
Abstract
Bifidobacteria resident in the gastrointestinal tract (GIT) are subject to constantly changing environmental conditions, which require rapid adjustments in gene expression. Here, we show that two predicted LacI-type transcription factors (TFs), designated AraQ and MalR1, are involved in regulating the central, carbohydrate-associated metabolic pathway (the so-called phosphoketolase pathway or bifid shunt) of the gut commensal Bifidobacterium breve UCC2003. These TFs appear to not only control transcription of genes involved in the bifid shunt and each other, but also seem to commonly and directly affect transcription of other TF-encoding genes, as well as genes related to uptake and metabolism of various carbohydrates. This complex and interactive network of AraQ/MalR1-mediated gene regulation provides previously unknown insights into the governance of carbon metabolism in bifidobacteria.
Similar content being viewed by others


Introduction
The microbial communities within the human gastrointestinal tract (GIT), commonly referred to as the gut microbiota, have enjoyed intense scientific scrutiny and are predominantly made up of members of the bacterial phyla Bacteroidetes, Firmicutes, Proteobacteria and Actinobacteria1,2. Of particular interest is the genus Bifidobacterium, a member of the Actinobacteria phylum, that represent Gram-positive, non-motile, non-spore forming, saccharoclastic anaerobic bacteria that commonly colonize the gut of a wide variety of animals, including birds, insects and mammals3. Several bifidobacterial strains/species are designated as probiotic, which means that when such bacteria are administered in adequate and viable numbers, they confer a health benefit to their (human) host4. These benefits include, among others, modulation of the host immune response5, mitigation of lactose intolerance, the ability to lower serum cholesterol levels in humans6,7,8, pathogen inhibition/exclusion9 and prophylaxis of tumour growth in certain cancers10,11,12. Bifidobacteria are among the first and predominant colonisers of the infant GIT, and persist throughout adulthood though at lower relative abundance (compared to the infant gut), with a further reduction in the elderly gut13,14,15.
Approximately 8–15% of the bifidobacterial coding capacity is dedicated to carbohydrate uptake and metabolism, depending on species and functional gene assignment16,17. These estimates are consistent with the well-documented ability of bifidobacteria to utilise a diverse array of carbohydrates16,18. This remarkable genetic dedication to a particular metabolic behaviour probably reflects the importance of carbohydrate utilisation for bifidobacterial gut colonization and persistence. Just as important is their ability to control expression of the genes involved in the associated metabolic pathways in order to prevent wasting energy and resources on something which is unnecessary or inefficient. This is a widespread phenomenon, exemplified by the lac operon of Escherichia coli, which is only switched on if the substrate lactose is present and if a better (i.e. more energy generating) substrate such as glucose is absent19.
Additionally, carbon flux control enables bifidobacteria to balance energy and biosynthetic demands, thereby allowing optimal growth and redox balance management, as based on environmental conditions. This control is of crucial importance to bifidobacteria, as carbohydrate metabolism is their sole mechanism to produce ATP by substrate phosphorylation20, while metabolic intermediates of carbon metabolism (such as pyruvate) are used as precursors for a range of biosynthetic processes. Carbohydrate metabolism of bifidobacteria is performed by a rather unique pathway, the so-called phosphoketolase pathway or bifid shunt, which allows this taxonomic group to produce more energy in the form of ATP from carbohydrates than fermentative pathways employed by other bacteria21. From 1 mol of glucose the bifid shunt theoretically yields 2.5 mol ATP, along with 1.5 mol of acetate and 1 mol of lactate22, whereas for example homofermentative lactic acid bacteria (LAB) produce 2 mol of ATP and 2 mol of lactic acid per mol of glucose23. This superior ATP production ability is due to the presence of a phosphoketolase called X5P/F6P phosphoketolase (XFPK, encoded by xfpK) which is unique in exhibiting comparable affinities for both xylulose 5-P (X5P) and fructose 6-P (F6P)24. The bifid shunt has been investigated in terms of its enzymatic functions in animal-derived species, such as Bifidobacterium globosum25,26, and it has been established that the bifid shunt is indeed the sole carbohydrate metabolism pathway to satisfy all energy demands in Bifidobacterium animalis subsp. lactis Bb-12 when grown on either glucose or lactose24. All members of the Bifidobacteriaceae family are said to possess xfpK homologs, and thus employ the bifid shunt27. Phylogenetic and protein modelling studies have recently suggested that the bifidobacterial XFPK enzyme is related to that found in members of the Coriobacteriales order and that xfpK was horizontally transferred between (an ancestor of) these two groups21,28,29. Another interesting difference between bifidobacteria (and now possibly Coriobacteriales) and other bacteria is that they lack a number of enzymes, such as phosphofructokinase, which are crucial to control central carbohydrate metabolism30.
Various carbohydrate utilisation pathways in bifidobacteria have been characterised31,32,33,34,35,36,37,38,39. Several carbohydrate metabolism-related LacI-type regulators are known in Bifidobacterium breve UCC200334,35,36,37 and are typically involved in local (i.e. the gene encoding the LacI-type regulator is located in the vicinity of the genes it regulates) transcriptional regulation of genes that are responsible for the metabolism of a particular carbohydrate. These LacI-type transcription factors (TFs) typically limit (and are then called repressors), yet may sometimes enhance (then designated as activators), access or progression of the RNA polymerase to the promoter region of the regulated gene. This ability to act as a repressor or activator depends on a number of factors such as the location of the Transcription Factor Binding Site (TFBs) of the TF with respect to the -10/-35 sequences of the promoter region and the presence or absence of effector molecules, which may bind to the TF to alter its conformation and consequently its DNA-binding ability (See Fig. 1 Panel 3 for an example of a LacI TFBS). The binding state of the TF must be in accordance with the external and/or cytoplasmic environment, and, as mentioned, bacteria achieve this through effector molecules. In the case of carbohydrate utilisation such effector molecules may be a carbohydrate or metabolic intermediate.

Fragmentation analysis and AraQ/MalR1 binding. Panel (1) depicts the promoter region of pyk (Bbr_0757) used for fragmentation analysis. −35 and −10 sites are indicated in red and the ATG start codon is indicated in green. The TFBS is indicated in bold. The fragment including the TFBS (Frag. 1) is underlined in blue, the fragment excluding the TFBS (Frag. 2) is underlined in yellow, while the full fragment is referred to as IR. Panel (2) EMSA to investigate AraQ and MalR1 abilities to bind to Bbr_0757 promoter region fragments (IR, Frag 1 and Frag 2). All reactions contain 0.5 nM Ird labelled DNA and 150 nM of either AraQ or MalR1 protein, while negative reactions contain 0 nM protein. Uncropped gel image available in Supplemental Fig. S5. Panel (3), (A) Illustrates consensus AraQ/MalR1 binding motif across the Bifidobacteriaceae family (B) Depicts the binding motif of AraQ/MalR1 in B. breve. (C) Depicts the AraQ/MalR1 binding motif for core members of the regulon.
It had previously been predicted that the B. breve UCC2003-encoded LacI-type regulators (termed MalR1, MalR2, MalR3, MalR5 and MalR6) are involved in the metabolism of starch-like carbohydrates, while AraQ was envisaged to be responsible for transcriptional control of genes encoding a number of bifid shunt enzymes40. We show here that two of the previously identified LacI-type TFs appear to be directly involved in transcriptionally regulating genetic components of the bifid shunt, as well as genes involved in a range of specific carbohydrate metabolic pathways, thereby revealing a previously undiscovered global carbohydrate control network, which is predicted to operate in many bifidobacterial species.
Results
Identification and genetic analysis of araQ and malR1 on the B. breve UCC2003 genome
This study was initially aimed at the functional characterization of six previously described LacI-type transcription factors (TFs), namely AraQ, MalR1, MalR2, MalR3, MalR5 and MalR6 along with their proposed regulation of central metabolic pathways and malto-oligosaccharide utilisation pathways in B. breve UCC200340. In the course of our experiments we found that MalR1 was in fact functionally more similar to AraQ and for this reason our experimental scope was narrowed to specifically characterise AraQ and MalR1. The encoded products of araQ and malR1 represent proteins of 371 and 338 amino acids, respectively, which exhibit 26% overall identity to each other (91% query coverage). Their respective DNA binding domains (encompassing residues 11–57 for AraQ and residues 3–47 for MalR1) exhibit a higher level of identity at 49% (93% query coverage) (BlastP). This may be a reflection of the conserved nature of the DNA-binding helix-turn-helix regions of LacI-type proteins in general41, coupled to the finding that, as will be outlined below, AraQ and MalR1 recognize (near) identical operator sequences.
In vitro characterisation of the regulons of AraQ & MalR1 by EMSA analysis
In order to assess AraQ and MalR1 interaction with their possible operator sequences, electrophoretic mobility shift assays (EMSAs) were carried out using DNA fragments encompassing upstream regions of genes corresponding to the previously predicted AraQ and MalR1 regulons40, and to a number of (differentially transcribed) genes that were identified in microarray analyses involving insertion mutants of araQ and malR1 (discussed below). Obtaining purified and active AraQ or MalR1 protein from B. breve initially proved to be troublesome, in line with similar issues observed for other bifidobacterial LacI-type proteins34,35,36,37,42. We adapted our original protocol so as to allow purification of active (i.e. DNA-binding competent) AraQ and MalR1 (see Materials and Methods), which were tested for their ability to bind to DNA sequences encompassing the operator sequence or transcription factor binding site (TFBS) previously predicted for AraQ40. The ability of AraQ and MalR1 to bind DNA fragments was graded utilising a non-linear, arbitrary scale (see Materials and Methods). This EMSA analysis revealed that AraQ and MalR1 bind with varying degrees of affinity to the promoter regions of 23 and 20 genes, respectively, out of a total of 33 DNA regions tested. Figure 2 presents an example of complete DNA fragment binding, while additional examples can be found in Supplemental Fig. S1. Table 1 summarises all obtained EMSA results, while Fig. 3 is a schematic representation of the results obtained in this study. Interestingly, some of the promoter regions to which AraQ/MalR1 did not appear to bind, are associated with genes that display differential transcription in the microarray analysis of the corresponding araQ/malR1 mutants (see below), indicating that such differential transcription reflects indirect regulatory effects.

EMSA analysis with AraQ/MalR1. EMSA analysis carried out with increasing concentrations (0, 10, 50, 100, 150, 250 nM) of purified AraQ (panel A) or MalR1 (panel B) incubated with 0.5 nM Ird-labelled DNA fragment encompassing the Bbr_1233 (gap) promoter region. Uncropped gel image available in Supplemental Fig. S4.

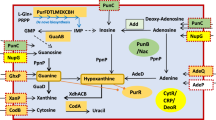
Schematic of AraQ and MalR1 effects on Central metabolism. Schematic of predicted central metabolism steps for carbohydrate metabolism in B. breve UCC2003. The XFPK enzymatic steps are indicated by an asterisk. As determined by EMSA analysis in Table 1, enzymatic steps which are under the regulation of AraQ and MalR1 are indicated by a red or blue dot, respectively. Abbreviation: Agl, alpha glucosidase; Bgl, beta gluosidase; GlkA, glucokinase; CldC, beta glucosidase; ApuB, amylopullunase; MalQ, glucanotransferase; Rk, ribokinase; Gpi, glucose- 6-p isomerase; Tal, transaldolase; Tkt, transketolase; R5PI, ribose-5-P isomerase; R5PR, ribose-5-P reductase; GADPH, glyceraldehyde-3-phosphate dehydrogenase; Xfpk, Xylose-5-P/Fructose-6-P phosphoketolase; AckA, acetate kinase; Ald2, alcohol dehydrogenase 2; Pgk, phosphoglycerate kinase; Gpm, phophoglycerate mutase; Eno, enolase; Pyk, pyruvate kinase; Pfl, pyruvate formate lyase; ldh2, lactate dehydrogenase 2.
Six of the genes/gene clusters whose promoter regions were clearly shown to be bound by AraQ and MalR1 encode enzymes that are part of the bifid shunt as illustrated in the schematic representation of central metabolism in Fig. 3. Several of these genes are essential for growth on glucose43 (Table 1) and are also highly transcribed during logarithmic growth on glucose as a sole carbon source44. The ability of AraQ to bind to the promoter regions of these six genes/gene clusters involved in central metabolism as well as those of certain genes involved in the metabolism of starch-like/-derived carbohydrates is consistent with previous in silico predictions40. EMSA analyses furthermore demonstrated that, similar to AraQ, MalR1 binds to promoter regions of genes/gene clusters involved in central carbon metabolism along with genes involved in the metabolism of starch-like/derived carbohydrates (and that the deduced AraQ and MalR1 regulons therefore largely overlap). The ability of MalR1 to transcriptionally control genes that are required for the metabolism of starch-like/-derived carbohydrates is consistent with in silico predictions40. However, MalR1’s presumed ability to control genes involved in central carbon metabolism was an unexpected finding, as these genes were not predicted to be members of MalR1’s predicted regulon40.
Based on their binding behaviour AraQ and MalR1 appear to have a similar affinity for the six promoter regions of the central metabolism genes (Table 1 and Fig. 3). Furthermore, AraQ and MalR1 were both found to be capable of binding their own and each other’s presumed promoter regions. This finding suggests that these genes are subject to interactive autoregulation, in which they not only control the transcription of their own genes, but also that of the other. Furthermore, as based on their binding behaviour, AraQ and MalR1 appear to control transcription of the LacI-type-encoding genes MalR2, MalR3, MalR5, MalR6 and CarD-like TF (Table 1).
Determination of the operator sequences recognized by AraQ and MalR1
As shown above, EMSA analyses employing AraQ and MalR1 clearly demonstrate their in-vitro affinity for various promoter regions, including those that correspond to several central metabolic genes. In order to identify the TFBSs that are recognized by either AraQ or MalR1, fragmentation analysis was carried out (Fig. 1). This analysis was performed employing the promoter regions of the following genes: eno (encoding enolase and corresponding to locus tag Bbr_0725) and pyk (encoding pyruvate kinase and corresponding to locus tag Bbr_0757). These genes were selected as they which hold key roles in central carbon metabolism (Fig. 3). DNA fragments encompassing various sections of the promoter region upstream of either eno or pyk were subjected to EMSA analysis employing purified AraQ or MalR1. These analyses revealed that a 47 and a 48-bp DNA segment of the eno and pyk promoter regions, respectively, each harbour an operator site for both AraQ and MalR1 (Fig. 1). Importantly, both of these segments contain previously predicted candidate AraQ binding sites40, specifically the 47-bp segment of DNA located upstream of the eno gene contains a 20 bp imperfect palindrome CATTGTGAGC><GCTCACATCA (with “><” indicating the center of the palindrome), while the 48-bp DNA segment located upstream of the pyk gene contains a very similar 20 bp palindrome CAGTGTGAGC><GCTCACAACA.
Next, bioinformatic analysis through the use of a Position Weight Matrix (PWM) approach was employed to identify additional genes which may be regulated by AraQ and MalR1 in B. breve. For this task two PWM’s were used (see Materials and Methods section), resulting in the identification of several additional genes/gene clusters that appear to contain this TFBS in their predicted promoter region (though this will require experimental verification). The full list of genes assigned to the AraQ/MalR1 regulon is present in Supplemental Table S1. The majority of these regulated genes are involved in central carbohydrate metabolism and were previously classified as components of the global AraQ regulon in Bifidobacteriaceae45. Some B. breve-specific genes of the AraQ/MalR1 regulon include Bbr_0037/0038 (predicted anhydrase/alkyl hydroperoxide reductase), Bbr_0221-Bbr_0222 (iron-uptake system BfeU-BfeO)46,47, Bbr_0603 (predicted cholate transporter Ctr), Bbr_1316 and Bbr_1685 (hypothetical MFS transporters) (Supplemental Table S1). The use of a modified PWM which was designed based only on sequences of promoter regions with which AraQ/MalR1 had a medium or high affinity did not significantly change the regulon composition. Specifically, additional potential TFBSs were found in (intergenic) promoter regions of Bbr_0117/Bbr_0118 (glucosidase/sugar-binding protein), Bbr_0176 (penicillin binding protein), and Bbr_0921 (long-chain fatty acid-CoA ligase) (Supplemental Table S1; the derived AraQ/MalR1 binding motifs as based on these predicted sites are presented in Fig. 1, Panel 3).
Some divergence was identified between AraQ and MalR1 specificity for certain TFBSs, although for both TFs the affinity for these is rather low (Table 1). The construction of a PWM specific for either AraQ or MalR1 was not possible, the primary reason for this is that TFBSs of differentially bound genes contribute significantly less to the overall weight matrix than those associated with central metabolism (such as eno and pyk), which form the core of the regulon. Furthermore, this analysis found that AraQ and MalR1 bind with the highest affinity to promoter regions of genes involved in central metabolism. This observed high affinity may be explained by the conserved G, C, and A in positions 9, 12 and 15, respectively, of the AraQ/MalR1 binding motif (Fig. 1, Panel 3).
Interestingly, the deduced TFBSs of AraQ and MalR1 are located upstream of recently assigned −10/−35 promoter sites of six bifid shunt-associated genes/gene clusters (Table 1 and Supplemental Table S3)24. This TFBS location relative to the −10/−35 sites is therefore typical of a transcriptional activator and in this context it should be noted that all genes encoding the bifid shunt enzymes exhibit a high level of transcription when grown on glucose24,44. Further analysis showed that the TFBSs of AraQ/MalR1 are not located at a particular distance from these promoter sequences: Supplemental Table S3 illustrates that the AraQ/MalR1 TFBS is located 16 and 15 bp upstream of the −35 site in the promoter regions of eno and pyk, and 33, 34 and 35 bp upstream of the −35 site of pfl, icfA and gap, respectively. Therefore, the position of the AraQ/MalR1 TFBS relative to the −35 site implies that AraQ/MalR1 bind in multiples of one helical turn plus one half (i.e. 1.5, 3.5 or 6.5 helical turns) away from the transcriptional start sites (TSS). Finally, analysis of the deduced TSS with respect to the location of the TFBSs of AraQ/MalR1 upstream of carD, glgP and cldE revealed that the TFBS is overlapping with predicted −35/−10 regions, indicating that AraQ/MalR1 in these cases act as repressors, being more typical of LacI-type TFs48.
Attempts to identify effector molecules for AraQ and MalR1
Bioinformatic and EMSA analyses allowed the deduction of the AraQ/MalR1 regulons and associated operator sequences. We next wanted to gain further insight into the nature of the environmental/metabolic signals that impact on the recognition ability and/or binding affinity of AraQ and MalR1 to their target DNA sequences. For this purpose, EMSA analyses were performed whereby AraQ or MalR1 was incubated with one of its binding targets, in this case the araQ DNA promoter region, in the presence of each of a variety of possible (available/relevant) effectors (Supplemental Table S4). These effectors were selected for two main reasons: (i) they occupy key positions within the bifid shunt (e.g. Phosphoenolpyruvate, D-erythrose-4-Phosphate and Glyceraldehyde 3-phosphate), which may allow them to act as cues to provide information on the metabolic status of the cell, and (ii) they are known to act as effector molecules in other organisms45,49. The results obtained indicate that under the employed experimental conditions none of the effector molecules tested in this study consistently decreased/increased AraQ or MalR1 ability to bind to DNA fragment encompassing the araQ promoter region (example effector EMSA can be found in Supplemental Fig. S2). It is possible that the in vitro experimental set up did not allow for the identification of the effector molecule(s), or perhaps that the molecule is the product of another metabolic pathway outside of central carbon metabolism.
Transcriptome analysis of B. breve UCC2003-araQ and B. breve UCC2003-malR1
In order to further investigate the role of araQ and malR1 in controlling central carbon metabolism, insertional mutants were created in each of these genes in B. breve UCC2003, resulting in strains B. breve UCC2003-araQ and B. breve UCC2003-malR1 (respectively; see Materials and Methods). These two mutants were then employed to assess transcriptional changes due to either of these mutations using microarray analysis.
The transcriptome of B. breve UCC2003-araQ, when compared to that of B. breve UCC2003 (Supplemental Tables S5 and S6), revealed that 32 genes were transcriptionally up-regulated and 45 were down-regulated above a fold change of 2.0, p-value < 0.001 (see materials and methods for further details). Furthermore microarray data revealed that, when compared to B. breve UCC2003, the previously characterised malto-oligosaccharide uptake system (Bbr_0118- Bbr_0121)50 is significantly up-regulated in B. breve UCC2003-araQ (4.4, 4.4, 4.5 and 3.9-fold change respectively; p < 0.001). In silico analysis via a PWM-based approach identified AraQ/MalR1 TFBSs upstream of the above-mentioned gene cluster, the location of these TFBSs indicates that AraQ/MalR1 act as repressors of the Bbr_0118-Bbr_0121 gene cluster. Similarly, malR3 (Bbr_0122; specifying a LacI type transcriptional regulator) and apuB (Bbr_0123; encoding an amylopullulanase) were up regulated 3.7 and 4.3-fold, respectively (p < 0.001) in B. breve UCC2003-araQ as compared with the wild type. rbsD (Bbr_1416), encoding a D-ribose pyranase, which is located in a ribose uptake cluster was also upregulated 2-fold (p < 0.001), PWM approach also identified the AraQ/MalR1 TFBS upstream of this cluster.
Most bifid shunt-associated genes whose promoter regions contain binding sites for AraQ/MalR1 were not significantly altered in the transcriptional analysis employing B. breve UCC2003-araQ or B. breve UCC2003-malR1 (when compared to the transcriptome of wild type B. breve UCC2003; Supplemental Tables S5 and S6). This may be due to compensatory activity of each of the two regulators, in which AraQ will functionally compensate for loss of MalR1 (in B. breve UCC2003-malR1) and vice versa (for strain B. breve UCC2003-araQ). Attempts to create a double mutant of both araQ and malR1 were unsuccessful, indicating that these two regulators form an essential transcriptional circuit of the bifid shunt genes.
The transcriptome of B. breve UCC2003-malR1 revealed, when compared to that of the B. breve UCC2003 WT (Supplemental Tables S7 and S8), just a single gene, malE1 (Bbr_0118), to be transcriptionally up-regulated above a fold change of 2.0, while no genes were found to be significantly (fold change of >2.0) down-regulated in the transcriptomic analysis of the B. breve UCC2003-malR1 mutant. In silico analysis via a PWM-based approach identified a MalR1 TFBS upstream of malE1 (Bbr_0118), where the location of this TFBS indicates that MalR1 acts as a repressor of this gene. Two genes, apuB and malG1, that form part of the predicted malR1-governed regulon40, were up-regulated to a lower, yet significant degree (1.8 and 1.3-fold change, respectively; P < 0.001). Notably, some of the central metabolic genes were found to be up-regulated at a rather modest, yet statistically significant level (i.e. xfp, eno, tkt, pfl and ldh, whose transcription was determined to be increased by 1.5, 1.3, 1.3, 1.2 and 1.2-fold, respectively; p-values < 0.004).
Distribution of araQ and malR1 homologues among Bifidobacteriaceae members
In order to estimate conservation of the proposed AraQ/MalR1-mediated regulatory circuit in other bifidobacterial species and their close relatives, we analysed the distribution of the araQ and malR1 genes in a set of 70 Bifidobacteriaceae genomes. This was carried out by aligning the 70 Bifidobacteriaceae genomes using a PATtyfam approach and the BLAST tool, followed by alignment with MUSCLE (see materials and methods for more details). The two regulator-encoding genes showed somewhat different patterns of distribution; araQ was fully conserved among all currently known species of the genus Bifidobacterium, while missing in genomes of closely related genera such as Scardovia and Pseudoscardovia (overall occurrence of araQ was determined to be 94%), whereas malR1 was conserved in 50 genomes (overall occurrence 71%) (Supplemental Table S9). Interestingly, the absence of araQ in several Scardovia and Pseudoscardovia genomes coincides with the absence the arabinose utilization operon araBDA, which supports our previous hypothesis that AraQ evolved from a local regulator of arabinose metabolism in an ancestor Bifidobacteriaceae species to a global regulator of central carbohydrate metabolism in Bifidobacterium and Gardnerella by regulon expansion51.
A phylogenetic tree illustrating this information was also was built using IQ-TREE52 utilising an ultrafast bootstrap with 1000 replicates. BglR (Bbr_1659) is a distant LacI family regulator from B. breve UCC2003, this TF was employed as an out group as its function is predicted to be different from those of MalR1 and AraQ (Supplemental Fig. S3).
AraQ has been identified in distant lineages of Actinobacteria (functioning as a local TF)45, while orthologs of malR1 (as well as the other malR genes) are confined to genera within the Bifidobacteriaceae family (Supplemental Table S9), implying that MalR1 is unique to this particular taxon and emerged after AraQ in the process of evolution. Although it is likely that MalR1 shares the same regulatory function as AraQ in many bifidobacteria by recognizing the same binding motif, this consideration is based on both the ubiquitous pattern of malR1 distribution (especially when compared with genes of other MalR regulators, which are mosaically distributed among analysed strains and exhibit considerably lower prevalence), along with experimental validation through EMSA analysis. Thus, the possibility that MalR1 operates differently in other bifidobacteria or closely related genera cannot be entirely excluded. We were also unable to identify conserved joint AraQ/MalR1 binding sites in Gardnerella vaginalis ATCC 14019 using a PWM approach, the only analysed strain that lacks araQ yet possesses malR1. This observation suggests that in this bacterium MalR1 recognizes a different binding motif, and presumably works as a local TF regulating maltose/maltodextrin utilization.
Survival of B. breve UCC2003-araQ and B. breve UCC2003-malR1 when exposed to porcine bile
Several publications have reported that the (in)ability to survive bile stress is linked with shifts in the expression of genes involved in central metabolism and with the concentration of its resulting end products in various Bifidobacterium species53,54,55,56. For this reason, B. breve UCC2003 WT, B. breve UCC2003-araQ and B. breve UCC2003-malR1 mutants were analysed for their ability to grow in the absence (control) as compared to the presence of 1% porcine bile with OD600nm readings taken at 24-hour post inoculation. Figure 4 displays the fold reduction in OD600nm following 24-hour growth in the presence of 1% porcine bile as compared to the control. A significant fold reduction in OD600nm readings was observed for all three strains when grown in the presence of porcine bile, as compared to OD600nm readings without porcine bile. However, the fold reduction in growth of B. breve UCC2003-araQ and B. breve UCC2003-malR1 mutants was more pronounced compared to the wild type strain B. breve UCC2003: in the presence of 1% porcine bile, the B. breve UCC2003-araQ and B. breve UCC2003-malR1 mutants demonstrated a fold reduction of 11.1 (p-value < 0.010) and 6.4 (p-value < 0.010) in OD600nm readings, respectively, whereas the fold reduction for the control strain B. breve UCC2003 was 3.5.

Phenotypic Growth Assay. Analysis of the growth of B. breve UCC2003 WT (WT), B. breve UCC2003-araQ (ΔaraQ) and B. breve UCC2003-malR1 (ΔmalR1) when grown in the presence of 1% porcine bile. The values are expressed as fold decrease in OD600nm values between growth on 0 and 1% bile. Error bars for each represent the standard deviation calculated from three replicates.
Discussion
The distal gut does not supply bacteria with a reliable carbon source; available carbohydrates are dependent on many factors such as host diet and other (competing) bacteria present in the GIT. Therefore, it is necessary for bifidobacteria to be capable of utilising a multitude of carbon sources and to do so efficiently in order to be competitive. Gene regulation is one way in which bifidobacteria ensure that their energy generation is in line with available carbohydrate substrates, and that energy and biosynthetic demands are kept in balance.
Previous analysis of the AraQ regulatory network in 39 bifidobacterial strains predicted that the conserved core of its regulon comprises genes involved in central carbohydrate metabolism45. In the current study, we experimentally validated these predictions. Additionally, it appears that AraQ and MalR1 together impose global shifts in gene transcription in B. breve UCC2003 through their apparent ability to regulate a number of TFs (i.e. CarD, MalR2, MalR3 and MalR5) and consequently the regulons of these TFs. AraQ and MalR1 were also found to control the transcription of key genes of the bifid shunt in B. breve UCC2003 (Fig. 3). Furthermore, AraQ and MalR1 appear to regulate transcription of their own genes (auto-regulation), while also regulating each other’s transcription. This mechanism of regulating central carbon metabolism with two LacI-type TFs is reminiscent of that of a previously described system in Corynebacterium glutamicum57,58, which utilises GntR1 and GntR2 to regulate many of its genes involved in central metabolism. From the current analysis AraQ and MalR1 are deduced to repress transcription of a number of genes related to sugar uptake, and to activate transcription of genes related to central carbon metabolism, while the opposite was seen for GntR1 and GntR258. Initially, we believed that the functional redundancy of malR1 was the product of a gene duplication event, as was predicted for gntR1 and gntR258. However, GntR1 and GntR2 exhibit 78% amino acid similarity, while AraQ and MalR1 display just 26% similarity. A higher level of similarity would be more in line with a (recent) gene duplication event. Additionally, it is also apparent from the phylogenetic analysis that AraQ and MalR1 form two non-orthologous groups (Fig. S3).
Analysis of araQ and malR1 distribution across a set of 70 Bifidobacteriaceae genomes revealed slightly different patterns of distribution with araQ present in 94% and malR1 present in 71% of these genomes tested. This distribution may be explained in a number of ways, with one possible evolutionary scenario being motif switching whereby MalR1 evolved to recognise the binding motif of AraQ. While duplicating the role of AraQ, the function of a maltose/maltodextrin utilization regulator, in that case, may have been transferred to other MalR paralogs. Additionally, it seems that in some bifidobacteria of insect/bird/marmoset origin malR1 was lost probably due to different selective pressures experienced by these microorganisms. A PWM approach was also utilised to identify to identify additional genes which may be regulated by AraQ and MalR1 in B. breve. In some cases, AraQ/MalR1 PWMs failed to identify TFBSs of genes not involved in central metabolism to which recombinant TFs showed weak or medium affinity. This contradiction can be explained either by: (i) limitations of the EMSA approach, namely, non-specific binding of AraQ/MalR1 to sites of other TFs from the LacI family present in promoter regions of these genes; (ii) limitations of the PWM approach, specifically insufficient sensitivity of designed matrices.
The binding sites of AraQ/MalR1, where AraQ/MalR1 are believed to function as activators, were found to bind at a distance of a multiple of one turn plus one half, away from the TSS. This is consistent with findings for another LacI-type family member, CcpA, which may act as a repressor or activator48, and which binds 14 bp upstream of the −35 site when functioning as an activator. These authors suggested that CcpA binds to the same side of the helix face as the position of the −35 site. The AraQ/MalR1 TFBS was found (in some cases) to be located at a similar distance upstream of the −35 site implying that AraQ/MalR1 may also bind on the same side of the helix face as the position of the −35 site. One important feature of LacI-type TFs is that they can alter the local DNA structure48, which in this case may enhance the affinity of the RNA polymerase holoenzyme to its cognate −35/−10 sites.
Transcriptional analysis for B. breve UCC2003-araQ vs the B. breve UCC2003 WT strain demonstrated that transcription of a number of genes is repressed by AraQ, whereas genes where AraQ is predicted to act as a transcriptional activator (based on the location of the TFBS in relation to the −35 site) showed no significant change in expression. This result is in line with our predictions as insertional mutagenesis should reduce the amount of active AraQ protein in the cell, therefore the expression of those genes which AraQ activates should remain unchanged.
The biological significance of a bacterium producing two distinct proteins which appear to have overlapping functions is currently unknown. We hypothesize that there may be a link between the presence of these two proteins and the stress response of the bacterium. Several publications have reported that the (in)ability to survive bile stress is linked with shifts in the expression of genes involved in central metabolism and with the concentration of its resulting end products in various Bifidobacterium species53,54,55,56. For this reason, growth assays were carried out on B. breve UCC2003-araQ and B. breve UCC2003-malR1, along with the wild type control strain B. breve UCC2003, when exposed to porcine bile (Fig. 4). This analysis revealed that there is a statistically significant drop in viable cell count for both B. breve UCC2003-araQ and B. breve UCC2003-malR1, suggesting that this hypothesis may be correct yet requires further in-depth investigation. The mechanism behind this phenotype remains unclear, though one possible cause may be that B. breve UCC2003-araQ is unable to quickly adjust its central metabolism in an optimal way under this stress condition, as previously reported59. AraQ and MalR1 binding sites have also been identified upstream of a predicted choline transporter (Bbr_0603) and an iron uptake system (Bbr_0121-0122)46,47, indicating that these TFs control a number of genes involved in bile and other cell stress responses. AraQ and MalR1 ability to control central metabolism may also aid survival under bile stress conditions by increasing ATP production from the bifid shunt, while fine tuning the expression on essential central metabolic genes, a role which would benefit from the presence of two distinct TF proteins.
As with all in vitro assays, EMSA and transcriptomic analyses have their limitations and it is important to consider these factors on interpretation of the obtained results. Nonetheless, our findings indicate that AraQ and MalR1 play an important role in regulating the metabolic flux of central metabolism in a large proportion of the Bifidobacteriaceae, presumably contributing to metabolic agility and associated ability to adapt to stressful conditions. In conclusion, this work revealed a novel and apparently unique regulatory mechanism employed by most human-derived bifidobacteria. This regulatory system appears to be crucial to provide adaptability and competitiveness in the constantly changing environment of the gastrointestinal tract.
Methods
Bacterial strains and culture conditions
Bacterial strains and plasmids are listed in Table 2. B. breve strains were routinely grown at 37 °C in either de Man Rogosa and Sharpe medium (MRS medium; Difco, BD, Le Pont de Claix, France), modified de Man Rogosa and Sharpe (mMRS) medium made from first principles60, or reinforced clostridial medium (RCM; Oxoid Ltd., Basingstoke, Hampshire, UK) supplemented with 0.05% cysteine-HCl. Bifidobacteria were incubated anaerobically in a modular, atmosphere-controlled system (Davidson and Hardy, Belfast, Ireland). Escherichia coli strains were routinely grown in Luria Bertani (LB) broth at 37 °C with liquid cultures agitated at 120 rpm. Where appropriate growth medium was supplemented with tetracycline (Tet; 10 µg ml−1), chloramphenicol (Cm; 5 µg ml−1 for E. coli, 2.5 µg ml−1 for B. breve), ampicillin (Amp; 100 μg ml−1), erythromycin (Em; 100 µg ml−1) or kanamycin (Kan; 50 µg ml−1) for plasmid/strain selection and/or maintenance.
Nucleotide sequence analysis
Sequence data were obtained from the Artemis-mediated61 genome annotations of the B. breve UCC2003 genome sequence62. Database searches were carried out using non-redundant sequences accessible at the National Centre for Biotechnology Information internet site (http://www.ncbi.mlm.nih.gov) utilising the basic local alignment search tool (BLAST). Sequence analysis was performed employing the Seqbuilder and Seqman programs of the DNASTAR software package (DNASTAR, Madison, WI). Protein functions were assigned with the use of BLASTP, and homology detection and structure prediction by HMM-HMM comparison and HHpred63,64.
For comparative studies, a non-redundant set of 70 Bifidobacteriaceae genomes was created. The search for AraQ and MalR1 orthologs in these genomes was carried out using the PATtyfam approach and the BLAST tool integrated into the SEED genomic platform65. Sequences of orthologs were collected into one file and then aligned via MUSCLE66. The alignment was subsequently manually filtered to reduce the number of gaps. A phylogenetic tree was built using IQ-TREE52 with the following parameters: automatic substitution model selection, ultrafast bootstrap with 1000 replicates, and employing BglR (Bbr_1659), a distant LacI family regulator from B. breve UCC2003 with predicted different function, as an outgroup.
DNA manipulations
DNA manipulations were carried out based on previously published protocols67. Restriction enzymes and T4 DNA ligase were obtained from Roche Diagnostics (Basel, Switzerland), and were used according to the manufacturer’s instructions. PCRs were performed using either Q5® High-Fidelity DNA polymerase (New England Biolabs, Hertfordshire, UK) or Extensor Long Range PCR Enzyme master mix (Thermo Scientific, Glouchester, UK). Synthetic oligonucleotides were synthesized by Eurofins (Ebersberg, Germany) and are listed in supplemental Table S2 and S10. Ird-labelled synthetic oligonucleotides were provided by IDT (Integrated DNA technologies, Dresden, Germany) and are listed in Supplemental Table S2. PCR products were purified with the use of a High-Pure PCR product purification kit (Roche, Basel, Switzerland). Plasmid DNA was introduced into E. coli and B. breve by electroporation, and large-scale preparation of chromosomal DNA from B. breve was performed as described previously68. Plasmid DNA was obtained from B. breve and E. coli using the Roche High Pure plasmid isolation kit (Roche Diagnostics, Basel, Switzerland). An initial lysis step was performed using 30 mg ml−1 of lysozyme for 30 min at 37 °C as part of the plasmid purification protocol for B. breve.
Construction of B. breve UCC2003 insertion mutants
Internal fragments of Bbr_0411 (designated here as araQ; fragment used was 402 bp in length, representing codons 74 through to 208 of the 371 codons of this gene) and Bbr_1846 (designated here as malR1; fragment used was 504 bp in length, representing codons 102 through to 267 of the 338 codons of this gene) were amplified by PCR using B. breve UCC2003 chromosomal DNA as a template (primers employed are listed in Supplemental Table S10). Insertional mutagenesis was carried out as previously described69. The presence of the tetracycline resistance cassette was confirmed by colony PCR employing primer combination TetF and TetR, while site-specific recombination of potential Tet-resistant mutants was confirmed by colony PCR using a combination of the TetR primer and a primer located upstream of the recombination site in araQ or malR1 in the chromosome of B. breve UCC2003 (see Supplemental Table S10 for primer details). The confirmed insertional mutants within araQ and malR1 were designated here as B. breve UCC2003-araQ and B. breve UCC2003-malR1, respectively.
Microarray analysis
The transcriptome of B. breve UCC2003-araQ and B. breve UCC2003-malR1 was compared to the global gene expression patterns of B. breve UCC2003 (WT). Insertional mutants and the WT strain were cultivated in mMRS medium supplemented with 0.5% ribose until an OD600nm of ∼0.6 was achieved. We employed ribose for such transcriptome analyses as this sugar has been utilized previously as a reference carbohydrate for various metabolic studies37,70,71,72. Furthermore, transcriptome analysis for B. breve UCC2003-araQ and B. breve UCC2003 WT was carried out when these strains were grown on lactose as a sole carbon source, which generated similar results as compared to ribose-grown cells except for the expected sugar-specific differences. Cells were harvested by centrifugation at 10,000 rpm for 2 min at room temperature and immediately frozen at −80 °C prior to RNA isolation. DNA microarrays containing oligonucleotide primers representing each of the annotated genes on the genome of B. breve UCC2003 were designed by and obtained from Agilent Technologies (Palo Alto, CA, USA). Cell disruption, RNA isolation, RNA quality control, and cDNA synthesis and labelling were performed as described previously73. The labelled cDNA was hybridized using the Agilent Gene Expression hybridization kit (part number 5188-5242) as described in the Agilent Two-ColorMicroarrayBased Gene Expression Analysis v4.0 manual (G4140-90050).
Following hybridization, the microarrays were washed in accordance with Agilent’s standard procedures and scanned using an Agilent DNA microarray scanner (model G2565A). The generated scans were converted to data files with Agilent’s Feature Extraction software (v9.5). The DNA microarray data sets were processed as previously described53,74,75. Differential expression tests were performed with the Cyber-T implementation of a variant of the t-test76. A gene was considered to exhibit a significantly different transcription level relative to the control when p < 0.001 and an expression ratio of >2 or <0.25. The microarray data sets obtained in this study have been deposited in NCBI’s Gene Expression Omnibus database and are accessible through GEO series accession number GSE108949.
AraQ and MalR1 expression and purification
For the construction of plasmids pQE60 + araQ and pQE60 + malR1, DNA fragments encompassing araQ (corresponding to locus tag Bbr_0411) and malR1 (corresponding to locus tag Bbr_1846) were generated by PCR amplification employing chromosomal DNA of B. breve UCC2003 as a template, Q5 high-fidelity DNA polymerase, and primers araQ_F and araQ_R, and malR1_F and malR1_R, respectively (Supplemental Table S2). An in-frame His10-encoding sequence is contained within the 3’ end of the pQE60 construct to facilitate downstream protein purification. The araQ-encompassing PCR product was digested with NcoI and BglII, while the malR1-encompassing PCR product was digested with NcoI and BamHI. The digested PCR products were ligated into a similarly digested pQE60, an IPTG-inducible translational fusion plasmid. The ligation mixtures were introduced into E. coli XL1-Blue or E. coli EC101 by electro-transformation, and transformants were then selected on the basis of Ampicillin resistance (AmpR). The plasmid contents of a number of AmpR transformants were screened by restriction analysis, and the integrity of positively identified clones was verified by sequencing. One verified clone of plasmid pQE60 + araQ and pQE60 + malR1 (i.e. plasmid pQE60 in which either araQ or malR1, respectively, was cloned) (Table 2) was then selected for protein expression and purification purposes.
E. coli XL1-BLUE was utilised for heterologous expression of AraQ and MalR1. E. coli XL1-BLUE strains containing either pQE60 + araQ or pQE60 + malR1 were inoculated at 2% in LB medium and grown until an OD600nm of ∼0.5 was reached, at which point protein expression was induced by the addition of IPTG25. Following incubation for a further two hours cells were harvested by centrifugation and re-suspended in EMSA buffer (see below). Bacterial cells were disrupted by bead beating in a mini-bead beater (BioSpec Products, Bartlesville, OK, USA) using glass beads. Cellular debris was removed by centrifugation to produce a crude cell extract. Recombinant AraQ and MalR1 proteins, each tagged with an incorporated C-terminal His10 sequence, were purified from a crude cell extract using a nickel-nitrilotriacetic acid column (Qiagen, Hilden, Germany) according to the manufacturer’s instructions (QIAexpressionist, June 2003). Lysis, wash and elution buffers were supplemented with 10% glycerol as this considerably improved the (binding) activity of AraQ and MalR1. Elution fractions were analysed by SDS-polyacrylamide gel electrophoresis, as described previously77, on a 12.5% polyacrylamide gel. Following electrophoresis, gels were fixed and stained with Coomassie brilliant blue to identify fractions containing the purified protein. Colour Prestained Protein Standard, Broad Range (11–245 kDa) (New England BioLabs, Hertfordshire, UK) was used to estimate the molecular weights of the purified proteins. Protein was concentrated using Amicon® Ultra Filters from Merck Millipore and dialysed into EMSA binding buffer (80 mM Tris-HCl [pH 8.0], 20 mM MgCl2, 2 mM dithiothreitol [DTT], 4 mM EDTA, 400 mM KCl and 40% glycerol). Concentration of the purified AraQ/MalR1 protein and subsequent dialysis into EMSA binding buffer also improved the binding ability of these two LacI-type proteins. Protein concentrations were determined using the Qubit® fluorometer as per manufacturer’s instructions (Thermofisher scientific, Glouchester, UK). Purified protein was aliquoted and stored at -80 °C for subsequent use in EMSAs.
Electrophoretic mobility shift assay (EMSA)
Promoter regions of genes of interest were amplified by PCR utilising individual primer pairs, of which one or both were 5’ Ird-700-labelled (provided by IDT, Dresden, Germany) as listed in supplemental Table S3. Electrophoretic mobility shift assays (EMSAs) were performed as described previously78. All binding reactions were carried out with poly(dI-dC) (0.05 µg µl−1), DNA probe (0.5 nmol), BSA (0.2 µg µl−1), binding buffer (80 mM Tris-HCl [pH 8.0], 20 mM MgCl2, 2 mM dithiothreitol [DTT], 4 mM EDTA, 400 mM KCl and 40% glycerol) and water to a final volume of 20 μl. Binding reactions were typically carried out with 150 nM purified AraQ or MalR1 and 0.5 nM Ird-700-labelled DNA, and incubated for 20 min at 37 °C prior to loading onto a 6% non-denaturing polyacrylamide (PAA) gel prepared in TAE buffer (40 mM Tris-acetate [pH 8.0], 2 mM EDTA) and bound/unbound DNA fragments were then separated by electrophoresis on a 0.5X to 2.0X gradient of TAE at 100 V for 90 min in an Atto Mini PAGE system (Atto Bioscience and Biotechnology, Tokyo, Japan).
Signals and percentage binding inferred from the Integrated Intensity (II) were detected/calculated using an Odyssey infrared imaging system (Li-Cor Biosciences, United Kingdom, Ltd., Cambridge, United Kingdom), and images were electronically captured with the use of Odyssey software v3.0. The ability of AraQ and MalR1 to bind DNA fragments was assessed employing an arbitrary, non-linear scale which spanned from no affinity, low, medium to high affinity (and annotated as −, +, ++, +++, respectively). This non-linear scale was correlated to the percentage of DNA which was bound as compared to unbound target DNA within a given reaction with – representing no DNA bound, + represents up to 15% DNA bound, ++ represents 15% – 50% DNA bound and +++ represents 50–100% DNA bound. In some cases, when a DNA fragment was shown to exhibit binding affinity, the binding site was more precisely determined by generating sub-fragments by PCR, followed by EMSAs as described above. To identify possible effectors for AraQ or MalR1, 10 mM of a particular compound (Supplemental Table S4) was added to the binding reaction mixture.
Motif searches
Motif searches were carried out using Position Weight Matrix (PWM) approach as described previously40. Several PWMs were implemented: (i) a PWM used for the AraQ regulon reconstruction in bifidobacteria based on a previous study40; (ii) a new AraQ/MalR1 PWM which was built using the EMSA data as a training set (based on promoter regions with which AraQ/MalR1 had a medium affinity (++) or high affinity (+++). A graphical representation of the identified motifs was obtained using WebLOGO software79.
Phenotypic analysis on bile
In order to assess the effects of bile on growth, B. breve UCC2003 and its isogenic derivatives B. breve UCC2003-araQ and B. breve UCC2003-malR1 were inoculated at 1% (v/v) from stock into mMRS medium supplemented with 1% maltose and 0.5% (v/v) L-cysteine HCl and were cultured overnight under anaerobic conditions at 37 °C. The above strains were then inoculated at 1% (v/v) into mMRS medium containing 1% maltose, 0.5% (v/v) L-cysteine HCl along with increasing concentration of porcine bile at 0, 1 and 2% (v/v). mMRS without the addition of a carbohydrate source served as a negative control. Bacteria were evaluated for their ability to grow in the presence of bile with Optical density (OD600nm) readings taken at 10 and 24 hr post-inoculation. Samples were assessed as biologically independent triplicates. Statistical analysis was carried out using the Microsoft Excel Data Analysis ToolPak. The statistical significance was determined for the two mutants, by comparing the difference in their OD600nm readings as compared with the WT control when exposed to bile.
Data availability
All data generated and/or analysed during this study are included in this article and its Supplementary Information files.
References
Shoaie, S. et al. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci Rep 3, 2532, https://doi.org/10.1038/srep02532 (2013).
Arumugam, M. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011).
Lugli, G. A. et al. Tracking the Taxonomy of the Genus Bifidobacterium Based on a Phylogenomic Approach. Appl Environ Microbiol 84, https://doi.org/10.1128/AEM.02249-17 (2018).
Hill, C. et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol 11, 506–514, https://doi.org/10.1038/nrgastro.2014.66 (2014).
Fanning, S. et al. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proceedings of the National Academy of Sciences of the United States of America 109, 2108–2113, https://doi.org/10.1073/pnas.1115621109 (2012).
Ooi, L. G. & Liong, M. T. Cholesterol-lowering effects of probiotics and prebiotics: a review of in vivo and in vitro findings. Int J Mol Sci 11, 2499–2522, https://doi.org/10.3390/ijms11062499 (2010).
Russell, D. A., Ross, R. P., Fitzgerald, G. F. & Stanton, C. Metabolic activities and probiotic potential of bifidobacteria. Int J Food Microbiol 149, 88–105, https://doi.org/10.1016/j.ijfoodmicro.2011.06.003 (2011).
Zanotti, I. et al. Evidence for cholesterol-lowering activity by Bifidobacterium bifidum PRL2010 through gut microbiota modulation. Appl Microbiol Biotechnol 99, 6813–6829, https://doi.org/10.1007/s00253-015-6564-7 (2015).
Trejo, F. M., Minnaard, J., Perez, P. F. & De Antoni, G. L. Inhibition of Clostridium difficile growth and adhesion to enterocytes by Bifidobacterium supernatants. Anaerobe 12, 186–193, https://doi.org/10.1016/j.anaerobe.2006.03.004 (2006).
Sivan, A. et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089, https://doi.org/10.1126/science.aac4255 (2015).
Kim, S. W. et al. Bifidobacterium lactis inhibits NF-kappaB in intestinal epithelial cells and prevents acute colitis and colitis-associated colon cancer in mice. Inflammatory bowel diseases 16, 1514–1525, https://doi.org/10.1002/ibd.21262 (2010).
Uccello, M. et al. Potential role of probiotics on colorectal cancer prevention. BMC Surg 12(Suppl 1), S35, https://doi.org/10.1186/1471-2482-12-S1-S35 (2012).
Turroni, F. et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS One 7, e36957, https://doi.org/10.1371/journal.pone.0036957 (2012).
Arboleya, S., Watkins, C., Stanton, C. & Ross, R. P. Gut Bifidobacteria Populations in Human Health and Aging. Frontiers in Microbiology 7, 1204, https://doi.org/10.3389/fmicb.2016.01204 (2016).
Claesson, M. J. et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184, https://doi.org/10.1038/nature11319 (2012).
Pokusaeva, K., Fitzgerald, G. F. & van Sinderen, D. Carbohydrate metabolism in Bifidobacteria. Genes & nutrition 6, 285–306, https://doi.org/10.1007/s12263-010-0206-6 (2011).
Milani, C. et al. Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Scientific Reports 5, 15782, https://doi.org/10.1038/srep15782, https://www.nature.com/articles/srep15782#supplementary-information (2015).
Turroni, F. et al. Glycan Utilization and Cross-Feeding Activities by Bifidobacteria. Trends Microbiol 26, 339–350, https://doi.org/10.1016/j.tim.2017.10.001 (2017).
Lewis, M. The lac repressor. Comptes rendus biologies 328, 521–548, https://doi.org/10.1016/j.crvi.2005.04.004 (2005).
Gonzalez-Rodriguez, I. et al. Catabolism of glucose and lactose in Bifidobacterium animalis subsp. lactis, studied by 13C Nuclear Magnetic Resonance. Appl Environ Microbiol 79, 7628–7638, https://doi.org/10.1128/AEM.02529-13 (2013).
Scardovi, V. The fructose-6-phosphate shunt as a peculiar pattern of hexose degradation in the genus Bifidobacterium. Ann Microbiol Enzymol 15, 19–24 (1965).
Palframan, R., Gibson, G. R. & Rastall, R. A. Development of a quantitative tool for the comparison of the prebiotic effect of dietary oligosaccharides. Lett Appl Microbiol 37, 281–284, https://doi.org/10.1046/j.1472-765X.2003.01398.x (2003).
Breuer, U. Lactobacillus Molecular Biology From Genomics to Probiotics Ã…sa Ljungh and Torkel WadstrÃm (eds.), Caister Academic Press, Norfolk, UK, 2009, 206 pages ISBN: 978-1-904455-41-7. Engineering in Life Sciences 9, 404–404, https://doi.org/10.1002/elsc.200990012 (2009).
Fushinobu, S. Unique sugar metabolic pathways of bifidobacteria. Bioscience, biotechnology, and biochemistry 74, 2374–2384, https://doi.org/10.1271/bbb.100494 (2010).
Meile, L., Rohr, L. M., Geissmann, T. A., Herensperger, M. & Teuber, M. Characterization of the D-xylulose 5-phosphate/D-fructose 6-phosphate phosphoketolase gene (xfp) from Bifidobacterium lactis. Journal of Bacteriology 183, 2929–2936, https://doi.org/10.1128/JB.183.9.2929-2936.2001 (2001).
van den Broek, L. A., Hinz, S. W., Beldman, G., Vincken, J. P. & Voragen, A. G. Bifidobacterium carbohydrases-their role in breakdown and synthesis of (potential) prebiotics. Mol Nutr Food Res 52, 146–163, https://doi.org/10.1002/mnfr.200700121 (2008).
Lugli, G. A. et al. Comparative genomic and phylogenomic analyses of the Bifidobacteriaceae family. BMC genomics 18, 568, https://doi.org/10.1186/s12864-017-3955-4 (2017).
de Vries, W. & Stouthamer, A. H. Pathway of glucose fermentation in relation to the taxonomy of bifidobacteria. Journal of Bacteriology 93, 574–576 (1967).
Gupta, R. S., Nanda, A. & Khadka, B. Novel molecular, structural and evolutionary characteristics of the phosphoketolases from bifidobacteria and Coriobacteriales. PLoS One 12, e0172176, https://doi.org/10.1371/journal.pone.0172176 (2017).
Bondue, P. & Delcenserie, V. Genome of Bifidobacteria and Carbohydrate Metabolism. Korean J Food Sci Anim Resour 35, 1–9, https://doi.org/10.5851/kosfa.2015.35.1.1 (2015).
Trindade, M. I., Abratt, V. R. & Reid, S. J. Induction of sucrose utilization genes from Bifidobacterium lactis by sucrose and raffinose. Appl Environ Microbiol 69, 24–32 (2003).
De Bruyn, F., Beauprez, J., Maertens, J., Soetaert, W. & De Mey, M. Unraveling the Leloir pathway of Bifidobacterium bifidum: significance of the uridylyltransferases. Appl Environ Microbiol 79, 7028–7035, https://doi.org/10.1128/AEM.02460-13 (2013).
Gilad, O. et al. Combined transcriptome and proteome analysis of Bifidobacterium animalis subsp. lactis BB-12 grown on xylo-oligosaccharides and a model of their utilization. Appl Environ Microbiol 76, 7285–7291, https://doi.org/10.1128/aem.00738-10 (2010).
Pokusaeva, K. et al. Ribose utilization by the human commensal Bifidobacterium breve UCC2003. Microbial biotechnology 3, 311–323, https://doi.org/10.1111/j.1751-7915.2009.00152.x (2010).
Pokusaeva, K. et al. Cellodextrin utilization by bifidobacterium breve UCC2003. Appl Environ Microbiol 77, 1681–1690, https://doi.org/10.1128/AEM.01786-10 (2011).
O’Connell Motherway, M., Fitzgerald, G. F. & van Sinderen, D. Metabolism of a plant derived galactose-containing polysaccharide by Bifidobacterium breve UCC2003. Microbial biotechnology 4, 403–416, https://doi.org/10.1111/j.1751-7915.2010.00218.x (2011).
O’Connell, K. J. et al. Transcription of Two Adjacent Carbohydrate Utilization Gene Clusters in Bifidobacterium breve UCC2003 Is Controlled by LacI- and Repressor Open Reading Frame Kinase (ROK)-Type Regulators. Applied and Environmental Microbiology 80, 3604–3614, https://doi.org/10.1128/aem.00130-14 (2014).
Egan, M., O’Connell Motherway, M. & van Sinderen, D. A GntR-type transcriptional repressor controls sialic acid utilization in Bifidobacterium breve UCC2003. FEMS microbiology letters 362, https://doi.org/10.1093/femsle/fnu056 (2015).
Andersen, J. M. et al. Transcriptional analysis of oligosaccharide utilization by Bifidobacterium lactis Bl-04. BMC genomics 14, 312, https://doi.org/10.1186/1471-2164-14-312 (2013).
Khoroshkin, M. S., Leyn, S. A., Van Sinderen, D. & Rodionov, D. A. Transcriptional Regulation of Carbohydrate Utilization Pathways in the Bifidobacterium. Genus. Frontiers in microbiology 7, 120, https://doi.org/10.3389/fmicb.2016.00120 (2016).
Wintjens, R. & Rooman, M. in Journal of molecular biology 262, 294–313 (1996).
Law, J. et al. A system to generate chromosomal mutations in Lactococcus lactis which allows fast analysis of targeted genes. Journal of Bacteriology 177, 7011–7018 (1995).
Ruiz, L. et al. The essential genomic landscape of the commensal Bifidobacterium breve UCC2003. Sci Rep 7, 5648, https://doi.org/10.1038/s41598-017-05795-y (2017).
Bottacini, F. et al. Global transcriptional landscape and promoter mapping of the gut commensal Bifidobacterium breve UCC2003. BMC genomics 18, 991, https://doi.org/10.1186/s12864-017-4387-x (2017).
Schumacher, M. A., Seidel, G., Hillen, W. & Brennan, R. G. Structural mechanism for the fine-tuning of CcpA function by the small molecule effectors glucose 6-phosphate and fructose 1,6-bisphosphate. Journal of molecular biology 368, 1042–1050, https://doi.org/10.1016/j.jmb.2007.02.054 (2007).
Lanigan, N., Bottacini, F., Casey, P. G., O’Connell Motherway, M. & van Sinderen, D. Genome-Wide Search for Genes Required for Bifidobacterial Growth under Iron-Limitation. Frontiers in microbiology 8, 964, https://doi.org/10.3389/fmicb.2017.00964 (2017).
Cronin, M., Zomer, A., Fitzgerald, G. F. & van Sinderen, D. Identification of iron-regulated genes of Bifidobacterium breve UCC2003 as a basis for controlled gene expression. Bioengineered bugs 3, 157–167, https://doi.org/10.4161/bbug.18985 (2012).
Grundy, F. J., Waters, D. A., Allen, S. H. & Henkin, T. M. Regulation of the Bacillus subtilis acetate kinase gene by CcpA. Journal of Bacteriology 175, 7348–7355 (1993).
Rezacova, P. et al. Crystal structures of the effector-binding domain of repressor Central glycolytic gene Regulator from Bacillus subtilis reveal ligand-induced structural changes upon binding of several glycolytic intermediates. Mol Microbiol 69, 895–910, https://doi.org/10.1111/j.1365-2958.2008.06318.x (2008).
O’Connell Motherway, M. et al. Characterization of ApuB, an Extracellular Type II Amylopullulanase from Bifidobacterium breve UCC2003. Appl Environ Microbiol 74, 6271–6279 (2008).
Arzamasov, A. A., van Sinderen, D. & Rodionov, D. A. Comparative Genomics Reveals the Regulatory Complexity of Bifidobacterial Arabinose and Arabino-Oligosaccharide Utilization. Frontiers in microbiology 9, 776–776, https://doi.org/10.3389/fmicb.2018.00776 (2018).
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic acids research 44, W232–W235, https://doi.org/10.1093/nar/gkw256 (2016).
van Hijum, S. A. et al. A generally applicable validation scheme for the assessment of factors involved in reproducibility and quality of DNA-microarray data. BMC genomics 6, 77, https://doi.org/10.1186/1471-2164-6-77 (2005).
Ruas-Madiedo, P., Hernandez-Barranco, A., Margolles, A. & de los Reyes-Gavilan, C. G. A bile salt-resistant derivative of Bifidobacterium animalis has an altered fermentation pattern when grown on glucose and maltose. Applied and environmental microbiology 71, 6564–6570, https://doi.org/10.1128/aem.71.11.6564-6570.2005 (2005).
Sanchez, B., de los Reyes-Gavilan, C. G. & Margolles, A. The F1F0-ATPase of Bifidobacterium animalis is involved in bile tolerance. Environmental microbiology 8, 1825–1833, https://doi.org/10.1111/j.1462-2920.2006.01067.x (2006).
Sanchez, B., Ruiz, L., de los Reyes-Gavilan, C. G. & Margolles, A. Proteomics of stress response in Bifidobacterium. Front Biosci 13, 6905–6919 (2008).
Auchter, M. et al. RamA and RamB are global transcriptional regulators in Corynebacterium glutamicum and control genes for enzymes of the central metabolism. J Biotechnol 154, 126–139, https://doi.org/10.1016/j.jbiotec.2010.07.001 (2011).
Frunzke, J., Engels, V., Hasenbein, S., Gatgens, C. & Bott, M. Co-ordinated regulation of gluconate catabolism and glucose uptake in Corynebacterium glutamicum by two functionally equivalent transcriptional regulators, GntR1 and GntR2. Mol Microbiol 67, 305–322 (2008).
Sanchez, B. et al. Adaptation and response of Bifidobacterium animalis subsp. lactis to bile: a proteomic and physiological approach. Applied and environmental microbiology 73, 6757–6767, https://doi.org/10.1128/aem.00637-07 (2007).
De Man, J. C., Rogosa, M. & Sharpe, M. E. A Medium for the Cultivation of Lactobacilli. Journal of Applied Bacteriology 23, 130–135, https://doi.org/10.1111/j.1365-2672.1960.tb00188.x (1960).
Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatics (Oxford, England) 16, 944–945 (2000).
O’Connell Motherway, M. et al. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proceedings of the National Academy of Sciences of the United States of America 108, 11217–11222, https://doi.org/10.1073/pnas.1105380108 (2011).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 25, 3389–3402 (1997).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Davis, J. J. et al. PATtyFams: Protein Families for the Microbial Genomes in the PATRIC. Database. Frontiers in microbiology 7, 118–118, https://doi.org/10.3389/fmicb.2016.00118 (2016).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32, 1792–1797, https://doi.org/10.1093/nar/gkh340 (2004).
Russell, S. J. D. W. Molecular Cloning: a laboratory manual. 3rd edn, (Cold Spring Harbor Laboratory Press, 2001).
O’Riordan, K. & Fitzgerald, G. F. Molecular characterisation of a 5.75-kb cryptic plasmid from Bifidobacterium breve NCFB 2258 and determination of mode of replication. FEMS microbiology letters 174, 285–294, https://doi.org/10.1111/j.1574-6968.1999.tb13581.x (1999).
O’Connell Motherway, M., O’Driscoll, J., Fitzgerald, G. F. & Van Sinderen, D. Overcoming the restriction barrier to plasmid transformation and targeted mutagenesis in Bifidobacterium breve UCC2003. Microbial biotechnology 2, 321–332, https://doi.org/10.1111/j.1751-7915.2008.00071.x (2009).
James, K., Motherway, M. O. C., Bottacini, F. & van Sinderen, D. Bifidobacterium breve UCC2003 metabolises the human milk oligosaccharides lacto-N-tetraose and lacto-N-neo-tetraose through overlapping, yet distinct pathways. Scientific Reports 6, 38560, https://doi.org/10.1038/srep38560, https://www.nature.com/articles/srep38560#supplementary-information (2016).
Egan, M., Jiang, H., O’Connell Motherway, M., Oscarson, S. & van Sinderen, D. Glycosulfatase-Encoding Gene Cluster in Bifidobacterium breve UCC2003. Applied and Environmental Microbiology 82, 6611-6623, https://doi.org/10.1128/aem.02022-16 (2016).
Egan, M., O’Connell Motherway, M., Ventura, M. & van Sinderen, D. Metabolism of sialic acid by Bifidobacterium breve UCC2003. Appl Environ Microbiol 80, 4414–4426, https://doi.org/10.1128/AEM.01114-14 (2014).
Zomer, A. et al. An interactive regulatory network controls stress response in Bifidobacterium breve UCC2003. Journal of bacteriology 191, 7039–7049, https://doi.org/10.1128/jb.00897-09 (2009).
van Hijum, S. A., de la Nava, G., Trelles, J., Kok, O. & Kuipers, J. O. P. MicroPreP: a cDNA microarray data pre-processing framework. Applied bioinformatics 2, 241–244 (2003).
Garcia de la Nava, J. et al. Engene: the processing and exploratory analysis of gene expression data. Bioinformatics (Oxford, England) 19, 657–658, https://doi.org/10.1093/bioinformatics/btg028 (2003).
Long, A. D. et al. Improved statistical inference from DNA microarray data using analysis of variance and a Bayesian statistical framework. Analysis of global gene expression in Escherichia coli K12. The Journal of biological chemistry 276, 19937–19944, https://doi.org/10.1074/jbc.M010192200 (2001).
Laemmli, U. K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 227, 680, https://doi.org/10.1038/227680a0 (1970).
Hamoen, L. W., Van Werkhoven, A. F., Bijlsma, J. J. E., Dubnau, D. & Venema, G. The competence transcription factor of Bacillus subtilis recognizes short A/T-rich sequences arranged in a unique, flexible pattern along the DNA helix. Genes Dev 12, 1539–1550 (1998).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome research 14, 1188–1190, https://doi.org/10.1101/gr.849004 (2004).
Maze, A., O’Connell-Motherway, M., Fitzgerald, G. F., Deutscher, J. & van Sinderen, D. Identification and characterization of a fructose phosphotransferase system in Bifidobacterium breve UCC2003. Appl Environ Microbiol 73, 545–553, https://doi.org/10.1128/AEM.01496-06 (2007).
Acknowledgements
This research was carried out with the support of the APC Microbiome Institute which is a research centre funded by Science Foundation Ireland (SFI), through the Irish Government’s National Development Plan. The authors (N.L. and D.V.S.) and their work were supported by SFI (Grant Numbers SFI/12/RC/2273-P1 and SFI/12/RC/2273-P2). E.K. and C.S. were supported by Food Institutional Research Measure (FIRM) (grant number: FIRM 11/F/023). A.A.A. and D.A.R. received support from the Russian Science Foundation (RSF) (Grant Number 19-14-00305).
Author information
Authors and Affiliations
Contributions
D.v.S., D.A.R., N.L. and E.K. conceived the experiments. N.L. and E.K. conducted the experiments. A.A.A. conducted the bioinformatic analysis. All authors analysed the results and contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lanigan, N., Kelly, E., Arzamasov, A.A. et al. Transcriptional control of central carbon metabolic flux in Bifidobacteria by two functionally similar, yet distinct LacI-type regulators. Sci Rep 9, 17851 (2019). https://doi.org/10.1038/s41598-019-54229-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54229-4
This article is cited by
-
Using fluorescent promoter-reporters to study sugar utilization control in Bifidobacterium longum NCC 2705
Scientific Reports (2022)
-
Carbohydrate-controlled serine protease inhibitor (serpin) production in Bifidobacterium longum subsp. longum
Scientific Reports (2021)
-
Genotyping and plant-derived glycan utilization analysis of Bifidobacterium strains from mother-infant pairs
BMC Microbiology (2020)
-
Strain-specific strategies of 2′-fucosyllactose, 3-fucosyllactose, and difucosyllactose assimilation by Bifidobacterium longum subsp. infantis Bi-26 and ATCC 15697
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
