
Abstract
The synthesis of polycyclic aromatic compounds generally requires stoichiometric oxidants or homogeneous metal catalysts, however, the risk of contamination of inorganic residues can affect their properties. Here we present a microwave (MW)-assisted platinum on beaded activated carbon (Pt/CB)-catalyzed C–C bond formation of diarylacetylenes and aromatic hydrocarbons under continuous-flow conditions. Various fused aromatic compounds were continuously synthesized via dehydrogenative C(sp2)–C(sp2) and C(sp2)–C(sp3) bond formation with yields of up to 87% without the use of oxidants and bases. An activated, local reaction site on Pt/CB in the flow reaction channel reaching temperatures of more than three hundred degrees Celsius was generated in the catalyst cartridge by selective microwave absorption in CB with an absorption efficiency of > 90%. Mechanistic experiments of the transformation reaction indicated that a constant hydrogen gas supply was essential for activating Pt. This is an ideal reaction with minimal input energy and no waste production.
Similar content being viewed by others

Introduction
Recently, π-conjugated polyaromatic compounds, including nanographenes and polycyclic aromatic hydrocarbons (PAHs), have attracted much attention owing to their attractive properties1,2,3,4. π-Aromatics have been utilized as organic electronic materials, such as organic field-effect transistors (OFET)5,6, organic solar cells (OSC)7,8, and organic light-emitting diodes (OLED)9,10,11,12,13,14,15, owing to their unique conjugation, conductivity, redox activity, and luminescent properties16. Moreover, the development of sensors for the microanalysis of explosive, high-energy aromatic nitro compounds (picric acid, TNT, etc.)17 is expected to utilize the quenching effect of luminescent π-aromatics based on π–π interactions.
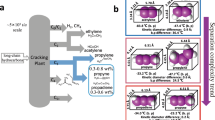
Polycyclic aromatic compounds are generally synthesized by various functionalization processes, such as oxidation, coupling, and/or cycloaromatization of alkanes and benzene, toluene, and xylenes (BTXs), derived from the catalytic reforming18,19 of petroleum (Fig. 1a)20. For example, several conventional methods for synthesizing phenanthrene derivatives, one of the smallest polycyclic aromatic hydrocarbons, have been developed (Fig. 1b). Photochemical-cyclization-oxidation reactions of diaryl ethylene derivatives under ultraviolet (UV) irradiation (i)21, called the Mallory reaction22 and transition-metal-catalyzed intramolecular cyclization reactions of styrene derivatives based on C–H/C–X (X = halogen) activation (ii)23,24 are well-developed methods for the synthesis of phenanthrene cores. Phenanthrene structures are also synthesized from biphenyls; for example, the acid-catalyzed intramolecular cyclization of 2-alkynyl biphenyl (iii)25,26, cross-coupling reactions of 2-halogenated biphenyl with alkynes (iv)27,28,29, and ring-closing metathesis of 2,2’-vinyl biphenyls (v)30,31. Itami et al. have developed various sophisticated molecular transformation methodologies for the synthesis of different π-aromatics, including phenanthrene derivatives32, based on transition-metal-catalyzed coupling and subsequent annulation reactions33,34. However, previously reported synthetic methodologies require stoichiometric amounts of oxidants and/or homogeneous metal catalysts; therefore, the risk of contamination of inorganic residues derived from catalysts and/or substrates into products is an issue that should be addressed to achieve super-high-purity organic electronic materials with excellent performance35,36,37. Alternatively, the use of heterogeneous metal catalysts could reduce metal contamination38 and the complicated workup process after the reaction39. Therefore, innovative and practical synthetic methodologies for π-aromatics based on heterogeneous catalysis are highly desirable.

a, b Conventional strategy for the synthesis of fused aromatic hydrocarbons. c Pt/CB-catalyzed dehydrogenation reaction of liquid organic hydrates. d, e MW-assisted Pt/CB-catalyzed cyclization reactions (this work).
Microwave (MW)-assisted organic synthesis (MAOS) has gained popularity in academic and industrial research because it can selectively and rapidly heat reactants40,41,42. The rapid and selective heating effects of MW often reduce the reaction time from an hour to minutes or even seconds, reducing the total reaction cost compared with conventional external heating methods41. However, despite the attractive advantages of MAOS, non-negligible side reactions often occur owing to the superabundant energy supply to the reaction materials (substrates, solvents, or products). For example, microwave flash pyrolysis (MFP)43,44, based on flash-flow pyrolysis (FFP)45 or flash-vacuum pyrolysis (FVP)46, is a known technique for instantaneously promoting various molecular transformations under high thermal conditions (typically 400–1100 °C). These methodologies can produce attractive intermediates and products by repeated cleavage reactions and the formation of various stable bonds. However, their application in organic synthesis is limited, and their selectivity and yields are often ignored in the case of functional materials43,44,47. Therefore, selective and well-controlled non-overheating irradiation of MW is essential to achieve efficient syntheses.
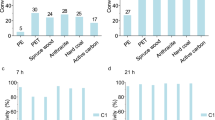
We have focused on the development of heterogeneous metal-catalyzed organic transformations under flow conditions48,49. In these studies, platinum on beaded activated charcoal (Pt/CB, CB; carbon bead) efficiently catalyzed the hydrogen extraction reactions of methylcyclohexanes (MCH)50 and 2-propanols (2-PrOH)51, the so-called liquid organic chemical hydrides (LOCHs)52,53, under single-mode MW irradiation conditions (Fig. 1c). These reactions proceeded under only 10–15 W MW irradiation, creating a local high-temperature reaction field on Pt/CB, using a selective MW-absorption effect (energy absorption of >90%) in beaded activated charcoal (Fig. 2)54. The flow system contributed to the smooth supply of reactants into the Pt/CB-packed cartridge and the immediate discharge of products from the local high-temperature reaction field to prevent side reactions55.

Temperature transition of 5% Pt/CB under 10 W single mode-MW irradiation after 0, 2, and 12 min.
Taking advantage of the MW-assisted dehydrogenation (hydrogen generation) reaction of LOCHs, this study aimed to hydrogenate multiple bonds. An MCH solution of diphenylacetylene was pumped into a 10 W MW-irradiated 5% Pt/CB-packed catalyst cartridge. Phenanthrene and diphenylethane were obtained in yields of 49 and 40%, respectively (Fig. 1d). To the best of our knowledge, this direct transformation56 has not been reported thus far. Based on these unexpected but excellent results, the MW-mediated and Pt-catalyzed redox-neutral C–C bond formation of diarylacetylenes and dehydrogenative coupling of aromatic hydrocarbons without activated functional groups is developed for the first time (Fig. 1e). Unlike existing microwave-assisted organic synthesis in which reagents and solvents are heated by multi-frequency MW irradiation, in the present Pt/CB catalyst continuous flow reaction, a single-frequency 10 W MW was selectively absorbed (>90%) by the CB to generate a localized high-temperature reaction field at 5% Pt/CB inside the catalyst cartridge. In addition, continuous-flow reaction systems minimize non-negligible side reactions and byproducts. Thus, the present MW-assisted continuous-flow reaction exhibits high energy efficiency and can potentially be an environmentally benign chemical transformation method-, allowing input energy to be utilized without waste.
Results and discussion
Effects of platinum group metals and CB of catalysts
Initially, the effect of the catalyst using a single-mode MW flow apparatus was investigated (Table 1). A solution of 1a (0.5 mmol) in MCH (0.25 M) flowed into the MW (10 W)-irradiated catalyst (80 mg)-packed glass cartridge at a flow rate of 0.25 mL/min. The 5% Pt/CB-catalyzed C–C bond formation reaction efficiently afforded 2a in 42% yield, as well as 21% 5a (entry 1). Catalyst efficiencies did not significantly change using 5% Pt/CB supported on different particle sizes of carbon beads (particle sizes: ~0.3 mm for 5% Pt/CB-SP and ~0.6 mm for 5% Pt/CB-LP, entries 2 and 3 vs. entry 1). The temperature of the 5% Pd/CB-packed catalyst cartridge (~270–360 °C measured with an infrared thermometer) and the reaction mixture at the cartridge outlet (~80–110 °C by a thermocouple) for Entries 1–3 also exhibited a similar trend. These results indicate that particle size does not affect the MW absorption efficiency. Furthermore, semi-hydrogenated cis and trans-stilbene were simultaneously observed in the reaction mixtures. When beaded SiO2 or Al2O3 was used instead of beaded carbon, the support dramatically reduced the MW energy absorption, and most of 1a remained unchanged (entries 4 and 5). Palladium and rhodium were also ineffective as metal catalysts for MW-assisted cyclization reactions and afforded only the hydrogenated products (entries 6 and 7). Moreover, it was evident that CB without a metal catalyst had nearly no catalytic activity (Entry 8). This result strongly suggests that the present cyclization reaction does not proceed via flash pyrolysis but via MW-mediated Pt catalysis. The cyclization reaction using the platinum on activated carbon (powdered 5% Pt/C)-packed catalyst cartridge was also tested. 5% Pt/C in the cartridge was excessively heated during 10 W MW irradiation, and the cyclization reaction was dramatically suppressed due to a decrease in catalyst activity (entry 9 and Supplementary Fig. 7 for the excessively heated and red-colored 5% Pt/C).
Effect of temperatures in the catalyst cartridge
The product ratios at various temperatures (220–420 °C) in the catalyst cartridge were further studied under MW-intensity-controlled isothermal conditions (Fig. 3). A solution of 1a (0.5 mmol) in MCH (0.05 M) was introduced into a 5% Pt/C (80 mg)-packed cartridge at a flow rate of 0.5 mL/min under MW-irradiation. 1a (dark blue bar) was completely consumed to afford cyclized phenanthrene (2a, red bar) and hydrogenated diphenylethane (5a, pale blue bar) in a 39:44 ratio at 220 °C. As the temperature increased from 220 to 370 °C, 5a gradually decreased, and conversely, the yield of 2a increased to 57%, accompanied by a small amount of cis- and trans-alkenes (3a, green bar, and 4a, violet bar, respectively). When the catalyst cartridge was heated at 420 °C, 4% of 1a was detected in the product and the yield of 2a was reduced to 46%. These trends indicate that hydrogenation of 1a to form 5a and cyclization to 2a proceeded in a similar ratio at a relatively low temperature (220 °C). However, the MW-assisted Pt/CB-catalyzed cyclization reaction (generation of 2a) was dominant over the hydrogenation of 1a, 3a, and 4a as the temperature was gradually increased from 220 to 370 °C. In contrast, at an extremely high temperature (420 °C), the reactant diphenylacetylene (1a) and yields of semi-hydrogenated alkenes (3a and 4a) increased. Therefore, the progress of the MW-assisted Pt/CB-catalyzed cyclization reaction is suppressed at 420 °C. Because MCH and 2-PrOH are completely vaporized at 420 °C, and the dehydrogenation reaction proceeds rapidly, the contact efficiency of the reaction mixture with the catalyst bed is probably poor owing to the significantly shortened residence time.

Composition of the substrate (1a) and products (2a–5a) under MW-assisted flow, isothermal conditions. Yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane (0.5 mmol) as an internal standard.
Effects of solvent and co-solvent
Subsequently, the solvent and co-solvent effects on the reaction were investigated (Table 2). A solution (0.05 M) of 1a (0.5 mmol) was pumped into the 5% Pt/CB (80 mg)-packed glass cartridge at 320 °C at a flow rate of 0.5 mL min−1 under MW (10 W)-irradiation. When MCH was used as the flow solvent, 2a formed in 52% yield, whereas 2a generated in 1% yield in toluene (entries 1 vs. 2). H2 gas may play an essential role in the progress of the MW-assisted C–C bond formation reaction because it is generated when using MCH as a flow solvent by MW-mediated dehydrogenative aromatization46.
When 2-PrOH was used as the flow solvent, the reaction efficiency of the MW-assisted Pt/CB-catalyzed cyclization reaction was significantly reduced (the yield of 2a was 6%), and the hydrogenated product (5a) was dominant (44% yield), and 24% of unreacted 1a remained (entry 3). However, under MW irradiation, by preheating the catalyst cartridge to 200 °C before flowing the 1a solution in 2-PrOH, 2a generated in 79% yield (entry 4). Because of the high loss factor (tanδ) of 2-PrOH, the irradiated MW was absorbed by CB in the catalyst cartridge and by 2-PrOH. Therefore, CB did not reach a sufficient temperature for the cyclization reaction40. Therefore, in addition to the Pt metal, activated carbon, and MW, the temperature is an essential factor for efficient cyclization. The combined use of MCH with 2-PrOH, acetone, or 2-butanol (2-BuOH) as a co-solvent resulted in a high reaction efficiency, affording 2a in 82%, 87%, and 85% yields, respectively (entries 5–7). In contrast, the addition of 1-BuOH as a co-solvent led to a lower cyclization yield (19%), whereas the hydrogenated product (5a) obtained 72% yield. Carbon monoxide (CO) generated by the decarbonylation of 1-BuOH via 1-PrCHO produced by Pt/CB-catalyzed dehydrogenation may block the active sites of the Pt metal by coordination. Generated CO was detected by GC-TCD (Supplementary Fig. 8). C–C bond formation proceeded smoothly when t-BuOH or 2-butanone was used as the co-solvent (entries 9 and 10). Other co-solvents, such as tetrahydrofuran, cyclopentyl methyl ether, ethanol, 2-pentanol, or 3-pentanol, did not significantly enhance the yield of 2a. The effect of the MCH/2-PrOH ratio and the flow rate was also studied, and experimental results are provided in Supplementary Tables 1–3.
Effect of hydrogen gas
Next, MW flow reactions were conducted using a solution of 3a (0.5 mmol, green bar) in toluene (0.05 M) under various gas flow conditions to obtain further insight into the role of H2 gas (80 mL min−1) (Fig. 4). The target phenanthrene (2a, red bar) was obtained only under H2 gas conditions. Conversely, the corresponding isomerization product (4a, violet bar) was obtained under O2 or N2 gas conditions in 15 and 61% yields, respectively, similar to the control experiment in Fig. 4 (Control). Therefore, it is evident that H2 gas efficiently activates the Pt metal57 and promotes cyclization. It is also revealed that the reaction efficiency was improved by increasing hydrogen gas addition (Supplementary Table 4).

Composition of the substrate (1a) and products (2a–5a) under MW-assisted and hydrogen, oxygen, or nitrogen gas flow conditions and control experiment. Yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane (0.5 mmol) as an internal standard.
Mechanistic investigations
To gain further insight into the MW-assisted C–C bond formation reaction mechanism, 2a–5a were each used as a substrate in the reaction (Table 3 and Supplementary Tables 4 and 5). 2a–5a in MCH/2-PrOH (2/1, 0.05 M) were pumped into the catalyst cartridge under the same flow conditions shown in Table 2, entry 5. Cis and trans-stilbenes (3a and 4a) and diphenylethane (5a) were similarly annulated to afford 2a in 82%, 71%, and 82% yields, respectively (entries 2–4). When 2a was used as the substrate, 3a, 4a, and 5a were not obtained, and 91% of the unchanged 2a was recovered (Fig. 5a). Moreover, the dehydrogenation of 5a was confirmed to proceed to production 2a and 4a under the same flow reaction conditions (Fig. 5b). Notably, diphenylacetylene (1a) was not observed in any case.

a The stability of 2a under MW-assisted C–C bond formation reaction conditions. b MW-assisted C–C bond formation reaction using 5a as a starting material. c Proposed reaction pathways.
Based on these results, reaction mechanisms were proposed for the Pt-catalyzed redox-neutral C–C bond formation of diphenylacetylene under MW irradiation (Fig. 5c). Using the H2 gas generated by the Pt-catalyzed dehydrogenation of MCH under MW irradiation, 1a was simultaneously semi-hydrogenated by the 5% Pt/CB catalyst to 3a. Subsequently, the MW-mediated 5% Pt/CB-catalyzed dehydrogenative coupling of 3a proceeded to afford the thermodynamically stable phenanthrene 2a. A portion of 3a was isomerized to 4a at equilibrium, and alkenes (3a and 4a) were further hydrogenated to the corresponding alkane (5a). Because the isomerization and hydrogenation of 3a and 4a, as well as the dehydrogenation of 5a, are in equilibrium under MW irradiation conditions using the 5% Pt/CB-packed catalyst cartridge, 3a was regenerated from 4a and 5a during the reaction process. It is possible that cycloaromatization via the formation of dihydrophenanthrene (A) and subsequent dehydrogenation (5a to 2a) proceeded simultaneously.
Scope of substrates
Next, the redox-neutral (oxidant-free) C–C bond formation reaction of various diarylacetylenes and aromatic hydrocarbons was conducted under isothermal or definite MW-irradiation conditions (Fig. 6). Asymmetrical 4-tert-butyl (tBu)- or 3-methyl (Me)-substituted diphenyl acetylenes (1b and 1c) were cyclized to the corresponding phenanthrene derivatives (2b and 2c) in 66 and 50% yields, respectively (entries 1 and 2). C–C bond formation of 4,4’-, 3,3’-, or 2,2’-dimethyldiphenylacetylenes (1d, 1e, and 1f) also proceeded to afford the corresponding phenanthrenes (2d, 2e, and 2f) in 43–85% yields (entries 3–5). Although the cyclization of 4,4’-ditertiarybutyldiphenylacetylene (1g) and 2,2’,4,4’-tetramethyldiphenylacetylenes (1h) was relatively difficult owing to the low solubility of both the substrate and product. This caused physical adsorption on the CB catalyst support, and cyclized products (2g and 2h) were obtained in 26 and 27% yields, respectively (entries 6 and 7). The solubility of products is essential, and thereby, 1-naphtylphenylacetylene (1i), 2-naphtylphenylacetylene (1j), and 1,4-bis(phenylethynyl)benzene (1k) were transformed to chrysene (2i), benz[a]anthracene (2j), and dibenz[a,h]anthracene (2k), which are low solubility materials, in MCH/2-PrOH in relatively low yields [24% (47% based on consumed starting material), 8% (14% based on consumed starting material), and 12% yields, respectively] (entries 8–10). The MW-assisted Pt/CB-catalyzed reaction was efficient for the dehydrogenative intramolecular coupling of various aromatic hydrocarbons. Decreasing the flow rate was effective in the case of the cyclization of diphenylmethane (6), and the yield of fluorene (7) was increased up to 77% (entry 12 vs. 13). Triarylmethanes (8 and 10) was also annulated to afford corresponding benzo[a]fluorenes (9) and 9-phenylfluorene (11) in each 23% yields (entries 14 and 15). Fluoranthene (13) and triphenylene (15) formed in 42 and 19% yields (84 and 49%, respectively, based on the consumed starting materials) from 1-phenylnaphthalene (12) and o-terphenyl (14), respectively (entries 1658 and 1714,15,59). Furthermore, the present MW-assisted C–C bond formation reaction could be applied to intramolecular dehydrogenative C(sp3)–H–C(sp2)–H coupling (cyclization) reactions without oxidants, radical-generating reagents or photosensitizers60,61,62,63,64. 2-Methylbiphenyl (16) and 2,6-dimethylbiphenyl (17) were transformed into the corresponding fluorene (7) and 4-methylfluorene (18) in 55 and 57% yields (99 and 83%, respectively, based on the consumed starting materials) (entries 18 and 19). 1,3-Dimethyl-5-(2-phenylethynyl)benzene (1l) and 1-(o-biphenylyl)-2-phenylethyne (1m) were transformed into 2-methyl-4H-cyclopenta[d,e,f]phenanthrene (2l) and benzo[b]fluoranthene (2m), respectively, via two sequential redox-neutral dehydrogenative cyclization reactions (entries 20 and 21). Although the starting materials were not completely converted by the catalyst in many cases, it is noteworthy that the yields of the MW-assisted Pt/CB-catalyzed cyclization products based on the consumed starting materials were relatively high (entries 2, 6–8 and 12–21).

a The yield was determined by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard. b The yield based on the consumed starting material is in parentheses. c The starting material was completely consumed. d The flow rate was 0.25 mL min−1.
Reuse of 5% Pt/CB
The reuse of a 5% Pt/CB-packed cartridge was examined under the MW-assisted C–C bond formation reaction conditions. Equal volumes of MCH/2-PrOH solution of 1a have flowed twice into a 5% Pt/CB-packed cartridge. In the second flow, the yield of 2a was slightly decreased, and the formation of diphenylethane (5a), in which the alkyne of 1a was hydrogenated, increased (Fig. 7a, First flow vs. Second flow). Before and after use, the physical properties of 5% Pt/CB in the catalyst cartridge were evaluated by scanning transmission electron microscopy (STEM) and X-ray photoelectron spectroscopy (XPS). A mixture of small Pt clusters of approximately 1–3 nm and relatively large Pt clusters of roughly 1 µm was loaded on 5% Pt/CB before use (Fig. 7b, c), whereas sintered Pt clusters of 2–4 µm were observed after the MW-assisted continuous-flow reaction (Fig. 7d, e). XPS analysis indicated that the ratio of Pt(0) to Pt(II) in the 5% Pt/CB catalyst did not change before and after use (Fig. 7f, g). Therefore, the particle size change of Pt loaded on CB may affect the reaction efficiency of the present cyclization reaction.

a The reuse test of 5% Pt/CB under MW-assisted C–C bond formation reaction using 1a as a starting material. b–e STEM image of 5% Pt/CB before and after use. f, g XPS spectra of 5% Pt/CB before and after use.
Conclusion
In this study, MW-assisted and Pt/CB-catalyzed C–C bond formation of diarylacetylenes under continuous-flow conditions was developed. A highly activated local and high-temperature reaction field was generated on Pt/CB in the catalyst cartridge by selective (>90%) MW energy absorption in CB. Therefore, the novel transformation proceeded under redox-neutral conditions without any bases or oxidants. Various mechanistic experiments revealed that H2 gas plays an essential role in this transformation. Firstly, in the activation of the Pt metal. Secondly, in the promotion of the elimination of the reaction solution from the catalyst cartridge to control the residence time, preventing the generation of undesirable byproducts. In the reaction cartridge, Pt/CB-catalyzed hydrogenation of diphenylacetylenes (1) and cis or trans-diphenylethylenes (3 and 4), isomerization of diphenylethylenes, and dehydrogenation of diphenylethanes (5) should occur in equilibrium. Thermodynamically stable phenanthrene derivatives (2) were produced by a Pt/CB-catalyzed dehydrogenative C–C coupling reaction during a short MW irradiation period in the cartridge. Furthermore, this MW-assisted cyclization reaction was applicable to dehydrogenative C(sp2)–C(sp2) and C(sp2)–C(sp3) bond formation reactions for the synthesis of fused aromatics, such as chrysene, fluorene, ben-zo[a]fluorene, fluoranthene, and triphenylene. This Pt/CB-catalyzed continuous-flow reaction exhibited high energy efficiencies; that is, only 10–20 W of MW energy was required to obtain hundreds of degrees at a local high-temperature reaction field in the flow reaction cartridge. Notably, pre-functionalization of the starting material, such as oxidation, halogenation, or metalation, is not required. Therefore, the MW-assisted reaction is an environmentally ideal transformation methodology, and the input energy can be accumulated without waste. These findings may contribute to the development of a new generation of chemical technology and the crisis of organic semiconductor shortage caused by COVID-19. Investigation of the detailed reaction mechanism of Pt/CB-catalyzed C–H activation under MW-assisted conditions and improvement of MW flow devices aiming for their practical applications are ongoing.
Methods
General methods
For Instrumentation and Materials, see Supplementary Method 1. For Additional experiments concerning optimization of the reaction conditions, the effect of external addition of hydrogen gas, gas detection experiment, and additional mechanistic investigations related to Table 3, see Supplementary Methods 4–7.
General procedures for MW-assisted and Pt/CB-catalyzed cyclization reactions
See below and Supplementary Method 8.
The reaction using EYELA, MR-2G-100 (Condition A)
The entire flow path was fitted with the 5% Pt/CB (80.0 mg)-packed EYELA reaction tube and filled with a mixed solvent of MCH/2-PrOH (2/1). The mixed solvent was pumped through the reaction tube at a flow rate of 0.5 mL min−1 at 320 °C under a maximum of 20 W MW irradiation for 5 min. A solution of 1 in the mixed solvent (0.05 M) was pumped into the reaction tube, and then the solution vessel was sequentially rinsed with the mixed solvents four times (1 mL ×3, then 20 mL ×1) using a pump. The MW irradiation was then stopped, and ethyl acetate/toluene (1/1, 40 mL) or dichloromethane (40 mL) was pumped to further wash the entire flow path. The entire reaction mixture and washing solution were collected and concentrated in vacuo, dissolved in deuterated chloroform (CDCl3), and analyzed by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane (52.5 µL, 0.5 mmol) as an internal standard.
The reaction using SAIDA, FMR-100 (Condition B)
The entire flow path was fitted with the 5% Pt/CB (80.0 mg)-packed SAIDA reaction tube and filled with a mixed solvent of MCH/2-PrOH (2/1). The mixed solvent was pumped through the reaction tube at a flow rate of 0.5 mL min−1 under a maximum of 10 W MW irradiation for 5 min. A solution of 1 in the mixed solvent (0.05 M) was pumped into the reaction tube, and then the solution vessel was sequentially rinsed with the mixed solvents four times (1 mL ×3, then 20 mL ×1) using a pump. The MW irradiation was then stopped, and ethyl acetate/toluene (1/1, 40 mL) or dichloromethane (40 mL) was pumped to further wash the entire flow path. The whole reaction mixture and washing solution were collected and concentrated in vacuo, dissolved in deuterated chloroform (CDCl3), and analyzed by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane (52.5 µL, 0.5 mmol) as an internal standard.
The reaction using EYELA, MR-2G-100 under external addition of hydrogen gas conditions (Condition C)
The entire flow path was fitted with the 5% Pt/CB (80.0 mg)-packed EYELA reaction tube and filled with toluene. Toluene (0.5 mL min−1) and hydrogen gas (80 mL min−1) were pumped through the reaction tube at a flow rate of 0.5 mL/min at 320 °C under a maximum of 20 W MW irradiation for 5 min. A solution of 3a in toluene (0.05 M) was pumped into the reaction tube, and then the solution vessel was sequentially rinsed with toluene four times (1 mL ×3, then 20 mL ×1) using a pump. The MW irradiation was then stopped, and ethyl acetate/toluene (1/1, 40 mL) was pumped to further wash the entire flow path. The whole reaction mixture and washing solution were collected and concentrated in vacuo, dissolved in deuterated chloroform (CDCl3), and analyzed by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane (52.5 µL, 0.5 mmol) as an internal standard.
MW flow devices (SAIDA and EYELA), catalyst cartridges, and peripheral devices
See Supplementary Method 2, Supplementary Figs. 1–6.
Preparation of substrates
See Supplementary Method 9, Supplementary Figs. 9–11.
Spectroscopic data of products
Data availability
The authors declare that the data supporting the findings of this study are available within the paper or its Supplementary Information files and from the corresponding author upon request.
References
Cornil, J., Beljonne, D., Calbert, J.-P. & Brédas, J.-L. Interchain interactions in organic π-conjugated materials: impact on electronic structure, optical response, and charge transport. Adv. Mater. 13, 1053–1067 (2001).
Beaujuge, P. M. & Reynolds, J. R. Color control in π-conjugated organic polymers for use in electrochromic devices. Chem. Rev. 110, 268–320 (2010).
Zhang, D.-Q. et al. Organic π-Conjugated Molecules for Organic Semiconductors and Photovoltaic Materials. In Organic Chemistry—Breakthroughs and Perspectives (eds Ding, K. & Dai, L.-X.) 759−780 (Wiley-VCH Verlag & Co. KGaA, New York, 2012).
Matsuoka, W., Ito, H., Sarlah, D. & Itami, K. Diversity-oriented synthesis of nanographenes enabled by dearomative annulative π-extension. Nat. Commun. 12, 3940 (2021).
Wu, W., Liu, Y. & Zhu, D. π-Conjugated molecules with fused rings for organic field-effect transistors: design, synthesis and applications. Chem. Soc. Rev. 39, 1489–1502 (2010).
Wang, C., Dong, H., Hu, W., Liu, Y. & Zhu, D. Semiconducting π-conjugated systems in field-effect transistors: a material odyssey of organic electronics. Chem. Rev. 112, 2208–2267 (2012).
Ameri, T., Dennler, G., Lungenschmied, C. & Brabec, C. J. Organic tandem solar cells: a review. Energy Environ. Sci. 2, 347–363 (2009).
Ameri, T., Li, N. & Brabec, C. J. Highly efficient organic tandem solar cells: a follow up review. Energy Environ. Sci. 6, 2390–2413 (2013).
Tang, C. W. & VanSlyke, S. A. Organic electroluminescent diodes. Appl. Phys. Lett. 51, 913–195 (1987).
C, A., Pahlevani, M. & Welch, G. C. Organic light emitting diodes (OLEDs) with slot-die coated functional layers. Mater. Adv. 33, 628–645 (2021).
Geffroy, B., Roy, P. L. & Prat, C. Organic light-emitting diode (OLED) technology: materials, devices and display technologies. Polym. Int. 55, 572–582 (2006).
Hung, H.-W., Yokoyama, N., Yahiro, M. & Adachi, C. Low driving voltage organic light emitting diode using phenanthrene oligomers as electron transport layer. Thin Solid Films 516, 8717–8720 (2008).
Kabe, R., Notsuka, N., Yoshida, K. & Adachi, C. Afterglow organic light-emitting diode. Adv. Mater. 28, 655–660 (2016).
Laschat, S. et al. Discotic liquid crystals: from tailor-made synthesis to plastic electronics. Angew. Chem. Int. Ed. 46, 4832–4887 (2007).
Osawa, T. et al. Wide-range 2D lattice correlation unveiled for columnarly assembled triphenylene hexacarboxylic esters. Angew. Chem. Int. Ed. 51, 7990–7993 (2012).
Forrest, S. R. The path to ubiquitous and low-cost organic electronic appliances on plastic. Nature 428, 911–918 (2004).
Shanmugaraju, S. & Mukherjee, P. S. π-Electron rich small molecule sensors for the recognition of nitroaromatics. Chem. Commun. 51, 16014–16032 (2015).
Davis, R. J. & Derouane, E. G. A non-porous supported-platinum catalyst for the aromatization of n-hexane. Nature 349, 313–315 (1991).
Ahuja, R. et al. Catalytic dehydroaromatization of n-alkanes by pincer-ligated iridium complexes. Nat. Chem. 3, 167–171 (2011).
Harvey, R. G. Polycyclic Aromatic Hydrocarbons Ch. 3 (Wiley-VCH, New York, 1997)
Parker, C. O. & Spoerri, P. E. Photochemical conversion of stilbene to phenanthrene. Nature 166, 603 (1950).
Lvov, A. G. Switching the Mallory reaction to synthesis of naphthalenes, benzannulated heterocycles, and their derivatives. J. Org. Chem. 85, 8749–8759 (2020).
Campeau, L., Parisien, M., Jean, A. & Fagnou, K. Catalytic direct arylation with aryl chlorides, bromides, and iodides: intramolecular studies leading to new intermolecular reactions. J. Am. Chem. Soc. 128, 581–590 (2006).
Harrowven, D. C., Guy, I. L. & Nanson, L. Efficient phenanthrene, helicene, and azahelicene syntheses. Angew. Chem. Int. Ed. 45, 2242–2245 (2006).
Hu, B.-L., Pi, S.-S., Qian, P.-C., Li, J.-H. & Zhang, X.-G. Palladium-catalyzed iodine-mediated electrophilic annulation of 2-(1-alkynyl)biphenyls with disulfides. J. Org. Chem. 78, 1300–1305 (2013).
Hossain, M. L. et al. Synthesis of phenanthrenes through copper-catalyzed cross-coupling of N-tosylhydrazones with terminal alkynes. J. Org. Chem. 79, 8689–8699 (2014).
Larock, R. C., Doty, M. J., Tian, Q. & Zenner, J. N. Synthesis of polycyclic aromatic hydrocarbons by the Pd-catalyzed annulation of alkynes. J. Org. Chem. 62, 7536–7537 (1997).
Wang, C., Rakshit, S. & Glorius, F. Palladium-catalyzed intermolecular decarboxylative coupling of 2-phenylbenzoic acids with alkynes via C−H and C−C bond activation. J. Am. Chem. Soc. 132, 14006–14008 (2010).
Matsumoto, A., Ilies, L. & Nakamura, E. Phenanthrene synthesis by iron-catalyzed [4 + 2] benzannulation between alkyne and biaryl or 2-alkenylphenyl Grignard reagent. J. Am. Chem. Soc. 133, 6557–6559 (2011).
Iuliano, A., Piccioli, P. & Fabbri, D. Ring-closing olefin metathesis of 2,2‘-divinylbiphenyls: a novel and general approach to phenanthrenes. Org. Lett. 6, 3711–3714 (2004).
Walker, E. R., Leung, S. Y. & Barrett, A. G. M. Studies towards the total synthesis of Sch 56036; isoquinolinone synthesis and the synthesis of phenanthrenes. Tetrahedron Lett. 46, 6537–6540 (2005).
Li, Y., Yagi, A. & Itami, K. Synthesis of sterically hindered 4,5-diarylphenanthrenes via acid-catalyzed bisannulation of benzenediacetaldehydes with alkynes. Chem. Sci. 10, 5470–5475 (2019).
Ito, H., Ozaki, K. & Itami, K. Annulative π-extension (APEX): rapid access to fused arenes, heteroarenes, and nanographenes. Angew. Chem. Int. Ed. 56, 11144–11164 (2017).
Ito, H., Segawa, Y., Murakami, K. & Itami, K. Polycyclic arene synthesis by annulative π-extension. J. Am. Chem. Soc. 141, 3–10 (2019).
Krebs, F. C., Nyberg, R. B. & Jørgensen, M. Influence of residual catalyst on the properties of conjugated polyphenylenevinylene materials: palladium nanoparticles and poor electrical performance. Chem. Mater. 16, 1313–1318 (2004).
Kuwabara, J., Yasuda, T., Takase, N. & Kanbara, T. Effects of the terminal structure, purity, and molecular weight of an amorphous conjugated polymer on its photovoltaic characteristics. ACS Appl. Mater. Interfaces 8, 1752–1758 (2016).
Lupina, G. et al. Residual metallic contamination of transferred chemical vapor deposited graphene. ACS Nano 9, 4776–4785 (2015).
Liu, S.-Y. et al. Pd/C as a clean and effective heterogeneous catalyst for C–C couplings toward highly pure semiconducting polymers. Macromolecules 45, 9004–9009 (2012).
Chen, D. & Ray, A. K. Removal of toxic metal ions from wastewater by semiconductor photocatalysis. Chem. Eng. Sci. 56, 1561–1570 (2001).
Kappe, C. O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 43, 6250–6284 (2004).
Kappe, C. O. Microwave dielectric heating in synthetic organic chemistry. Chem. Soc. Rev. 37, 1127–1139 (2008).
Gao, Y., Remón, J. & Matharu, A. S. Microwave-assisted hydrothermal treatments for biomass valorization: a critical review. Green Chem. 23, 3502–3525 (2021).
Cho, H. Y., Ajaz, A., Himali, D., Waske, P. A. & Johnson, R. P. Microwave flash pyrolysis. J. Org. Chem. 74, 4137–4142 (2009).
Ajaz, A. et al. Microwave flash pyrolysis: C9H8 interconversions and dimerisations. Aust. J. Chem. 67, 1301–1308 (2014).
Cantillo, D., Sheibani, H. & Kappe, C. O. Flash flow pyrolysis: mimicking flash vacuum pyrolysis in a high-temperature/high-pressure liquid-phase microreactor environment. J. Org. Chem. 77, 2463–2473 (2012).
Wentrup, C. Flash vacuum pyrolysis: techniques and reactions. Angew. Chem. Int. Ed. 56, 14808–14835 (2017).
Zelinsky, N. D., Titz, I. & Gaverdowskaja, M. Bildung von kondensierten ringsystemen bei der dehydrogenisations-katalyse. Chem. Ber. 59, 2590–2593 (1926).
Yamada, T. et al. Highly selective hydrogenative conversion to the tertiary, secondary, and primary amines of nitriles under flow reaction conditions. ChemSusChem 15, e202102138 (2022).
Monguchi, Y., Ichikawa, T., Yamada, T., Sawama, Y. & Sajiki, H. Continuous-flow Suzuki–Miyaura and Mizoroki–Heck reactions under microwave heating conditions. Chem. Rec. 19, 3–14 (2019).
Ichikawa, T. et al. Microwave-mediated site-selective heating of spherical-carbon-bead-supported platinum for the continuous, efficient catalytic dehydrogenative aromatization of saturated cyclic hydrocarbons. ACS Sustain. Chem. Eng. 7, 3052–3061 (2019).
Ichikawa, T. et al. Microwave-mediated continuous hydrogen abstraction reaction from 2-PrOH catalyzed by platinum on carbon bead. Catalysts 9, 655 (2019).
Zhu, Q.-L. & Xu, Q. Liquid organic and inorganic chemical hydrides for high-capacity hydrogen storage. Energy Environ. Sci. 8, 478–512 (2015).
Preuster, P., Papp, C. & Wasserscheid, P. Liquid organic hydrogen carriers (LOHCs): toward a hydrogen-free hydrogen economy. Acc. Chem. Res. 50, 74–85 (2017).
Horikoshi, S., Osawa, A., Sakamoto, S. & Serpone, N. Control of microwave-generated hot spots. part V. mechanisms of hot-spot generation and aggregation of catalyst in a microwave-assisted reaction in toluene catalyzed by Pd-loaded AC particulates. Appl. Catal., A 460−461, 52–60 (2013).
Martina, K., Cravotto, G. & Varma, R. S. Impact of microwaves on organic synthesis and strategies toward flow processes and scaling up. J. Org. Chem. 86, 13857–13872 (2021).
John, S. E., Tokala, R., Kaki, V. R. & Shankaraiah, N. Expedition to phenanthrene nucleus: a two-decade research on bench. Asian J. Org. Chem. 10, 2105–2136 (2021).
Kubas, G. J. Activation of dihydrogen and coordination of molecular H2 on transition metals. J. Organomet. Chem. 751, 33–49 (2014).
Sawama, Y., Monguchi, Y. & Sajiki, H. Efficient H-D exchange reactions using heterogeneous platinum-group metal on carbon-H2-D2O system. Synlett 23, 959–972 (2012).
Thivasasith, A., Maihom, T., Pengpanich, S., Limtrakul, J. & Wattanakit, C. Insights into the reaction mechanism of n-hexane dehydroaroma-tization to benzene over gallium embedded HZSM-5: effect of H2 incorporated on active sites. Phys. Chem. Chem. Phys. 21, 5359–5367 (2019).
Rice, J. E. & Cai, Z.-W. A palladium-catalyzed intramolecular arene-triflate coupling for the synthesis of fluoranthenes and benzofluoranthenes. Tetrahedron Lett. 33, 1675–1678 (1992).
Copeland, P. G., Dean, R. E. & McNeil, D. The cyclodehydrogenation of o-terphenyl and 1,2′-biphenylyl-3,4-dihydronaphthalene. J. Chem. Soc. 1687–1689 https://pubs.rsc.org/en/content/articlelanding/1960/jr/jr9600001687 (1960).
Shaw, M. H., Shurtleff, V. W., Terrett, J. A., Cuthbertson, J. D. & Macmillan, D. W. C. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science 352, 1304–1308 (2016).
Dutta, U., Maiti, S., Bhattacharya, T. & Maiti, D. Arene diversification through distal C(sp2)−H functionalization. Science 372, d5992 (2021).
Rogge, T. et al. C–H activation. Nat. Rev. Methods Primers 1, 43 (2021).
Acknowledgements
This study was partially funded by a Grant-in-Aid from the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Number 20H03367) for H.S., the New Energy and Industrial Technology Development Organization (NEDO, Project code: P19004), and the Research Foundation for the Electrotechnology of Chubu (Grant Number R-02103) for T.Y. We thank N. E. Chemcat Corporation for the kind gift of heterogeneous platinum group metal catalysts. We also thank Tokyo Rikakikai Co., Ltd. and SAIDA FDS Inc. for lending MW flow devices and constructive suggestions. We also thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conceptualization of the research. T.Y., W.T., N.S., S.O., Y.A., T.M., T.I., and H.S. contributed to the investigation of the research. Y.M. and T.Y. (Yoshimura) contributed to the development of microwave flow devices and catalyst cartridges. M.Y. contributed to the development and investigation of catalysts. T.Y. and H.S. contributed to the funding acquisition. T.Y. wrote the original draft, and H.S. reviewed and edited the draft. H.S. orchestrated the whole study as a project administrator. All authors acquired research data and approved the version of the paper to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Xingquan He, Jun Tang, and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamada, T., Teranishi, W., Sakurada, N. et al. Microwave-assisted C–C bond formation of diarylacetylenes and aromatic hydrocarbons on carbon beads under continuous-flow conditions. Commun Chem 6, 78 (2023). https://doi.org/10.1038/s42004-023-00880-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-023-00880-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
