM.
Similar content being viewed by others

Introduction
Organic photovoltaics (OPVs) have greatly progressed in recent decades, and currently exceed power conversion efficiencies (PCE) of 10%, a value which is considered commercially viable1,2,3. To enhance the PCE further for mass production, optimization of the photophysical process during OPV operation is necessary.
In OPVs, photogenerated excitons are dissociated by the photovoltaic energy gap (EPVG), which is a potential difference between the highest occupied molecular orbital (HOMO) level of a donor (HOMOdonor) and the lowest unoccupied molecular orbital (LUMO) level of an acceptor (LUMOacceptor), and then the charges are drifted by the built-in potential induced from the work function difference between electrodes. Therefore, understanding the energy level alignments at the electrode/organic and organic/organic interfaces is essential to build a strategy for highly efficient OPVs4,5,6,7. For these, accurate measurements of the charge transport levels and EPVG are of great importance. Despite such importance, there have been no direct measurements on the EPVG at polymeric junction, which should be followed by the accurate measurements on the energy level alignment at the junction. Inaccurate presumption from separate energy levels of each constituent at the junction could give over/underestimated results. The interface effects such as an interface dipole (eD) and charge transfer must be considered for the true EPVG8,9,10.
The accurate measurements of such energy levels could play a crucial role in EPVG model itself. The actual VOC deviates from the EPVG by an empirical value11,12,13,14. This could be due to the missing parameters in the EPVG model or inaccurate EPVG value for a given junction. It cannot be concluded that which one is the main factor for the deviation unless the EPVG is measured accurately. The same is true for the exciton binding energy (Eexc) of a polymer. It was suggested that the lower Eexc generates the higher photocurrent with theoretical calculations15. However, only inaccurate values of Eexc have been reported without comparison between the optical gap and transport gap. We will resolve these issues in the result section below.
In addition, the free energy of charge separation (GCS) is a very recent issue related to the exciton dissociation at the interfaces. Jonathan et al. suggested that the optimum GCS value exists to maximize the JSC4,16,17. However, it still remains as an open problem because no experimental evidence for GCS has been reported at the polymeric junction due to the technical issue discussed below. Collectively, it is highly necessary to measure the energy levels accurately at the polymeric junction to give deep insight into the core OPV-parameters such as EPVG, Eexc, GCS and so on.
To determine the true energy level alignments in OPVs, there are two key requirements. (1) Measurements of charge transport level in a solid state: The energy level measurements in a liquid state, e.g. cyclic voltammetry, cannot provide true charge transport levels associated with the solid state device due to ignorance of the screening effect by surrounding molecules/polymers in a solid state. (2) Consideration of an interface dipole and energy level relaxation across the interface: It is well known that the Schottky-Mott limit is not always valid at organic interfaces, and thus the charge injection/extraction barriers in OPVs cannot be estimated accurately from individual measurements of each layer. Therefore, in situ photoelectron spectroscopy (PES) with stepwise deposition-measurement procedures are indispensable18,19.
For the interfaces in small molecule OPVs, stepwise measurements are feasible since thermal evaporation enables incremental deposition of organic molecules5,20. However, it is challenging to study the energy level alignments at polymeric interfaces prepared by solution process due to technical limitations. It is often impossible to trace the evolution of energy levels from the stepwise formation of polymeric interfaces. This is because the polymer solution used to form an upper layer could dissolve the preformed underlying-layer. Therefore, spin-coating is not an adequate method for film preparation to investigate the interfacial energy level alignments. Due to such limitations, the true electronic structure and its evolution at the polymer donor/acceptor “interface” have not been shown. To overcome these limitations of spin-coating methods, we adopted a vacuum electrospray deposition (VESD) method to form the polymeric thin film in a stepwise manner (see the experimental section and previous report)21,22,23. The VESD OPVs have shown almost identical device performance with the spin-coated OPVs, ensuring high film quality with the VESD method for device fabrication21,24. By VESD, the HOMO and LUMO energy levels, the interface dipole (eD), and the band bending (VB) of polymers in bulk heterojunction (BHJ) devices can be studied.
In BHJ OPVs, poly(4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl-alt-3-fluoro-2-[(2-ethylhexyl)carbonyl] thieno[3,4-b]thiophene-4,6-diyl) (PTB7) is a promising donor material recording the remarkable PCE because of its superior optoelectronic properties25,26. The low-band gap polymer PTB7 is well-suited to [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) which absorbs a broad range of light. However, the true energy level alignments at the PTB7:PC71BM interface have not been measured previously due to the inherent problems in spin-coating deposition as aforementioned.
In this study, we determined the complete energy level alignments at the PTB7/PC71BM interface and related interfaces. In situ ultraviolet and X-ray photoelectron spectroscopy (UPS and XPS) measurements were performed to determine the valence density of states of PTB7 and PC71BM. Inverse photoelectron spectroscopy (IPES) was used to measure their transport band gap (Et), and thus the true electron transport level (LUMO). Theoretical calculations using density functional theory (DFT) were performed and the calculated results were compared with the measured UPS and IPES spectra. Finally, the PTB7/PC71BM interface was compared with a poly(3-hexylthiophene-2,5-diyl) (P3HT)/[6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) interface, which is a former popular donor/acceptor junction. From the measured energy level alignments, critical parameters of OPVs such as EPVG, Eexc and Gcs were accurately evaluated to understand the OPV operation in depth. Finally brief strategy to improve OPV performance will be suggested.
Results and Discussion
The donor/acceptor junction was prepared on PEDOT:PSS since the most widely used OPV structure is ITO/PEDOT:PSS/Active layer/Cathode27,28,29. Yang et al. reported that BHJ films show phase separation and form microscopic junctions of donor and acceptor30,31,32. This junction was simulated with the bilayer structure of PTB7/PC71BM to determine the energy level alignments across the junction. Step-by-step deposition and in situ photoelectron spectroscopy (PES) measurements for PTB7 and PC71BM were performed to study the interfacial energy level alignments. Prior to the in situ PES measurements, we compared the morphology and electronic structure of the spin-coated and VESD films to ensure consistent film quality. For that, atomic force microscope (AFM) and PES measurements on each bulk film were conducted (Supplementary Figs S1 and S2). Both films showed quite similar morphology and the same electronic structure, and from this we conclude that interfaces prepared by VESD are identical with the spin-coated films.
PEDOT:PSS/PTB7 and PEDOT:PSS/P3HT interface
First, we considered the interface of PEDOT:PSS/PTB7. Figure 1 shows the UPS spectra of (a) the normalized secondary electron cutoff (SEC) region, (b) the background-removed HOMO region and (c) the magnified HOMO region of the PEDOT:PSS/PTB7 (0.3, 0.9, 2.0, 2.5, 5.6, 7.0 nm) sample. At the 0.3 nm-deposition step, the HOMO level of PTB7 is observed at 0.45 eV below the Fermi level (c). The SEC of PEDOT:PSS is observed at 4.80 eV in kinetic energy scale, indicating the work function (WF) of PEDOT:PSS (a). At the first PTB7 deposition step, the SEC abruptly shifts toward lower kinetic energies by 0.20 eV, then gradually shifts further toward lower kinetic energies by 0.35 eV. The HOMO onset also shifts by 0.35 eV, implying the VB in the PTB7 side. The change of WF (ΔWF) contains both contributions of VB and eD; thus they are related33:

UPS spectra of PEDOT:PSS/PTB7 interface.
UPS spectra of (a) the normalized secondary electron cutoff region, (b) the background-removed HOMO region and (c) magnified HOMO region of the PEDOT:PSS/PTB7 (0.3, 0.9, 2.0, 2.5, 5.6, 7.0 nm).

To separate the contributions of VB and eD, the thickness dependent evolution of the WF and PTB7 HOMO are depicted in Fig. 2. The VB and eD of PTB7 were evaluated to be 0.35 eV and 0.20 eV, respectively. The high work function of PEDOT:PSS induces the VB and electron transfer from PTB7 to PEDOT:PSS, and thus increases electron carrier in PEDOT:PSS and hole carrier density in PTB75,34. Increased hole carrier density induces the VB, and this bending direction assists the hole extraction from PTB7 to PEDOT:PSS.

Energy level change of PEDOT:PSS/PTB7 interface.
Work function and PTB7 HOMO onset evolutions as a function of PTB7 film thickness.
XPS core level spectra of polymers were cross-checked with the energy level shifts measured from UPS spectra. To evaluate the peak shift, we deconvoluted each spectrum while keeping system constraints. We first analysed each bulk (pristine) spectrum of S 2p of PTB7 (topmost) and PEDOT:PSS (bottommost). The pristine PTB7 S 2p core level was observed at 163.3 eV and 165.1 eV with a spin-orbit splitting of 1.8 eV35. In the bottom of the Fig. 3a, we also deconvoluted the S 2p spectrum with three components (PEDOT, PSSH and PSS−-Na+) to evaluate the peak shift accurately. We used fitting constraints of PEDOT:PSS from previous reports such as the intensity ratio and energetic distance between each peak (PEDOT, PSSH and PSS−-Na+) during the fitting process36,37. As a result, we observed the 0.3 eV peak shift of PTB7 and PEDOT:PSS in the S 2p orbital, which coincides well with the HOMO level shifts measured from UPS spectra within the margin of error. However, the F 1s peak maintained the same position during PTB7 deposition.

XPS spectra of PEDOT:PSS/PTB7 interface.
Measured XPS spectra of (a) S 2p and (b) F 1s of the PEDOT:PSS/PTB7 (0.3, 0.9, 2.0, 2.5, 5.6, 7.0 nm).
To understand the different core level shifts in S 2p and F 1s, the electronic structures of PTB7 were calculated using DFT. To simulate long polymer chains, we modeled 8-mer of PTB7 unit structure (Supplementary Fig. S4). Figure 4 shows the calculated (a) electrostatic potential map, (b) HOMO and (c) LUMO wave function of PTB7 8-mer. The electrostatic potential map shows the charge distribution of PTB7. In Fig. 4a, a low (red) and high (blue) electrostatic potential indicates the electron rich and deficient regions, respectively. The F atom (red-dashed circle) withdraws electrons in the vicinity due to its high electronegativity, which is seen in the electrostatic potential map (a). However, F hardly contributes to the HOMO (b) and LUMO (c) because of the very strong C-F bonding due to the high electronegativity of F. This strong bonding induces localized electronic states which are energetically far below (above) the delocalized HOMO (LUMO) state. Thus, the contribution of F to the HOMO and LUMO is negligible. Since the electron transfer occurs predominantly from the HOMO level of PTB7 to the Fermi level of PEDOT:PSS, the intact F 1s position can be understood with the localized C-F bonding state during the interface charge transfer process.

DFT calculation.
Calculated (a) Electrostatic potential map, (b) HOMO wave function and (c) LUMO wave function of a monomer unit on PTB7 (scale bar: hartree units). Dashed red circles indicate the F atom in PTB7. (Atomic colour code: grey for C, red for O, yellow for S, cyan for F, white for H).
We measured the interface between PEDOT:PSS and P3HT as a non-push-pull donor counterpart. Figure 5 shows the UPS spectra of (a) the normalized SEC region, (b) the background-removed HOMO region and (c) magnified HOMO region of the PEDOT:PSS/P3HT (0.3, 0.6, 1.0, 2.0, 5.0, 7.0 nm) sample. At the 0.3 nm-deposition step, the HOMO level of P3HT is observed at 0.35 eV below the Fermi level. As the film thickens, the HOMO of P3HT gradually shifts toward higher binding energies by 0.57 eV. The SEC of PEDOT:PSS is observed at 4.75 eV in kinetic energy and shifts to lower kinetic energy by 1.10 eV, indicating WF reduction. In the same manner, we evaluated VB,P3HT and eD by 0.57 eV and 0.53 eV, respectively. Detailed energy level alignments of each interface will be discussed below.

UPS spectra of PEDOT:PSS/P3HT interface.
UPS spectra of (a) the normalized secondary electron cutoff region, (b) the background-removed HOMO region and (c) magnified HOMO region of the PEDOT:PSS/P3HT (0.3, 0.6, 1.0, 2.0, 5.0, 7.0 nm).
PTB7/PC71BM and P3HT/PC61BM interfaces
Another important interface in BHJ is the donor/acceptor interface, PTB7/PC71BM and P3HT/PC61BM, and we continued PC71BM and PC61BM deposition on the final PTB7 and P3HT layers explained in section A, respectively. Figure 6 shows the UPS spectra of (a) the normalized SEC region, (b) the background-removed HOMO region and (c) magnified HOMO region of the PTB7 (7 nm)/PC71BM (0.4, 0.6, 0.9, 1.3, 2.5, 5.0 nm) sample. There is no SEC shift during the PC71BM deposition, which implies that the WF is intact as 4.25 eV. The bulk HOMO level of PTB7 and PC71BM appears at 0.80 and 1.60 eV, respectively. To determine the HOMO onset of PTB7 and PC71BM in the middle of PC71BM deposition, the HOMO spectra were deconvoluted with two pristine bulk PTB7 (bottommost spectrum) and PC71BM spectra (topmost spectrum). The intensity of deconvoluted PTB7 and PC71BM spectra was estimated using the electron effective attenuation length (EAL) of PC71BM, which was obtained from the attenuation of S 2p of PTB7 (detailed evaluation is in supplementary information). The summation of two bulk spectra reproduced the measured spectra quite well without any additional components. This indicates the absence of molecular orbital hybridization and chemical reactions, which is consistent with the XPS spectra (Supplementary Fig. S3). Furthermore, both HOMO and SEC positions were maintained intact during the PC71BM deposition, meaning the absence of both charge transfer and eD. It concludes that PTB7 and PC71BM have the same charge neutrality level5,38,39,40.

UPS spectra of PTB7/PC71BM interface.
UPS spectra of (a) the normalized secondary electron cutoff, (b) the background removed HOMO and (c) magnified HOMO region of the PTB7/PC71BM (0.4, 0.6, 0.9, 1.3, 2.5, 5.0 nm).
The interfacial electronic structure of PTB7/PC71BM is compared with P3HT/PC61BM as a non-push-pull counterpart. Figure 7 shows the UPS spectra of (a) the SEC region, (b) HOMO region and (c) magnified HOMO region of P3HT (7.0 nm)/PC61BM (0.2, 0.5, 1.1 and 2.2 nm). As shown in Fig. 7a, a significant SEC shift is observed from 3.65 eV to 4.10 eV. From the SEC shift and VB, a large eD of 0.35 eV (= 0.45–0.10 eV) exists, which is caused by a large charge-neutrality level difference between P3HT and PC61BM. In Fig. 7c, the bulk HOMO level of P3HT and PC61BM appears at 0.92 eV and 1.92 eV. We also deconvoluted the HOMO spectra in the same manner. As shown in Fig. 7c, the HOMO of P3HT shifts toward lower binding energies by 0.10 eV, indicating the VB. For the origin of this VB, two scenarios are plausible: (1) electron attraction of PC61BM from P3HT and subsequent induced polarization of charge densities or (2) local changes in the inter-ring torsional angle of P3HT at the interface due to the interaction with PC61BM15. As a result, the P3HT/PC61BM interface forms a more complex junction than the PTB7/PC71BM interface.

UPS spectra of P3HT/PC61BM interface.
UPS spectra of (a) the normalized secondary electron cutoff, (b) the background-removed HOMO region and (c) magnified HOMO region of the P3HT/PC61BM (0.2, 0.5, 1.1, 2.2 nm).
Transport band gap determination
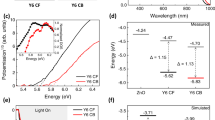
Figure 8 shows the combined UPS and IPES spectra of bulk (a) PTB7 and (c) PC71BM and their comparison with calculated spectrum (b) PTB7 and (d) PC71BM. The calculated spectrum was rigidly shifted to match the measured HOMO and LUMO position41,42,43. In both HOMO and LUMO regions, the DOS of measured and calculated spectra match well, which ensures reliable determination of the HOMO and LUMO onsets regardless of spectral noise, particularly for the IPES. As a result, the transport band gap (Et, PTB7 = 2.25 eV, Et, PC71BM = 1.90 eV), ionization energy (IEPTB7 = 5.05 eV, IEPC71BM = 5.85 eV) and electron affinity (EAPTB7 = 2.80 eV, EAPC71BM = 3.95 eV) were determined. From these values, the exciton binding energy (Eexc) of each material was determined, which was evaluated from the following equation8,44,45.

UPS and IPES spectra of single layer.
(a) UPS and IPES spectra of the measured HOMO and LUMO region of PTB7, (b) comparison between UPS-IPES spectrum and the calculated DOS of PTB7. UPS and IPES spectra of (c) the measured HOMO and LUMO region of the PC71BM and (d) comparison between UPS-IPES spectrum and the calculated DOS of PC71BM.

where Et is the transport band gap and Eopt is the optical band gap. Eopt of each material was determined from previous reports46,47,48,49. Ram et al. reported a relatively smaller Eexc of PTB7 and thus suggested easy exciton dissociation in PTB715. However, current results show the same Eexc of P3HT and PTB7. Therefore, the interfacial electronic structure is more important than the Eexc of the bulk donor for efficient exciton dissociation. The measured and evaluated IE, EA, Et and Eexc are listed in Table 1.
Energy level alignment
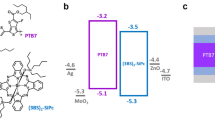
Collecting all the WF and HOMO level changes during the interface formation of ITO/PEDOT:PSS/PTB7/PC71BM and ITO/PEDOT:PSS/P3HT/PC61BM, the energy level diagrams are drawn in Fig. 9. The LUMO levels were determined by measured Et values (2.25 eV for PTB7 and 1.90 eV for PC71BM) and the reported Et values (2.52 eV for P3HT and 2.00 eV for PC61BM)50. As shown in Fig. 9ab, the hole extraction barriers from donor to PEDOT:PSS are 0.45 eV for PTB7 and 0.35 eV for P3HT. PEDOT:PSS strongly pulls electrons from both polymers due to its high WF, which induces band bending of VB,PTB7 = 0.35 eV and VB,P3HT = 0.57 eV. The eD at both interfaces of PEDOT:PSS/PTB7 and PEDOT:PSS/P3HT indicates that the high WF of PEDOT:PSS induces the energy level pinning at both interfaces. The pinning threshold of P3HT (Ep+ = 3.9 eV) matches well with previous reports6,51. The pinning threshold of PTB7 can be estimated to be less than 4.80 eV based on the eD at the PEDOT:PSS/PTB7 interface.

Energy level diagrams.
Energy level diagrams of (a) ITO/PEDOT:PSS/PTB7/PC71BM and (b) ITO/PEDOT:PSS/P3HT/PC61BM. Фh and eD are the hole extraction barrier and interface dipole, respectively. ΨITO, ΨPEDOT:PSS, ΨPTB7, ΨPC71BM, ΨP3HT and ΨPC61BM are the work functions of ITO, PEDOT:PSS, PTB7, PC71BM, P3HT and PC61BM, respectively. Comparing the diagrams, we observed the strong interface dipole between P3HT and PC61BM. Schematic image of (c) PC71BM and PTB7 and (d) PC61BM and P3HT interfaces.
In Fig. 9a, the PTB7/PC71BM interface shows a flat band alignment and no interfacial potential change. However, the P3HT/PC61BM interface shows a large eD due to Fermi level pinning of PCBM (Ep− = 4.3 eV)6,52. In the P3HT:PC61BM BHJ film, the eD’s are randomly distributed in 3-dimensional space, which gives random local electric fields, and thus it does not affect the built-in potential. However, it does gives local singularities in potential distribution and may assist or hinder exciton dissociation and charge transport according to its direction. Therefore it exerts a strong influence on the EPVG at D/A interfaces.
The EPVG of PTB7/PC71BM and P3HT/PC61BM is evaluated as energetic difference between HOMOdonor and LUMOacceptor thus 1.10 eV (0.80 + 0.30 eV) and 0.90 eV (0.82 + 0.08 eV) for P3HT/PC61BM, respectively. This accords well with the fact that the PTB7/PC71BM OPVs have higher VOC than the P3HT/PC61BM OPVs by about 0.1~0.2 eV53,54,55. These findings not only show the direct evidence of correlation between VOC and EPVG but also explain the role of eD in EPVG, i.e. the eD can change the EPVG significantly. Therefore, the eD at the interface should be considered rigorously to determine the EPVG accurately.
Another interesting issue in BHJ OPVs is the ΔGCS during the charge separation process. It is reported that ΔGCS acts as the driving force to dissociate the exciton at the donor/acceptor interfaces16. Thus, a high ΔGCS leads to more efficient exciton dissociation and enhances the PCE. However, too large a ΔGCS creates parasitic energy losses and limits the PCE reversely. Generally, the ΔGCS is calculated by the abbreviated Rehm-Weller equation17.

where the Es is the singlet excited state energy, which is derived from the Eopt46,47,48,49. Until now, ΔGCS was estimated from the IEDonor and EAacceptor measured from individual film without any consideration of electronic polarization (solid state effect) at the donor-acceptor interface. Therefore, it would under/over-estimate the ΔGCS inherently. In this study, ΔGCS is evaluated as 0.53 eV for PTB7/PC71BM and 1.00 eV for P3HT/PC61BM including all local interfacial potentials such as VB and eD. Too large a ΔGCS of P3HT/PC61BM induces large parasitic energy losses and induces negative effects on exciton dissociation, limiting the PCE significantly. Jonathan et al. reported that an ideal value of ΔGCS and the optical band gap of the donor is about 0.4 eV and 1.6 eV, respectively16. Current measurements show that the ΔGCS and optical band gap of PTB7/PC71BM system is quite close to the optimum values. Absence of eD, which hinders charge separation in P3HT/PC61BM, and proper ΔGCS at the PTB7/PC71BM interface are the origin of high PCE of PTB7 in conjunction with its excellent light absorption. Too large a ΔGCS and singular potential due to eD at the P3HT interfaces have a negative effect on device performance. All these factors are determined from the interfacial electronic structure of donor and acceptor materials. These results demonstrate the role of eD in determination of EPVG and the correlation between VOC and EPVG.
Therefore, we suggest the strategy for the high performance OPVs: (1) proper combination of donor and acceptor materials should be chosen based on their charge neutrality levels. Improper level alignment may cause the potential discontinuity due to the eD, which impedes the charge separation and increases the interface recombination. (2) ΔGCS should be optimized to dissociate exciton efficiently without parasitic energy losses.
Conclusion
In this study, in situ UPS, IPES and XPS measurements were performed to figure out the pristine valence density of states of PEDOT:PSS, PTB7, PC71BM and their interfacial energy level alignments. As a result, the Et (Et, PTB7 = 2.25 eV, Et, PC71BM = 1.90 eV), ionization energy (IEPTB7 = 5.05 eV, IEPC71BM = 5.85 eV) and electron affinity (EAPTB7 = 2.80 eV, EAPC71BM = 3.95 eV) were obtained. Large band bending is observed in PTB7 on the PEDOT:PSS layer, which reduces the hole extraction barrier at the PEDOT:PSS/PTB7 interface, and the same is true for PEDOT:PSS/P3HT. Interfacial dipoles and strong band bending are not observed at the PTB7/PC71BM interface, meaning negligible charge exchanges between PTB7 and PC71BM. In contrast, a large interface dipole is observed between P3HT and PC61BM. In addition, the EPVG between a donor and an acceptor (1.10 eV for PTB7/PC71BM and 0.90 eV for P3HT/PC61BM) were also measured. The ΔGCS at the interface of PTB7/PC61BM and P3HT/PC71BM were determined as 0.53 eV and 1.00 eV, respectively. A not too large ΔGCS reduces parasitic energy losses and thus enhances the exciton dissociation, which is the reason why the PTB7/PC71BM OPV shows high PCE. Current analysis on the energy level alignment with the emphasis on detailed interface dipole and band bending could suggest a proper material combination for high performance BHJ OPVs.
Methods
Sample preparation
PTB7 (Sigma Aldrich), PEDOT:PSS (1.3 wt% dispersion in H2O, Sigma Aldrich) and PC71BM (Luminescence Technology Corp.) were used as received. PTB7 and PC71BM were dissolved in chlorobenzene and each solution was stirred at room temperature for 24 hours before use. The concentration of both solutions for the VESD was 1 mg ml−1. ITO substrates were sequentially cleaned by ultrasonication with deionized water, detergent, acetone, and methanol and dried with N2 gas.
Vacuum electrospray deposition
PTB7, PC71BM, P3HT and PC61BM were deposited with a bias of 1.5 kV between the capillary out of the syringe and the inlet of the vacuum system. The injection speed of solution was uniformly controlled by a syringe pump (1 μl s−1). The deposition rate was monitored by measuring the ionic current of the injected solute into the vacuum system. All parameters were optimized to obtain a uniform thin film with fine thickness control down to sub-nm scales.
Photoelectron spectroscopy measurement
The electronic structure of ITO/PEDOT:PSS/PTB7/PC71BM was investigated with in situ UPS and XPS. PC71BM, PTB7 and PEDOT:PSS were deposited on an ITO substrate in a stepwise manner. The sample was transferred to the analysis chamber at each deposition step under UHV condition below 10−8 Torr to obtain UPS and XPS spectra. The base pressure of the analysis and preparation chamber was 5.0 × 10−9 and 2.0 × 10−8 Torr, respectively. A PHI-5700 spectrometer was used to analyse the kinetic energy of photoelectrons with an Al Kα X-ray (1486.6 eV) and a He І (21.22 eV) ultraviolet source. The spectrometer was calibrated with respect to the binding energy of Au 4f (84.0 eV), using its Fermi step as a reference. The uncertainty of our UPS measurements estimated from the broadening of the Au Fermi step was 0.09 eV. A sample bias of −10 V was applied during UPS measurements to obtain the true SEC. To determine the HOMO onset accurately, Shirley-type background was removed from the measured HOMO region spectrum. The film thicknesses and deposition rates were monitored by the attenuation ratio of In 3d intensity of the ITO substrate as well as the ionic current measurements during the VESD deposition process.
DFT calculations
A Becke-style three-parameter exchange and Lee-Yang-Parr correlation hybrid functional (B3LYP) and a split basis set of 6-31G(d) were used in the GAUSSIAN 09 software package. We increased the number of PTB7 monomers to simulate the PTB7 polymer. The effect of the number of a monomer on the frontier orbitals was tested from a monomer to 8-mer unit. The side chains were truncated to a methyl group for simplicity. As shown in supplementary Fig. S4, 8-mer is a reasonable choice and the generated DOS well-matched with the experimental spectrum (Fig. 8b). We also tested the side chain effect by comparing the complete side chain with a truncated side chain, which indicates that this truncation does not affect the calculated DOS near frontier orbitals. This coincides well with previous reports56,57,58.
Morphology
Morphological images were obtained by non-contact mode atomic force microscopy measurements (Park system).
Additional Information
How to cite this article: Park, S. et al. The origin of high PCE in PTB7 based photovoltaics: proper charge neutrality level and free energy of charge separation at PTB7/PC71BM interface. Sci. Rep. 6, 35262; doi: 10.1038/srep35262 (2016).
References
Chen, J. D. et al. Single‐Junction Polymer Solar Cells Exceeding 10% Power Conversion Efficiency. Adv. Mater. 27, 1035–1041 (2015).
Liao, S. H. et al. Single junction inverted polymer solar cell reaching power conversion efficiency 10.31% by employing dual-doped zinc oxide nano-film as cathode interlayer. Sci. Rep. 4, 6813 (2014).
Zhou, H. et al. Polymer Homo‐Tandem Solar Cells with Best Efficiency of 11.3%. Adv. Mater. 27, 1767–1773 (2015).
Scharber, M. C. et al. Design rules for donors in bulk‐heterojunction solar cells—Towards 10% energy‐conversion efficiency. Adv. Mater. 18, 789–794 (2006).
Vazquez, H., Gao, W., Flores, F. & Kahn, A. Energy level alignment at organic heterojunctions: Role of the charge neutrality level. Phys. Rev. B 71, 041306 (2005).
Xu, Z., Chen, L.-M., Chen, M. H., Li, G. & Yang, Y. Energy level alignment of poly(3-hexylthiophene):([6,6]-phenyl C61 butyric acid methyl ester bulk heterojunction. Appl. Phys. Lett. 95, 13301 (2009).
Tada, A., Geng, Y., Wei, Q., Hashimoto, K. & Tajima, K. Tailoring organic heterojunction interfaces in bilayer polymer photovoltaic devices. Nat. Mater. 10, 450–455 (2011).
Hiroyuki, Y. Low Energy Inverse Photoemission Study on the Electron Affinities of Fullerene Derivatives for Organic Photovoltaic Cells. J. Phys. Chem. C. 118, 24377−24382 (2014).
Potscavage Jr, W. J., Sharma, A. & Kippelen, B. Critical interfaces in organic solar cells and their influence on the open-circuit voltage. Acc. Chem. Res. 42, 1758–1767 (2009).
Stevens, D. M., Speros, J. C., Hillmyer, M. A. & Frisbie, C. D. Relationship between Diode Saturation Current and Open Circuit Voltage in Poly(3-alkylthiophene) Solar Cells as a Function of Device Architecture, Processing Conditions, and Alkyl Side Chain Length. J. Phys. Chem. C. 115, 20806–20816 (2011).
Vandewal, K. et al. The relation between open‐circuit voltage and the onset of photocurrent generation by charge‐transfer absorption in polymer: fullerene bulk heterojunction solar cells. Adv. Funct. Mater. 18, 2064–2070 (2008).
Gadisa, A., Svensson, M., Andersson, M. R. & Inganäs, O. Correlation between oxidation potential and open-circuit voltage of composite solar cells based on blends of polythiophenes/fullerene derivative. Appl. Phys. Lett. 84, 1609–1611 (2004).
Veldman, D., Meskers, S. C. & Janssen, R. A. The Energy of Charge‐Transfer States in Electron Donor–Acceptor Blends: Insight into the Energy Losses in Organic Solar Cells. Adv. Funct. Mater. 19, 1939–1948 (2009).
Brabec, C. J. et al. Origin of the open circuit voltage of plastic solar cells. Adv. Funct. Mater. 11, 374–380 (2001).
Bhatta, R. S. & Tsige, M. Chain length and torsional dependence of exciton binding energies in P3HT and PTB7 conjugated polymers: A first-principles study. Polymer. 55, 2667–2672 (2014).
Servaites, J. D., Savoie, B. M., Brink, J. B., Marks, T. J. & Ratner, M. A. Modeling geminate pair dissociation in organic solar cells: High power conversion efficiencies achieved with moderate optical bandgaps. Energy Environ. Sci. 5, 8343–8350 (2012).
Holcombe, T. W. et al. Steric control of the donor/acceptor interface: Implications in organic photovoltaic charge generation. J. Am. Chem. Soc. 133, 12106–12114 (2011).
Hill, I., Rajagopal, A., Kahn, A. & Hu, Y. Molecular level alignment at organic semiconductor-metal interfaces. Appl. Phys. Lett. 73, 662–664 (1998).
Oehzelt, M., Koch, N. & Heimel, G. Organic semiconductor density of states controls the energy level alignment at electrode interfaces. Nat. Commum. 5, 4174 (2014).
Braun, S., Salaneck, W. R. & Fahlman, M. Energy-level alignment at organic/metal and organic/organic interfaces. Adv. Mater. 21, 1450–1472 (2009).
Park, S., Lee, Y. & Yi, Y. Vacuum-integrated electrospray deposition for highly reliable polymer thin film. Rev. Sci. Instrum. 83, 105106 (2012).
Lee, J. H. et al. Interface formation between ZnO nanorod arrays and polymers (PCBM and P3HT) for organic solar cells. J. Phys. Chem. C. 116, 26342–26348 (2012).
Wang, W., Alsmeier, J. H., Wolak, M. & Schlaf, R. Determination of the charge neutrality level of poly(3-hexylthiophene). J. Chem. Phys. 138, 054705 (2013).
Shah, S. K., Abbas, M., Ali, M., Hirsch, L. & Gunnella, R. Optimal construction parameters of electrosprayed trilayer organic photovoltaic devices. J. Phys. D. 47, 045106 (2013).
Lu, L. & Yu, L. Understanding low bandgap polymer PTB7 and optimizing polymer solar cells based on it. Adv. Mater. 26, 4413–4430 (2014).
Liu, S. et al. Enhanced efficiency of polymer solar cells by adding a high-mobility conjugated polymer. Energy Environ. Sci. 8, 1463–1470 (2015).
He, Z., Wu, H. & Cao, Y. Recent advances in polymer solar cells: Realization of high device performance by incorporating water/alcohol‐soluble conjugated polymers as electrode buffer layer. Adv. Mater. 26, 1006–1024 (2014).
Yip, H.-L. & Jen, A. K.-Y. Recent advances in solution-processed interfacial materials for efficient and stable polymer solar cells. Energy Environ. Sci. 5, 5994–6011 (2012).
Choi, H. et al. Versatile surface plasmon resonance of carbon-dot-supported silver nanoparticles in polymer optoelectronic devices. Nat. Photonics. 7, 732–738 (2013).
Yang, X. et al. Nanoscale morphology of high-performance polymer solar cells. Nano Lett. 5, 579–583 (2005).
Yao, Y., Hou, J., Xu, Z., Li, G. & Yang, Y. Effects of solvent mixtures on the nanoscale phase separation in polymer solar cells. Adv. Funct. Mater. 18, 1783 (2008).
Collins, B. A. et al. Absolute measurement of domain composition and nanoscale size distribution explains performance in PTB7: PC71BM Solar Cells. Adv. Energy Mater. 3, 65–74 (2013).
Schlaf, R., Parkinson, B., Lee, P., Nebesny, K. & Armstrong, N. HOMO/LUMO alignment at PTCDA/ZnPc and PTCDA/ClInPc heterointerfaces determined by combined UPS and XPS measurements. J. Phys. Chem. B. 103, 2984–2992 (1999).
Kahn, A., Zhao, W., Gao, W., Vázquez, H. & Flores, F. Doping-induced realignment of molecular levels at organic–organic heterojunctions. Chem. Phys. 325, 129–137 (2006).
Kim, S. H. et al. Auger electron nanoscale mapping and x-ray photoelectron spectroscopy combined with gas cluster ion beam sputtering to study an organic bulk heterojunction. Appl. Phys. Lett. 104, 243303 (2014).
Greczynski, G. et al. Photoelectron spectroscopy of thin films of PEDOT:PSS conjugated polymer blend: a mini-review and some new results. J. Electron. Spectrosc. 121, 1–17 (2001).
Hwang, J., Amy, F. & Kahn, A. Spectroscopic study on sputtered PEDOT· PSS: Role of surface PSS layer. Org. Electron. 7, 387–396 (2006).
Ishii, H., Sugiyama, K., Ito, E. & Seki, K. Energy level alignment and interfacial electronic structures at organic/metal and organic/organic interfaces. Adv. Mater. 11, 605–625 (1999).
Vázquez, H., Dappe, Y., Ortega, J. & Flores, F. Energy level alignment at metal/organic semiconductor interfaces: “Pillow” effect, induced density of interface states, and charge neutrality level. J. Chem. Phys. 126, 144703 (2007).
Liu, F. et al. Understanding the morphology of PTB7: PCBM blends in organic photovoltaics. Adv. Energy Mater. 4 (2014).
Cornil, J. et al. Valence electronic structure of π-conjugated materials: Simulation of the ultraviolet photoelectron spectra with semiempirical Hartree-Fock approaches. Chem. Mater. 11, 2436–2443 (1999).
Hill, I., Kahn, A., Cornil, J., Dos Santos, D. & Brédas, J. L. Occupied and unoccupied electronic levels in organic π-conjugated molecules: comparison between experiment and theory. Chem. Phys. Lett. 317, 444–450 (2000).
Lee, H. et al. Hole injection enhancement of a single-walled carbon nanotube anode using an organic charge-generation layer. Carbon. 71, 268–275 (2014).
Brédas, J. L., Cornil, J. & Heeger, A. J. The exciton binding energy in luminescent conjugated polymers. Adv. Mater. 8, 447–452 (1996).
Djurovich, P. I., Mayo, E. I., Forrest, S. R. & Thompson, M. E. Measurement of the lowest unoccupied molecular orbital energies of molecular organic semiconductors. Org. Electron. 10, 515–520 (2009).
Liao, S. H., Jhuo, H. J., Cheng, Y. S. & Chen, S. A. Fullerene Derivative‐Doped Zinc Oxide Nanofilm as the Cathode of Inverted Polymer Solar Cells with Low‐Bandgap Polymer (PTB7‐Th) for High Performance. Adv. Mater. 25, 4766–4771 (2013).
Heeney, M. et al. Regioregular poly(3-hexyl)selenophene: A low band gap organic hole transporting polymer. Chem. Commun. 5061–5063 (2007).
Bright, W. et al. Nanoscale phase separation and high photovoltaic efficiency in solution-processed, small-molecule bulk heterojunction solar cells. Adv. Funct. Mater. 19, 3063–3069 (2009).
Steffan, C. et al. A photophysical study of PCBM thin film. Chem. Phys. Lett. 445, 276–280 (2007).
Guan, Z. L., Kim, J. B., Loo, Y. L. & Kahn, A. Electronic structure of the poly(3-hexylthiophene):indene-C60 bisadduct bulk heterojunction. J. Appl. Phys. 110, 043719 (2011).
Tengstedt, C. et al. Fermi-level pinning at conjugated polymer interfaces. Appl. Phys. Lett. 88, 053502 (2006).
Kim, J. H., Hong, J. A., Kwon, D. G., Seo, J. & Park, Y. Energy level alignment in polymer organic solar cells at donor-acceptor planar junction formed by electrospray vacuum deposition. Appl. Phys. Lett. 104, 163303 (2014).
Li, G. et al. High efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends. Nat. Mater. 4, 864–868 (2005).
Yang, Y. M. et al. High-performance multiple-donor bulk heterojunction solar cells. Nat. Photonics. 9, 190–198 (2015).
Yang, Q. D. et al. Probing the Energy Level Alignment and the Correlation with Open-Circuit Voltage in Solution-Processed Polymeric Bulk Heterojunction Photovoltaic Devices. ACS Appl, Mater. Inter. 8, 7283–7290 (2016).
Bhatta, R. S., Perry, D. S. & Tsige, M. Nanostructures and Electronic Properties of a High-Efficiency Electron-Donating Polymer. J. Phys. Chem. A. 117, 12628–12634 (2013).
Khoshkholgh, M. J., Marsusi, F. & Abolhassani, M. R. Density functional theory investigation of opto-electronic properties of thieno [3, 4-b] thiophene and benzodithiophene polymer and derivatives and their applications in solar cell. Spectrochim. Acta. Mol. 136, 373–380 (2015).
Niklas, J. et al. Highly-efficient charge separation and polaron delocalization in polymer–fullerene bulk-heterojunctions: A comparative multi-frequency EPR and DFT study. Phys. Chem. Chem. Phys. 15, 9562–9574 (2013).
Acknowledgements
This work was supported by Nano-Material Technology Development Program and a research project of the National Research Foundation of Korea (Grant No. 2015R1C1A1A01055026 and 2012M3A7B4049801), and Samsung Display Company.
Author information
Authors and Affiliations
Contributions
S.P. wrote the manuscript; prepared the samples for PES, IPES experiments; collected and analysed all of the data presented in this manuscript under the supervision of H.L. and Y.Y. J.J. and M.K. performed and analysed the PES and IPES experiments. G.H. performed the simulation of PTB7. All authors commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Park, S., Jeong, J., Hyun, G. et al. The origin of high PCE in PTB7 based photovoltaics: proper charge neutrality level and free energy of charge separation at PTB7/PC71BM interface. Sci Rep 6, 35262 (2016). https://doi.org/10.1038/srep35262
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35262
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
