
Abstract
Coronavirus disease 2019 (COVID-19) predisposes patients to thrombotic and thromboembolic events, owing to excessive inflammation, endothelial cell activation and injury, platelet activation and hypercoagulability. Patients with COVID-19 have a prothrombotic or thrombophilic state, with elevations in the levels of several biomarkers of thrombosis, which are associated with disease severity and prognosis. Although some biomarkers of COVID-19-associated coagulopathy, including high levels of fibrinogen and d-dimer, were recognized early during the pandemic, many new biomarkers of thrombotic risk in COVID-19 have emerged. In this Consensus Statement, we delineate the thrombotic signature of COVID-19 and present the latest biomarkers and platforms to assess the risk of thrombosis in these patients, including markers of platelet activation, platelet aggregation, endothelial cell activation or injury, coagulation and fibrinolysis as well as biomarkers of the newly recognized post-vaccine thrombosis with thrombocytopenia syndrome. We then make consensus recommendations for the clinical use of these biomarkers to inform prognosis, assess disease acuity, and predict thrombotic risk and in-hospital mortality. A thorough understanding of these biomarkers might aid risk stratification and prognostication, guide interventions and provide a platform for future research.
Similar content being viewed by others
Introduction
In patients with coronavirus disease 2019 (COVID-19), the conventional coagulation abnormalities mimic those of other systemic coagulopathies that are associated with severe infections, such as disseminated intravascular coagulation, but the disease also has some distinct features1,2. Deep-vein thrombosis and pulmonary embolism are frequently found in patients with COVID-19; the incidence depends on whether routine (ultrasonography) screening is instituted and the severity of the disease, with a higher prevalence in patients in the intensive care unit (ICU)3,4,5. In a meta-analysis of 49 studies, including 18,093 patients, the reported pooled incidence of venous thromboembolism (VTE) was 17.0%, with an incidence of 33.0% in studies that used systematic screening (28% of studies) and an incidence of 9.8% in those relying on clinical diagnosis6. Large, randomized, controlled trials in hospitalized patients with COVID-19, which did not screen for VTE, have found a lower incidence of VTE of 6–10% when prophylactic-dose anticoagulation was used and an incidence of 4–8% when treatment-dose anticoagulation was used7,8,9. In patients with COVID-19 in the ICU, systematic reviews suggest an incidence of VTE of 28%, although the latest reviews suggest an incidence of 19–24% when clinical diagnosis is used and 36–46% when routine screening is used10,11.
Superficial-vein thrombosis and catheter-related thrombosis in patients with COVID-19 are also frequently reported12. Akin to venous thrombosis, arterial thrombosis can affect all organs, with ischaemic stroke, systemic arterial embolism, acute coronary syndrome, and limb and mesenteric ischaemia reported to occur in 1–5% of patients with COVID-19 (refs13,14,15,16,17). Furthermore, evidence of in situ microvascular thrombus formation, particularly in the pulmonary circulation, has been reported18,19.
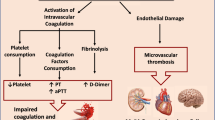
In addition to disease severity, the occurrence of thrombosis and the elevated levels of coagulation markers are important determinants of prognosis in patients with COVID-19 (refs20,21). Early in the pandemic, hospitalized patients with COVID-19 were often observed to have derangements in the levels of coagulant biomarkers, including fibrinogen, d-dimer and activated partial thromboplastin time (aPTT), and routine measurement of these biomarkers was recommended22,23,24. However, a number of other markers of coagulation have emerged that have helped to refine our understanding of the thrombotic signature of COVID-19 (Fig. 1). In addition to helping to understand the pathophysiological processes underlying COVID-19 and its complications, assessment of these markers, including with the use of pathway-specific platforms (Box 1), can identify individuals who are at higher risk of thrombosis and potentially guide thromboprophylaxis and treatment approaches.

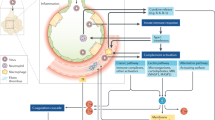
Following entry of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) into the host endothelial cell by binding to the angiotensin-converting enzyme 2 (ACE2) receptor, the expression and enzymatic activity of ACE2 are reduced, resulting in increased vascular permeability and tissue factor (TF) expression in subendothelial cells, as well as in leukocytes and platelets, which triggers coagulation. ACE2 can exert antithrombotic effects through various mechanisms, including the renin–angiotensin pathway, in which angiotensin I is converted by angiotensin-converting enzyme to angiotensin II (Ang II), which is then broken down by ACE2 to angiotensin 1–7 (Ang 1–7). Reduction in the expression of ACE2 leads to an increase in the level of Ang II, which stimulates the expression of plasminogen activator inhibitor 1 (PAI-1) in various cells, including smooth muscle cells, endothelial cells and adipocytes. The increase in PAI-1 levels results in hypofibrinolysis. Endothelial cell activation or dysfunction results in a generalized inflammatory state, characterized by high levels of inflammatory cytokines, release of von Willebrand factor (vWF) and increased endothelial cell-surface expression of adhesion molecules such as P-selectin, promoting thrombus formation and leukocyte recruitment. Inflammation is an important driver of thrombosis, through several mechanisms. Inflammatory cytokines and viral-specific Toll-like receptors (TLRs) induce TF expression in monocytes, resulting in activation of the coagulation cascade. Platelet activation by TLR signalling results in increased platelet reactivity and platelet aggregation. Activation of neutrophils results in the release of neutrophil extracellular traps (NETosis), leading to activation of coagulation and providing a scaffold for the adhesion of platelets, red blood cells and platelet-adhesion molecules. In parallel, activation of coagulation via TF also results in thrombin generation and the formation of fibrin, which allows crosslinking of platelets and other cellular constituents and results in occlusive thrombus formation. CCL2, CC-motif chemokine 2; COVID-19, coronavirus disease 2019; CRP, C-reactive protein; DAMP, damage-associated molecular pattern; FIXa, activated factor IX; FVIIa, activated factor VII; FVIIIa, activated factor VIII; FXa, activated factor X; HIF, hypoxia-inducible factor; ICAM, intercellular adhesion molecule; RANTES, regulated on activation, normal T expressed and secreted; TFPI, tissue factor pathway inhibitor; TMPRSS, transmembrane serine protease 2; TNF, tumour necrosis factor; tPA, tissue plasminogen activator; VCAM, vascular cell adhesion molecule.
In this Consensus Statement, we describe the thrombotic signature of COVID-19 and present the latest biomarkers and platforms to assess the risk of thrombosis, on the basis of a review of the available clinical evidence. We include markers of platelet activation, platelet aggregation, endothelial cell activation or injury, coagulation and fibrinolysis as well as biomarkers of the newly recognized post-vaccine thrombosis with thrombocytopenia syndrome. We review the evidence that supports the use of the available biomarkers in guiding prognosis, including disease acuity, and in predicting thrombotic risk and in-hospital mortality. We make evidence-based consensus recommendations for the clinical use of these markers in patients with COVID-19, according to the latest available clinical data and expert review (Tables 1,2).
Methods for consensus recommendations
We conducted a search of the literature on biomarkers associated with thrombosis in patients with COVID-19. The evidence-based literature was synthesized using the PubMed, Embase and Cochrane library databases, with no restriction on language. In addition, the reference lists of selected articles were checked for further relevant articles. Case reports, animal studies, comments and author replies were excluded. Given that a systematic review of all the literature on potential biomarkers was not logistically feasible, we aimed to synthesize studies that identified serological or cellular markers that correlated with clinical outcomes up to October 2021. Because the literature search yielded a very large number of studies (>100,000), we prioritized reporting of larger studies over smaller ones. Systematic reviews and meta-analyses were reported where available and, again, larger cohorts were prioritized over smaller, preliminary reviews.
All the authors reviewed all sections of the manuscript, participated in the Delphi process (conducted virtually) and judged the available tests of thrombosis, leading to the consensus recommendations. These recommendations were formulated after a review of the available evidence synthesized by the experts. Each author participated in the final evaluation of each method, and reconsideration of their own judgements was incorporated into the Delphi consensus process. There were no disagreements among the authors.
This Consensus Statement summarizes the final conclusions and recommendations agreed by the expert panel, on the basis of the best available evidence and expert opinion, and the recommendations are graded according to the Oxford Centre for Evidence-Based Medicine (OCEBM) level of evidence25. In particular, the evidence base was evaluated against the diagnostic criteria in the OCEBM for “is this diagnostic or monitoring test accurate?” and the screening criteria for “is this (early detection) test worthwhile?”, with grading levels 1–5.
Soluble biomarkers of inflammation, coagulation and immune system activation
In a minority of patients, infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) leads to a severe multisystem disease characterized largely by inflammatory and thrombotic processes that damage host tissues. The cumulative evidence now suggests that the extent to which specific pathways are triggered determines, or at least predicts, a more severe COVID-19 trajectory. In this context, several biomarkers associated with specific inflammatory and coagulation pathways have been identified that correlate with, or that predict progression to, severe COVID-19. In this section, we review the biomarkers that are relevant to thrombotic risk in COVID-19.
IL-6 and C-reactive protein
The cytokine IL-6 is a pleiotropic mediator of inflammation and a central stimulus in the acute-phase response. Data support an important role for IL-6 in the pathological inflammatory response that drives severe COIVD-19, because high serum levels of IL-6 are prognostic of more severe outcomes in hospitalized patients with COVID-19 (ref.26), and IL-6 receptor blockade improves survival in this group27. Therefore, IL-6 is a biomarker of severe COVID-19 that is also important in its pathogenesis. However, although IL-6 is prognostic of more severe outcomes in COVID-19 (ref.26), IL-6 cannot be used to discriminate between COVID-19 and other causes of severe illness (similar to other biomarkers discussed below), because a meta-analysis of 19 studies (1,245 patients) showed that although IL-6 levels were elevated in patients with severe COVID-19, levels were significantly lower than in non-COVID-19-related sepsis and acute respiratory distress syndrome28. Overall, the data support the use of IL-6 levels as a predictive biomarker of more severe outcomes in hospitalized patients with COVID-19, but a specific link with the development of thrombosis has not been established.
C-reactive protein (CRP) is an established marker of acute inflammation across many disease states. CRP is produced in the liver in response to inflammatory cytokines, especially IL-6. Levels of both CRP and IL-6 are increased in patients with COVID-19 and are positively correlated with disease severity and mortality28,29,30. A meta-analysis of 20 studies, including 4,843 patients with COVID-19, found a fourfold increased risk of poor outcomes in patients with COVID-19 who had elevated CRP levels (pooled OR 3.97, 95% CI 2.89–5.45, P < 0.00001)31. In a large analysis of 2,782 patients with COVID-19 from a health system in New York City, USA, >97% of patients were found to have elevated CRP levels at presentation, and high baseline CRP levels were associated with progression to critical illness, acute kidney injury, VTE and all-cause mortality32. Elevated IL-6 levels measured at hospital presentation were associated with progression to critical illness and with all-cause mortality in patients with COVID-19 and CRP levels either above or below the median, suggesting additional prognostic information from the measurement of IL-6 levels, although the relationship to the occurrence of thrombotic events was not specifically examined32. Overall, studies have shown a positive correlation between CRP levels and COVID-19 severity and, in contrast to IL-6, CRP levels have been shown to be predictive of the risk of thrombosis32.
d-Dimer
d-Dimer is the principal breakdown fragment of fibrin and is used as a biomarker of coagulation and fibrinolysis33. In the setting of COVID-19, d-dimer has been widely investigated, and levels have been reported to be elevated in most patients hospitalized with COVID-19 (ref.34). Levels of d-dimer peak approximately 5 days into hospitalization and are higher in patients with critical illness or in those who subsequently died29,35,36. An in-depth investigation of d-dimer levels in 2,377 hospitalized patients with COVID-19 found a stepwise association between baseline and peak d-dimer levels and critical illness, thrombosis, acute kidney injury and all-cause mortality34. d-Dimer levels could be used to screen for thrombosis, with higher levels of d-dimer found in patients with COVID-19 and VTE than in those without VTE, both in multiple individual studies and in a systematic review of 47 studies37. Another review of 71 studies suggested that d-dimer levels could be used to identify patients with COVID-19 who needed CT pulmonary angiography to diagnose pulmonary embolism, using a d-dimer cut-off level of ≥1,000 μg/l38.
In a retrospective analysis of outcomes in 195 patients with COVID-19 admitted to the ICU, in which two of the five ICUs managed patients with a protocol-based escalation of anticoagulation on the basis of d-dimer levels, d-dimer-driven anticoagulation was associated with a reduction in mortality and renal failure events39. In a prospective study in 803 patients hospitalized with COVID-19, all receiving anticoagulant thromboprophylaxis, a protocolized escalation of anticoagulation dose on the basis of a combination of illness severity, body weight and d-dimer level was associated with reductions in mortality (6.3% versus 11.8%; P = 0.02) and thrombotic events (4.4% versus 10.7%; P = 0.002) compared with patients treated off-protocol40. Several trials investigating anticoagulation in patients with COVID-19 have used d-dimer levels in the inclusion criteria41,42. The multiplatform ATTACC trial9 tested the effect of therapeutic anticoagulation with heparin in 2,219 hospitalized patients with COVID-19 with either high (twice or more than twice the upper limit of the normal range) or low (less than twice the upper limit of the normal range) d-dimer levels and found a benefit in both groups. However, the probability of superiority of using therapeutic-dose heparin over usual-care thromboprophylaxis was 97.3% in the high d-dimer cohort compared with 92.9% in the low d-dimer cohort9. In another large, open-label, randomized, controlled trial in 615 hospitalized patients with COVID-19 and d-dimer levels above the normal range at presentation to hospital, therapeutic anticoagulation did not improve clinical outcomes, regardless of the actual level of d-dimer at enrolment7. Overall, a majority of studies support the use of d-dimer levels as a marker of thrombosis.
Coagulation activation markers
More specific information about the coagulation pathways can be obtained by measuring biomarkers that indicate the concentrations of specific protease inhibitors or activation peptides. In COVID-19, coagulopathy is thought to be driven by cellular tissue factor (TF) expression, which, as a plasma marker, is detectable on extracellular vesicles. The TF–factor VIIa complex engages the extrinsic coagulation route, resulting directly (or indirectly via factor IX) in factor X activation and subsequent prothrombin conversion to thrombin. The activation of prothrombin yields an F1+2 fragment that can be quantified in the blood. The generation of thrombin can be indirectly probed by measuring thrombin–antithrombin complexes. Levels of both F1+2 and thrombin–antithrombin complexes were elevated in patients with acute respiratory distress syndrome (with or without COVID-19) and were further increased during extracorporeal membrane oxygenation43. Elevated F1+2 levels were associated with increased risk of clinical deterioration44,45, and increased levels of thrombin–antithrombin complexes persisted despite detectable anti-Xa activity during thromboprophylaxis46. Using markers for activation of the contact and intrinsic systems, Busch and colleagues showed that COVID-19 severity-dependent increases in plasma kallikrein levels (a group of serine proteases that indicate contact system activation) are linked to increments in markers of downstream activation of factor IX, factor XI and prothrombin47. These changes in contact and intrinsic system coagulation were closely associated with the activation of neutrophils and the complement system47. These data support the hypothesis that contact activation due to persistent NETosis, and complement activation might be an important accelerator of thromboinflammation in patients with COVID-19 (ref.48) and provide potential therapeutic targets49.
Tests of coagulation
COVID-19-associated coagulopathy is characterized by a near-normal platelet count and prothrombin time in the majority of patients, and uniformly high d-dimer and fibrinogen levels50,51. By contrast, disseminated intravascular coagulation, a consumptive coagulopathy, which is observed only occasionally in patients with severe COVID-19, is characterized by a decreased platelet count, elevated d-dimer levels, prolonged prothrombin time and decreased fibrinogen levels51, and was mainly reported at the start of the pandemic. As in other severe diseases, the occurrence of laboratory features of disseminated intravascular coagulation is associated with a poor prognosis in patients with COVID-19 (ref.52).
Prothrombin time
Prothrombin time, which assesses the extrinsic coagulation pathway, measures ex vivo the time taken for plasma to clot following the addition of an excess of thromboplastin plus an optimal concentration of calcium. Although routine checking of prothrombin time to assess for COVID-19-associated coagulopathy was recommended early in the pandemic23, this parameter is normal or near-normal in most patients with COVID-19, with prolonged prothrombin times occasionally reported in those with severe disease51,53.
Activated partial thromboplastin time
The aPTT is usually normal in patients with COVID-19 and is not associated with disease severity54. Prolongation of the aPTT might indicate a clotting factor deficiency, the presence of specific coagulation inhibitors (such as antibodies to factor VIII) or a laboratory artefact due to an antibody that interferes with phospholipids (such as lupus anticoagulant). In patients with COVID-19, artefactual aPTT prolongation has been documented secondary to the presence of lupus-like inhibitors or the increased prevalence of heparin resistance due to high fibrinogen and factor VIII levels55.
Anti-Xa assay
The anti-Xa assay is used to monitor the effect of treatment with low-molecular-weight heparin, unfractionated heparin or factor Xa inhibitors. The anti-Xa assay correlates better with the unfractionated heparin concentration than either the activated clotting time or the aPTT. In this functional assay, citrated platelet-poor plasma is mixed with a known amount of factor Xa and, depending on the level of factor Xa inhibition, a clotting-based factor Xa assay or an assay with a specific chromogenic substrate is used to measure the residual factor Xa levels. The residual factor Xa level is directly related to the concentration of unfractionated heparin or low-molecular-weight heparin. Therefore, although not recommended for routine monitoring, assessing the effect of low-molecular-weight heparin with the use of the anti-Xa assay in patients with severe renal impairment is mentioned in clinical guidelines for the management of coagulopathy and thrombosis in patients with COVID-19 (ref.55), with a recommended target range of anti-Xa levels of 0.3–0.7 IU/ml. A retrospective study in 56 patients with COVID-19 showed that personalization of the dose of low-molecular-weight heparin on the basis of an anti-Xa assay was independently associated with a lower risk of COVID-19-related death (OR 0.040, P = 0.043)56.
Thrombin generation
Thrombin generation can be assessed using commercially available, ex vivo assays (such as the Calibrated Automated Thrombogram; Diagnostica Stago) and in vivo by measuring prothrombin F1+2 levels. In the ex vivo assays, thrombin generation is measured after stimulation of the plasma with TF in order to assess the potential to form thrombin. Given that most hospitalized patients with COVID-19 will be receiving anticoagulant thromboprophylaxis, thrombin generation can be assessed in the presence of polybrene or heparinase as a neutralizing agent57. Analysis of thrombin generation was further modified for the COVID-19-associated coagulopathy by addition of ellagic acid or PPP-Reagent HIGH (platelet-poor plasma (PPP) with a high concentration of TF) as triggers of thrombin generation to enable measurements in plasma containing heparins and very high levels of fibrinogen58.
Although some studies have shown similar levels of thrombin generation in patients with COVID-19 as in healthy controls, others have shown elevated thrombin generation in patients with severe COVID-19, despite anticoagulant treatment45,59,60. Peak levels of thrombin were associated with poor outcomes in a retrospective study in 99 patients with COVID-19 (ref.44). In another study in 127 patients with COVID-19, a longer lag time until thrombin generation and a lower endogenous thrombin potential were associated with an increased risk of disseminated intravascular coagulation59. In this study, an increased ratio of d-dimer to endogenous thrombin potential indicated a higher risk of major adverse events59. Daily analysis of thrombin generation (in conjunction with viscoelastic tests) might be helpful to tailor antithrombotic medication in patients with COVID-19 (ref.61). One study showed dose-dependent heparin effects on thrombin generation parameters, including endogenous thrombin potential and peak thrombin level62. Another study showed persistently increased thrombin generation in convalescent patients at a median follow-up of 68 days after SARS-CoV-2 infection63.
Viscoelastic assays
In contrast to conventional plasma-based assays that reflect a specific pathway of coagulation, viscoelastic assays such as thromboelastography (TEG; Haemonetics) and rotational thromboelastometry (ROTEM; Instrumentation Laboratories) provide a global assessment of dynamic changes in clot characteristics, from the initiation of clot formation to platelet–fibrin clot generation, stability and lysis in whole blood. In ROTEM, the tension that is generated following platelet–fibrin clot formation in a cup is recorded using a pin suspended in the cup. This tension is recorded as ‘clot strength/amplitude’ and displayed on a time versus amplitude graph (Fig. 2). The new point-of-care, cartridge-based TEG 6-s assay uses microfluidic resonance frequency technology to assess real-time clot characteristics and provides similar information on clot tensile strength to that obtained by ROTEM64.

Rotational thromboelastometry tracings from a healthy individual (part a) and a critically ill patient with coronavirus disease 2019 (COVID-19) (part b). The patient with COVID-19 has more rapid coagulation, as shown by the shortened clotting time (X) and clot formation time (Y), increased maximum amplitude, greater fibrin clot strength (maximum clot firmness; MCF) and the complete absence of fibrinolysis (‘fibrinolysis shutdown’). LY30, lysis achieved by 30 min after clotting time.
These assays are more sensitive to unfractionated heparin, low-molecular-weight heparin and direct thrombin inhibitors and less sensitive to warfarin and direct factor Xa inhibitors. In patients with COVID-19, viscoelastic assays can be used to assess hypercoagulability. In patients with COVID-19, various groups have noted a shorter time for initial clot generation (shorter reaction time, R) despite widespread use of anticoagulants; more rapid clot formation, as evidenced by an increased ‘angle’, indicating elevated fibrinogen concentration and/or function; increased clot strength, indicated by an increased maximum amplitude (with the use of TEG) or maximum clot firmness (MCF) (with the use of ROTEM); and increased clot stability, as demonstrated by prolonged or absent lysis54.
Multiple studies have performed viscoelastic tests in patients with COVID-19. These studies were either prospective or retrospective and generally enrolled 20–50 patients, mainly those in the ICU. Although definitions and cut-off values vary, a consistent theme is the detection of hypercoagulability marked by reduced clot formation time and increased fibrin clot strength (demonstrated by increased maximal amplitude or MCF), despite anticoagulant prophylaxis53,65,66. The platelet–fibrin clot strength and fibrin clot strength were reported to be better for discriminating between patients with COVID-19 and patients with pneumonia but without COVID-19 than standard COVID-19 biomarkers such as CRP and d-dimer levels67. In patients monitored with ROTEM during ICU admission, >80% of EXTEM and FIBTEM MCF values (which measure the external part of coagulation and elasticity of the fibrin-based clot, respectively) remained above the upper reference value during 6 weeks of follow-up68. After discharge from the ICU, complete normalization of viscoelastic testing at 3 and 6 months was shown in limited case series69,70. A systematic review that included data on a total of 1,063 hospitalized patients with COVID-19 showed that studies using viscoelastic assays, mainly ROTEM, did not demonstrate a retrospective relationship between clot formation characteristics and the occurrence of VTE65. Conversely, a systematic review of 15 studies, mainly small observational cohort studies in patients with COVID-19 in the ICU, showed that TEG can be used to predict thrombotic complications71. In a study of 119 hospitalized patients with COVID-19, a fibrin clot strength of >40 mm measured with TEG was independently associated with a more than threefold increase in the risk of the composite end point of thrombotic events and death67. Based on the measurement of reaction time in the TEG 6-s assay, an inadequate anticoagulant response was noted in patients with COVID-19 treated with enoxaparin compared with those receiving heparin prophylaxis54,72. These data provide preliminary evidence supporting the use of ROTEM and TEG for diagnostic and prognostic purposes, as well as to personalize anticoagulant therapy in patients with COVID-19, although prospective validation is needed (Table 1).
Tests of fibrinolysis
Impaired fibrinolysis is central to the coagulopathy observed in patients with COVID-19 (ref.73). The balance between coagulation and fibrinolysis is lost in patients with COVID-19. Profound hypercoagulability, which is exacerbated by a state of fibrinolysis shutdown, seems to be mediated by the overexpression of plasminogen activator inhibitor 1 (PAI-1) and thrombin-activatable fibrinolysis inhibitor (also known as carboxypeptidase B2). Despite the hypofibrinolytic state, the overall processes of clot formation and lysis are augmented in patients with COVID-19, explaining the elevated levels of d-dimer observed in these patients73.
Viscoelastic assays
Hypofibrinolysis and fibrinolysis shutdown, based on the estimation of lysis at a fixed time in viscoelastic assays, have been reported in patients with COVID-19 and correlate with the severity of COVID-19 and COVID-19-related adverse outcomes74,75,76. The degree of clot lysis detected at 30 min after maximal clot strength is achieved (LY30) is a measure of the efficiency of fibrinolysis. In one study, complete fibrinolysis shutdown, defined as an LY30 of 0% on TEG and a d-dimer level >2.6 μg/ml, was seen in 57% of patients with COVID-19 in the ICU, and was associated with the development of renal failure, VTE and other thrombotic events, which were seldom observed in patients with COVID-19 without fibrinolysis shutdown74. Indeed, LY30 was predictive of the occurrence of VTE in patients with COVID-19, with an area under the receiver operating characteristic curve (AUC) of 0.742 (P = 0.022). Among 52 patients with COVID-19 in the ICU, all receiving anticoagulant thromboprophylaxis, a hypercoagulable state with fibrinolysis shutdown was demonstrated in 31% and was associated with the occurrence of VTE77. In another prospective study in 40 patients with COVID-19 admitted to the ICU, substantially impaired fibrinolysis was detected in all patients, but this impairment was more pronounced in patients with subsequent thromboembolic complications78. Furthermore, the researchers showed that assessment of maximum lysis in the EXTEM test resulted in an AUC of 0.8 (95% CI 0.7–0.9, P = 0.001) for the prediction of thromboembolic events, whereas assessing maximum lysis in INTEM (which measures the intrinsic part of clotting) resulted in an AUC of 0.79 (95% CI 0.6–0.9, P = 0.002), with the combination of d-dimer and EXTEM maximum lysis resulting in an AUC of 0.92 (95% CI 0.8–1.0)78. In another prospective study, hypofibrinolysis was found in all 24 patients with COVID-19 who were tested in the ICU53. In a retrospective study in 40 patients with COVID-19 in the ICU, testing with ROTEM showed that maximum lysis at 60 min significantly decreased over time from admission to hospital through to day 10 (ref.79). Similarly, fibrinolysis shutdown, defined as having an EXTEM maximum lysis of <3.5% in ROTEM, was seen in 44% of 25 patients with COVID-19 in the ICU and was associated with a 73% incidence of thrombotic complications75. In patients monitored with ROTEM during ICU admission, t-PA-ROTEM (an assay that also probes fibrinolysis) showed a persistent hypofibrinolytic pattern68. These observations suggest that viscoelastic assays are useful as prognostic methods to detect fibrinolysis shutdown and to predict adverse outcomes in patients with COVID-19.
Soluble markers of fibrinolysis
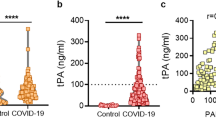
A central mechanism for hypofibrinolysis is the elevation of plasma PAI-1 levels, secondary to the release of PAI-1 by endothelial cells and activated platelets80. Additional mechanisms include elevated plasma levels of thrombin-activatable fibrinolysis inhibitor and protein C inhibitor81. This hypofibrinolytic environment is evident despite raised tissue plasminogen activator (tPA) levels, emphasizing the strength of the anti-fibrinolytic response to SARS-CoV-2 infection80.
Two crucial questions arise: do changes in the fibrinolytic system have a prognostic value and can they alter patient management? An early case–control study demonstrated that plasma levels of PAI-1, tPA and thrombin-activatable fibrinolysis inhibitor are elevated in patients with SARS-CoV-2 infection and are also significantly higher in those who required admission to an ICU (n = 48) compared with those who did not (n = 30)82. In a larger study of 118 patients with COVID-19, elevated plasma PAI-1 levels were associated with more severe disease83, but the wide range observed made PAI-1 an inaccurate prognostic marker. However, the researchers found a possible prognostic utility for plasma tPA levels, although large-scale studies are warranted before firm conclusions can be drawn. A systematic review and meta-analysis of 17 small, mainly retrospective studies, including three that assessed tPA levels, four that measured PAI-1 levels and three that assessed soluble thrombomodulin levels (an integral, membrane-bound glycoprotein expressed on the surface of endothelial cells that is an anticoagulant and antifibrinolytic factor), found that elevations in each of these markers was associated with adverse prognosis in patients with COVID-19 (ref.84). A small study (n = 50) suggested that soluble thrombomodulin levels might be predictive of clinical outcomes in patients with COVID-19, with levels of >3.26 ng/ml associated with increased in-hospital mortality (HR 5.9, 95% CI 1.9–18.4) or longer inpatient stay85, and increased levels have also been shown in convalescent patients (median 68 days after infection)63. Elevated thrombomodulin levels might be a marker of endothelial dysfunction, an important abnormality in COVID-19, as discussed below.
Platelet count and function
Numerous studies have assessed platelet count and markers of platelet reactivity and activation in patients with COVID-19. The potential utility of these biomarkers is summarized in Table 2.
Platelet count
Many studies have compared platelet counts in patients with COVID-19 and those in healthy controls, and have generally shown that the most severe manifestations of COVID-19 were associated with lower platelet counts than milder forms of the disease32,86,87, and this association was supported by the results of a meta-analysis88. Moreover, a platelet count of <100 × 109/l was much more common in those with critical COVID-19 than in those with severe or moderate disease89, with serial measurements indicating that the difference in platelet count between non-survivors and survivors increases during hospitalization90. The immature platelet fraction has also shown some utility in predicting outcomes such as length of hospital stay and admission to the ICU, including in fairly large cohort studies91,92,93. Mean platelet volume, a biomarker of platelet hyperactivity, has also been found to be associated with disease severity and the occurrence of thrombosis in patients with COVID-19 in small studies and in a meta-analysis of 15 studies, with the presence of larger, more immature platelets being associated with increased risk of thrombosis94,95.
Platelet activation
Post-mortem studies in individuals who died from COVID-19 have shown a high incidence of platelet–fibrin microthrombi in organs such as the lungs, heart, liver and brain96,97,98. Platelet aggregates and granules have been observed in blood films and might indicate disease severity; in one study, films from all patients with COVID-19 who required admission to the ICU showed platelet aggregates and granules, whereas these features were not seen in patients managed outside the ICU setting99. Electron microscopy has demonstrated platelet clumping, membrane fragmentation and degranulation100,101.
After activation, platelets express P-selectin, which binds to and activates leukocytes and is cleaved to a soluble form102. In small case–control studies in <50 patients each, platelet P-selectin expression was consistently significantly higher in patients with COVID-19 than in healthy controls86,103,104,105,106 and in patients with severe COVID-19 than in those with mild or asymptomatic disease107. Circulating levels of soluble P-selectin were significantly higher in patients with COVID-19 who required admission to the ICU than in healthy controls, but not higher than those in patients with COVID-19 who did not require admission to the ICU in one study85, although higher in patients who required admission to the ICU than in those who did not in another study108. Soluble P-selectin levels were found to be a reasonable predictor of intubation and death in patients with COVID-19 (ref.109). In a post-hoc analysis of samples from >300 patients with COVID-19, plasma P-selectin level at hospital admission was strongly associated with a subsequent diagnosis of VTE and was independent of disease severity110.
Platelet activation also leads to surface expression of CD40 ligand (CD40L), which is subsequently cleaved to a soluble form that can stimulate leukocytes and stabilize platelet aggregates. In case–control studies including 50–100 patients, circulating soluble CD40L levels were significantly increased in patients with COVID-19 compared with the levels in healthy controls85,108,111. Analysis of 100 hospitalized patients with COVID-19 found that soluble CD40L levels were independently associated with the composite of thrombosis or death, but not with thrombosis alone95, whereas another cross-sectional study in 68 patients with COVID-19 found no difference in soluble CD40L levels in relation to disease severity85. Higher levels of platelet cytosolic calcium and phosphatidylserine externalization have also been associated with an increased likelihood of admission to the ICU in patients with COVID-19 (ref.112). A very small study showed higher platelet expression of glycoprotein Ib and glycoprotein IX in eight patients with COVID-19 than in healthy controls113. Another study of 36 patients with COVID-19 indicated that markers of platelet activation have returned to normal 2–3 months after discharge from hospital103.
In studies including >100 patients, levels of urinary 11-dehydro-thromboxane B2, a marker of thromboxane A2 generation that occurs after platelet activation, were higher in patients with COVID-19 than in patients with non-COVID-19 pneumonia72, and in patients with COVID-19 complicated by adverse events than in those with less severe disease114. Similarly, serum thromboxane B2 levels were significantly higher in patients with severe disease than in those with mild or asymptomatic COVID-19 or in healthy volunteers in one study107, but not in another111. A retrospective assessment showed that raised serum thromboxane B2 levels were independently associated with the occurrence of thrombosis in 100 patients hospitalized with COVID-19 (ref.95).
The SARS-CoV-2 virus and antibodies to SARS-CoV-2 might also have direct roles in platelet activation. In one study, immune complexes containing recombinant SARS-CoV-2 spike protein and anti-spike IgG increased platelet-mediated thrombosis in vitro after administration of von Willebrand factor (vWF), especially in response to high shear stress, when the glycosylation of the IgG Fc domain was modified to correspond with the aberrant glycosylation previously identified in patients with severe COVID-19 (ref.115).
It has generally been accepted that SARS-CoV-2 requires host cell expression of angiotensin-converting enzyme 2 (ACE2) to facilitate entry into the cell and of transmembrane protease serine 2 (TMPRSS2) to prime the spike protein116. Various studies have found no evidence of ACE2 or TMPRSS2 expression in platelets104,111,117. Conversely, another group observed ‘robust’ expression of both proteins118, also demonstrating that binding of SARS-CoV-2 to platelets upregulated a range of processes relating to platelet activation and aggregation. Furthermore, SARS-CoV-2-associated mRNA was detected in the platelets of two out of 25 patients with COVID-19, suggesting that platelets might allow entry of the virus independent of ACE2 (ref.104). A study analysing platelet RNA showed the presence of fragmented SARS-CoV-2 viral genome in the platelets of all 17 patients with COVID-19 who were tested119. Furthermore, platelets were shown to internalize SARS-CoV-2 virions rapidly, regardless of the presence of ACE2, leading to programmed cell death and the release of extracellular vesicles119. Finally, active virus uptake has been demonstrated in megakaryocytes120.
Platelet aggregation
Studies investigating platelet aggregation in patients with COVID-19 are heterogeneous. In a study in 54 patients with COVID-19, of whom ~10% received aspirin and none received a P2Y12 inhibitor, ADP-induced platelet aggregation, as assessed by light transmission aggregometry, was increased in those admitted to the ICU compared with those who were not, whereas no significant difference was noted between patients with COVID-19 who were not admitted to the ICU and healthy controls108. In another study, in which 41 patients with COVID-19 received no antiplatelet therapy, platelet aggregation in response to a range of agonists was significantly increased compared with that in healthy volunteers and was most marked in those admitted to the ICU104. A further study found greater platelet responses in patients hospitalized with COVID-19 than in healthy controls120. Whether any patients in these studies were receiving unfractionated heparin, which can potentially confound findings by increasing platelet aggregation121, was not reported. By contrast, another small study utilizing multiplate multiple electrode aggregometry with ADP, arachidonic acid and thrombin receptor-activating peptide (TRAP) as agonists, showed no significant difference in platelet aggregation between 27 patients with COVID-19 receiving mechanical ventilation and 12 healthy controls; although ADP-induced aggregation was lower in patients with COVID-19, this difference was no longer significant after adjusting for sex122. Antiplatelet therapy was not fully described in this study, making interpretation of the results problematic. Another study showed significantly lower platelet reactivity in patients with COVID-19 than in healthy controls when assessed by multiple electrode aggregometry using arachidonic acid and TRAP, but not ADP, as agonists123. A study of TEG platelet mapping in 24 hospitalized patients with COVID-19 showed ADP-induced responses in the high–normal range, which was not related to disease severity124. Another study in which 100 hospitalized patients with COVID-19 had TEG platelet mapping on admission to hospital showed that platelet hyper-reactivity was associated thrombotic and/or ischaemic complications, and antiplatelet treatment guided by TEG platelet mapping was associated with a significant reduction in mortality125, although this protocol was not randomized. Assessment using the Platelet Function Analyser PFA-100 in 20 hospitalized patients with COVID-19 who were not treated with antiplatelet therapy showed mean platelet aggregation values in the normal range, similar to those in healthy controls105. A small study using the Total Thrombus Formation Analysis System in hospitalized patients with COVID-19 demonstrated impaired platelet thrombus formation in the early phase of the disease compared with later stages of the disease and with platelet thrombus formation in healthy controls, independent of illness severity126.
Platelet-omics (transcriptomics, proteomics and metabolomics)
A study of the platelet transcriptome in hospitalized patients with COVID-19 compared with that in healthy controls found significant differences in mRNA expression of 3,325 genes, including many relevant to protein ubiquitination, antigen presentation and mitochondrial dysfunction104. Of note, levels of the mRNA for interferon-induced transmembrane protein 3, which has an important role in limiting viral infectivity, were significantly upregulated104. Another study showed broad differences in serum proteomics and metabolomics between patients with severe COVID-19 and control individuals, including a striking downregulation of 15 out of 17 proteins involved in platelet degranulation, particularly the chemokines pro-platelet basic protein and platelet factor 4 (PF4)127. A retrospective study in 3,915 patients with COVID-19 demonstrated a distinct transcriptomic profile that was characteristic of prothrombotic large and immature platelets and further showed that the interaction between SARS-CoV-2 and megakaryocytes alters the platelet transcriptome, and the effects of SARS-CoV-2 are distinct from those of the coronavirus that causes the common cold120.
Platelet–leukocyte interactions
Levels of platelet–monocyte and platelet–neutrophil aggregates are increased in the blood of patients with COVID-19 (ref.105), especially those who require hospitalization128, with the highest levels observed in patients treated in the ICU107, indicating that platelet–leukocyte aggregates might be a surrogate marker for the severity of COVID-19. Plasma P-selectin levels in patients with COVID-19 are predictive of subsequent VTE, independent of disease severity110. Moreover, platelet engulfment by white blood cells has been detected in blood from patients with COVID-19 (ref.105). In a small study in 35 patients with COVID-19, platelet adhesion to monocytes was shown to be associated with monocyte TF expression, and platelets from individuals with COVID-19 could trigger TF expression by normal monocytes107. Moreover, in this study, monocyte TF expression correlated with d-dimer levels107. Together, these observations support a role for platelet-induced TF expression in COVID-19 thrombosis.
Endothelial cell activation and injury
Endothelial dysfunction, in particular the consequent effects on coagulation and angiogenesis, seems to be a crucial mediator of pathogenesis in COVID-19 (ref.129). In patients with severe COVID-19, a relationship exists between the extent of endotheliopathy and the magnitude of the immune inflammatory response, as well as the extent of organ dysfunction, even after adjustment for other risk factors such as age, sex and BMI130. The addition of plasma from critically ill patients with COVID-19 to cultured pulmonary microvascular endothelial cells in vitro triggered endothelial damage131, suggestive of immune-mediated endotheliopathy. Incubation of microvascular endothelial cells with platelet releasates isolated from patients with COVID-19 induced an inflammatory hypercoagulable endotheliopathy, demonstrating an under-appreciated role of platelets in the pathogenesis of COVID-19-induced endothelial activation, which was not seen with the use of control platelet releasates from healthy individuals132. ACE2 is highly expressed on endothelial cells133, and even more so in tissues from patients with COVID-19 than in those from patients with influenza or from healthy individuals134. Although ACE2 is considered to be essential for entry of SARS-CoV-2 into host cells, further research is needed into whether direct infection is the most common trigger of endothelial cell activation or whether paracrine pathways are the dominant driver of endothelial cell inflammation135.
In the section above on soluble markers of fibrinolysis, the roles of tPA, PAI-1 and thrombomodulin, which are also markers of endothelial dysfunction, have been discussed, and we discuss vWF below.
von Willebrand factor
vWF is a large glycoprotein, ranging in size from 600,000 to 20 million daltons, that is synthesized by megakaryocytes and vascular endothelial cells and which actively participates in platelet adhesion to injured, disrupted or activated vascular endothelial cells, high shear stress-associated platelet aggregation, inflammation and immune activation136. Plasma concentrations of vWF are primarily determined by its synthesis and release from vascular endothelial cells after activation or injury137. Other stimuli for vWF release include hypoxaemia, angiogenesis, inflammation and tissue injury. The greater the stimulus for virus-induced cytotoxicity, pro-inflammatory cytokine production, cellular injury linked to cell-free nucleic acids and impaired vWF clearance mechanisms, the higher the level of vWF measured in peripheral blood138. Fluid shear stress dynamically regulates vWF by promoting aggregation of multiple vWF units, while at the same time reducing multimer size through force-dependent cleavage by ADAMTS13 (ref.139).
vWF is a common factor in three pathophysiological features of the acute phase of COVID-19: immunoinflammation, vasculopathy and coagulopathy. Several groups have found increased levels of vWF antigen, heightened vWF activity, including increased binding to collagen, and an increased ratio of vWF antigen to ADAMTS13 activity in patients with COVID-19 (refs85,131). In a single-centre, cross-sectional study in 68 patients with COVID-19, of whom 48 were treated in the ICU, vWF antigen levels were markedly elevated, especially in patients in the ICU, and were found to correlate with mortality85. A reduction in ADAMTS13 activity relative to the level of vWF antigen suggests a processing abnormality that could contribute to altered vWF multimer profiles and a heightened prothrombotic state140,141,142. In small, cross-sectional studies of hospitalized patients with COVID-19, an increased ratio of vWF antigen to ADAMTS13 activity was strongly associated with disease severity and complications143,144. Although many cross-sectional cohort studies have shown a direct correlation between vWF levels and disease acuity, morbidity and in-hospital mortality87,141,145,146,147, the data to support the usefulness of vWF as a diagnostic or prognostic marker of thrombotic events are sparse147. Routine measurement of vWF levels in patients with moderate-to-severe COVID-19 at the time of hospitalization is not recommended, but future clinical trials on vWF inhibitors should be considered148,149.
Other markers of endothelial injury and pro-angiogenesis
In addition to the effects mediated by vWF, endothelial cell dysfunction induces hypercoagulation through PAI-1, soluble thrombomodulin and TF pathway inhibitor. Furthermore, elevated levels of tPA, the main pro-fibrinolytic serine protease, which is found on endothelial cells, its principal inhibitor PAI-1 and soluble thrombomodulin have been associated with adverse outcomes in patients with COVID-19, such as respiratory failure, multiorgan failure and death84,85,130. Furthermore, increased expression of pro-angiogenic factors, including vascular endothelial growth factor, hypoxia-inducible factor 1α, IL-6, tumour necrosis factor (TNF) receptor superfamily 1A and TNF receptor superfamily 12, have been found in both serum and lung biopsy samples from patients with COVID-19 (refs129,130,150). Moreover, hypercytokinaemia has been documented in patients with COVID-19, manifesting as raised levels of IL-1β, IL-7, IL-9, IL-10 and TNF150. In particular, elevated levels of IL-6 and TNF are a marker of adverse prognosis151 and are likely to contribute to endothelial dysfunction in patients with COVID-19 (ref.152).
Extracellular vesicles
Released by blood, vascular and other cells, extracellular vesicles circulate in the blood, where they have many functions. In their lipid bilayer, extracellular vesicles can contain proteins, metabolites and nucleic acids that are being eliminated or recycled, or acting as signalling molecules. Additionally, the lipid bilayer can contain membrane-associated proteins, including adhesion receptors such as integrins and selectins, which enable the extracellular vesicles to dock to and deliver their contents to the cell. Pro-coagulant extracellular vesicles, largely produced by endothelial cells and monocytes, express phosphatidylserine, TF and other coagulation factors. Classic activation of the complement system, which can be stimulated by antibodies to SARS-CoV-2 or other factors, triggers the release of extracellular vesicles153,154. When formed, extracellular vesicles can theoretically induce various events, such as coagulation, angiogenesis, cell survival and intercellular communication155,156. In 100 hospitalized patients with COVID-19, extracellular vesicle TF activity correlated with disease severity and thrombosis, prothrombin time and levels of d-dimer155, fibrinogen, plasmin–antiplasmin complexes, vWF, ADAMTS13, circulating leukocytes and inflammatory markers157. An observational study in 111 hospitalized patients with COVID-19 showed much higher extracellular vesicle TF activity in those with severe disease than in those with moderate disease, and extracellular vesicle TF levels strongly correlated with the occurrence of clinical thromboembolic events (AUC 0.851; P < 0.0001)157. Another observational study in 84 patients with COVID-19 showed that extracellular vesicle TF activity was markedly higher in the five patients with arterial or venous thrombosis than in those without158.
Both mean particle size and the volume of extracellular vesicles are higher in patients with severe COVID-19 than in those with milder disease159, and the extracellular vesicles are enriched in prothrombotic factors (TF, tPA and vWF) and TNF superfamily and IL-6 family members. Levels of platelet-derived extracellular vesicles are also higher in patients with COVID-19 who require hospitalization than in those who do not111,160. Importantly, no evidence from retrospective analyses indicates that extracellular vesicle levels are altered in patients with COVID-19 who receive anticoagulant or corticosteroid therapy155. Although studies of extracellular vesicles might provide insights into the pathogenesis of COVID-19, methods for the analysis of extracellular vesicles are not yet automated, making routine measurements in patients with COVID-19 challenging at this time.
Novel soluble biomarkers
Neutrophil extracellular traps
COVID-19 is characterized by a high prevalence of immunothrombotic complications11,18,161. Neutrophil extracellular traps (NETs) were first identified in 2010 as a pro-thrombotic scaffold and consist of neutrophil-derived chromatin associated with pro-coagulant proteins and antimicrobial proteins, such as myeloperoxidase (MPO) or neutrophil elastase162,163. A decade later, studies have shown that NET components are present abundantly in plasma, serum and post-mortem specimens from patients with COVID-19 (refs164,165). The detection of NETosis in patients with severe COVID-19 not only opened diagnostic and prognostic possibilities (with NET biomarkers), but also helped to define new therapeutic targets.
Autopsy specimens from patients who died from COVID-19 have revealed NET-containing microthrombi in many cases166,167,168,169,170. Histopathology of lungs and other organs consistently showed microvascular obstruction by aggregated NETs, associated with endothelial cell disruption. A prospective autopsy cohort study revealed neutrophilic plugs in 10 of 21 patients with COVID-19 (ref.96). Clinically, thromboembolic events had been diagnosed in 48% of these patients. NETs were often co-localized with platelets and detected in the lungs, heart, kidneys, liver, spleen and brain (median disease course 22 days). NETs have subsequently been found in approximately half of autopsy cases, suggesting that NETosis contributes to severe COVID-19.
Increased plasma or serum levels of cell-free DNA, MPO–DNA complexes, neutrophil elastase–DNA complexes and citrullinated histone 3 (H3Cit) have been found in various inflammatory disorders and, although their diagnostic utility might therefore be limited, these NET markers might be predictive of disease severity in patients with COVID-19. Interestingly, compared with levels of MPO–DNA or neutrophil elastase–DNA, levels of both cell-free DNA and H3Cit correlated more strongly with COVID-19 severity169. NET markers should preferably be measured in plasma rather than in serum, because neutrophils release NETs during clotting of the blood sample in vitro. Accordingly, cell-free DNA levels were consistently higher in serum than in plasma and showed greater variability169.
However, owing to better accessibility, NET markers have also been investigated in serum. Levels of cell-free DNA, MPO–DNA and H3Cit were increased twofold in patients with COVID-19 compared with healthy controls, with a correlation between concentration and disease severity164. Interestingly, serum from patients with COVID-19 induced NET formation in control neutrophils in vitro164. Higher levels of cell-free DNA, MPO–DNA and H3Cit were also associated with an increased risk of developing thrombotic events164.
In plasma, levels of MPO–DNA and H3Cit correlated strongly with COVID-19 severity and were also associated with thrombotic events in most studies103,168,170,171,172,173,174. These observational studies enrolled between 19 and 135 patients with COVID-19 at various disease stages. In vitro, plasma-derived antibodies from patients with COVID-19 induced NET formation in neutrophils isolated from healthy donors170. Moreover, NET release could also be induced by direct interaction between SARS-CoV-2 and neutrophils170. Cell-free DNA, H3Cit and neutrophil elastase concentrations in the plasma of patients with COVID-19 were associated with requirement for respiratory support and mortality172, indicating their potential prognostic utility. In critically ill patients with COVID-19, the level of cell-free DNA showed a rather weak negative correlation with oxygenation parameters, but its decrease over time predicted the number of ventilator-free days175. Together, these data show that NETosis is not disease-specific and, therefore, is not useful for assessing the presence of SARS-CoV-2 infection, but NET parameters do indicate COVID-19 severity. Their prognostic value should now be investigated in further studies.
Complement factors
Levels of several complement factors were found to be elevated in the plasma of patients with COVID-19 (C3 (ref.173), C5 (ref.173), C5a176,177,178,179 and soluble C5b-9 (refs177,178,179,180)) and were associated with disease severity. Subsequently, in an open-label, non-randomized clinical trial, the addition of the C5 inhibitor eculizumab to standard of care was compared with standard of care alone in 80 patients with severe COVID-19 who were treated in the ICU; eculizumab significantly improved 15-day survival and oxygenation181. In accordance with the consistently strong association between plasma complement factors and disease severity, a prospective observational study in 134 patients with COVID-19 showed increased activation of the complement system (higher levels of C5a and soluble C5b-9) in critically ill patients, suggesting prognostic utility182. A prospective cohort study in 219 patients with COVID-19 found elevated C5a levels in those who died183.
MicroRNAs
An early study found 35 microRNAs that were upregulated and 38 microRNAs that were downregulated in whole blood from patients with COVID-19 compared with healthy controls184. The most promising marker was hsa-miR-16-2-3p, with a 1.6-fold upregulation in patients with COVID-19 (ref.184). In a larger study in 84 patients with COVID-19, microRNA levels were correlated with disease severity, and specific circulating microRNA profiles (such as miR-148a-3p, miR-451a and miR-486-5p) seemed to have prognostic predictive utility185.
ACE2
ACE2 expression is upregulated in bronchoalveolar lavage fluid in some patients with COVID-19 (ref.186), and a case report found increased serum ACE2 levels in a patient with COVID-19 acute respiratory distress syndrome187. By contrast, a study in 85 patients showed no significant difference in serum ACE2 levels between SARS-CoV-2-positive patients and matched SARS-CoV-2-negative control patients with similar symptoms on presentation to hospital188, indicating that serum ACE2 level might not be useful as a diagnostic or prognostic marker for COVID-19.
HMGB1
Levels of the chromatin protein and transcription regulator high-mobility group box protein 1 (HMGB1) are elevated in the serum and plasma of patients with severe COVID-19 and are associated with an adverse prognosis189,190. HMGB1 induces the expression of ACE2 in alveolar epithelial cells, and genetic or pharmacological inhibition of HMGB1 was shown to block ACE2 expression189, indicating that HMGB1 inhibition might be a potential treatment strategy in patients with COVID-19.
Progranulin
Progranulin is predominantly expressed in epithelial cells, neurons and macrophages and promotes inflammation and cell proliferation. Progranulin was identified in a screen of serum protein expression levels performed in 85 all-comers to the emergency room with COVID-19 symptoms and either positive or negative SARS-CoV-2 status191. Progranulin levels were upregulated in patients with COVID-19 and were associated with adverse outcomes, suggesting prognostic utility.
Calprotectin
Levels of calprotectin (also known as S100A8/S100A9), a neutrophil cytosolic component, were found to be elevated in the serum and plasma from patients with COVID-19 compared with the levels in healthy individuals164,192,193,194,195. In these observational studies in up to 172 patients with COVID-19, calprotectin concentrations were associated with disease severity192,193,194,195 and thrombotic risk164, suggesting that calprotectin might be of prognostic utility, pending prospective validation. A retrospective study found that circulating calprotectin levels were significantly higher in 291 hospitalized patients with COVID-19 than in SARS-CoV-2-negative control patients with a similar clinical presentation and, among patients with COVID-19, calprotectin levels were significantly higher in those who developed thrombosis or critical illness132. After adjustment for age, sex, race/ethnicity, BMI, diabetes mellitus, chronic obstructive pulmonary disease or asthma, history of coronary artery disease or cancer, and antiplatelet or anticoagulant therapy, calprotectin levels were independently associated with thrombosis, with those in the highest quartile having a more than threefold increased risk of thrombosis.
Novel urinary markers
In addition to urinary 11-dehydro-thromboxane B2, patients with COVID-19 who at hospital admission had higher levels of 8-hydroxy-2′-deoxyguanosine (a proposed biomarker of DNA oxidative damage) and fatty acid-binding protein 1 (a more accurate indicator of acute kidney injury than serum creatinine levels) had longer durations of hospitalization and higher risks of thrombotic events and death than those with lower urinary levels of these biomarkers114.
Vaccine-induced thrombotic thrombocytopenia
Vaccine-induced thrombotic thrombocytopenia (VITT), also known as post-vaccine thrombosis with thrombocytopenia syndrome, has been described as an uncommon complication of COVID-19 vaccination administered 4–30 days before symptom onset196. The diagnosis requires the presence of venous or arterial thrombosis, most often splanchnic or cerebral venous sinus thrombosis, mild-to-severe thrombocytopenia and a positive enzyme-linked immunosorbent assay (ELISA) for PF4.
VITT is mostly commonly associated with the ChAdOx1 CoV-19 vaccine (manufactured by AstraZeneca and the University of Oxford) and the Ad26.COV2.S vaccine (manufactured by Janssen; Johnson & Johnson), that both contain recombinant adenoviral vectors. One report suggests that the incidence of VITT is 3.6 per 1 million vaccinated people in those who received who received the ChAdOx1 CoV-19 vaccine and 0.9 per 1 million vaccinated people in those who received the Ad26.COV2.S vaccine197. A single case of possible VITT related to the mRNA-1273 vaccine (manufactured by Moderna) has been published, although the diagnosis has been questioned198. VITT has not been reported with other adenoviral vaccines or with the other mRNA-based vaccine, BNT162b2 (manufactured by Pfizer–BioNTech).
The presentation and pathophysiology of VITT are highly similar to those of heparin-induced thrombocytopenia, with a similar presence of antibodies binding to PF4. These antibodies are IgG molecules that activate platelets via low-affinity platelet FcγIIa receptors, leading to extensive platelet activation and a hypercoagulable state that causes VTE.
Various haematological biomarkers are associated with the VITT syndrome, including platelet concentration, fibrin split products such as d-dimer and fibrinogen, antibodies to PF4 and whole-blood impedance aggregometry. Patients with VITT commonly have a platelet count of about 20 × 109/l, but a platelet count of <15 × 109/l is sufficient to consider a diagnosis of VITT in the presence of clinically evident arterial or venous thrombosis199. Very low platelet counts (<10 × 109/l) after vaccination suggest an alternative diagnosis of acute immune thrombocytopenic purpura, another rare post-vaccination complication that presents as bleeding rather than thrombosis200. In most patients with VITT, d-dimer concentrations are markedly elevated, but fibrinogen concentrations might be reduced in some patients, and other coagulation markers such as the international normalized ratio and aPTT tend to be normal201. ELISA revealed high levels of antibodies to PF4–polyanion complexes in almost every reported case of VITT, with optical densities of >2.0–3.0 (refs201,202). Because the turnaround time of PF4–heparin ELISAs can be slow, treatment might first be initiated in patients with thrombosis, thrombocytopenia and an elevated d-dimer levels, while waiting for the results of a PF4 ELISA200,203. Rapid non-ELISA assays for heparin-induced thrombocytopenia are not currently recommended for the diagnosis of VITT because of their poor diagnostic accuracy204.
Whole-blood impedance aggregometry using patient serum or plasma with donor platelets has also been proposed for the diagnosis of VITT202. In contrast to heparin-induced thrombocytopenia, in which platelet aggregation occurs only in the presence of a low concentration of heparin and is subsequently attenuated by a high concentration of heparin, patients with VITT have increased platelet aggregation in the absence of heparin but the addition of a high concentration of heparin does not attenuate the platelet aggregation202.
Discussion and future directions
The prothrombotic profile that is now a well-recognized hallmark of COVID-19 seems to be mediated by excessive inflammation, endothelial activation and injury, platelet activation, impaired or dysfunctional fibrinolysis, immune-related molecular events, and systemic hypercoagulability that is still to be fully characterized. These processes predispose patients to thrombotic and thromboembolic events that affect small, medium and large veins and, to a lesser extent, arteries.
Circulating biomarkers have been associated with early phase COVID-19 acuity and outcomes, including the occurrence of venous thrombosis, the need for treatment in the ICU or mechanical ventilation, and in-hospital mortality. The role of serial measurements and their relationships with longer-term outcomes, including post-acute sequelae of SARS-CoV-2 infection, are less clear.
The prognostic potential of measuring platelet count, the levels of d-dimer, CRP or IL-6 and vWF antigen or the ratio of vWF antigen to ADAMTS13 is strong and well-aligned with the severity of COVID-19 and prognosis. However, the evidence is based predominantly on observational, retrospective studies that used tests that are readily available in most hospital laboratories. Several other biomarkers require more specialized assays and testing platforms. These include, among others, markers of platelet activation, hypofibrinolysis and NETs. In a number of small, retrospective studies, these markers have shown an association with COVID-19 severity and prognosis, but require specialist laboratory expertise and are not easily measurable within a clinically relevant time frame. Automated, point-of-care testing might overcome this limitation, but instruments are not in widespread clinical use, and data supporting the utility in guiding prognosis with point-of-care techniques are scarce, although viscoelastic assays to personalize antithrombotic therapy for patients with COVID-19 in the ICU setting might be useful67,71.
In addition to the lack of prospective validation, for many biomarkers the optimal cut-off level that defines increased risk is unclear, and given that these biomarkers have generally been measured at a single time point, whether dynamic changes could be used to guide prognosis with changing clinical acuity is uncertain. Furthermore, most of these biomarkers have been measured in isolation, and their additive value in guiding prognosis in patients with COVID-19 is unknown. Finally, a very important limitation of grouping these findings together is that data were obtained during the first, second and third waves of the COVID-19 pandemic, during which both the predominant viral strains and the therapeutic approaches, notably the use of anticoagulation and anti-inflammatory treatments such as dexamethasone and tocilizumab, changed substantially.
A novel avenue is the use of proteomics in the setting of COVID-19, which has enabled the identification of large numbers of proteins associated with SARS-CoV-2 pathogenesis and clinical outcomes205. Using a machine learning-based pipeline, an early proteomics study from Wuhan, China, found that 11 host proteins and a set of biomarker combinations were predictive of COVID-19 outcomes206. These proteins and pathways relate to immune or inflammatory responses, platelet degranulation and coagulation, and metabolism, which are likely to contribute to pathogenesis. Another analysis used state-of-the-art proteomics to investigate the infection process of SARS-CoV-2 and its cellular targets207. Overall, proteomics is fostering a better understanding of the virus and its pathogenesis and identifying additional therapeutic targets.
A major unmet need is to understand the role of biomarkers in determining optimal treatment, including drug selection, dose and duration of administration. Prospective, randomized trials must include blood sampling, and targeted assays are needed to answer many unresolved questions about routine measurement in clinical practice. We have highlighted those biomarkers that have an established routine role in the clinic and others that have evident potential. We hope that this Consensus Statement will underpin future large, prospective studies to assess the prognostic and diagnostic value of these biomarkers, both alone and in combination. Additionally, studies on antithrombotic treatments (specifically antiplatelet and anticoagulant strategies) should assess the value of these biomarkers at baseline and over time to risk-stratify patients who might benefit most from these targeted approaches.
In addition to COVID-19-associated coagulopathy, the recognition of VITT has highlighted the need to identify individuals who are at risk of thrombotic events after vaccination. In addition to a heightened index of clinical suspicion, the prompt recognition of thrombosis with thrombocytopenia syndrome requires the measurement of platelet count and CRP levels and an ELISA for PF4. Future studies are needed to investigate whether biomarkers predicting thrombotic risk can be used to identify individuals who are at risk of VITT, not only to enable greater vigilance and possible thromboprophylaxis, but also when booster vaccinations are administered.
Conclusions
In addition to conventional tests of coagulation, several novel biomarkers and platforms are available to assess the risk of thrombosis and prognosis in patients with COVID-19. Although most have not been sufficiently tested or validated prospectively for adoption into routine clinical practice, growing clinical data support the use of these tests and markers to aid risk stratification and prognostication. Furthermore, these biomarkers could potentially guide therapy, and the current data should provide a platform to underpin future research to enable the optimization of treatment to reduce the risk of thrombosis in patients with COVID-19.
References
Iba, T., Levy, J. H., Levi, M., Connors, J. & Thachil, J. Coagulopathy of coronavirus disease 2019. Crit. Care Med. 48, 1358–1364 (2020).
Peyvandi, F. et al. Hemostatic alterations in COVID-19. Haematologica 106, 1472–1475 (2020).
Cui, S., Chen, S., Li, X., Liu, S. & Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thrombosis Haemost. 18, 1421–1424 (2020).
Klok, F. et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb. Res. 191, 148–150 (2020).
Poissy, J. et al. Pulmonary embolism in COVID-19 patients: awareness of an increased prevalence. Circulation 142, 184–186 (2020).
Jiménez, D. et al. Incidence of VTE and bleeding among hospitalized patients with coronavirus disease 2019: a systematic review and meta-analysis. Chest 159, 1182–1196 (2021).
Lopes, R. D. et al. Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated d-dimer concentration (ACTION): an open-label, multicentre, randomised, controlled trial. Lancet 397, 2253–2263 (2021).
REMAP-CAP Investigators et al.Therapeutic anticoagulation with heparin in critically ill patients with Covid-19. N. Engl. J. Med. 385, 777–789 (2021).
ATTACC Investigators et al.Therapeutic anticoagulation with heparin in noncritically ill patients with Covid-19. N. Engl. J. Med. 385, 790–802 (2021).
Mansory, E. M., Srigunapalan, S. & Lazo-Langner, A. Venous thromboembolism in hospitalized critical and noncritical COVID-19 patients: a systematic review and meta-analysis. TH Open 5, e286–e294 (2021).
Nopp, S., Moik, F., Jilma, B., Pabinger, I. & Ay, C. Risk of venous thromboembolism in patients with COVID-19: a systematic review and meta-analysis. Res. Pract. Thrombosis Haemost. 4, 1178–1191 (2020).
Kunutsor, S. K. & Laukkanen, J. A. Cardiovascular complications in COVID-19: a systematic review and meta-analysis. J. Infect. 81, e139–e141 (2020).
Klok, F. A. et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 191, 145–147 (2020).
Lodigiani, C. et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 191, 9–14 (2020).
Bilaloglu, S. et al. Thrombosis in hospitalized patients with COVID-19 in a New York City health system. JAMA 324, 799–801 (2020).
Helms, J. et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intens. Care Med. 46, 1089–1098 (2020).
Fraissé, M. et al. Thrombotic and hemorrhagic events in critically ill COVID-19 patients: a French monocenter retrospective study. Crit. Care 24, 275 (2020).
McFadyen, J. D., Stevens, H. & Peter, K. The emerging threat of (micro)thrombosis in COVID-19 and its therapeutic implications. Circ. Res. 127, 571–587 (2020).
O’Donnell, J. S., Peyvandi, F. & Martin-Loeches, I. Pulmonary immuno-thrombosis in COVID-19 ARDS pathogenesis. Intens. Care Med. 47, 899–902 (2021).
Tang, N., Li, D., Wang, X. & Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 18, 844–847 (2020).
Zhang, L. et al. Deep vein thrombosis in hospitalized patients with coronavirus disease 2019 (COVID-19) in Wuhan, China: prevalence, risk factors, and outcome. Circulation 142, 114–128 (2020).
Bonaventura, A. et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 21, 319–329 (2021).
Thachil, J. et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 18, 1023–1026 (2020).
Bikdeli, B. et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up: JACC state-of-the-art review. J. Am. Coll. Cardiol. 75, 2950–2973 (2020).
Centre for Evidence-Based Medicinb. OCEBM levels of evidence. University of Oxford https://www.cebm.ox.ac.uk/resources/levels-of-evidence/ocebm-levels-of-evidence (2021).
Valle, D. M. D. et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 26, 1636–1643 (2020).
Abani, O. et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet 397, 1637–1645 (2021).
Leisman, D. E. et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 8, 1233–1244 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Qin, C. et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 71, 762–768 (2020).
Malik, P. et al. Biomarkers and outcomes of COVID-19 hospitalisations: systematic review and meta-analysis. BMJ Evid. Based Med. 26, 107–108 (2021).
Smilowitz, N. R. et al. C-reactive protein and clinical outcomes in patients with COVID-19. Eur. Heart J. 42, 2270–2279 (2021).
Krychtiuk, K. A. et al. Biomarkers of coagulation and fibrinolysis in acute myocardial infarction: a joint position paper of the Association for Acute CardioVascular Care and the European Society of Cardiology Working Group on Thrombosis. Eur. Heart J. Acute Cardiovasc. Care 10, 343–355 (2020).
Berger, J. S. et al. Prevalence and outcomes of d-dimer elevation in hospitalized patients with COVID-19. Arterioscler. Thromb. Vasc. Biol. 40, 2539–2547 (2020).
Shah, S. et al. Elevated d-dimer levels are associated with increased risk of mortality in coronavirus disease 2019: a systematic review and meta-analysis. Cardiol. Rev. 28, 295–302 (2020).
Levi, M., Thachil, J., Iba, T. & Levy, J. H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 7, e438–e440 (2020).
Kollias, A. et al. Venous thromboembolism in COVID-19: a systematic review and meta-analysis. Vasc. Med. 26, 415–425 (2021).
Kwee, R. M., Adams, H. J. A. & Kwee, T. C. Pulmonary embolism in patients with COVID-19 and value of d-dimer assessment: a meta-analysis. Eur. Radiol. 31, 8168–8186 (2021).
Tassiopoulos, A. K. et al. d-dimer-driven anticoagulation reduces mortality in intubated COVID-19 patients: a cohort study with a propensity-matched analysis. Front. Med. 8, 631335 (2021).
Farrar, J. E. et al. Evaluation of a patient specific, targeted-intensity pharmacologic thromboprophylaxis protocol in hospitalized patients with COVID-19. J. Thromb. Thrombolysis https://doi.org/10.1007/s11239-021-02552-x (2021).
Sholzberg, M. et al. Effectiveness of therapeutic heparin versus prophylactic heparin on death, mechanical ventilation, or intensive care unit admission in moderately ill patients with covid-19 admitted to hospital: RAPID randomised clinical trial. BMJ 375, n2400 (2021).
Spyropoulos, A. C. et al. Efficacy and safety of therapeutic-dose heparin vs standard prophylactic or intermediate-dose heparins for thromboprophylaxis in high-risk hospitalized patients with COVID-19. JAMA Intern. Med. 181, 1612–1620 (2021).
Hékimian, G. et al. Extracorporeal membrane oxygenation induces early alterations in coagulation and fibrinolysis profiles in COVID-19 patients with acute respiratory distress syndrome. Thromb. Haemost. 121, 1031–1042 (2021).
Billoir, P. et al. Investigation of coagulation biomarkers to assess clinical deterioration in SARS-CoV-2 infection. Front. Med. 8, 670694 (2021).
Ranucci, M. et al. Covid-19-associated coagulopathy: biomarkers of thrombin generation and fibrinolysis leading the outcome. J. Clin. Med. 9, 3487 (2020).
Blasi, A. et al. In vitro hypercoagulability and ongoing in vivo activation of coagulation and fibrinolysis in COVID-19 patients on anticoagulation. J. Thromb. Haemost. 18, 2646–2653 (2020).
Busch, M. H. et al. Neutrophils and contact activation of coagulation as potential drivers of COVID-19. Circulation 142, 1787–1790 (2020).
Englert, H. et al. Defective NET clearance contributes to sustained FXII activation in COVID-19-associated pulmonary thrombo-inflammation. Ebiomedicine 67, 103382 (2021).
Lipcsey, M. et al. The outcome of critically ill COVID-19 patients is linked to thromboinflammation dominated by the kallikrein/kinin system. Front. Immunol. 12, 627579 (2021).
Iba, T., Warkentin, T. E., Thachil, J., Levi, M. & Levy, J. H. Proposal of the definition for COVID-19-associated coagulopathy. J. Clin. Med. 10, 191 (2021).
Leentjens, J., Haaps, T. F., van, Wessels, P. F., Schutgens, R. E. & Middeldorp, S. COVID-19-associated coagulopathy and antithrombotic agents — lessons after 1 year. Lancet Haematol. 8, e524–e533 (2021).
Polimeni, A. et al. Differences in coagulopathy indices in patients with severe versus non-severe COVID-19: a meta-analysis of 35 studies and 6427 patients. Sci. Rep. 11, 10464 (2021).
Panigada, M. et al. Hypercoagulability of COVID-19 patients in intensive care unit: a report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 18, 1738–1742 (2020).
Gurbel, P. A., Tantry, U. S. & Storey, R. F. International COVID-19 Thrombosis Biomarkers Colloquium: COVID-19 diagnostic tests. J. Thromb. Thrombolysis 52, 992–998 (2021).
Flaczyk, A. et al. Comparison of published guidelines for management of coagulopathy and thrombosis in critically ill patients with COVID 19: implications for clinical practice and future investigations. Crit. Care 24, 559 (2020).
Trunfio, M. et al. Anti-Xa monitoring improves low-molecular-weight heparin effectiveness in patients with SARS-CoV-2 infection. Thromb. Res. 196, 432–434 (2020).
Hardy, M. et al. Management of the thrombotic risk associated with COVID-19: guidance for the hemostasis laboratory. Thrombosis J. 18, 17 (2020).
Berg, T. W. V. D. et al. Suggestions for global coagulation assays for the assessment of COVID-19 associated hypercoagulability. Thromb. Res. 201, 84–89 (2021).
Morena-Barrio, M. D. L. et al. Prognostic value of thrombin generation parameters in hospitalized COVID-19 patients. Sci. Rep. 11, 7792 (2021).
Campello, E. et al. Thrombin generation in patients with COVID-19 with and without thromboprophylaxis. Clin. Chem. Lab. Med. 59, 1323–1330 (2021).
Hardy, M. et al. Prothrombotic disturbances of hemostasis of patients with severe COVID-19: a prospective longitudinal observational study. Thromb. Res. 197, 20–23 (2020).
Chistolini, A. et al. Effect of low or high doses of low-molecular-weight heparin on thrombin generation and other haemostasis parameters in critically ill patients with COVID-19. Br. J. Haematol. 190, e214–e218 (2020).
Fogarty, H. et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J. Thromb. Haemost. 19, 2546–2553 (2021).
Gurbel, P. A. et al. First report of the point-of-care TEG: a technical validation study of the TEG-6S system. Platelets 27, 642–649 (2016).
Bareille, M. et al. Viscoelastometric testing to assess hemostasis of COVID-19: a systematic review. J. Clin. Med. 10, 1740 (2021).
Tsantes, A. E. et al. COVID-19 infection-related coagulopathy and viscoelastic methods: a paradigm for their clinical utility in critical illness. Diagnostics 10, 817 (2020).
Gurbel, P. A. et al. Thrombogenicity markers for early diagnosis and prognosis in COVID-19: a change from the current paradigm? Blood Coagul. Fibrin 32, 544–549 (2021).
Hulshof, A.-M. et al. Serial EXTEM, FIBTEM, and tPA rotational thromboelastometry observations in the Maastricht Intensive Care COVID cohort — persistence of hypercoagulability and hypofibrinolysis despite anticoagulation. Front. Cardiovasc. Med. 8, 654174 (2021).
Magomedov, A. et al. Viscoelastic testing reveals normalization of the coagulation profile 12 weeks after severe COVID-19. Sci. Rep. 11, 13325 (2021).
Hulshof, A. et al. Hemostasis and fibrinolysis in COVID-19 survivors 6 months after intensive care unit discharge. Res. Pract. Thromb. Haemost. 5, e12579 (2021).
Hartmann, J., Ergang, A., Mason, D. & Dias, J. D. The role of TEG analysis in patients with COVID-19-associated coagulopathy: a systematic review. Diagnostics 11, 172 (2021).
Gurbel, P. A. et al. Bedside thromboelastography to rapidly assess the pharmacodynamic response of anticoagulants and aspirin in COVID-19: evidence of inadequate therapy in a predominantly minority population. J. Thromb. Thrombolysis 51, 902–904 (2021).
Meizoso, J. P., Moore, H. B. & Moore, E. E. Fibrinolysis shutdown in COVID-19: clinical manifestations, molecular mechanisms, and therapeutic implications. J. Am. Coll. Surg. 232, 995–1003 (2021).
Wright, F. L. et al. Fibrinolysis shutdown correlation with thromboembolic events in severe COVID-19 infection. J. Am. Coll. Surg. 231, 193–203.e1 (2020).
Creel-Bulos, C. et al. Fibrinolysis shutdown and thrombosis in a COVID-19 ICU. Shock 55, 316–320 (2020).
Ibañez, C. et al. High D dimers and low global fibrinolysis coexist in COVID19 patients: what is going on in there? J. Thromb. Thrombolysis 51, 308–312 (2021).
Salem, N. et al. Thromboelastography findings in critically ill COVID-19 patients. J. Thromb. Thrombolysis 51, 961–965 (2021).
Kruse, J. M. et al. Thromboembolic complications in critically ill COVID-19 patients are associated with impaired fibrinolysis. Crit. Care 24, 676 (2020).
Pavoni, V. et al. Evaluation of coagulation function by rotation thromboelastometry in critically ill patients with severe COVID-19 pneumonia. J. Thromb. Thrombolysis 50, 281–286 (2020).
Kwaan, H. C. & Lindholm, P. F. The central role of fibrinolytic response in COVID-19–a hematologist’s perspective. Int. J. Mol. Sci. 22, 1283 (2021).
Whyte, C. S., Morrow, G. B., Mitchell, J. L., Chowdary, P. & Mutch, N. J. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J. Thromb. Haemost. 18, 1548–1555 (2020).
Nougier, C. et al. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J. Thromb. Haemost. 18, 2215–2219 (2020).
Zuo, Y. et al. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 11, 1580 (2021).
Andrianto, Al-Farabi, M., Nugraha, R., Marsudi, B. & Azmi, Y. Biomarkers of endothelial dysfunction and outcomes in coronavirus disease 2019 (COVID-19) patients: a systematic review and meta-analysis. Microvasc. Res. 138, 104224 (2021).
Goshua, G. et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 7, e575–e582 (2020).
Canzano, P. et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic. Transl. Sci. 6, 202–218 (2021).
Bazzan, M. et al. Low ADAMTS 13 plasma levels are predictors of mortality in COVID-19 patients. Intern. Emerg. Med. 15, 861–863 (2020).
Lippi, G., Plebani, M. & Henry, B. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin. Chim. Acta 506, 145–148 (2020).
Liao, D. et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID-19: a retrospective cohort study. Lancet Haematol. 7, e671–e678 (2020).
Ouyang, S.-M. et al. Temporal changes in laboratory markers of survivors and non-survivors of adult inpatients with COVID-19. BMC Infect. Dis. 20, 952 (2020).
Welder, D. et al. Immature platelets as a biomarker for disease severity and mortality in COVID-19 patients. Br. J. Haematol. 194, 530–536 (2021).
Cohen, A. et al. Immature platelets in patients with Covid-19: association with disease severity. J. Thromb. Thrombolysis 52, 708–714 (2021).
Incir, S. et al. Immature platelet fraction: is a novel early predictive marker for disease severity in patients with Covid-19 pneumonia? Turkish J. Biochem. 46, 359–366 (2021).
Daniels, S., Wei, H. & Denning, D. W. Platelet size as a predictor for severity and mortality in COVID-19 patients: a systematic review and meta-analysis. Preprint at medRxiv https://doi.org/10.1101/2021.07.15.21260576 (2021).
Barrett, T. J. et al. Platelet and vascular biomarkers associate with thrombosis and death in coronavirus disease. Circ. Res. 127, 945–947 (2020).
Schurink, B. et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe 1, e290–e299 (2020).
Hooper, J. E. et al. A postmortem portrait of the coronavirus disease 2019 (COVID-19) pandemic: a large multi-institutional autopsy survey study. Arch. Pathol. Lab. Med. 145, 529–535 (2021).
Dolhnikoff, M. et al. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 18, 1517–1519 (2020).
Rampotas, A. & Pavord, S. Platelet aggregates, a marker of severe COVID-19 disease. J. Clin. Pathol. 74, 750–751 (2020).
Venter, C. et al. Erythrocyte, platelet, serum ferritin, and P-selectin pathophysiology implicated in severe hypercoagulation and vascular complications in COVID-19. Int. J. Mol. Sci. 21, 8234 (2020).
Litvinov, R. I. et al. Altered platelet and coagulation function in moderate-to-severe COVID-19. Sci. Rep. 11, 16290 (2021).
Parker, W. A. & Storey, R. F. Antithrombotic therapy for patients with chronic coronary syndromes. Heart 107, 925–933 (2021).
Petito, E. et al. Neutrophil more than platelet activation associates with thrombotic complications in COVID-19 patients. J. Infect. Dis. 223, 933–944 (2021).
Manne, B. et al. Platelet gene expression and function in patients with COVID-19. Blood 136, 1317–1329 (2020).
Taus, F. et al. Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler. Thromb. Vasc. Biol. 40, 2975–2989 (2020).
McCafferty, C. et al. Increased platelet activation in SARS-CoV-2 infected non-hospitalised children and adults, and their household contacts. Br. J. Haematol. 195, 90–94 (2021).
Hottz, E. D. et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 136, 1330–1341 (2020).
Campo, G. et al. Over time relationship between platelet reactivity, myocardial injury and mortality in patients with SARS-CoV-2-associated respiratory failure. Platelets 32, 560–567 (2021).
Yatim, N. et al. Platelet activation in critically ill COVID-19 patients. Ann. Intensive Care 11, 113 (2021).
Fenyves, B. G. et al. Plasma P-selectin is an early marker of thromboembolism in COVID-19. Am. J. Hematol. 96, E468–E471 (2021).
Zaid, Y. et al. Platelets can associate with SARS-CoV-2 RNA and are hyperactivated in COVID-19. Circ. Res. 127, 1404–1418 (2020).
Althaus, K. et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood 137, 1061–1071 (2020).
Bongiovanni, D. et al. SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis. 12, 50 (2021).
Tantry, U. S. et al. First experience addressing the prognostic utility of novel urinary biomarkers in patients with COVID-19. Open Forum Infect. Dis. 8, ofab274 (2021).
Bye, A. P. et al. Aberrant glycosylation of anti-SARS-CoV-2 IgG is a pro-thrombotic stimulus for platelets. Blood 138, 1481–1489 (2021).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
Campbell, R. A., Boilard, E. & Rondina, M. T. Is there a role for the ACE2 receptor in SARS-CoV-2 interactions with platelets? J. Thromb. Haemost. 19, 46–50 (2021).
Zhang, S. et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 13, 120 (2020).
Koupenova, M. et al. SARS-CoV-2 initiates programmed cell death in platelets. Circ. Res. 129, 631–646 (2021).
Barrett, T. J. et al. Platelets contribute to disease severity in COVID-19. J. Thromb. Haemost. 19, 3139–3153 (2021).
Storey, R. F., May, J. A. & Heptinstall, S. Potentiation of platelet aggregation by heparin in human whole blood is attenuated by P2Y12 and P2Y1 antagonists but not aspirin. Thromb. Res. 115, 301–307 (2005).
Heinz, C. et al. Greater fibrinolysis resistance but no greater platelet aggregation in critically ill COVID-19 patients. Anesthesiology 134, 457–467 (2021).
Bertolin, A. J. et al. Platelet reactivity and coagulation markers in patients with COVID-19. Adv. Ther. 38, 3911–3923 (2021).
Bliden, K. et al. Heightened platelet function: an unrecognized component of the covid hypercoagulability state [abstract]. Circulation 142 (Suppl. 3), 16676 (2020).
Hranjec, T. et al. Integral use of thromboelastography with platelet mapping to guide appropriate treatment, avoid complications, and improve survival of patients with coronavirus disease 2019-related coagulopathy. Crit. Care Explor. 2, e0287 (2020).
Ghirardello, S. et al. Assessment of platelet thrombus formation under flow conditions in adult patients with COVID-19: an observational study. Thromb. Haemost. 121, 1087–1096 (2021).
Shen, B. et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 182, 59–72.e15 (2020).
Léopold, V. et al. Platelets are hyperactivated but show reduced glycoprotein VI reactivity in COVID-19 patients. Thromb. Haemost. 121, 1258–1262 (2021).
Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 383, 120–128 (2020).
Dupont, A. et al. Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscler. Thromb. Vasc. Biol. 41, 1760–1773 (2021).
Rauch, A. et al. Endotheliopathy is induced by plasma from critically-ill patients and associated with organ failure in severe COVID-19. Circulation 142, 1881–1884 (2020).
Barrett, T. J. et al. Platelets amplify endotheliopathy in COVID-19. Sci. Adv. 7, eabh2434 (2021).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Maccio, U. et al. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. Ebiomedicine 63, 103182 (2021).
McCracken, I. R. et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation 143, 865–868 (2021).
Ruggeri, Z. Role of von Willebrand factor in platelet thrombus formation. Ann. Med. 32, 2–9 (2000).
Huisman, B., Hoore, M., Gompper, G. & Fedosov, D. A. Modeling the cleavage of von Willebrand factor by ADAMTS13 protease in shear flow. Med. Eng. Phys. 48, 14–22 (2017).
Becker, R. C. COVID-19-associated vasculitis and vasculopathy. J. Thromb. Thrombolysis 50, 499–511 (2020).
Lynch, C. J., Lane, D. A. & Luken, B. M. Control of VWF A2 domain stability and ADAMTS13 access to the scissile bond of full-length VWF. Blood 123, 2585–2592 (2014).
Pascreau, T. et al. ADAMTS 13 deficiency is associated with abnormal distribution of von Willebrand factor multimers in patients with COVID-19. Thromb. Res. 204, 138–140 (2021).
Vassiliou, A. G. et al. ICU admission levels of endothelial biomarkers as predictors of mortality in critically ill COVID-19 patients. Cells 10, 186 (2021).
Cristofaro, R. et al. Marked von Willebrand factor and factor VIII elevations in severe acute respiratory syndrome coronavirus-2-positive, but not severe acute respiratory syndrome coronavirus-2-negative, pneumonia: a case–control study. Blood Coagul. Fibrinolysis 32, 285–289 (2021).
Henry, B. M. et al. ADAMTS13 activity to von Willebrand factor antigen ratio predicts acute kidney injury in patients with COVID-19: evidence of SARS-CoV-2 induced secondary thrombotic microangiopathy. Int. J. Lab. Hematol. 43, 129–136 (2021).
Mancini, I. et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 19, 513–521 (2021).
Ladikou, E. E. et al. Von Willebrand factor (vWF): marker of endothelial damage and thrombotic risk in COVID-19? Clin. Med. 20, e178–e182 (2020).
Doevelaar, A. A. et al. von Willebrand factor multimer formation contributes to immunothrombosis in coronavirus disease 2019. Crit. Care Med. 49, e512–e520 (2021).
Rauch, A. et al. Coagulation biomarkers are independent predictors of increased oxygen requirements in COVID-19. J. Thromb. Haemost. 18, 2942–2953 (2020).
Zhu, S. et al. Potent and rapid reversal of the von Willebrand factor inhibitor aptamer BT200. J. Thromb. Haemost. 18, 1695–1704 (2020).
Nimjee, S. M. et al. Preclinical development of a vWF aptamer to limit thrombosis and engender arterial recanalization of occluded vessels. Mol. Ther. 27, 1228–1241 (2019).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Ruan, Q., Yang, K., Wang, W., Jiang, L. & Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intens. Care Med. 46, 846–848 (2020).
Teuwen, L.-A., Geldhof, V., Pasut, A. & Carmeliet, P. COVID-19: the vasculature unleashed. Nat. Rev. Immunol. 20, 389–391 (2020).
Yang, Y. et al. Autoantibodies against human epithelial cells and endothelial cells after severe acute respiratory syndrome (SARS)-associated coronavirus infection. J. Med. Virol. 77, 1–7 (2005).
Inal, J. COVID-19 comorbidities, associated procoagulant extracellular vesicles and venous thromboembolisms: a possible link with ethnicity? Br. J. Haematol. 190, e218–e220 (2020).
Rosell, A. et al. Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality — brief report. Arterioscler. Thromb. Vasc. Biol. 41, 878–882 (2021).
Yuana, Y., Sturk, A. & Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 27, 31–39 (2013).
Guervilly, C. et al. Dissemination of extreme levels of extracellular vesicles: tissue factor activity in patients with severe COVID-19. Blood Adv. 5, 628–634 (2021).
Krishnamachary, B. et al. Extracellular vesicle-mediated endothelial apoptosis and EV-associated proteins correlate with COVID-19 disease severity. J. Extracell. Vesicles 10, e12117 (2021).
Krishnamachary, B., Cook, C., Kumar, A., Spikes, L., Chalise, P. & Dhillon, N. K. Extracellular vesicle-mediated endothelial apoptosis and EV-associated proteins correlate with COVID-19 disease severity. J. Extracell. Vesicles 10, e12117 (2021).
Cappellano, G. et al. Circulating platelet-derived extracellular vesicles are a hallmark of Sars-Cov-2 infection. Cells 10, 85 (2021).
Fernández-Ruiz, I. Microthrombi cause cardiac injury in COVID-19. Nat. Rev. Cardiol. 18, 306–306 (2021).
Fuchs, T. A. et al. Extracellular DNA traps promote thrombosis. Proc. Natl Acad. Sci. USA 107, 15880–15885 (2010).
Brühl, M.-L. V. et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209, 819–835 (2012).
Zuo, Y. et al. Neutrophil extracellular traps in COVID-19. JCI Insight 5, e138999 (2020).
Stark, K. & Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 18, 666–682 (2021).
Nicolai, L. et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation 142, 1176–1189 (2020).
Barnes, B. J. et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J. Exp. Med. 217, e20200652 (2020).
Middleton, E. A. et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 136, 1169–1179 (2020).
Leppkes, M. et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. Ebiomedicine 58, 102925 (2020).
Veras, F. et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 217, e20201129 (2020).
Ouwendijk, W. J. et al. Neutrophils set extracellular traps to injure lungs in COVID-19. J. Infect. Dis. 223, 1503–1505 (2021).
Ng, H. et al. Circulating markers of neutrophil extracellular traps are of prognostic value in patients with COVID-19. Arterioscler. Thromb. Vasc. Biol. 41, 988–994 (2021).
Zhang, Y. et al. Carboxypeptidase B blocks ex vivo activation of the anaphylatoxin–neutrophil extracellular trap axis in neutrophils from COVID-19 patients. Crit. Care 25, 51 (2021).
Guéant, J. et al. Blood myeloperoxidase-DNA, a biomarker of early response to SARS-CoV-2 infection? Allergy 76, 892–896 (2021).
Huckriede, J. et al. Evolution of NETosis markers and DAMPs have prognostic value in critically ill COVID-19 patients. Sci. Rep. 11, 15701 (2021).
Carvelli, J. et al. Association of COVID-19 inflammation with activation of the C5a–C5aR1 axis. Nature 588, 146–150 (2020).
Cugno, M. et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J. Autoimmun. 116, 102560 (2021).
Cugno, M. et al. Complement activation in patients with COVID-19: a novel therapeutic target. J. Allergy Clin. Immun. 146, 215–217 (2020).
Holter, J. C. et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl Acad. Sci. USA 117, 25018–25025 (2020).
Skendros, P. et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Invest. 130, 6151–6157 (2020).
Annane, D. et al. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: a proof-of-concept study. Eclinicalmedicine 28, 100590 (2020).
Ma, L. et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci. Immunol. 6, eabh2259 (2021).
Bruin, S. D. et al. Clinical features and prognostic factors in Covid-19: a prospective cohort study. Ebiomedicine 67, 103378 (2021).
Li, C., Hu, X., Li, L. & Li, J. Differential microRNA expression in the peripheral blood from human patients with COVID-19. J. Clin. Lab. Anal. 34, e23590 (2020).
Gonzalo-Calvo, Dde et al. Circulating microRNA profiles predict the severity of COVID-19 in hospitalized patients. Transl. Res. 236, 147–159 (2021).
Garvin, M. R. et al. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 9, e59177 (2020).
Nagy, B. et al. A dramatic rise in serum ACE2 activity in a critically ill COVID-19 patient. Int. J. Infect. Dis. 103, 412–414 (2020).
Rieder, M. et al. Serum ACE-2, angiotensin II, and aldosterone levels are unchanged in patients with COVID-19. Am. J. Hypertens. 34, hpaa169 (2020).
Chen, R. et al. HMGB1 as a potential biomarker and therapeutic target for severe COVID-19. Heliyon 6, e05672 (2020).
Sivakorn, C. et al. High mobility group Box 1 and interleukin 6 at intensive care unit admission as biomarkers in critically ill COVID-19 patients. Am. J. Trop. Med. Hyg. 105, 73–80 (2021).
Rieder, M. et al. Serum protein profiling reveals a specific upregulation of the immunomodulatory protein progranulin in COVID-19. J. Infect. Dis. 223, 775–784 (2021).
Bauer, W. et al. Outcome prediction by serum calprotectin in patients with COVID-19 in the emergency department. J. Infect. 82, 84–123 (2020).
Guadiana Romualdo, L. G.de et al. Circulating levels of GDF-15 and calprotectin for prediction of in-hospital mortality in COVID-19 patients: a case series. J. Infect. 82, e40–e42 (2020).
Shi, H. et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J. Leukoc. Biol. 109, 67–72 (2021).
Silvin, A. et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell 182, 1401–1418.e18 (2020).
Furie, K. L. et al. Diagnosis and management of cerebral venous sinus thrombosis with vaccine-induced thrombotic thrombocytopenia. Stroke 52, 2478–2482 (2021).
Bikdeli, B. et al. Cerebral venous sinus thrombosis in the US population, after adenovirus-based SARS-CoV-2 vaccination, and after COVID-19. J. Am. Coll. Cardiol. 78, 408–411 (2021).
Sangli, S. et al. Thrombosis with thrombocytopenia after the messenger RNA-1273 vaccine. Ann. Intern. Med. 174, 1480–1482 (2021).
Greinacher, A. et al. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N. Engl. J. Med. 384, 2092–2101 (2021).
American Society of Hematology. Thrombosis with thrombocytopenia syndrome (also termed vaccine-induced thrombotic thrombocytopenia). https://www.hematology.org/covid-19/vaccine-induced-immune-thrombotic-thrombocytopenia (2021).
Pavord, S. et al. Clinical features of vaccine-induced immune thrombocytopenia and thrombosis. N. Engl. J. Med. 385, 1680–1689 (2021).
Schultz, N. H. et al. Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N. Engl. J. Med. 384, 2124–2130 (2021).
Nazy, I. et al. Recommendations for the clinical and laboratory diagnosis of VITT against COVID-19: communication from the ISTH SSC subcommittee on platelet immunology. J. Thromb. Haemost. 19, 1585–1588 (2021).
Vayne, C. et al. PF4 immunoassays in vaccine-induced thrombotic thrombocytopenia. N. Engl. J. Med. 385, 376–378 (2021).
Arthur, L. et al. Cellular and plasma proteomic determinants of COVID-19 and non-COVID-19 pulmonary diseases relative to healthy aging. Nat. Aging 1, 535–549 (2021).
Shu, T. et al. Plasma proteomics identify biomarkers and pathogenesis of COVID-19. Immunity 53, 1108–1122.e5 (2020).
Stukalov, A. et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 594, 246–252 (2021).
Acknowledgements
M.Y.C. acknowledges grants from the Ministry of Health, Singapore (CIRG17may014 and MOH-000280). G.V. acknowledges grant PGC2018-094025-B-I00 from the Spanish Ministry of Science, Innovation and Universities. L.B. acknowledges grants from the Spanish Ministry of Science, Innovation and Universities (PID 2019-107160RB-I00) and from Institute of Health Carlos III (CIBERCV CB16/11/00411 and Red TerCel RD16/0011/0018) cofounded by FEDER; Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat Catalunya (2017 SGR 1480) and Fundacion Investigacion Cardiovascular-Fundacion Jesus Serra.
Author information
Authors and Affiliations
Contributions
The authors contributed substantially to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
P.A.G. has received consulting fees and/or honoraria from Bayer, Hikari Dx, Janssen, Medicure, Otitopic, UpToDate and US WorldMeds, and institutional research grants from Amgen, Bayer, Haemonetics, Idorsia, Instrumentation Laboratories, Janssen, the NIH, Medicure, Otitopic and US WorldMeds. H.t.C. is a shareholder of Coagulation Profile and a consultant for Alveron. R.C.B. is a scientific advisor for Basking Biosciences. The other authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Cardiology thanks José Luis Ferreiro; John Hwa, who co-reviewed with Sean Gu; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gorog, D.A., Storey, R.F., Gurbel, P.A. et al. Current and novel biomarkers of thrombotic risk in COVID-19: a Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nat Rev Cardiol 19, 475–495 (2022). https://doi.org/10.1038/s41569-021-00665-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-021-00665-7
This article is cited by
-
Children with severe neurological symptoms associated with SARS-CoV-2 infection during Omicron pandemic in China
Pediatric Research (2024)
-
Biomarkers of coagulation, endothelial, platelet function, and fibrinolysis in patients with COVID-19: a prospective study
Scientific Reports (2024)
-
Antiphospholipid syndrome, thrombosis, and vaccination in the COVID-19 pandemic
Rheumatology International (2024)
-
Coagulation biomarkers and coronavirus disease 2019 phenotyping: a prospective cohort study
Thrombosis Journal (2023)
-
Changes and significance of vascular endothelial injury markers in patients with diabetes mellitus and pulmonary thromboembolism
BMC Pulmonary Medicine (2023)
