
Abstract
SLL-039 (N-cyclopropylmethyl-7α−4′-(N’-benzoyl) amino-phenyl-6,14-endoethano-tetrahydronorthebaine) and SLL-1206 (N-cyclopropylmethyl-7α−3′-(p-methoxybenzyl) amino-phenyl-6,14-endoethano-tetrahydronorthebaine) are two 4,5-epoxymorphinan-based high selective κ receptor agonists that we recently discovered. In the present study we characterized their pharmacological properties in comparison with arylacetamide-based typical κ agonist U50,488H. We showed that both SLL-039 and SLL-1206 produced potent and long-lasting antinociceptive actions in three different rodent models of pain via activation of κ opioid receptor. In hot-plate assay, the antinociceptive potency of SLL-039 and SLL-1206 increased about 11-and 17.3-fold compared to U50,488H and morphine, respectively, with ED50 values of 0.4 mg/kg. Following repeated administration, SLL-1206, SLL-039, and U50,488H all developed analgesic tolerance tested in hot-plate assay. U50,488H and SLL-039 produced antipruritic effects in a dose-dependent manner, whereas SLL-1206 displayed some antipruritic effects only at very low doses. In addition, SLL-1206 was capable of decreasing morphine-induced physical dependence. More importantly, SLL-039 and SLL-1206 at effective analgesic doses did not cause sedation and conditioned place aversion (CPA), whereas U50,488H did. In comparison with SLL-039, SLL-1206 caused similar antinociceptive responses, but fewer sedation and CPA. In conclusion, our results suggest that SLL-039 and SLL-1206 have potential to be developed as novel analgesic agents, and 4,5-expoxymorphinan scaffold is an attractive structure for the development of selective κ agonists with fewer side effects.
Similar content being viewed by others
Introduction
Classified opioid receptors are mainly categorized into three types: μ-, κ-, and δ-opioid receptors [1]. The most prescribed opioids such as morphine, fentanyl and codeine mainly bind to μ-opioid receptor to promote potent analgesia, but also cause severe addiction risk [2], respiratory depression [3, 4] and constipation [5]. The prevalence of these opioids use and abuse has led to a serious national crisis and is generally known as the opioid epidemic. It has been long recognized that like μ-opioid receptor, activation of κ-opioid receptor (KOR) also produces analgesic effects, but with low abuse potential [6]. In addition to providing potent pain-relieving effects, activation of KOR also produces anti-rewards effects as well. Both nonhuman primates and rats studies demonstrate that κ agonists can effectively attenuate morphine or cocaine-induced tolerance [7], conditioned place preference [8], self-administration [9], and relapse [10]. Therefore, KOR emerged as an attractive target for development of safer pain therapeutics, and much effort was devoted. U50,488H was the first synthetic arylacetamide as high selective KOR agonist [11, 12]. After that, several arylacetamides including enadoline [13, 14] and spiradoline [15] were synthesized and entered into clinical trials for the treatment of pain. However, the clinical developments were failed at phase II trials due to their severe psychotomimetic effects, sedation, and dysphoria [14, 16]. Salvinorin A, a neoclerdane diterpene selective KOR agonist, was structurally different from U50,488H. But like U50,488H, salvinorin A produced antinociceptive [17] and anti-addiction effects [18, 19] and also elicited severe depressive-like effects [20]. Nalfurafine (TRK820), a 4,5-epoxymorphinan derivative, was identified as a potent and centrally acting KOR agonist [21,22,23]. Preclinical pain studies showed that nalfurafine produced potent antinociception similar to other KOR agonists [24, 25]. Interestingly, unlike U50,488H and salvinorin A, nalfurafine did not induce aversion or dysphoria at therapeutic doses [26,27,28,29,30], but still caused the incidence of sedation [24]. Nalfurafine was finally approved in 2009 in Japan for the treatment of uremic pruritis [27,28,29], and was currently the only selective κ agonist in clinical use.
Nalfurafine’s properties that maintain appropriate analgesia but produce fewer side effects encourage us to pursue further investigation for the development of KOR agonists with nalfurafine similar structures as novel analgesic drugs. Based on extensive structure–activity relationship analysis and molecular modeling, our researches have led to a series of KOR ligands with nalfurafine 4,5-epoxymorphinan scaffold [31,32,33,34,35,36] navigated by the Message-Address Concept. We originally synthesized orvinol analogs from thebaine and obtained SLL-004C (7α−4′-Amino-phenyl-6α,14α-endo-etheno-tetrahydrothebaine), which produced weak affinity and efficacy to κ opioid receptor with Ki value of 7.89 μM [36]. Then, with 7α-phenyl-6,14-endoetheno-tetrahydrothebaine as a precursor, the morphinan scaffold was optimized, and we identified SLL-039 (N-cyclopropylmethyl-7α−4′-(N’-benzoyl) amino-phenyl-6,14-endoethano-tetrahydronorthebaine) and SLL-1206 (N-cyclopropylmethyl-7α−3′-(p-methoxybenzyl) amino-phenyl-6,14-endoethano-tetrahydronorthebaine) as high selective potent κ receptor agonists. In vitro studies showed that SLL-039 exhibited high selective binding affinity at the KOR with Ki value of 0.47 nM (κ/μ = 683, κ/δ = 284) and agonistic activity with EC50 of 2.0 nM [34]. In comparison with SLL-039, SLL-1206 showed similar binding affinity (Ki = 7.1 nM) and agonistic potency (EC50 = 4.3 nM) at the KOR (Qian He, Wei Li and Li-ming Shao, Department of Medicinal Chemistry, School of Pharmacy, Fudan University), but relatively lower selectivity (κ/μ = 264, κ/δ = 62). The pharmacological characterization of these compounds are not fully identified yet. Therefore, we aimed to determine SLL-039 and SLL-1206 for in vivo behavioral properties, while also assessed whether such 4,5-expoxymorphinan scaffold-based compounds might serve as promising candidates for pain management with fewer adverse effects as compared with conventional KOR agonist U50,488H.
Materials and methods
Chemicals
SLL-1206 and SLL-039 were prepared by Department of Medicinal Chemistry, School of Pharmacy, Fudan University. Nor-binaltorphimine (nor-BNI) was purchased from Abcam (Ab120078, Cambridge, MA, USA). β-funaltrexamine hydrochloride (β-FNA) was purchased from Tocris Bioscience (0926, Bristol, UK). (±)-U-50488 hydrochloride (U50,488H) was purchased from Tocris Bioscience (0495, Bristol, UK). Naloxone hydrochloride was purchased from Tocris Bioscience (0599, Bristol, UK). Chloroquine diphosphate salt was purchased from Sigma (C6628, Louis, MO, USA). Morphine hydrochloride was purchased from Northeast Pharmaceutical Group Shenyang No.1 Pharmaceutical Co. Ltd. (181102-2, Shenyang, China).
Apparatus
The intelligent hot plate instrument was purchased from Shandong Academy of Medical Sciences (YLS-6B, Jinan, Shandong, China). The rotorod system was purchased from Sumtor (JLBehv-RRTG-5, Wuxi, Jiangsu, China). The CPA and CPP equipment were purchased from Jiliang (JLBehv-CPPg-4, Shanghai, China)
Animals
CD1 strain mice (male, 30–35 g) were obtained from Beijing Weitonglihua Laboratory Animal Technology Co., Ltd. C57BL/6J mice, ICR and Kunming strain mice (18–20 g) were obtained from Beijing Huafukang Biotechnology Co., Ltd. Animals were housed in Laboratory Animal Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences (Shanghai, China). Five mice per cage were housed in a temperature-controlled room (24 ± 2 °C) on a 12 h light/12 h dark cycle. Mice were free for food and water. All animal treatments were strictly in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Bioethics Committee of the Shanghai Institute of Materia Medica (Approval No. 2019-12-LJG-46, 2019-12-LJG-47, 2018-08-LJG-38).
Hot plate test
The experiment was carried out according to our previous study [6] to assess thermal pain with C57BL/6 J and ICR mice. The YLS-6B intelligent hot plate instrument was equipped with automatic recording system. The hot plate temperature was set at 55 °C to record the latency time (the time to licking of the hind paw or to an escape jump). Before drug administration, sensitivity and stability of mice for nociceptive response were measured for three times. The mice with latency time of less than 5 s or more than 30 s were removed from further studies. The cut-off time of 60 s was set to minimize tissue damage. Vehicle, U50,488H (7.0 mg/kg, i.p.), SLL-039 (0.2–1.0 mg/kg, i.p.) or SLL-1206 (0.125–2.0 mg/kg, i.p.) was administered and the latency time was measured. Analgesia rate was calculated as:
The antinociceptive ED50 value was obtained as the dose that produced 50% analgesia.
Formalin test
The experiment was carried out according to our previous study [37] to assess acute chemical pain (phase I) and inflammatory pain (phase II) with C57BL/6J and ICR mice. After acclimation, mice were injected with 20 μL 1.0% formaldehyde solution into the plantar of right hind paw. The mice were observed for 1 h after injection, the first 10 min was phase I, and the last 50 min was phase II. The time each mouse licked the formalin injected paw was recorded during phase I and phase II, respectively. Vehicle, SLL-039 (0.2–2.0 mg/kg, i.p.) or SLL-1206 (0.03125–1.0 mg/kg, i.p.) was administered 2 or 3 h before the plantar injection. Analgesia rate was calculated as:
The antinociceptive ED50 value was obtained as the dose that produced 50% analgesia.
Abdominal constriction test
The experiment was carried out according to our previous study [6] to assess visceral pain with C57BL/6J and ICR mice. The abdominal constriction was defined as a wave of contraction of the abdominal musculature followed by extension of the hind limbs. Abdominal constriction was induced by 0.6% acetic acid (10 mL/kg body weight, i.p.), and the number of writhing was recorded for 20 min. Vehicle, SLL-039 (0.05–0.3 mg/kg, i.p.) or SLL-1206 (0.0625–1.0 mg/kg, i.p.) was administered 2 or 3 h before the test. Analgesia rate was calculated as:
The antinociceptive ED50 value was obtained as the dose that produced 50% analgesia.
Agonist effects of SLL-1206
The experiment was carried out according to our previous study [34] with ICR mice. The κ opioid receptor antagonist (nor-BNI, 10 mg/kg, i.p., −24 h), μ opioid receptor antagonist (β-FNA, 10 mg/kg, i.p., −24 h) or saline was pretreated to determine the opioid receptor profile of SLL-1206 in vivo. Then, mice received SLL-1206 (1.0 mg/kg, i.p.) or saline administration. The antinociception was assessed 3 h after SLL-1206 injection using the abdominal constriction test.
Antinociceptive tolerance assay
The experiment was carried out according to our previous study [7] with C57BL/6J and ICR mice. Briefly, a 3-day consecutive administration paradigm was applied to induce antinociceptive tolerance. U50,488H (2.0–8.0 mg/kg, i.p.), SLL-039 (0.25–1.0 mg/kg, i.p.) or SLL-1206 (0.125–2.0 mg/kg, i.p.) was administered twice on day 1 and 2 with 7 h interval, and once on day 3. The antinociceptive tolerance was evaluated on day 4 using hot plate test. Measurements were performed 15 min or 4 h after administration of U50,488H, SLL-039, or SLL-1206 respectively. The dose which produced near 80% antinociceptive efficacy was chosen for vehicle-treat and test. Thus, the dose of U50,488H, SLL-039 and SLL-1206 was 8.0 mg/kg, 1.0 mg/kg and 2.0 mg/kg, respectively. Tolerant ED50 values were the dose that results in 50% reduction of the antinociceptive efficacy compared with the vehicle pretreated mice.
Scratching test
The experiment was carried out according to our previous study [38] with C57BL/6 J and ICR mice. Mice were habituated to clear acrylic testing boxes for 1 h prior to the start of the experiment. Mice were injected with U50,488H (0.5–3.0 mg/kg, i.p., −15 min), SLL-039 (0.1–1.0 mg/kg, i.p., −2 h) or SLL-1206 (0.001–1.0 mg/kg, i.p., −3 h), then received chloroquine diphosphate (4 mL/kg, 50 μL, s.c. neck) injection to induce acute pruritus. Immediately after that, the number of scratching bouts are recorded for 30 min. One bout of scratching was defined as a lifting of either hind paw to scratch the injection region of body and then replacing it back to the floor. The relative % reduction of scratching was calculated as:
The anti-scratching ED50 value was obtained as the dose that produced 50% anti-scratching effect.
Rotorod test
The experiment was carried out according to our previous study [39] to assess sedation effects with ICR mice. The rotorod system was equipped with automatic recording system (the test time and acceleration time was 240 s; the minimum rotation speed was 4 r/min; the maximum rotation speed was 40 r/min). The mice were trained and screened to maintain on the rotorod system for more than 200 s. Prior to drug administration, the mice were tested three times and the average of the results was used as the basal level. Vehicle, SLL-039 (0.5–4.0 mg/kg, i.p.) or SLL-1206 (0.5–32.0 mg/kg, i.p.) was administered 2 or 3 h before rotorod test. U50,488H (5.0 mg/kg, i.p.) was administered 15 min before the test. The time duration for each mouse to fall was recorded.
Conditioned place aversion (CPA) test
CPA is a rodent model of dysphoria. The experiment was carried out according to the study [40] with CD1 mice. The CPA equipment was equipped with computerized automonitoring system (Jiliang, Shanghai, China), with dividing into two parts by partition (18.0 cm × 14.5 cm × 13.5 cm each). The right side is white with red light and the left side is plaid with blue light. The process consisted three stages: pre-test, conditioning, post-test. Pre-test: Mice were allowed to move freely between the two sides of the apparatus for 15 min, and the time spent in each side was recorded. The preference of mice to both sides of the apparatus was analyzed. Mice that showed a strong preference for one side (>80% of the session, 720 s) were eliminated. Conditioning: This stage lasted for 6 days, during which the mice were injected with saline (10 mL/kg, i.p.) in the morning and placed in the non-preferred side for 30 min. After 5 h, the mice which were injected with saline (10 mL/kg, i.p.), U50,488H (5.0 mg/kg, i.p.), SLL-039 (0.5–2.0 mg/kg, i.p.) or SLL-1206 (0.4–16.0 mg/kg, i.p.) were placed in the preferred side for 30 min. Vehicle, SLL-039 or SLL-1206 was administered 2 or 3 h in advance. U50,488H was administered 15 min in advance. Post-test: The effect of the compounds on dysphoria in mice was detected on the post-test day. At this stage, the mice were allowed to move freely between the two sides of the apparatus for 15 min, and the time on each side was recorded. The preference of mice to both sides of the apparatus was analyzed, and compared with pretest state.
Physical dependence assay
The experiment was carried out according to our previous study [6] with Kunming mice. Morphine was administered twice a day (10 mL/kg, s.c.) with 6 h interval. Morphine dose was increased progressively from 20 to 100 mg/kg over a period of 7 days, i.e., 1st (20 and 40 mg/kg, s.c.), 2nd (60 and 80 mg, s.c.), and remained a dose of 100 mg/kg from 3rd to 7th. Four hours after the last morphine injection, mice were given naloxone (3.0 mg/kg, s.c.) to induce physical dependence withdrawal reaction. The number of jump, diarrhea, teeth chattering, paw tremor of mice in each group was recorded within 30 min. Body weight was measured before and 60 min after naloxone administration. To investigate the effects of SLL-1206 on morphine-induced physical dependence, vehicle or SLL-1206 (0.4–3.8 mg/kg, i.p.) was administered 3 h before daily morphine administration.
Morphine-induced conditioned place preference (CPP)
The experiment was carried out according to our previous study [8] with Kunming mice. The CPP equipment was equipped with computerized automonitoring system (Jiliang, Shanghai, China). The equipment was divided into two parts by partition (30 cm × 30 cm × 40 cm each) with sides painted black and white, and both sides have the same brightness of red light. The process consisted three stages: pre-test, conditioning, post-test. Pre-test: On day 1-3, mice were allowed to move freely between the two sides of the apparatus for 15 min, and the time spent in each side was recorded. The preference of mice to both sides of the apparatus was analyzed. Mice that showed a strong preference for one side (>80% of the session, 720 s) were eliminated. Conditioning: This stage lasted for 6 days, during which the mice were injected with saline (10 mL/kg, i.p.) in the morning and placed in the preferred side for 50 min. After 5 h, the mice were injected with morphine (4.0 mg/kg, i.p.) or saline (10 mL/kg, i.p.) and placed in the non-preferred side for 50 min. To detect the effect of SLL-1206 on morphine-induced CPP, mice were injected with SLL-1206 (2.0–7.0 mg/kg, i.p.) or saline (10 mL/kg, i.p.) 3 h before daily morphine administration during the conditioning session. Post-test: The effect of SLL-1206 on morphine-induced CPP was detected on the post-test day. The mice were allowed to move freely between the two sides of the apparatus for 15 min, the time spent in each side was recorded. The preference of mice to both sides of the apparatus was analyzed, and compared with pre-test state.
Statistical analysis
Data were analyzed by GraphPad Prism 6.0, and presented as mean ± SEM. Significance was determined by using one-way ANOVA followed by Bonferoni post-hoc test. P < 0.05 was considered significant.
Results
The antinociceptive effects of SLL-039 and SLL-1206
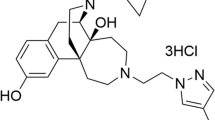
Fig. 1 depicts the structure of SLL-039, SLL-1206, and U50,488H. SLL-039 and SLL-1206 are 4,5-expoxymorphinan based κ agonists, and U50,488H is arylacetamide based κ agonist. The antinociceptive properties were characterized by three different pain models, including the hot-plate test, the abdominal constriction test, and the formalin test. A traditional graded dose–responses procedures were conducted to obtain the antinociceptive ED50 values. The results were presented in Fig. 2 and Table 1. The hot-plate test was applied to evaluate thermal pain, and we found that both SLL-039 and SLL-1206 dose-dependently increased withdrawal latency and produced antinociceptive effects (Fig. 2a and b) with ED50 values of 0.4 mg/kg, which increased 11.0-fold compared to U50,488H and 17.3-fold compared to morphine (Table 1). Abdominal constriction test was applied to evaluate visceral pain. In the abdominal constriction test, SLL-039 and SLL-1206 dose-dependently reduced acetic acid-induced writhing behavior and produced antinociceptive effects (Fig. 2c and d) with ED50 values of 0.1 mg/kg and 0.2 mg/kg, respectively. The antinociceptive potency of SLL-039 increased 9.0-fold compared to U50,488H and 8.0-fold compared to morphine, and SLL-1206 increased 4.5-fold compared to U50,488H and 4.0-fold compared to morphine (Table 1). Formalin test was applied to evaluate chemical pain (phase I) and inflammatory pain (phase II). As shown in Fig. 2e and f, in initial acute phase I of the formalin test, both SLL-039 and SLL-1206 did not have significant analgesic effects. In phase II, SLL-039 and SLL-1206 dose-dependently decreased licking time and produced antinociceptive effects in inflammatory pain with ED50 values of 0.6 mg/kg and 0.1 mg/kg, respectively. The antinociceptive potency of SLL-039 increased 5.2-fold compared to morphine, SLL-1206 increased 4.0-fold compared to U50,488H and 31.0-fold compared to morphine (Table 1).

The molecular formula of SLL-039 is C37H40N2O4 (Molecular weight: 576), the molecular formula of SLL-1206 is C38H44N2O4 (Molecular weight: 592). SLL-039 and SLL-1206 are 4,5-expoxymorphinan based κ agonist, and U50, 488H is arylacetamide based κ agonist

The anti-thermal pain effects of SLL-039 and SLL-1206 were evaluated in the hot plate test. After injected with various doses of SLL-039 (a) or SLL-1206 (b), the latency time was measured 4 h later, and the antinociception (%) was calculated. SLL-039 (a) and SLL-1206 (b) dose-dependently induced anti-thermal pain effects in hot plate test. The anti-visceral pain effects of SLL-039 and SLL-1206 were evaluated in the acid writhing test. After injected with various doses of SLL-039 (−2 h) or SLL-1206 (−3 h), 0.6% acetic acid was injected (10 mL/kg body weight, i.p.). The number of writhes was counted for 20 min and the antinociception (%) was calculated. SLL-039 (c) and SLL-1206 (d) dose-dependently induced anti-visceral pain effects in acid writhing test. The anti-chemical pain and anti-inflammatory pain activities of SLL-039 and SLL-1206 were evaluated in the formalin test. After injected with various doses of SLL-039 (e) or SLL-1206 (f), 2 or 3 h later mice were injected with 20 μL 1.0% formaldehyde solution into the plantar of right hind paw and the licking time was measured for 60 min, and the antinociception (%) was calculated. SLL-039 and SLL-1206 dose-dependently induced anti-inflammatory pain in phase II. Data were analyzed by one-way ANOVA followed by Bonferoni post-hoc test. Data are presented as mean ± SEM from at least six mice. *P < 0.05, **P < 0.01 vs. saline
The antinociceptive duration of SLL-039 and SLL-1206
The duration of action was determined by the hot-plate test. Doses for near 80% analgesic rate were chosen: SLL-039 1.0 mg/kg, SLL-1206 2.0 mg/kg, and U50,488H 7.0 mg/kg. As shown in Fig. 3a and b, both SLL-039 and SLL-1206 produced a longer duration (near 24 h), with the action peaked at 2–4 h. In contrast, the duration of U50,488H at 7.0 mg/kg was about 2 h.

After injected with U50,488H (7.0 mg/kg), SLL-039 (1.0 mg/kg) (a) or SLL-1206 (2.0 mg/kg) (b), the antinociceptive effect was measured at various time. Both SLL-039 (a) and SLL-1206 (b) produced a longer duration. Data were presented as mean ± SEM from at least eight mice
Agonist effects of SLL-1206
Our recent in vitro binding assay showed that SLL-1206 exerted high κ receptor binding affinity with Ki value of 7.1 nM and κ agonist activity with EC50 value of 4.3 nM. To identify whether the antinociception of SLL-1206 was mediated by KOR, animals were pretreated with selective opioid receptor antagonists including KOR antagonist nor-BNI (10 mg/kg) and μ antagonist β-FNA (10 mg/kg). We found that nor-BNI, but not β-FNA significantly inhibited SLL-1206-produced antinociception (P < 0.01) (Fig. 4). We previously demonstrated that nor-BNI significantly suppressed SLL-039-induced antinociceptive effects [34]. Taken together, both SLL-039 and SLL-1206 induced potent and long lasting antinociceptive action, and both effects were mediated by activation of KOR.

Mice were pretreated with κ antagonist nor-BNI (10 mg/kg) or μ antagonist β-FNA (10 mg/kg) for 24 h, then injected with SLL-1206. After 3 h, the acetic acid was injected and the number of writhes was counted for 20 min, and the antinociception (%) was calculated. Nor-BNI and β-FNA did not influence basal pain threshold. Nor-BNI, but not β-FNA significantly inhibited SLL-1206-produced antinociception. Data were analyzed by one-way ANOVA followed by Bonferroni post-hoc test. Data are presented as mean ± SEM from at least eight mice. **P < 0.01 vs control. ##P < 0.01 vs SLL-1206
Antinociceptive tolerance potential of SLL-039, SLL-1206, and U50,488H
We examined the potential ability of U50,488H, SLL-039 and SLL-1206 to develop analgesia tolerance. As shown in Fig. 5, after treated with various doses of U50,488H (2.0–8.0 mg/kg), SLL-039 (0.25–1.0 mg/kg) and SLL-1206 (0.125–2.0 mg/kg), all three compounds-induced analgesic effects were decreased, indicative of developing analgesia tolerance. The ratios of the ED50 of tolerance to that of antinociception were calculated, and the larger ratio means less potential to develop tolerance. The ratios were as follows: SLL-039, 1.8; SLL-1206, 1.8; U50,488H, 1.6 (Table 2), which suggest that SLL-039 and SLL-1206 have similar or relative less potential to develop tolerance in comparison with U50,488H.

Mice were administered various indicated doses of the compounds or vehicle for 3 days. On day 4, the antinociceptive actions were measured as an injection of U50,488H (8.0 mg/kg, i.p.) (a), SLL-039 (1.0 mg/kg, i.p.) (b) or SLL-1206 (2.0 mg/kg, i.p.) (c). All three compounds-induced analgesia tolerance dose dependently. Data were analyzed by one-way ANOVA followed by Bonferoni post-hoc test. Each value represents the mean ± SEM from at least seven mice. **P < 0.01 vs. repeated vehicle + U50,488H. #P < 0.05, ##P < 0.01 vs repeated vehicle + SLL-039. &P < 0.05 vs repeated vehicle + SLL-1206
The anti-itch effects of SLL-039, SLL-1206, and U50,488H
Itch is defined as an unpleasant sensation that elicits scratching behaviors. Activation of KOR exhibits anti-itch effects in both animals and humans [41, 42]. Thus we examined the anti-itch effects of U50,488H, SLL-039, and SLL-1206. As shown in Fig. 6, U50,488H and SLL-039 dose-dependently inhibited chloroquine diphosphate-induced scratching behavior with ED50 values of 1.2 mg/kg and 0.3 mg/kg, respectively. Whereas, SLL-1206 showed low anti-scratching behaviors with maximal effects of 52% at 0.016 mg/kg (Fig. 6c).

Animals were injected with U50,488H (−15 min, a), SLL-039 (−2 h, b) or SLL-1206 (−3 h, c), then administered chloroquine diphosphate (4.0 mg/mL, 50 μL, s.c., neck). The bouts of scratching were counted for 30 min, and the anti-scratch effects were calculated. U50,488H and SLL-039 dose-dependently inhibited chloroquine diphosphate-induced scratching behavior, SLL-1206 obtained a bell shaped dose-response curve. Data were analyzed by one-way ANOVA followed by Bonferroni post-hoc test. Data are presented as mean ± SEM from at least six mice. **P < 0.01 vs saline
The sedative effects of SLL-039 and SLL-1206
Kappa agonists always produce undesirable side effects such as sedation, which limits the therapeutic use for pain management [14, 16]. Rotorod test was performed to study the sedative deficits of SLL-039 and SLL-1206 in comparison with U50,488H, which has been demonstrated to produce sedation at the doses producing analgesic effects. As shown in Fig. 7, U50,488H at a dose of 5.0 mg/kg (near antinociceptive ED50) produced significant sedation (P < 0.01), whereas, both SLL-039 and SLL-1206 at 0.5 mg/kg (near antinociceptive ED50) did not. Furthermore, SLL-039 at a dose of 1.0 mg/kg (2.5× antinociceptive ED50) did not produce significant sedation (Fig. 7a). SLL-1206, at 16.0 mg/kg (40× antinociceptive ED50) still did not produce sedation (Fig. 7b). The sedative side effects were ranked as follows: U50,488H > SLL-039 > SLL-1206.

Mice were injected with saline or various doses of SLL-039 (−2 h), SLL-1206 (−3 h), or U50,488H (−15 min), and put on the rotorod apparatus. The time of each mice stayed on the rotorod was recorded. Data were analyzed by one-way ANOVA followed by Bonferroni’s post-poc test. Data are presented as mean ± SEM from at least nine mice. *P < 0.05, **P < 0.01 vs saline
The dysphoric effects of SLL-039 and SLL-1206
Dysphoric effect is another undesirable side effect produced by activation of KOR [43], and it can be measured by conditioned place aversion (CPA), a Pavlovian conditioning paradigm. The experimental timeline was presented in Fig. 8a. As shown in Fig. 8b and c, U50,488H at 5.0 mg/kg (near antinociceptive ED50) produced significant CPA behavior in mice (P < 0.01). However, both SLL-039 and SLL-1206 at 0.4–0.5 mg/kg (near antinociceptive ED50) did not. SLL-039 at a dose of 2.0 mg/kg (5× antinociceptive ED50) produced CPA behavior (P < 0.01) (Fig. 8b). SLL-1206 at 16.0 mg/kg (40× antinociceptive ED50) did not cause significant CPA behaviors (Fig. 8c). Although SLL-1206 exhibited a trend of CPA, it did not reach statistical significance. The side effects of dysphoria were ranked as follows: U50,488H > SLL-039 > SLL-1206.

a The schedule of the test. On day 1, mice were subjected for pre-test. On day 2–7, mice were injected with saline or various doses of b SLL-039 (−2 h), c SLL-1206 (−3 h) or U50,488H (−15 min) before each 30-min conditioning. On day 8, the time spending in each side was recorded and the CPA score was calculated. Data were analyzed by one-way ANOVA followed by Bonferroni post hoc test. Value data were presented as mean ± SEM from at least ten mice. **P < 0.01 vs saline
Effects of SLL-1206 on morphine-induced physical dependence and conditioned place preference (CPP) behavior
Kappa agonists have known anti-addiction effects [44, 45], we further determine the inhibitory effects of SLL-1206 on morphine-induced physical dependence and CPP behaviors. As shown in Fig. 9, the withdrawal reactions were precipitated by injection of naloxone after continuous treatment with morphine. SLL-1206 significantly decreased withdrawal symptoms including body weight loss (Fig. 9a), jump (Fig. 9b), teeth chattering (Fig. 9c), diarrhea (Fig. 9d) and paw tremor (Fig. 9e). However, SLL-1206 was not able to significantly decrease morphine-induced CPP behavior (Fig. 10).

Mice were treated with SLL-1206 (0.4–3.8 mg/kg, i.p.) or saline (10 mL/kg, i.p.) 3 h prior progressively increasing doses of morphine treatment for 7 days. Morphine withdrawal reactions were precipitated by naloxone (3.0 mg/kg, s.c.). Body weight loss was measured initially and 60 min after the naloxone injection (a). The number of jumping (b), teeth chattering (c), diarrhea (d), paw tremor (e) was measured for 30 min after naloxone injection. SLL-1206 decreased the physical dependence induced by chronic morphine application. Data were analyzed by one-way ANOVA followed by Bonferroni post hoc test. Data were presented as the mean ± SEM from at least eight animals. *P < 0.05, **P < 0.01 vs control. #P < 0.05, ##P < 0.01 vs morphine

a The schedule of the test. b On day 1–3, mice were subjected for pre-test. On day 4–9, mice were treated with SLL-1206 (2.0–7.0 mg/kg, i.p.) or saline (10 mL/kg, i.p.) 3 h prior morphine (4.0 mg/kg, i.p.) before each 50-min conditioning. On day 11, The residence time of the mice in both sides of the apparatus was recorded, and the CPP score was calculated. Morphine induces significant CPP. SLL-1206 had no antagonistic effect on morphine induced CPP (b). Data were analyzed by one-way ANOVA followed by Bonferroni post hoc test. Data were presented as the mean ± SEM from at least eight animals. **P < 0.01 vs control
Discussion
The present study was undertaken to study the pharmacology of SLL-039 and SLL-1206, and compared with that of typical KOR agonist U50,488H. The major finding of this study was that both SLL-039 and SLL-1206 produced more potent antinociceptive effects but much lower sedation and aversion than U50,488H. SLL-039 and SLL-1206 show a separation of analgesia from classic psychoactive effects of KOR activation, suggesting that SLL-039 and SLL-1206 have potential to be developed as novel therapeutic agents for the treatment of pain.
SLL-039 and SLL-1206 selectively bind to KOR with Ki value of 0.5 nM, 7.1 nM respectively, and stimulated [35S]GTPγS binding with EC50 value of 2.0 nM, 4.3 nM, respectively [34] (Qian He, Wei Li and Li-ming Shao, Department of Medicinal Chemistry, School of Pharmacy, Fudan University). In the present study, we found that SLL-039 and SLL-1206 produced similar degree of antinociceptive activities with long duration, which were much more potent than U50,488H and morphine. Both SLL-039- and SLL-1206-produced antinociception was via κ specific manner [34]. The data suggested a correlation between the potency of SLL-039 and SLL-1206 in vitro [35S]GTPγS binding assay and in vivo antinociceptive studies, and our observations were consistent with prior findings [6, 7, 37] that activation of KOR produced antinociceptive effects. Previous studies also revealed that KOR activation induced potent anti-scratching effects, and there is a strong correlation between the potency of antinociceptive effects and anti-scratching behaviors. However, this correlation was maintained in this study only for U50,488H and SLL-039, but not for SLL-1206. SLL-1206 produced potent antinociception, but showed low activity in countering chloroquine diphosphate-induced scratching at unusual low doses. Thus, SLL-039 and SLL-1206 showed similar antinociceptive activity, but had different anti-scratching effects. Similar to U50,488H, SLL-039 and SLL-1206 had potential to develop antinociceptive tolerance [46, 47]. SLL-1206 also showed activity to decrease morphine withdrawal-induced physical dependence, which was in accord with previous study of Tao et al. [6], supporting that κ agonist might have therapeutic benefits for attenuating abuse-related behaviors, which might be achieved by suppressing dopamine release [44, 48]. Thus, like classical arylacetamides U50,488H, morphinan-based κ agonists SLL-039 and SLL-1206 exhibited similar pharmacological properties including antinociception and tolerance, but low in anti-scratching profile.
A growing body of evidence suggests that KOR agonists are effective analgesics with low abuse potential, but can cause severe sedation and dysphoric side effects [14, 16, 43]. By applying rotorod test and CPA test, we found that at the effective analgesia doses, both SLL-039 and SLL-1206 did not cause sedation and aversion. U50,488H, at antinociceptive ED50 value, caused significant sedation and dysphoric effect, and the adverse effect of U50,488H was consistent with published reports [40]. These data indicate that SLL-039 and SLL-1206 produced much lower side effects than U50,488H, and there is dose separation between analgesia and sedation/aversion for SLL-039 and SLL-1206. We further found that SLL-039 induced sedation and aversion when increasing dose up to 2.0 mg/kg (5× analgesic ED50), while SLL-1206 did not even at 16.0 mg/kg (40× analgesic ED50). Thus, in contrast to SLL-039, SLL-1206 elicited less adverse effects. When exposed to increasing doses or long-term treatment of SLL-1206, sedation or aversion may still exist. Nalfurafine is another 4,5-expoxymorphinan derivate. It was found that nalfurafine, at the dose of 20 μg/kg (3.4× analgesic ED50), did not cause aversion, but produced sedation [25]. Compound 42B, a 3-dehydroxy analog of nalfurafine, at the effective analgesic dose ranges, did not produce aversion, but caused sedation [25]. The data indicate that the pharmacological profiles of morphinan-based compounds SLL-039, SLL-1206, nalfurafine and 42B are different from that of arylacetamide U50,488H, with less side effects, suggesting that 4,5-expoxymorphinan scaffold is a promising scaffold for novel analgesic drugs discovery. On the other hand, these different morphinan-based compounds also show individual differences, specifically individual’s inability to dissociate analgesia from sedation. It is believed that G protein-mediated signaling pathway mediated KOR-induced analgesic effects, anti-scratch effects and CPA, but β-arrestin 2-mediated signaling is involved in sedation/motor incoordination [49]. Nalfurafine and 42B are G protein-biased ligands acting as full KOR agonists, whether SLL-039 and SLL-1206 show similar G protein biases needs further investigation. However, it should be noted that recent studies demonstrated that bias factors in vitro could not fully predict in vivo pharmacological properties [25]. In addition, there was evidence in the literature indicating that mTOR and p38 MAPK signaling pathways were implicated in KOR-mediated aversion. We found that U50,488H, SLL-039 and SLL-1206 showed differential ability to cause CPA. It would be interesting to study the signaling pathways differentially impacted to identify the underlying mechanism by which the compounds lack of aversion.
References
Stevens CW, Brasel CM, Mohan S. Cloning and bioinformatics of amphibian mu, delta, kappa, and nociceptin opioid receptors expressed in brain tissue: evidence for opioid receptor divergence in mammals. Neurosci Lett. 2007;419:189–94.
Rodriguez RE. Morphine and microRNA activity: is there a relation with addiction? Front Genet. 2012;3:223.
Abushanab D, Alsoukhni O, AbouNahia F, Al-Badriyeh D. Clinical and economic analysis of morphine versus fentanyl in managing ventilated neonates with respiratory distress syndrome in the intensive care setting. Clin Ther. 2019;41:714–27 e8.
Kiyatkin EA. Respiratory depression and brain hypoxia induced by opioid drugs: Morphine, oxycodone, heroin, and fentanyl. Neuropharmacology. 2019;151:219–26.
Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid-induced constipation in patients with noncancer pain. N Engl J Med. 2014;370:2387–96.
Tao YM, Li QL, Zhang CF, Xu XJ, Chen J, Ju YW, et al. LPK-26, a novel kappa-opioid receptor agonist with potent antinociceptive effects and low dependence potential. Eur J Pharmacol. 2008;584:306–11.
Wang YJ, Tao YM, Li FY, Wang YH, Xu XJ, Chen J, et al. Pharmacological characterization of ATPM [(-)−3-aminothiazolo[5,4-b]-N-cyclopropylmethylmorphinan hydrochloride], a novel mixed kappa-agonist and mu-agonist/-antagonist that attenuates morphine antinociceptive tolerance and heroin self-administration behavior. J Pharmacol Exp Ther. 2009;329:306–13.
Sun JF, Wang YH, Chai JR, Li FY, Hang A, Lu G, et al. Pharmacological characterization and therapeutic potential for the treatment of opioid abuse with ATPM-ET, an N-ethyl substituted aminothiazolomorphinan with kappa agonist and mu agonist/antagonist activity. Eur J Pharmacol. 2014;740:455–63.
Freeman KB, Naylor JE, Prisinzano TE, Woolverton WL. Assessment of the kappa opioid agonist, salvinorin A, as a punisher of drug self-administration in monkeys. Psychopharmacology. 2014;231:2751–8.
Heinsbroek JA, Furbish AB, Peters J. A single, extinction-based treatment with a kappa opioid receptor agonist elicits a long-term reduction in cocaine relapse. Neuropsychopharmacology. 2018;43:1492–7.
Lahti RA, VonVoigtlander PF, Barsuhn C. Properties of a selective kappa agonist. U-50,488H Life Sci. 1982;31:2257–60.
Vonvoigtlander PF, Lahti RA, Ludens JH. U-50,488: a selective and structurally novel non-Mu (kappa) opioid agonist. J Pharmacol Exp Ther. 1983;224:7–12.
Pande AC, Pyke RE, Greiner M, Wideman GL, Benjamin R, Pierce MW. Analgesic efficacy of enadoline versus placebo or morphine in postsurgical pain. Clin Neuropharmacol. 1996;19:451–6.
Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology. 2001;157:151–62.
Vonvoigtlander PF, Lewis RA. Analgesic and mechanistic evaluation of spiradoline, a potent kappa opioid. J Pharmacol Exp Ther. 1988;246:259–62.
Rimoy GH, Wright DM, Bhaskar NK, Rubin PC. The cardiovascular and central nervous system effects in the human of U-62066E. A selective opioid receptor agonist. Eur J Clin Pharmacol. 1994;46:203–7.
Paton KF, Biggerstaff A, Kaska S, Crowley RS, La Flamme AC, Prisinzano TE, et al. Evaluation of biased and balanced salvinorin A analogs in preclinical models of pain. Front Neurosci. 2020;14:765.
Morani AS, Kivell B, Prisinzano TE, Schenk S. Effect of kappa-opioid receptor agonists U69593, U50488H, spiradoline and salvinorin A on cocaine-induced drug-seeking in rats. Pharmacol Biochem Behav. 2009;94:244–9.
Morani AS, Schenk S, Prisinzano TE, Kivell BM. A single injection of a novel κ opioid receptor agonist salvinorin A attenuates the expression of cocaine-induced behavioral sensitization in rats. Behav Pharmacol. 2012;23:162–70.
Taylor GT, Manzella F. Kappa opioids, salvinorin A and major depressive disorder. Curr Neuropharmacol. 2016;14:165–76.
Endoh T, Tajima A, Izumimoto N, Suzuki T, Saitoh A, Suzuki T, et al. TRK-820, a selective kappa-opioid agonist, produces potent antinociception in cynomolgus monkeys. Jpn J Pharmacol. 2001;85:282–90.
Nakao K, Hasebe K, Yoshikawa S, Ikeda K, Hirakata M, Miyamoto Y, et al. Pharmacological effects of nalfurafine hydrochloride, a kappa-opioid receptor agonist. Nihon Shinkei Seishin Yakurigaku Zasshi. 2010;30(5-6):185-91.
Suzuki T, Izumimoto N, Takezawa Y, Fujimura M, Togashi Y, Nagase H, et al. Effect of repeated administration of TRK-820, a kappa-opioid receptor agonist, on tolerance to its antinociceptive and sedative actions. Brain Res. 2004;995:167–75.
Endoh T, Matsuura H, Tajima A, Izumimoto N, Tajima C, Suzuki T, et al. Potent antinociceptive effects of TRK-820, a novel kappa-opioid receptor agonist. Life Sci. 1999;65:1685–94.
Cao D, Huang P, Chiu YT, Chen C, Wang H, Li M, et al. Comparison of pharmacological properties between the kappa opioid receptor agonist nalfurafine and 42B, its 3-dehydroxy analogue: disconnect between in vitro agonist bias and in vivo pharmacological effects. ACS Chem Neurosci. 2020;11:3036–50.
Kozono H, Yoshitani H, Nakano R. Post-marketing surveillance study of the safety and efficacy of nalfurafine hydrochloride (Remitch(®) capsules 2.5 μg) in 3,762 hemodialysis patients with intractable pruritus. Int J Nephrol Renovasc Dis. 2018;11:9–24.
Kumagai H, Ebata T, Takamori K, Miyasato K, Muramatsu T, Nakamoto H, et al. Efficacy and safety of a novel ĸ-agonist for managing intractable pruritus in dialysis patients. Am J Nephrol. 2012;36:175–83.
Kumagai H, Ebata T, Takamori K, Muramatsu T, Nakamoto H, Suzuki H. Effect of a novel kappa-receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a phase III, randomized, double-blind, placebo-controlled study. Nephrol Dial Transplant. 2010;25:1251–7.
Wikström B, Gellert R, Ladefoged SD, Danda Y, Akai M, Ide K, et al. Kappa-opioid system in uremic pruritus: multicenter, randomized, double-blind, placebo-controlled clinical studies. J Am Soc Nephrol. 2005;16:3742–7.
Liu JJ, Chiu YT, DiMattio KM, Chen C, Huang P, Gentile TA, et al. Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in κ opioid aversion. Neuropsychopharmacology. 2019;44:939–49.
Li W, Long JD, Qian YY, Long Y, Xu XJ, Wang YJ, et al. The Pharmacological heterogeneity of nepenthone analogs in conferring highly selective and potent κ-opioid agonistic activities. ACS Chem Neurosci. 2017;8:766–76.
Liu X, Jiang S, Kong L, Ye R, Xiao L, Xu X, et al. Exploration of the SAR connection between morphinan- and arylacetamide-based κ opioid receptor (κOR) agonists using the strategy of bridging. ACS Chem Neurosci. 2021;12:1018–30.
Sun HJ, Wang YH, Yuan CM, Kong LH, Xu XJ, Wang YJ, et al. 7β-Methyl substituent is a structural locus associated with activity cliff for nepenthone analogues. Bioorg Med Chem. 2018;26:4254–63.
Xiao L, Wang Y, Zhang M, Wu W, Kong L, Ma Y, et al. Discovery of a highly selective and potent κ opioid receptor agonist from N-cyclopropylmethyl-7α-phenyl-6,14-endoethanotetrahydronorthebaines with reduced central nervous system (CNS) side effects navigated by the message-address concept. J Med Chem. 2019;62:11054–70.
Li W, Tang Y, Zheng YL, Qiu ZB. Molecular modeling and 3D-QSAR studies of indolomorphinan derivatives as kappa opioid antagonists. Bioorg Med Chem. 2006;14:601–10.
Qi H, Wei LI, Qiu Y, Cui YY, Ma J, Gao XL, et al. Synthesis and evaluation of κ-opioid receptor agonistic activity and antinociceptive effect of novel morphine analogues, 7α-phenyl-6α,14α-endo-etheno-tetrahydrothebaine with substituted o-, m- and p-amino group. Med Chem Res. 2011;20:1364–70.
Zhang LS, Wang J, Chen JC, Tao YM, Wang YH, Xu XJ, et al. Novel kappa-opioid receptor agonist MB-1C-OH produces potent analgesia with less depression and sedation. Acta Pharmacol Sin. 2015;36:565–71.
Lu YC, Wang YJ, Lu B, Chen M, Zheng P, Liu JG. ACC to dorsal medial striatum inputs modulate histaminergic itch sensation. J Neurosci. 2018;38:3823–39.
Wang YH, Chai JR, Xu XJ, Ye RF, Zan GY, Liu GYK, et al. Pharmacological characterization of dezocine, a potent analgesic acting as a kappa partial agonist and mu partial agonist. Sci Rep. 2018;8:14087.
Liu JJ, Sharma K, Zangrandi L, Chen C, Humphrey SJ, Chiu YT, et al. In vivo brain GPCR signaling elucidated by phosphoproteomics. Science. 2018;360:eaao4927.
Inan S, Cowan A. Antipruritic effects of kappa opioid receptor agonists: evidence from rodents to humans. Handb Exp Pharmacol. 2020. https://doi.org/10.1007/164_2020_420.
Togashi Y, Umeuchi H, Okano K, Ando N, Yoshizawa Y, Honda T, et al. Antipruritic activity of the kappa-opioid receptor agonist, TRK-820. Eur J Pharmacol. 2002;435:259–64.
Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, et al. Kappa opioid receptor-induced aversion requires p38 MAPK activation in VTA dopamine neurons. J Neurosci. 2015;35:12917–31.
Kivell BM, Ewald AW, Prisinzano TE. Salvinorin A analogs and other κ-opioid receptor compounds as treatments for cocaine abuse. Adv Pharmacol. 2014;69:481–511.
Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology. 2010;210:121–35.
Zhou JJ, Bian JS, Pei JM, Wu S, Li HY, Wong TM. Role of protein kinase C-epsilon in the development of kappa-opioid receptor tolerance to U50,488H in rat ventricular myocytes. Br J Pharmacol. 2002;135:1675–84.
He L, Fong J, von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108:271–82.
Devine DP, Leone P, Wise RA. Mesolimbic dopamine neurotransmission is increased by administration of mu-opioid receptor antagonists. Eur J Pharmacol. 1993;243:55–64.
White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, et al. The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther. 2015;352:98–109.
Acknowledgements
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDA12040319 to JGL) and National Natural Science Foundation of China (Grant 81773710 to YJW and 82030112 to JGL), and the Youth Innovation Promotion Association of the Chinese Academy of Sciences (Grant 2017334 to YJW).
Author information
Authors and Affiliations
Contributions
JGL, YJW, WL, and LMS designed the experiments. YYW and YM performed the experiments with the assistance of SYY, JRC, and JC. LHK and XL synthesized compounds. YYW, YM, and YJW performed the statistical data analysis. YYW and YJW written this manuscript. JGL, WL, and LMS revised this manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Wei, Yy., Ma, Y., Yao, Sy. et al. Novel selective κ agonists SLL-039 and SLL-1206 produce potent antinociception with fewer sedation and aversion. Acta Pharmacol Sin 43, 1372–1382 (2022). https://doi.org/10.1038/s41401-021-00761-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00761-x
