
Abstract
The gastrointestinal tract plays a role in the development and treatment of metabolic diseases. During a meal, the gut provides crucial information to the brain regarding incoming nutrients to allow proper maintenance of energy and glucose homeostasis. This gut–brain communication is regulated by various peptides or hormones that are secreted from the gut in response to nutrients; these signaling molecules can enter the circulation and act directly on the brain, or they can act indirectly via paracrine action on local vagal and spinal afferent neurons that innervate the gut. In addition, the enteric nervous system can act as a relay from the gut to the brain. The current review will outline the different gut–brain signaling mechanisms that contribute to metabolic homeostasis, highlighting the recent advances in understanding these complex hormonal and neural pathways. Furthermore, the impact of the gut microbiota on various components of the gut–brain axis that regulates energy and glucose homeostasis will be discussed. A better understanding of the gut–brain axis and its complex relationship with the gut microbiome is crucial for the development of successful pharmacological therapies to combat obesity and diabetes.
Similar content being viewed by others

Introduction
The prevalence of metabolic diseases, such as obesity and type 2 diabetes (T2D), is steadily rising worldwide, due in large part to a westernized lifestyle that favors increased energy intake of highly palatable foods coupled with decreased energy expenditure. In the United States, almost three-quarters of adults are considered overweight and 43% are obese, nearly tripling the prevalence of obesity observed 40 years ago1. Similarly, the worldwide prevalence of diabetes has more than doubled in the past 40 years, reaching 9.3% in 20192. The health care cost associated with diabetes is equal to that of obesity, as are the rates of comorbidities and mortality3. Despite a dire need for the prevention and amelioration of these metabolic diseases, few successful therapeutic options exist. Although highly invasive, the most successful current treatment for obesity and diabetes is bariatric surgery4, highlighting the major impact of the gastrointestinal (GI) tract on energy and glucose homeostasis. In line with this, some of the more promising pharmacological therapies also involve modulation of the GI tract. For example, glucagon-like peptide-1 (GLP-1) agonists and dipeptidyl peptidase-4 inhibitors used for T2D treatment originate from gut-derived signaling mechanisms5. Even metformin, the most prescribed drug for T2D, acts at least partly via the gut to lower blood glucose via gut–brain–liver signaling mechanisms that lower hepatic glucose production or via alterations in gut microbiota and bile acid signaling6,7,8. Intertwined with the targeting of the GI tract for metabolic disease therapy, studies suggest that manipulations of the gut microbiome can directly impact host metabolic homeostasis9 and influence the success of other treatment options, such as gastric bypass surgery and metformin6,8,10. Taken together, these findings highlight the function of the GI tract in the maintenance of metabolic diseases and emphasize its role as a therapeutic target.
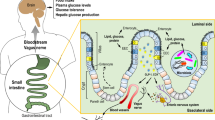
The gut–brain axis is a bidirectional hormonal and neural signaling pathway connecting the gut and the brain. As depicted in Fig. 1, several mechanisms link the gut to the brain in the regulation of metabolic homeostasis. Classically, signals originating from the gut in response to an influx of nutrients during a meal are relayed to the brain, informing the central nervous system (CNS) about meal size11 and composition12,13. The brain, mainly the hypothalamus, integrates these gut-derived signals, along with others, to coordinate the regulation of food intake, energy expenditure, and glucose homeostasis14,15. Enteroendocrine cells (EECs), specialized neuroendocrine cells of the intestinal epithelium, mediate this postprandial gut feedback, as they sense the luminal milieu and release gut peptides that influence metabolic homeostasis, such as GLP-1 and cholecystokinin (CCK)16. Nutrient-induced gut peptides can act in a paracrine fashion by activating vagal neurons that innervate tissue near the intestinal epithelium and signal to the brain or in an endocrine fashion targeting the brain and other peripheral organs involved in maintaining metabolic homeostasis17. Despite the established significance of postprandial gut–brain signaling in energy and glucose homeostasis, recent work highlights the physiological and neural complexity of this negative feedback and questions this gut peptide-mediated vagal signaling. In the current review, we will provide an overview of how the gut–brain axis contributes to metabolic homeostasis, highlighting some of the novel ideas as to how this gut–brain communication is mediated. Furthermore, this traditional, nutrient-induced, neural, and hormonal gut–brain signaling pathway fails to incorporate the role of the gut microbiome in mediating gut-derived feedback mechanisms. Given the high numbers of microbial cells and genes18, it is now evident that the gut microbiome can impact glucose and energy homeostasis via numerous mechanisms, including both impacting host gut–brain signaling and initiating direct communication to the brain via microbe-derived metabolites19. This review will highlight the current ideas as to how the gut microbiome incorporates into host gut–brain signaling and the impact of gut bacteria on host energy and glucose homeostasis via CNS signaling pathways.

Specialized intestinal epithelial cells, enteroendocrine cells (EECs), neuropod cells, and enterochromaffin cells (ECs), secrete gut peptides, including GLP-1, CCK, GIP, and PYY, on the basolateral side. These gut peptides are released in close proximity to vagal afferent neurons innervating the intestinal mucosa and activate these neurons. Vagal afferent neurons send signals to the nucleus tractus solitarius (NTS), which can send signals to higher-order brain regions, such as the arcuate nucleus (ARC). Vagal afferent neurons are also activated via the enteric nervous system, which can be activated by the release of gut-derived neurotransmitters, such as 5-HT, from ECs and intraganglionic laminar endings (IGLEs) sensing intestinal stretch. In addition, gut peptides are able to enter the circulation and carry signals directly to the NTS or activate splanchnic nerve endings to signal to the brain via spinal afferent neurons.
Neuronal gut–brain connection
Gut–brain signaling is a bidirectional avenue of communication. Signals from the brain communicate to the gut mainly through the autonomic nervous system and the hypothalamic–pituitary axis to regulate many physiological processes (reviewed elsewhere20). Neural signaling from the gut to the brain, however, is mediated by vagal and spinal afferent neurons21,22. Spinal afferents innervate the intestine, but their role in energy and glucose homeostasis appears minimal23,24, although a more recent study revealed a role for spinal afferents in mediating intestinal sensing of glucose22. In addition, the enteric nervous system (ENS), which is a recognized “second brain” that can regulate many GI functions independent of CNS action, can also act as an intermediate in relaying gut-derived information to the CNS25. The following section will focus on the role of the vagus nerve and ENS in regulating metabolic homeostasis.
Vagus nerve
The vagus nerve is aptly nicknamed the “wanderer nerve” due to long extensions originating from the brainstem and innervating many of the visceral organs. Within the gut, vagal afferent endings are distributed in different layers, with many innervating the lamina propria of the GI tract near EECs26,27. Vagal afferent neurons (VANs) innervate the entire GI tract, although recent studies have discovered a distinct heterogeneity of VANs, with different subgroups more densely populating different anatomical locations of the GI tract27,28,29,30,31,32. The gut stimulates VANs in two main ways: via chemoreception or mechanostimulation32. Vagal mucosal endings are better situated to sense chemical stimuli, while sensing of GI stretching and distention is accomplished by intraganglionic laminar endings (IGLEs) that act as mechanoreceptors26,29,30. Important for the regulation of energy and glucose homeostasis, a high number of VANs contain receptors for gut peptides released by EECs, and the physiological function of these VANs has been recently elucidated via new advances in the ability to characterize and target specific neural populations. Indeed, recent studies have found that over 10 distinct cell types likely reside in the nodose ganglion according to the expression profile of various proteins and receptors, but the results are sometimes contradictory28,31,32. As such, one paper detailed over 15 different subtypes of vagal neurons that innervate the viscera31, while another study demonstrated 12 unique clusters specifically originating from the GI tract28. This newly recognized heterogeneity in vagal neuron protein expression likely impacts the functions of specific neurons, as detailed below, and requires further investigation. However, it is recognized that vagal terminals activated within the GI tract send signals up to the brain; the signals are processed within cell bodies contained in the nodose ganglia and ultimately reach the nucleus tractus solitarius (NTS) and the area postrema within the hindbrain. From the hindbrain, the information can be relayed to various local brain regions, including the vagal dorsal motor nucleus and parabrachial nucleus, and eventually to higher-order brain centers, such as the hypothalamus. In particular, signals from the gut can activate specialized neurons of the arcuate nucleus (ARC) that regulate energy and glucose homeostasis, including agouti-related peptide/neuropeptide Y orexigenic neurons and pro-opiomelanocortin/cocaine and amphetamine-regulated transcript anorexigenic neurons22,33,34,35. Importantly, the left and right branches of the vagal nerve are not symmetric and are unique in where they innervate within the GI tract and where they terminate in the hindbrain28. For example, the common hepatic branch of the vagus diverges from the left subdiaphragmatic vagus trunk to innervate the intestines and liver, while both the left and right trunks contain celiac branches that innervate the distal intestine36. Accordingly, recent research demonstrates distinct differences in the function of neurons within the left vs. right nodose ganglia in the regulation of energy homeostasis, as the right nodose ganglia of mice mediate the rewarding response to nutrients, while the left nodose ganglia control satiation independent of reward37.
The vagus nerve relays information regarding a meal to the brain to regulate both energy and glucose homeostasis13. Indeed, VANs respond to nutrients sensed in the gut, as vagal afferent activity is increased during infusion of either carbohydrates, amino acids, or fatty acids into the small intestine12,38,39,40. Likewise, c-fos, a marker of neuronal activity, is increased in the NTS, where vagal afferents terminate, and in the nodose ganglia after different nutrient loads41. It is possible that this is due to the nutrient-induced release of gut peptides (see the section below); however, using genetic approaches to target specific neurons, a recent study challenges that idea32. Williams et al. demonstrated that contrary to expectations, subsets of VANs containing GLP-1 receptor (GLP-1R) detected stomach and intestinal stretch and, interestingly, had no impact on nutrient sensing, which was instead regulated by G protein-coupled receptor 65 (GPR65)-positive neurons32. Conversely, Bai et al. demonstrated no effect of optogenetic or chemogenetic activation of GPR65-positive cells on food intake and only a mild effect via GLP-1R-positive cells28. Instead, only activation of Oxtr-containing neurons, exclusively intestinal IGLEs that detect stretch, potently suppressed food intake, while various subtypes of mucosal ending neurons, which are classically thought to sense gut peptides, had no effect28. However, gut peptide signaling could influence IGLE neuronal signaling, as these neurons express gut peptide receptors. Despite this, the salient role of overall vagal signaling in energy homeostasis has been established, as surgical or chemical ablation of vagal afferent signaling decreases acute sensitivity to intestinal nutrients, leading to increased food intake and reduced NTS neural activation23,24,42,43,44. In addition, chronic stimulation of VANs reduces food intake and weight gain in rats45,46,47 and is associated with weight loss in humans48, highlighting the long-term physiological relevance of vagal signaling.
In addition to food intake, vagal afferents also mediate the ability of intestinal nutrients to regulate hepatic glucose production. Infusion of lipids and carbohydrates into the small intestine initiates a gut–brain–liver axis that is dependent on vagal signaling to lower hepatic glucose production and improve glucose tolerance49. Interestingly, this effect was mediated by the release of CCK and GLP-1 and subsequent activation of their receptors, possibly on VANs (see “Hormonal gut-brain signaling” section). However, as mentioned previously for vagal afferent regulation of food intake, recent work has failed to demonstrate an effect of food intake via direct gut peptide signaling on VAN-localized receptors. Given the increased ability to identify and target specific neural populations, future studies will need to more comprehensively address the effects of IGLEs and mucosal ending VANs on both energy and glucose homeostasis. Indeed, it is puzzling why only IGLE neurons detecting intestinal distention induce satiation, while mucosal ending VANs that contain gut peptide receptors and are ideally situated in close proximity to EECs would not be involved in metabolic homeostasis.
Although it is possible that vagal afferents are activated directly within the GI tract, several studies have highlighted the role of vagal nutrient sensing in the portal vein50,51. For example, glucose is absorbed by the intestine and enters the portal vein, where glucose sensing increases hepatic vagal signaling to the NTS52 and subsequently to the hypothalamus53 to decrease food intake54,55 and regulate glycemia56,57. Furthermore, the GI tract generates glucose via gluconeogenesis, and it has been shown that both proteins and fiber increase gluconeogenic enzymes in intestinal epithelial cells. This increase in intestinal gluconeogenesis leads to portal glucose sensing to regulate energy homeostasis and improve insulin sensitivity58. Although not fully understood, nutrients likely first activate CNS mechanisms to induce intestinal gluconeogenesis via release of vasoactive intestinal peptide by the ENS. These peptides then increase intestinal glucose-6-phosphotase, ultimately leading to activation of a portal glucose sensor that signals to the brain59. However, this portal glucose sensor may still require gut input, as these effects appear to be mediated by vagal GLP-1R activation60,61,62.
Interestingly, impaired vagal signaling may play a role in obesity, as decreased vagal activation to both mechanical stimuli and satiety-inducing signals is characteristic of a high-fat diet (HFD)63,64,65,66. One study suggests that the decrease in vagal signaling seen with HFD feeding is due to upregulation of K+ channels in the nodose ganglia, increasing hyperpolarization of VANs67, while other studies have implicated changes in the gut microbiota as a contributor to vagal afferent desensitization during HFD feeding (see the “Gut microbiota–brain connection” section). For a more comprehensive review on impaired gut signaling during obesity, see ref. 68.
Enteric nervous system
The ENS is an independently functioning neural system innervating the entire GI tract and containing two bundles of ganglia: one underneath the epithelial layer of the gut (submucosal plexus) and the other between the smooth muscle layers of the gut (myenteric plexus)25. One of the main roles of the ENS is mechanoreception and stimulation of the smooth muscle surrounding the gut69,70, providing autonomous control of GI transit and motility. In regard to mediating energy or glucose homeostasis, neurons of the myenteric plexus of the ENS are activated via duodenal glucose infusion71 and by hormones released in response to nutrient sensing in the gut, such as GLP-1 from EECs and serotonin (5-HT) from enterochromaffin cells (ECs)71,72,73,74. Indeed, enteric neurons express GLP-1Rs, and primary cultures of myenteric neurons have increased neuronal activation following GLP-1 treatment73. Given that vagal afferents innervate the ENS, it is plausible that the ENS can relay local intestinal signaling mechanisms to the CNS. For example, enteric neurons express glutamate transporters and are in close proximity to VANs expressing glutamate-sensitive receptors75. Although VANs are activated by glutamate76,77,78, the link between glutamate release by the ENS and the stimulation of VANs has yet to be proven76,77,78. Nonetheless, GLP-1-induced gastric emptying and insulin secretion depend upon ENS GLP-1R sensitivity and subsequent relay to the hindbrain via enteric nitric oxide (NO) production72. Interestingly, GLP-1 resistance during HFD feeding reduces enteric activation and NO production, which is due to changes in the gut microbiota (see the “Gut microbiota–brain connection” section)72. Along these lines, increased duodenal contractility by the ENS is characteristic of T2D and likely contributes to metabolic dysfunction. Treatment with galanin or apelin, which are both peptides released in the intestine, results in decreased duodenal contraction via the ENS, which subsequently signals to the hypothalamus, leading to increased hypothalamic NO release and whole-body glucose utilization79,80. Taken together, evidence suggests that the ENS may mediate gut–brain signaling that regulates metabolic homeostasis.
Hormonal gut–brain signaling
The GI tract is a highly specialized organ system that is sophisticated beyond its functions of digestion and absorption. Indeed, while the gut is responsible for providing the host with required nutrients from the diet, it is also home to hormonal, neural, and microbial systems that can have significant impacts on the host19. Despite the small percentage (~1%) of intestinal epithelial cells classified as EECs, they constitute the largest endocrine organ in the body and are present throughout the entire GI tract, increasing in density distally81. Most EECs are considered open-type cells, resembling a bottleneck shape, with an apical membrane in direct contact with the intestinal lumen, as well as a basolateral membrane in close proximity to blood vessels and neurons that innervate the intestinal mucosa and come in close contact with the epithelial layer82. EECs contain peptides (also sometimes termed hormones) in secretory granules, and upon chemical, mechanical, or neural stimulation, an influx of intracellular calcium stimulates the secretion of gut peptides through the basolateral membrane and into the extracellular space17. EECs are classically characterized based on the gut hormones they secrete, such as L-cells (GLP-1), I-cells (CCK), and K-cells (GIP)83. However, recent advances in EEC-specific transgenic mouse models and single cell RNA sequencing (RNA-seq) studies suggest that EECs exhibit a complex phenotype with multiple hormones expressed in specific EECs and that the similarity of subtypes depends more on EEC location81. For example, one study examined cells expressing preproglucagon, a precursor of GLP-1, in the upper small intestine of mice and found three separate clusters of cells with overlapping but distinct hormonal profiles84. Interestingly, the clusters differed not only in hormonal expression but also in the expression of receptors and ion channels, highlighting the potential specificity of targeting these unique EEC clusters84. More recently, intestinal organoid models and real-time EEC reporter mice have demonstrated the complexity and heterogeneity of EEC differentiation and gene expression85,86. Similar to previous reports, many EECs were multihormonal81,87, and many subtypes within EECs may not be distinct lineages but rather are separate stages in EEC maturation86. For example, as cells with high levels of Gcg expression decreased, a concomitant rise in cells expressing Cck was observed at the same timepoint, indicating a hormonal shift in the same EECs85,86. This unique maturation of EECs is unlike the more traditional maturation of enterocytes, which move from the crypts up the villi88. Importantly for the translational impact of potential EEC-targeted therapies, comparisons between murine and human EECs via RNA-seq have established that receptors expressed in murine models are largely equivalent to those seen in humans89. However, despite this similarity, whether human EECs follow a similar shift in uniqueness during various maturation stages remains to be assessed. Nonetheless, these studies taken together challenge the classical idea of EEC cell types and indicate that EECs are a rare and complex class of cells that secrete peptides based largely on their location and maturation within the gut81,84,89,90,91.
Given their open-type nature, EECs are uniquely suited to act as initial messengers regarding incoming nutrients. This allows the host to prepare for an influx of nutrients to maintain energy and glucose homeostasis, and interestingly, obesity and HFD feeding are associated with impaired postprandial gut–brain signaling, highlighting the pathophysiological impact of these signaling pathways (for extensive review, see, ref. 92). EECs highly express chemosensory machinery compared to non-EEC intestinal epithelial cells, including nutrient-specific G protein-coupled receptors (GPCRs) and nutrient transporters, enabling them to sense and respond to the intestinal milieu17. The specific mechanisms regulating nutrient-induced gut peptide release from EECs are still under investigation but appear more complicated than originally hypothesized17. Although EECs might sense nutrients via activation of specific GPCRs on the apical side, where they are in direct contact with the luminal environment, elegantly performed ex vivo and in vitro studies suggest that many nutrients, including fatty acids, may be detected on the basolateral surface17,93,94.
There are over 20 identified gut peptides within the GI tract, many of which play a key role in energy and glucose homeostasis, with the following sections focusing on the incretin hormones GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), CCK, and several others.
In addition to the more traditionally characterized EECs that secrete gut peptides, recent studies have discovered the presence of specialized EECs with long cytoplasmic processes on the basal side that directly signal primary afferent neurons innervating the gut95. These “neuropods” synapse and thus establish direct connections with vagal afferents, enteric glia, and efferent fibers, releasing neurotransmitters such as glutamate. Many EECs have neuropods and are heterogeneous depending on the GI tract region. First discovered in distal intestinal peptide YY (PYY)-containing EECs95, neuropod cells in the intestine can sense sugars and transduce a signal to the brainstem within milliseconds via a single synapse96. This rapid signal, mediated by neuropod cell release of glutamate onto VANs, provides nearly instantaneous spatial and temporal information on a meal, which can then be followed by more traditional gut peptide–vagal afferent signaling pathways outlined below95,96.
Incretin hormones: GLP-1 and GIP
The incretin hormones GLP-1 and GIP are secreted by EECs in response to ingested nutrients and stimulate insulin secretion97,98. Although GLP-1 expression is highest in the large intestine, a large fraction of GLP-1-secreting EECs populate the small intestine16. Direct nutrient stimulation may induce the initial release of GLP-1 from small intestinal EECs, while large intestinal EECs likely release GLP-1 via a neural hormonal reflex or from microbe-derived metabolites, although this remains to be explored93,99 (see the “Gut microbiota–brain connection” section). As such, GLP-1 is present in circulation at low levels in fasting humans and begins to increase in the first few minutes after nutrient ingestion, peaking ~60–90 min postprandially at ~15 pmol/L of active GLP-197,100. The release of GLP-1 from nutrient stimulation is ultimately dependent upon calcium influx, but macronutrients likely differ in their signaling mechanisms (Fig. 2)101,102. For example, glucose-stimulated GLP-1 release requires sodium/glucose cotransporter-1 induction of inward currents, initiating depolarization of EECs and calcium influx to release GLP-1103,104,105. Lipids, on the other hand, likely activate long-chain fatty acid (LCFA) receptors (GPR40 and GPR120) on the basolateral side, indicating that absorption and chylomicron formation are prerequisites106,107,108,109. The LCFA receptors GPR120 and GPR40 are colocalized in GLP-1-containing cells. However, GPR40 but not GPR120 knockout mice exhibit impaired GLP-1 secretion from dietary fat, and only GPR40 agonists induce GLP-1 release in vitro and in vivo106,110,111,112. In addition to LCFAs, 2-monoacylglycerol, the other component of triglycerides, can also induce GLP-1 release via G protein-coupled receptor 119113,114,115. Amino acid stimulation of GLP-1 is not fully understood but likely involves calcium-sensing receptor (CaSR) and peptide transporter 1 (PepT1)116,117.

The different macronutrients act through alternate pathways to elicit gut peptide release. Fatty acids can signal through multiple receptors on both the apical and basolateral membranes. Signaling at the basolateral membrane requires the uptake and packaging of fats into chylomicrons in enterocytes followed by the release and breakdown of these chylomicrons at the basolateral surface. Fatty acids bind their receptors on enteroendocrine cells, which activate a downstream signaling cascade leading to the fusion of gut peptide–containing vesicles and the release of their contents at the basolateral membrane. Glucose sensing occurs at the apical membrane of an EEC and requires uptake into the cell, along with Na+, via SGLT-1. Na+ entry into the EEC causes depolarization and subsequent activation of Ca2+ channels, resulting in vesicle fusion and gut peptide release. Amino acid signaling in the enteroendocrine cell involves the uptake of peptides and Na+ via PepT1 at the apical membrane. This Na+ may depolarize the cells, but research is still needed to determine its exact mechanism of action. Once a peptide enters the cell, it is broken down into amino acids, which are transported out of the cell through the basolateral membrane, where they can activate CaSR, leading to Ca2+ release and vesicle fusion. CaSR may also be present at the apical membrane, but research is still needed to elucidate the exact mechanism of protein-induced gut peptide release.
Oral glucose administration results in up to three times more insulin being released than direct β-cell glucose sensing, highlighting the glucoregulatory impact of glucose-induced GLP-1 and GIP release from EECs118,119. Although β-cells express GLP-1R, and it is likely that GLP-1 induces insulin secretion directly in the pancreas120, GLP-1 has an extremely short half-life of ~1–2 min; thus, only ~12% of gut-derived GLP-1 enters the systemic circulation intact121,122,123. This highlights the possibility that GLP-1 could activate VANs that innervate either the gut or the hepatoportal region to signal to the brain and back down to the pancreas via vagovagal reflexes to induce insulin secretion124. Indeed, the common hepatic branch, as well as celiac and gastric branches, have all been implicated in contributing to the glucoregulatory effects of gut GLP-162,125,126, although selective knockdown of GLP-1R in nodose ganglia does not impair oral glucose tolerance or glucose-stimulated insulin secretion, similar to genetic knockout of GLP-1R in vagal neurons127,128. As such, selective restoration of islet and pancreatic duct GLP-1R was sufficient to improve impaired oral glucose tolerance in global GLP-1R knockout mice, although there was no difference in glucose-stimulated insulin release129. Thus, the extent to which the gut–brain axis contributes to the incretin effect of GLP-1 remains to be determined. Despite this, GLP-1 can impact glucose homeostasis via a gut–brain neuronal axis, independent of changes in insulin secretion.
As described earlier, infusion of both lipids and glucose into the small intestine leads to an improvement in glucose tolerance via a reduction in hepatic glucose production. Intestinal glucose infusion increases portal levels of GLP-1, and coinfusion of exendin-9, a GLP-1R antagonist, abolishes the ability of both intestinal fatty acids and glucose infusion to lower hepatic glucose production130,131. In addition, intestinal lipid infusion lowers hepatic glucose production via a vagal afferent and subsequent hepatic vagal efferent pathway49. Thus, endogenous GLP-1 likely acts on GLP-1R expressed in vagal afferents128. Indeed, nodose GLP-1R lentivirus-mediated knockdown increases postprandial glycemia128, and Glp1rΔPhox2b−/− mice, which exhibit selective knockout of GLP-1R in the nodose ganglia, midbrain, and hindbrain, exhibit increased fasting levels of glucose, both highlighting a role for GLP-1 vagal afferent signaling in mediating glucose homeostasis127. However, it is possible that GLP-1 acts on other local cell types that can subsequently activate VANs, such as the enteric neurons described earlier71,72,73,74. Given the short half-life of endogenous GLP-1, local vagal signaling likely mediates the effect of intestinal GLP-1 release on food intake as well123.
In addition to a glucoregulatory effect, exogenous GLP-1 and GLP-1R agonists reduce food intake132,133,134. However, caution must be taken when examining exogenous gut peptide studies, as many times the dose is supraphysiological, and while there may be therapeutic relevance, it may not be an accurate representation of endogenous gut peptide action. Thus, throughout the manuscript (as with GLP-1 glucoregulatory function above), we aim to highlight studies that directly examine endogenous gut peptide action via receptor knockdown or antagonism. However, when possible, we identified the dosage of exogenous gut peptide administration and whether it is a plausible representation of endogenous levels. For example, capsaicin treatment, which destroys some vagal afferents, abolishes the suppressive effect of low levels of GLP-1 administration (6 and 120 pmol/kg)135. Furthermore, vagotomy abolishes the effect of GLP-1 on food intake, while more selective subdiaphragmatic vagal deafferentation attenuates the inhibitory effect of higher doses of intraperitoneal GLP-1 (100 nmol/kg)136. Long-term administration of exendin-9 abolishes the feeding-suppressive effect of small intestinal lipid infusion, indicating a role of nutrient-induced endogenous GLP-1 in food intake130,137. As such, selective vagal afferent GLP-1R knockdown in rats results in increased meal size and gastric emptying. However, there is no overall impairment in long-term energy balance, similar to Glp1rΔPhox2b−/− mice, which have similar body weight, adiposity, and long-term food intake to controls on either chow or HFD127. Thus, although it is likely that endogenous GLP-1 signals via a vagal gut–brain axis to lower meal size, the impact of this signaling pathway on overall energy homeostasis may be limited.
GIP acts on the pancreas, stimulating the release of insulin and glucagon to regulate blood glucose138,139. However, the effect of GIP on energy homeostasis is contradictory. In addition to GIP’s role in promoting lipid storage in adipocytes140, the GIP receptor (GIPR) is found in neurons of the ARC, dorsomedial nucleus (DMH), and paraventricular nucleus of the hypothalamus141. Thus, GIP could act in an endocrine fashion to impact food intake. Some studies suggest that central GIP signaling induces body weight gain and adiposity, possibly via induction of neural leptin resistance142. In humans, GIP secretion is positively correlated with obesity100,143,144. Furthermore, increases in GIP are seen with HFD consumption in humans and rodents; in humans, this occurs even before changes in body weight145,146. In line with this, some research suggests that antagonism of GIP receptors is beneficial for decreased weight gain147,148 and that CNS GIPR knockout mice are protected from diet-induced obesity149. On the other hand, more recent work suggests that GIP analogs lower body weight via GIPR agonism150,151. Activation of hypothalamic GIPR cells reduces food intake, while the suppressive effects of central and peripheral administration of acyl-GIP on food intake are abolished in CNS GIPR knockout mice149. Thus, while direct gut–brain signaling of GIP contributes to energy homeostasis, it is still unclear whether endogenous GIP is beneficial, and this unresolved discrepancy remains puzzling and requires further investigation152.
CCK
CCK is released from EECs of the upper small intestine in response to fats and proteins. GPR40 mediates fatty acid sensing for CCK release, whereas protein sensing is mediated by CaSR in the proximal small intestine153,154,155. Traditionally, EECs are thought to release CCK through GPR40 activation, which induces cellular depolarization through intracellular calcium store release by inositol triphosphate–dependent mechanisms17. However, similar to the release of GLP-1, there may be a role for cluster of differentiation 36-mediated lipid absorption, chylomicron formation, and basolateral EEC activation in CCK release (Fig. 2)156. In healthy humans, CCK released from EECs enters the circulation and peaks in concentration from 6 to 15 pmol/L at ~90–120 min postprandial depending on fat content and meal form157,158.
CCK is a peptide differentially cleaved from pro-CCK by prohormone convertases into various forms depending on cleavage length, which have been isolated in the GI tracts of various species159, each thought to exert slightly different effects on energy homeostasis160. For example, while CCK-8, the most abundantly utilized form, reduced meal size when given exogenously (at various nmol/kg doses), CCK-58, the major endocrine form in rats and humans, reduced meal size and increased the interval time between meals161. Furthermore, the effects of the various forms of CCK, namely, CCK-8, 33, and 58, differ depending on their site of action in the gastrointestinal tract162,163,164. However, in studies, the most frequently administered exogenous form is CCK-8165, despite it not being the major endocrine form. Unfortunately, it is extremely difficult to measure the different forms, and most studies cannot differentiate the concentration of specific CCK forms166,167,168,169. Thus, moving forward, future studies should attempt to be more specific in the cleavage form when utilizing CCK in their hypothesis and experiments, as the effects of the different forms differ greatly. Nonetheless, endogenous and exogenous CCK (mostly CCK-8) acts on VANs to decrease meal size170,171. VANs innervating the GI tract contain CCK-1 receptor (CCK-1R) mRNA and respond to CCK-8 ex vivo12,14,172,173,174. Low levels of exogenous CCK-8 administration (4 ng/kg/min IV) have also been shown to decrease food intake in both lean and obese humans175, and intraperitoneal injection of CCK-8 decreases food intake in rats in a dose-dependent manner176. Slightly higher doses of CCK-8 administered via intraperitoneal injection (~1–10 µg/kg) activate vagal afferents and increase neuronal activation in the NTS where VANs terminate, while vagotomy and capsaicin treatment abolish the suppressive effects of CCK-8 administration177,178. Furthermore, knockdown of CCK-1R in vagal afferents via nodose ganglia lentiviral injection abolishes the ability of exogenous CCK-8 (2.5 µg/kg) to lower food intake179. Given that exogenous CCK, at potentially similar levels to postprandial endogenous CCK, activates vagal signaling pathways to lower food intake, it is likely that endogenous CCK mediates the suppressive effects of nutrients.
Lipids potently increase CCK release180,181, and administration of CCK-1R antagonists blocks the satiating effects of intestinal lipid infusion in both rodents and humans169,182. In support of a role for vagus-mediated CCK signaling, peripheral, but not central, antagonism of CCK-1R blocks the inhibitory effects of intestinal oleate and attenuates hindbrain activation183. The ability of lipids to induce CCK release and activate VANs to lower food intake may be due to chylomicron formation and subsequent activation of sensory machinery on the basolateral side of EECs, as mentioned previously. For example, coinfusion of lipids with a lipase inhibitor blocks the ability of lipids to increase CCK166,167,168,169,184,185 and ultimately suppress food intake168,169. Furthermore, treatment with Pluronic L-81, a surfactant that inhibits chylomicron formation, significantly attenuates the anorectic effects of intraduodenal lipids and attenuates both CCK release and celiac and cervical vagal afferent activation following lipid infusion40,109,156,186. Similarly, in humans, infusion of fatty acids with a chain length of less than C10, which do not assemble in chylomicrons but directly diffuse out of enterocytes, fails to reduce energy intake and hunger and does not increase circulating CCK levels169.
In addition to lipids, intestinal protein infusion significantly reduces food intake187 and increases CCK release. Several studies suggest that similar to lipids, reductions in food intake via intestinal protein in rodents are due to CCK release and subsequent activation of VANs, as (1) intestinal protein infusion increases CCK in rodents and humans188,189; (2) CCK-1R knockout rats exhibit a blunted response to peptone190; (3) peripheral chemical CCK antagonism abolishes the suppressive effects of peptone191; (4) intestinal peptones induce vagal afferent firing192; (5) capsaicin, a neurotoxin that selectively destroys small unmyelinated primary sensory neurons, abolishes the suppressive effect of amino acid L-phenylalanine43; and (6) vagotomy reduces the suppressive effect of L-phenylalanine193. Taken together, the evidence indicates that CCK vagal signaling likely mediates the inhibitory effects of food intake via intestinal nutrients.
Although the glucoregulatory effects of CCK are less studied than the suppression of food intake, CCK-8 can activate a neuronal signaling axis to lower hepatic glucose production194. Small intestinal lipid infusion lowers hepatic glucose production via a vagal gut–brain–liver axis that is dependent upon CCK-1R signaling, although recent evidence suggests that GLP-1R signaling also contributes to the effect130,194. Nonetheless, direct administration of CCK-8 also lowers hepatic glucose production during a glucose clamp195. Furthermore, in a more physiological setting, CCK-1R antagonism during a fasting-refeeding paradigm results in hyperglycemia, highlighting the glucoregulatory relevance of CCK-8 signaling194. In addition, CCK signaling may contribute to the pathophysiology of diabetes, as short-term HFD consumption abolished the ability of intralipid or low levels of exogenous CCK-8 (35 pmol/kg/min) to lower glucose production in rats during a glucose clamp49,194. However, upper small intestinal activation of protein kinase A (PKA) bypasses CCK resistance in regulating glucose homeostasis, although the role of PKA in feeding regulation remains unclear196. It is likely that CCK-1R signaling activates vagal afferents at least partly via PKA, as (1) CCK-1R activation increases cyclic adenosine monophosphate197, (2) PKA is necessary for CCK to activate central afferent terminals172, and (3) duodenal PKA activation leads to vagal afferent firing196. Overall, there is strong evidence suggesting that CCK can impact energy and glucose homeostasis by activating a gut–brain vagal pathway.
Other gut-derived peptides
The previously mentioned gut peptides (GLP-1, GIP, and CCK) have been extensively studied for their role in metabolic homeostasis. However, the GI tract secretes numerous other peptides in response to ingested nutrients that may also play a role in energy and glucose homeostasis. PYY is a peptide released from colonic EECs in response to nutrients, with PYY3-36 being the most prevalent form of this gut peptide in the circulation198,199. The predominant role of PYY is thought to be the maintenance of energy intake, as it is a salient signal of satiety, although some studies have suggested a role for PYY in glucose homeostasis and insulin sensitivity200,201,202. The mechanism mediating the ability of PYY to regulate food intake is contentious but likely involves alterations in gastric emptying, vagal neuronal signaling to the hypothalamus200,203,204, and direct endocrine activation of the ARC200.
Oxyntomodulin (OXM) is a gut peptide produced by the colonic EECs that acts via GLP-1Rs and glucagon receptors205,206 to reduce body weight207,208 and improve glucose metabolism209,210. OXM exerts its homeostatic effects on energy balance by increasing energy expenditure via the glucagon receptor and decreasing energy intake, likely through hypothalamic and area postrema activation by GLP-1R signaling207,211. The mechanism for action for regulating glucose metabolism of OXM remains elusive but might involve a similar pathway to GLP-1 since it activates the same receptor, although OXM is still able to improve glucose metabolism in mice lacking GLP-1R211.
Ghrelin and nesfatin are gut peptides secreted primarily from gastric endocrine cells212,213,214 that induce opposing effects on energy homeostasis and metabolism. Ghrelin, commonly called the “hunger hormone,” is a potent stimulus for food intake in both humans215 and animals216,217; consistent with this, circulating levels are elevated during fasted and calorie-restricted states and decreased upon feeding216,218. Both central and peripheral administration of ghrelin increases food intake, body weight, and adiposity in rodents216,217,219. In addition, peripheral ghrelin administration at a dose almost double physiological levels (5 pmol/kg/min) increased acute food intake by nearly 30% in humans215. Ghrelin likely exerts these effects through both suppression of VAN firing and endocrine signaling in the ARC. In support of ghrelin’s proposed actions through VANs, ghrelin receptors are expressed on VANs, and subdiaphragmatic or gastric vagotomy abolishes increased food intake following peripherally administered ghrelin (1.5 nmol)220. In addition to regulating hunger, one study has strongly implicated peripheral ghrelin as a regulator of fasting blood glucose levels; global ghrelin receptor knockout mice exhibit low fasting blood glucose levels, which are increased when ghrelin receptor expression is selectively restored in nodose ganglia221. However, ghrelin receptors are expressed in the ARC222, and ghrelin can cross the blood–brain barrier223, although ghrelin transport from the blood to the brain might be too low to exert effects223. Therefore, it is hypothesized that ghrelin synthesized within the hypothalamus, as opposed to gut-derived ghrelin, is the mediator of central orexigenic effects of ghrelin, although further research is needed224.
In contrast to ghrelin, gastric endocrine cells also release an anorexigenic peptide, nesfatin214,225. Nesfatin is cleaved into three different forms: nesfatin-1, nesfatin-2, and nesfatin-3226. Of these, only nesfatin-1 has been established as a modulator of food intake226 and glucose homeostasis in rodents227. One study demonstrated that intraperitoneal injection of nesfatin-1 decreased food intake in a dose-dependent manner in mice and that c-Fos expression was significantly increased in the NTS, suggesting the role of VANs in this effect228. However, nesfatin-1 crosses the blood–brain barrier in a non-saturable fashion229,230. As such, centrally, but not peripherally, administered nesfatin-1 reproducibly causes reductions in food intake in rodents231,232. Furthermore, 2 weeks of subcutaneous infusion of nesfatin-1 improves glucose tolerance and insulin sensitivity in chow-fed and HFD-fed mice227, likely due to the direct action of nesfatin-1 on beta cells and not gut–brain signaling, similar to other peptides, such as GLP-1 and CCK227,233,234. However, circulating levels of nesfatin are generally not known; thus, despite the therapeutic potential, the effect and mechanism of endogenous nesfatin is still poorly understood.
Insulin-like peptide 5 (Insl5) is an orexigenic peptide released by EECs of the colon. As such, Insl5 plasma concentrations are increased in calorie-restricted states and suppressed during refeeding235. Likewise, Insl5 is decreased in energy-rich states such as high-fat feeding236. Due to the close proximity of EECs that produce Insl5 and enteric neurons expressing its receptor (relaxin/insulin-like family peptide receptor 4)235, Insl5 likely increases food intake via peripheral neuron signaling to the CNS. In support of this, peripheral, but not central, administration of high levels of Insl5 elicits an increase in food intake235. Consistent with this, Insl5 increases food intake in a dose-dependent manner in nonfasted control mice, but these effects are not preserved in relaxin/insulin-like family peptide receptor 4 knockout mice, receptors expressed on enteric and vagal afferent neurons but not in the hypothalamus235. However, peripheral Insl5-induced activation of brain areas associated with regulating food intake has yet to be established. Relatively little is known yet regarding the recently discovered Insl5. Therefore, future studies are warranted to ascertain whether Insl5 meets the necessary criteria to be considered a regulator of feeding given that the exogenous studies mentioned above utilize high doses of Insl5, although the initial work is promising. For an extensive review of additional neuroendocrine gut peptides regulating food intake, see, ref. 237.
Gut microbiota–brain connection
The gut microbiota consists of over 10 trillion diverse microbes, increasing in density distally through the GI tract238. The host and gut microbes exist in a symbiotic relationship, in which the host creates a nutrient-rich environment for microbes, and the microbes, in turn, impact host physiology, immunology, and metabolism239. Germ-free (GF) mice, devoid of any microbiome, can provide valuable insight into the impact of the gut microbiome on host metabolism. GF mice have reduced adiposity and increased insulin sensitivity, despite increased food intake240, and are protected against diet-induced obesity241. GF mice exhibit altered gut–brain metabolic signaling, with changes in EEC number, as well as differences in intestinal nutrient-sensing machinery and nutrient-induced gut peptide release compared to conventional mice242. Transcriptomic analysis revealed significant differences in small intestinal GLP-1-secreting L-cells from GF versus conventional mice, including increased GLP-1 content in GF mice, which was rapidly altered following conventionalization, implicating a strong role of the gut microbiota in regulating EEC physiology243. In line with this, treatment of cultured cells with several different bacteria decreased GLP-1 mRNA but increased fatty acid sensor GPR120 expression, which induces GLP-1 release244.
In addition to impaired EEC signaling and gut peptide release, GF models have impaired ENS activation compared with conventionalized and specific-pathogen-free models245,246. In GF mice and in antibiotic-treated mice, which have a very depleted gut microbiota, enteric neuron number and activation are reduced72,246,247, likely via impairments in Toll-like receptor 4 (TLR4) signaling. TLR4 is expressed on the ENS, recognizes microbial byproducts such as lipopolysaccharide (LPS), and when activated, initiates a downstream signaling cascade to increase inflammatory cytokine production and immune system activation247,248. In addition, ENS maturation and function may be regulated by the production of 5-HT by the gut microbiota249. Regardless of the mechanism, these impairments in ENS signaling could lead to alterations in the gut–brain axis that regulate energy homeostasis, as GF mice are GLP-1 resistant, demonstrating reduced activation of brainstem neurons. Importantly, colonization of GF mice with a healthy gut microbiota restores neuronal GLP-1 and ENS signaling pathways in the gut; however, this effect is abolished in mice recolonized with a microbiome from diabetic mice72. These studies not only indicate a role for the gut microbiota in impacting gut–brain neuronal signaling, but also suggest that differential gut microbiomes could have varying effects.
Metabolic diseases are associated with altered microbial functionality, composition, and diversity250,251,252. Transplanting the gut microbiota from either obese or lean mice or humans into GF mice recapitulates the host phenotype, highlighting the potential causality of the gut microbiota in the development of metabolic dysregulation252,253. Furthermore, fecal microbiota transplant from lean donors to individuals with metabolic syndrome improves the insulin sensitivity of recipients254, demonstrating that altering the gut microbiota could improve metabolic outcomes. Alternatively, beneficially altering the gut microbiota through the use of prebiotics, nondigestible carbohydrates that promote the growth of beneficial bacteria, reduces body weight and adiposity, and improves glucose tolerance in rodents and humans255,256,257. Treatment with inulin or oligofructose increases the production and release of gut peptides, which is associated with reduced food intake and increased satiety258,259,260,261. Furthermore, the improvements in glucose homeostasis following prebiotic treatment are dependent on GLP-1R signaling, implicating that prebiotic-induced alterations in the microbiome restore the gut–brain axis257. In addition to increased secretion of gut peptides, prebiotic treatment improves gut barrier integrity under the conditions of HF feeding and obesity, ultimately reducing circulating LPS256. LPS translocation across the epithelium during high-fat feeding leads to metabolic endotoxemia and systemic inflammation262. In addition to acting on other metabolically relevant tissues, vagal afferents contain TLR4263, and LPS impairs gut–brain vagal signaling264,265. LPS reduces leptin-mediated activation of VANs, both in vitro and in vivo264,265. Interestingly, vagal CCK signaling is potentiated by vagal leptin signaling266,267, and diet-induced leptin resistance in VANs mediates impairments in vagal CCK action268. Thus, impaired CCK signaling in HFD-induced obesity may be due to increased LPS-driven leptin resistance at VANs. In line with this, chronic low-dose treatment with LPS increases food intake, decreases CCK-induced satiety, and causes vagal afferent leptin resistance in rats264. Therefore, it is possible that the gut microbiota can impact intestinal nutrient-sensing mechanisms (Fig. 3); however, most work to date has focused on the distal gut microbiota, while nutrient-sensing negative feedback takes place in the small intestine.

The microbiota produces several metabolites that impact gut–brain signaling directly and indirectly. The microbiome can directly influence nutrient receptor expression and gut peptide release from EECs and can influence the production of serotonin (5-HT) from ECs, which may alter ENS or vagal afferent activation. Microbially derived or modified metabolites also influence the gut–brain axis. Bile acids (BAs) can alter the expression of TGR5 and regulate gut peptide release from EECs, while SCFAs alter nutrient receptor expression and gut peptide production by EECs or activate vagal afferents directly. Alternatively, metabolites such as LPS, a product of pathogenic microbes, can impair gut–brain signaling by preventing the activation of vagal afferents or the ENS. Overall, the gut microbiome and its metabolites can alter gut–brain signaling to influence the activation of the NTS and ARC, which regulate food intake, energy expenditure, glucose disposal and production, and insulin secretion.
Recent work has highlighted the small intestinal microbiota as a major mediator of the gut–brain axis in regulating glucose homeostasis. A high-fat diet rapidly shifts the small intestinal gut microbiota, resulting in a drastic reduction in Lactobacillius130. In parallel with rapid shifts in the small intestinal microbiota, impairments in the regulation of glucose homeostasis via small intestinal lipid sensing occur following just 3 days of HFD feeding130. Interestingly, small intestinal microbiota transplant from rats fed either a healthy chow or a HFD restored small intestinal lipid sensing, improving whole-body glucose homeostasis, and vice versa130. Similarly, acute metformin treatment restores HFD-induced impairments in small intestinal glucose sensing that regulate hepatic glucose production. This is due to increased small intestinal sodium/glucose cotransporter 1 (SGLT1) expression and subsequent GLP-1 release following intestinal glucose infusion, which are mediated by metformin-induced alterations in the small intestinal microbiota131. In line with alterations of the small intestinal microbiota mediating nutrient-induced gut–brain signaling, direct small intestinal infusion of Lactobacillus gasseri restores intestinal lipid sensing49,130. Lactobacillus gasseri expresses bile salt hydrolase, and improvements in lipid sensing were dependent on reduced farnesoid-X receptor (FXR) signaling130, highlighting the impact of bile acids on gut–brain signaling mechanisms that regulate metabolic homeostasis.
Bile acids are produced in the liver and excreted into the small intestine to aid in fat digestion269. In addition, specific bile acids can act as signaling molecules by activating the transcription factor FXR or binding to G protein-coupled bile acid receptor-1 (TGR5), both of which are expressed in EECs270. As such, TGR5 activation stimulates secretion of GLP-1, while FXR activation reduces GLP-1 secretion, demonstrating a complex interplay between the differential bile acid pool composition and gut–brain signaling270,271. For example, high-fat feeding alters the small intestinal microbiota to increase intestinal, brain, and circulatory levels of taurine-conjugated bile acids, including and taurochenodeoxycholic acid (TCDCA), which is an FXR agonist272. Increased TCDCA leads to insulin resistance that is due to impaired insulin action in the dorsal vagal complex of the hindbrain from increased local TCDCA–FXR signaling272. Furthermore, at the intestinal level, TCDCA-mediated activation of FXR leads to impaired nutrient-sensing mechanisms that regulate glucose homeostasis273. Interestingly, transplant of the small intestinal microbiome from animals fed regular chow restores both central insulin and intestinal nutrient-sensing signaling mechanisms, highlighting the role of the gut microbiota in shaping bile acid pool composition272,273. However, the role of FXR in improving metabolic dysfunction is contentious, as intestine-specific FXR agonism improves the metabolic profiles of mice274,275 and increases the expression of TGR5276, which mediates bile acid-induced GLP-1 secretion from EECs277.
In addition to shaping the bile acid pool, the gut microbiota produces a substantial quantity of metabolites that can have a vast impact on the host. In the gut–brain axis specifically, short-chain fatty acids (SCFAs), butyrate, acetate, and propionate, produced from gut microbiota fermentation of nondigestible fiber, improve both energy and glucose homeostasis in rodent models of obesity and insulin resistance278,279. For example, chronic butyrate administration reduces food intake and body weight via a neural gut–brain axis280. SCFAs bind to GPR43 and GPR41 and olfactory receptor 558 localized on EECs281 and can regulate the number and activity of EECs282,283. Treatment of human and mouse small intestine organoid models with SCFAs increases the number of GLP-1-producing cells twofold284, and SCFA infusion activates colonic EECs to increase GLP-1 release in mice281. SCFA-induced GLP-1 and PYY release is at least partially dependent upon the GPR43 receptor pathway113,281,282,283. However, the exact mechanism of SCFA-induced gut peptide release is controversial, as SCFAs may activate EECs via basolateral sensing mechanisms, similar to LCFAs. For example, while luminal or vascular administration of butyrate during an isolated perfused colon study increased GLP-1 and PYY secretion93, only vascular administration of acetate induced GLP-1 and PYY secretion, with luminal acetate inducing only GLP-1 secretion93. Interestingly, although vascular SCFAs activate EECs, vascular administration of a GPR43 agonist did not induce gut peptide secretion, and vascular coadministration of butyrate with a GPR41 antagonist did not impede gut peptide secretion93. This suggests that GPR43 and GPR41 may not be part of the mechanism required for GLP-1 and PYY release from SCFAs; however, this study did not test luminal GPR43 or GPR41 effects.
In addition, given the close proximity of VANs, SCFAs could directly activate neurons that contain GPR41285. Indeed, while SCFAs infused into the intestinal lumen activate VANs projecting from the gut12 and intraperitoneal SCFA injection suppressed food intake through vagal afferent signaling, butyrate treatment directly activates isolated nodose ganglion neurons from mice12. Thus, given that oral, but not intravenous, administration of butyrate decreased food intake in a manner that is dependent on gut–brain signaling280, SCFAs may activate VANs either directly or indirectly via gut peptide release93,283. Despite these findings, endogenously produced acetate can also cross the blood–brain barrier and can directly activate energy-regulating neurons to decrease appetite286.
The gut microbiota is also a major regulator of 5-HT production. GF mice have decreased 5-HT levels compared to conventional mice, which is rescued following gut microbiota transplantation287. Improvements in 5-HT production occur by increasing EC expression of tryptophan hydroxylase, the rate-limiting enzyme in EC 5-HT production288, or by increasing microbial metabolites, such as SCFAs and BAs, that are known to stimulate EC 5-HT production74. ECs in the gut produce ~95% of the 5-HT in the body, which is presumed to act on 5-HT receptors expressed on local gut neurons289. 5-HT administered intravenously290 or intraluminally291 activates intestinal VANs and myenteric neurons292,293 in rats. This 5-HT-mediated vagal afferent activation increases c-Fos expression in the NTS and area postrema294, demonstrating the potential gut–brain relay of ECs. More recently, it was shown that ECs act as gut chemosensory cells, similar to other EECs such as GLP-1-secreting cells, and that they form synapse-like connections with primary afferent neurons295. Overall, there is extensive evidence indicating that the gut microbiota has a large impact on nutrient-induced gut–brain signaling that regulates metabolic homeostasis (Fig. 3). However, our current understanding likely represents a small proportion of the potential of the gut microbiota to impact metabolic homeostasis via gut–brain signaling, and this is a field that needs to continue to be explored. For example, in addition to 5-HT, bacteria have been found to produce a large number of hormones or neurotransmitters, including dopamine, leptin, and gamma-aminobutyric acid (GABA). Certain Bifidobacterium and Lactobacillus species can produce GABA296,297,298,299,300, and oral treatment with GABA or bacteria that produce GABA improves metabolic parameters in mice298,301. However, the impact of bacterially derived GABA, the primary inhibitory brain neurotransmitter, on the CNS is completely unknown.
Conclusion
Our understanding of how the GI tract contributes to disease, especially the development of obesity and diabetes, is still developing. As detailed in Fig. 1, there is a complex relationship between the gut and the brain. For example, EECs within the GI tract can sense nutrients and release a variety of gut peptides to influence both energy and glucose homeostasis. However, more work is needed to fully understand the mechanisms through which various nutrients activate EECs and release gut peptides (Fig. 2), the metabolic capabilities of specific peptides, and their mechanisms and sites of action. Traditionally, it was thought that gut peptides act on VANs to signal to the brain to mediate their metabolic effects, as detailed above for GLP-1 and CCK. However, recent work has implicated the ENS in mediating the effects of GLP-1, while the role of gut peptide-mediated VAN signaling is controversial. Indeed, with technological and methodological advancements, the intricacy of the gut–brain axis in regulating energy and glucose homeostasis is currently being uncovered. For example, although vagal afferents were once thought to be fairly homogenous in their signaling capacity and function, extensive tracing and single-cell sequencing studies have demonstrated their complexity and diversity28,31,32. Although these recent studies appear somewhat conflicting and contentious28,31,32, as more studies continue to map out the neuronal gut–brain signaling axis, they could ultimately lead to very precise therapeutics directly targeting specific neural subgroups within vagal afferents to treat specific physiological mechanisms, such as controlling food intake versus intestinal motility. Similarly, given the recent work characterizing the uniqueness and heterogeneity of EECs81,84,85,86,87, it may also be possible to target specific populations of EECs or even convert certain EECs into cells that secrete specific peptides302,303,304.
Along these lines, our understanding of the impact of the gut microbiota on host gut–brain signaling is still poorly understood, due in part to the current limitations in analyzing the gut microbiome and the plethora of metabolites it produces. In the current review, we detail the ability of the gut microbiome to impact small intestinal nutrient-sensing mechanisms that activate a gut–brain neuronal axis to regulate hepatic glucose production. However, given the potential similarities in gut–brain signaling pathways regulating food intake and hepatic glucose production (detailed above), it is likely that the small intestinal microbiota can also impact food intake and energy homeostasis, although this remains to be assessed. Furthermore, we highlight the strong impact of bacterially derived metabolites, mainly SCFAs and bile acids, on overall metabolic homeostasis (Fig. 3) via activation of EECs or peripheral neurons directly. However, these microbe–host interactions are likely the tip of the iceberg, as the bacteria in the gut contain numerous genes, likely corresponding to an extensive degree of functionality. Therefore, it is very plausible that there are many unrecognized mechanisms through which the gut microbiota could impact gut–brain signaling pathways. In addition to better elucidating the impact of the gut microbiota on host homeostasis, only a small amount has been done to tap into the therapeutic potential of the gut microbiota. For example, with advancements in molecular engineering, scientists are currently attempting to alter the gut microbiota into specific compositions using CRISPR/Cas9 technology. Furthermore, bacteria are being created that contain specific proteins or signaling molecules to achieve site-specific delivery of gut-targeted therapies that can impact metabolic homeostasis305,306,307. These advances could lead to novel therapeutics to target specific microbiota–gut–brain signaling pathways that regulate energy and glucose homeostasis.
Overall, the gut–brain axis holds significant promise as a target for treatment options addressing obesity and diabetes. The gut plays a crucial role in maintaining energy and glucose homeostasis through extensive relays to the brain to inform the energy-regulating centers about incoming nutrient compositions. Although this review focused on the ability of gut-derived signals to control food intake, there is evidence that the gut and gut microbiota can also impact energy expenditure, although this requires more investigation308,309,310. Importantly, obesity and diabetes are associated with impairments in gut–brain signaling68,311, and the success of both surgical and pharmacological treatments for both diseases is due at least in part to alterations in the gut–brain axis6,7,10,131. With advances in our understanding of how these specific gut-derived relay signals contribute to food intake, energy expenditure, and glucose homeostasis, the potential to target these pathways in the treatment of metabolic disease will grow.
References
Hales, C. M., Carroll, M. D., Fryar, C. D. & Ogden, C. L. Prevalence of obesity and severe obesity among adults: United States, 2017–2018. NCHS Data Brief, 1–8 (2020).
Saeedi, P. et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 157, 107843 (2019).
Leung, M. Y. M., Carlsson, N. P., Colditz, G. A. & Chang, S.-H. The burden of obesity on diabetes in the United States: medical expenditure panel survey, 2008 to 2012. Value Health 20, 77–84 (2017).
Sjöström, L. Bariatric surgery and reduction in morbidity and mortality: experiences from the SOS study. Int. J. Obes. 32, S93–S97 (2008).
Reid, T. Choosing GLP-1 receptor agonists or DPP-4 inhibitors: weighing the clinical trial evidence. Clin. Diabetes 30, 3 (2012).
Wu, H. et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 23, 850–858 (2017).
Duca, F. A. et al. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 21, 506–511 (2015).
Sun, L. et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 24, 1919–1929 (2018).
Mithieux, G. Gut Microbiota and host metabolism: what relationship. Neuroendocrinology 106, 352–356 (2018).
Ilhan, Z. E. et al. Temporospatial shifts in the human gut microbiome and metabolome after gastric bypass surgery. NPJ Biofilms Microbiomes 6, 12 (2020).
Iggo, A. Tension receptors in the stomach and the urinary bladder. J. Physiol. 128, 593–607 (1955).
Lal, S., Kirkup, A. J., Brunsden, A. M., Thompson, D. G. & Grundy, D. Vagal afferent responses to fatty acids of different chain length in the rat. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G907–G915 (2001).
de Lartigue, G. & Diepenbroek, C. Novel developments in vagal afferent nutrient sensing and its role in energy homeostasis. Curr. Opin. Pharmacol. 31, 38–43 (2016).
Schwartz, M. W., Woods, S. C., Porte, D., Seeley, R. J. & Baskin, D. G. Central nervous system control of food intake. Nature 404, 661–671 (2000).
Lam, C. K. L., Chari, M. & Lam, T. K. T. CNS regulation of glucose homeostasis. Physiol. (Bethesda) 24, 159–170 (2009).
Gribble, F. M. & Reimann, F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu. Rev. Physiol. 78, 277–299 (2016).
Gribble, F. M. & Reimann, F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat. Rev. Endocrinol. 15, 226–237 (2019).
Turnbaugh, P. J. et al. The human microbiome project. Nature 449, 804–810 (2007).
Bauer, P. V., Hamr, S. C. & Duca, F. A. Regulation of energy balance by a gut–brain axis and involvement of the gut microbiota. Cell. Mol. Life Sci. 73, 737–755 (2016).
Tsigos, C. & Chrousos, G. P. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 53, 865–871 (2002).
Berthoud, H.-R. The vagus nerve, food intake and obesity. Regul. Pept. 149, 15–25 (2008).
Goldstein, N. et al. Hypothalamic detection of macronutrients via multiple gut-brain pathways. Cell Metab. 33, 676–687.e5 (2021).
Brown, T. A. L., Washington, M. C., Metcalf, S. A. & Sayegh, A. I. The feeding responses evoked by cholecystokinin are mediated by vagus and splanchnic nerves. Peptides 32, 1581–1586 (2011).
Sclafani, A., Ackroff, K. & Schwartz, G. J. Selective effects of vagal deafferentation and celiac-superior mesenteric ganglionectomy on the reinforcing and satiating action of intestinal nutrients. Physiol. Behav. 78, 285–294 (2003).
Furness, J. B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 9, 286–294 (2012).
Berthoud, H. R. & Patterson, L. M. Anatomical relationship between vagal afferent fibers and CCK-immunoreactive entero-endocrine cells in the rat small intestinal mucosa. Acta Anat. 156, 123–131 (1996).
Berthoud, H. R., Kressel, M., Raybould, H. E. & Neuhuber, W. L. Vagal sensors in the rat duodenal mucosa: distribution and structure as revealed by in vivo DiI-tracing. Anat. Embryol. 191, 203–212 (1995).
Bai, L. et al. Genetic identification of vagal sensory neurons that control feeding. Cell 179, 1129–1143.e23 (2019).
Berthoud, H. R., Patterson, L. M., Neumann, F. & Neuhuber, W. L. Distribution and structure of vagal afferent intraganglionic laminar endings (IGLEs) in the rat gastrointestinal tract. Anat. Embryol. 195, 183–191 (1997).
Blackshaw, L. A. & Grundy, D. Effects of cholecystokinin (CCK-8) on two classes of gastroduodenal vagal afferent fibre. J. Auton. Nerv. Syst. 31, 191–201 (1990).
Kupari, J., Häring, M., Agirre, E., Castelo-Branco, G. & Ernfors, P. An atlas of vagal sensory neurons and their molecular specialization. Cell Rep. 27, 2508–2523.e4 (2019).
Williams, E. K. et al. Sensory neurons that detect stretch and nutrients in the digestive system. Cell 166, 209–221 (2016).
Fan, W., Boston, B. A., Kesterson, R. A., Hruby, V. J. & Cone, R. D. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385, 165–168 (1997).
Cowley, M. A. et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484 (2001).
Sohn, J.-W., Elmquist, J. K. & Williams, K. W. Neuronal circuits that regulate feeding behavior and metabolism. Trends Neurosci. 36, 504–512 (2013).
Berthoud, H.-R. & Neuhuber, W. L. Functional and chemical anatomy of the afferent vagal system. Auton. Neurosci. 85, 1–17 (2000).
Han, W. et al. A neural circuit for gut-induced reward. Cell 175, 665–678.e23 (2018).
Mei, N. Vagal glucoreceptors in the small intestine of the cat. J. Physiol. 282, 485–506 (1978).
Schwartz, G. J. & Moran, T. H. Duodenal nutrient exposure elicits nutrient-specific gut motility and vagal afferent signals in rat. Am. J. Physiol. 274, R1236–R1242 (1998).
Randich, A. et al. Responses of celiac and cervical vagal afferents to infusions of lipids in the jejunum or ileum of the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 278, R34–R43 (2000).
Emond, M., Schwartz, G. J. & Moran, T. H. Meal-related stimuli differentially induce c-Fos activation in the nucleus of the solitary tract. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R1315–R1321 (2001).
Lorenz, D. N. & Goldman, S. A. Vagal mediation of the cholecystokinin satiety effect in rats. Physiol. Behav. 29, 599–604 (1982).
Yox, D. P. & Ritter, R. C. Capsaicin attenuates suppression of sham feeding induced by intestinal nutrients. Am. J. Physiol. Regul. Integr. Comp. Physiol. 255, R569–R574 (1988).
Mönnikes, H. et al. Pathways of Fos expression in locus ceruleus, dorsal vagal complex, and PVN in response to intestinal lipid. Am. J. Physiol. Regul. Integr. Comp. Physiol. 273, R2059–R2071 (1997).
Yao, G. et al. Effective weight control via an implanted self-powered vagus nerve stimulation device. Nat. Commun. 9, 5349 (2018).
Bugajski, A. J. et al. Effect of long-term vagal stimulation on food intake and body weight during diet induced obesity in rats. J. Physiol. Pharmacol. 58(Suppl 1), 5–12 (2007).
Krolczyk, G. et al. Effects of continuous microchip (MC) vagal neuromodulation on gastrointestinal function in rats. J. Physiol. Pharmacol. 52, 705–715 (2001).
Burneo, J. G., Faught, E., Knowlton, R., Morawetz, R. & Kuzniecky, R. Weight loss associated with vagus nerve stimulation. Neurology 59, 463 (2002).
Wang, P. Y. T. et al. Upper intestinal lipids trigger a gut–brain–liver axis to regulate glucose production. Nature 452, 1012–1016 (2008).
Fujita, S. & Donovan, C. M. Celiac-superior mesenteric ganglionectomy, but not vagotomy, suppresses the sympathoadrenal response to insulin-induced hypoglycemia. Diabetes 54, 3258–3264 (2005).
Mithieux, G. et al. Portal sensing of intestinal gluconeogenesis is a mechanistic link in the diminution of food intake induced by diet protein. Cell Metab. 2, 321–329 (2005).
Adachi, A., Shimizu, N., Oomura, Y. & Kobáshi, M. Convergence of hepatoportal glucose-sensitive afferent signals to glucose-sensitive units within the nucleus of the solitary tract. Neurosci. Lett. 46, 215–218 (1984).
Shimizu, N., Oomura, Y., Novin, D., Grijalva, C. V. & Cooper, P. H. Functional correlations between lateral hypothalamic glucose-sensitive neurons and hepatic portal glucose-sensitive units in rat. Brain Res. 265, 49–54 (1983).
Langhans, W., Grossmann, F. & Geary, N. Intrameal hepatic-portal infusion of glucose reduces spontaneous meal size in rats. Physiol. Behav. 73, 499–507 (2001).
Tordoff, M. G. & Friedman, M. I. Hepatic portal glucose infusions decrease food intake and increase food preference. Am. J. Physiol. 251, R192–R196 (1986).
Burcelin, R., Dolci, W. & Thorens, B. Portal glucose infusion in the mouse induces hypoglycemia: evidence that the hepatoportal glucose sensor stimulates glucose utilization. Diabetes 49, 1635–1642 (2000).
Pal, A., Rhoads, D. B. & Tavakkoli, A. Effect of portal glucose sensing on systemic glucose levels in SD and ZDF rats. PLoS ONE 11, e0165592 (2016).
Soty, M., Gautier-Stein, A., Rajas, F. & Mithieux, G. Gut-brain glucose signaling in energy homeostasis. Cell Metab. 25, 1231–1242 (2017).
De Vadder, F., Plessier, F., Gautier-Stein, A. & Mithieux, G. Vasoactive intestinal peptide is a local mediator in a gut-brain neural axis activating intestinal gluconeogenesis. Neurogastroenterol. Motil. 27, 443–448 (2015).
Balkan, B. & Li, X. Portal GLP-1 administration in rats augments the insulin response to glucose via neuronal mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1449–R1454 (2000).
Burcelin, R., Da Costa, A., Drucker, D. & Thorens, B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes 50, 1720 (2001).
Vahl, T. P. et al. Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology 148, 4965–4973 (2007).
Daly, D. M., Park, S. J., Valinsky, W. C. & Beyak, M. J. Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J. Physiol. 589, 2857–2870 (2011).
Donovan, M. J., Paulino, G. & Raybould, H. E. Activation of hindbrain neurons in response to gastrointestinal lipid is attenuated by high fat, high energy diets in mice prone to diet-induced obesity. Brain Res. 1248, 136–140 (2009).
Duca, F. A., Zhong, L. & Covasa, M. Reduced CCK signaling in obese-prone rats fed a high fat diet. Horm. Behav. 64, 812–817 (2013).
Swartz, T. D., Duca, F. A. & Covasa, M. Differential feeding behavior and neuronal responses to CCK in obesity-prone and -resistant rats. Brain Res. 1308, 79–86 (2010).
Grabauskas, G. et al. High-fat diet-induced vagal afferent dysfunction via upregulation of 2-pore domain potassium TRESK channel. JCI Insight 4, e130402 (2019).
de Lartigue, G. Role of the vagus nerve in the development and treatment of diet-induced obesity. J. Physiol. 594, 5791–5815 (2016).
Kugler, E. M. et al. Mechanical stress activates neurites and somata of myenteric neurons. Front. Cell. Neurosci. 9, 342–342 (2015).
Mao, Y., Wang, B. & Kunze, W. Characterization of myenteric sensory neurons in the mouse small intestine. J. Neurophysiol. 96, 998–1010 (2006).
Vincent, K. M., Sharp, J. W. & Raybould, H. E. Intestinal glucose-induced calcium-calmodulin kinase signaling in the gut-brain axis in awake rats. Neurogastroenterol. Motil. 23, e282–e293 (2011).
Grasset, E. et al. A specific gut microbiota dysbiosis of type 2 diabetic mice induces GLP-1 resistance through an enteric NO-dependent and gut-brain axis mechanism. Cell Metab. 25, 1075–1090.e5 (2017).
Richards, P. et al. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 63, 1224 (2014).
Yano, J. M. et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161, 264–276 (2015).
Liu, M.-T., Rothstein, J. D., Gershon, M. D. & Kirchgessner, A. L. Glutamatergic enteric neurons. J. Neurosci. 17, 4764 (1997).
Kondoh, T., Mallick, H. N. & Torii, K. Activation of the gut-brain axis by dietary glutamate and physiologic significance in energy homeostasis. Am. J. Clin. Nutr. 90, 832S–837S (2009).
Niijima, A. Reflex effects of oral, gastrointestinal and hepatoportal glutamate sensors on vagal nerve activity. J. Nutr. 130, 971S–973S (2000).
Ritter, R. C. A tale of two endings: modulation of satiation by NMDA receptors on or near central and peripheral vagal afferent terminals. Physiol. Behav. 105, 94–99 (2011).
Fournel, A. et al. Apelin targets gut contraction to control glucose metabolism via the brain. Gut 66, 258–269 (2017).
Abot, A. et al. Galanin enhances systemic glucose metabolism through enteric Nitric Oxide Synthase-expressed neurons. Mol. Metab. 10, 100–108 (2018).
Engelstoft, M. S., Egerod, K. L., Lund, M. L. & Schwartz, T. W. Enteroendocrine cell types revisited. Curr. Opin. Pharmacol. 13, 912–921 (2013).
Eissele, R. et al. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur. J. Clin. Invest. 22, 283–291 (1992).
Furness, J. B., Rivera, L. R., Cho, H.-J., Bravo, D. M. & Callaghan, B. The gut as a sensory organ. Nat. Rev. Gastroenterol. Hepatol. 10, 729–740 (2013).
Glass, L. L. et al. Single-cell RNA-sequencing reveals a distinct population of proglucagon-expressing cells specific to the mouse upper small intestine. Mol. Metab. 6, 1296–1303 (2017).
Gehart, H. et al. Identification of enteroendocrine regulators by real-time single-cell differentiation mapping. Cell 176, 1158–1173.e16 (2019).
Beumer, J. et al. Enteroendocrine cells switch hormone expression along the crypt-to-villus BMP signalling gradient. Nat. Cell Biol. 20, 909–916 (2018).
Egerod, K. L. et al. A major lineage of enteroendocrine cells coexpress CCK, secretin, GIP, GLP-1, PYY, and neurotensin but not somatostatin. Endocrinology 153, 5782–5795 (2012).
Moor, A. E. et al. Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell 175, 1156–1167.e15 (2018).
Roberts, G. P. et al. Comparison of human and murine enteroendocrine cells by transcriptomic and peptidomic profiling. Diabetes 68, 1062–1072 (2019).
Grün, D. et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525, 251–255 (2015).
Habib, A. M. et al. Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 153, 3054–3065 (2012).
Page, A. J. Vagal afferent dysfunction in obesity: cause or effect. J. Physiol. 594, 5–6 (2016).
Christiansen, C. B. et al. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 315, G53–G65 (2018).
Kuhre, R. E. & Holst, J. J. Mechanisms underlying gut hormone secretion using the isolated perfused rat small intestine. JoVE 26, e58533 (2019).
Bohórquez, D. V. et al. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J. Clin. Invest. 125, 782–786 (2015).
Kaelberer, M. M. et al. A gut-brain neural circuit for nutrient sensory transduction. Science 361, eaat5236, https://doi.org/10.1126/science.aat5236 (2018).
Nauck, M. A. & Meier, J. J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 20, 5–21 (2018).
Drucker, D. J. The biology of incretin hormones. Cell Metab. 3, 153–165 (2006).
Rocca, A. S. & Brubaker, P. L. Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology 140, 1687–1694 (1999).
Vilsbøll, T., Krarup, T., Deacon, C. F., Madsbad, S. & Holst, J. J. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes 50, 609–613 (2001).
Parker, H. E., Reimann, F. & Gribble, F. M. Molecular mechanisms underlying nutrient-stimulated incretin secretion. Expert Rev. Mol. Med. 12, e1 (2010).
Ezcurra, M., Reimann, F., Gribble, F. M. & Emery, E. Molecular mechanisms of incretin hormone secretion. Curr. Opin. Pharmacol. 13, 922–927 (2013).
Gribble, F. M., Williams, L., Simpson, A. K. & Reimann, F. A novel glucose-sensing mechanism contributing to glucagon-like peptide-1 secretion from the GLUTag cell line. Diabetes 52, 1147–1154 (2003).
Parker, H. E. et al. Predominant role of active versus facilitative glucose transport for glucagon-like peptide-1 secretion. Diabetologia 55, 2445–2455 (2012).
Gorboulev, V. et al. Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61, 187 (2012).
Edfalk, S., Steneberg, P. & Edlund, H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57, 2280–2287 (2008).
Hirasawa, A. et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 11, 90–94 (2005).
Psichas, A., Larraufie, P. F., Goldspink, D. A., Gribble, F. M. & Reimann, F. Chylomicrons stimulate incretin secretion in mouse and human cells. Diabetologia 60, 2475–2485 (2017).
Lu, W. J. et al. Chylomicron formation and secretion is required for lipid-stimulated release of incretins GLP-1 and GIP. Lipids 47, 571–580 (2012).
Christensen, L. W., Kuhre, R. E., Janus, C., Svendsen, B. & Holst, J. J. Vascular, but not luminal, activation of FFAR1 (GPR40) stimulates GLP-1 secretion from isolated perfused rat small intestine. Physiol. Rep. 3, e12551 (2015).
Luo, J. et al. A potent class of GPR40 full agonists engages the enteroinsular axis to promote glucose control in rodents. PLoS ONE 7, e46300 (2012).
Iwasaki, K. et al. Free fatty acid receptor GPR120 is highly expressed in enteroendocrine K cells of the upper small intestine and has a critical role in GIP secretion after fat ingestion. Endocrinology 156, 837–846 (2015).
Psichas, A. et al. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int. J. Obes. 39, 424–429 (2015).
Hassing, H. A. et al. Oral 2-oleyl glyceryl ether improves glucose tolerance in mice through the GPR119 receptor. Biofactors 42, 665–673 (2016).
Hansen, K. B. et al. 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J. Clin. Endocrinol. Metab. 96, E1409–E1417 (2011).
Mace, O. J., Schindler, M. & Patel, S. The regulation of K- and L-cell activity by GLUT2 and the calcium-sensing receptor CasR in rat small intestine. J. Physiol. 590, 2917–2936 (2012).
Modvig, I. M., Kuhre, R. E. & Holst, J. J. Peptone-mediated glucagon-like peptide-1 secretion depends on intestinal absorption and activation of basolaterally located calcium-sensing receptors. Physiol. Rep. 7, e14056 (2019).
Elrick, H., Stimmler, L., Hlad, C. J. J. & Arai, Y. Plasma insulin response to oral and intravenous glucose administration. J. Clin. Endocrinol. Metab. 24, 1076–1082 (1964).
Perley, M. J. & Kipnis, D. M. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic subjects. J. Clin. Invest. 46, 1954–1962 (1967).
Leech, C. A. et al. Molecular physiology of glucagon-like peptide-1 insulin secretagogue action in pancreatic β cells. Prog. Biophys. Mol. Biol. 107, 236–247 (2011).
Hansen, L., Deacon, C. F., Orskov, C. & Holst, J. J. Glucagon-like peptide-1-(7-36)amide is transformed to glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 140, 5356–5363 (1999).
Holst, J. J. The physiology of glucagon-like peptide 1. Physiol. Rev. 87, 1409–1439 (2007).
Müller, T. D. et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 30, 72–130 (2019).
Muscogiuri, G., DeFronzo, R. A., Gastaldelli, A. & Holst, J. J. Glucagon-like peptide-1 and the central/peripheral nervous system: crosstalk in diabetes. Trends Endocrinol. Metab. 28, 88–103 (2017).
Ionut, V. et al. Hepatic portal vein denervation impairs oral glucose tolerance but not exenatide’s effect on glycemia. Am. J. Physio. l Endocrinol. Metab. 307, E644–E652 (2014).
Hayes, M. R. et al. The common hepatic branch of the vagus is not required to mediate the glycemic and food intake suppressive effects of glucagon-like-peptide-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1479–R1485 (2011).
Varin, E. M. et al. Distinct neural sites of GLP-1R expression mediate physiological versus pharmacological control of incretin action. Cell Rep. 27, 3371–3384.e3 (2019).
Krieger, J. P. et al. Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes 65, 34–43 (2016).
Lamont, B. J. et al. Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice. J. Clin. Invest. 122, 388–402 (2012).
Bauer, P. V. et al. Lactobacillus gasseri in the upper small intestine impacts an ACSL3-dependent fatty acid-sensing pathway regulating whole-body glucose homeostasis. Cell Metab. 27, 572–587.e6 (2018).
Bauer, P. V. et al. Metformin alters upper small intestinal microbiota that impact a glucose-SGLT1-sensing glucoregulatory pathway. Cell Metab. 27, 101–117.e5 (2018).
Baumgartner, I. et al. Hepatic-portal vein infusions of glucagon-like peptide-1 reduce meal size and increase c-Fos expression in the nucleus tractus solitarii, area postrema and central nucleus of the amygdala in rats. J. Neuroendocrinol. 22, 557–563 (2010).
Rüttimann, E. B., Arnold, M., Hillebrand, J. J., Geary, N. & Langhans, W. Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology 150, 1174–1181 (2009).
Gutzwiller, J. P. et al. Glucagon-like peptide-1: a potent regulator of food intake in humans. Gut 44, 81–86 (1999).
Imeryüz, N. et al. Glucagon-like peptide-1 inhibits gastric emptying via vagal afferent-mediated central mechanisms. Am. J. Physiol. 273, G920–G927 (1997).
Abbott, C. R. et al. The inhibitory effects of peripheral administration of peptide YY(3-36) and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal-brainstem-hypothalamic pathway. Brain Res. 1044, 127–131 (2005).
Dailey, M. J., Moghadam, A. A. & Moran, T. H. Jejunal linoleic acid infusions require GLP-1 receptor signaling to inhibit food intake: implications for the effectiveness of Roux-en-Y gastric bypass. Am. J. Physiol. Endocrinol. Metab. 301, E1184–E1190 (2011).
Adrian, T. E., Bloom, S. R., Hermansen, K. & Iversen, J. Pancreatic polypeptide, glucagon and insulin secretion from the isolated perfused canine pancreas. Diabetologia 14, 413–417 (1978).
Taminato, T., Seino, Y., Goto, Y., Inoue, Y. & Kadowaki, S. Synthetic gastric inhibitory polypeptide stimulatory effect on insulin and glucagon secretion in the rat. Diabetes 26, 480–484 (1977).
Ebert, R., Nauck, M. & Creutzfeldt, W. Effect of exogenous or endogenous gastric inhibitory polypeptide (GIP) on plasma triglyceride responses in rats. Horm. Metab. Res. 23, 517–521 (1991).
Adriaenssens, A. E. et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. 30, 987–996.e6 (2019).
Kaneko, K. et al. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. J. Clin. Invest. 129, 3786–3791 (2019).
Creutzfeldt, W., Ebert, R., Willms, B., Frerichs, H. & Brown, J. C. Gastric inhibitory polypeptide (GIP) and insulin in obesity: Increased response to stimulation and defective feedback control of serum levels. Diabetologia 14, 15–24 (1978).
Falko, J. M., Crockett, S. E., Cataland, S. & Mazzaferri, E. L. Gastric inhibitory polypeptide (GIP) stimulated by fat ingestion in man. J. Clin. Endocrinol. Metab. 41, 260–265 (1975).
Brøns, C. et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J. Physiol. 587, 2387–2397 (2009).
Suzuki, K. et al. Transcriptional regulatory factor X6 (Rfx6) increases gastric inhibitory polypeptide (GIP) expression in enteroendocrine K-cells and is involved in GIP hypersecretion in high fat diet-induced obesity. J. Biol. Chem. 288, 1929–1938 (2013).
Boylan, M. O., Glazebrook, P. A., Tatalovic, M. & Wolfe, M. M. Gastric inhibitory polypeptide immunoneutralization attenuates development of obesity in mice. Am. J. Physiol. Endocrinol. Metab. 309, E1008–E1018 (2015).
Miyawaki, K. et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 8, 738–742 (2002).
Zhang, Q. et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 33, 833–844.e5 (2021).
Kim, S. J. et al. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS ONE 7, e40156 (2012).
Mroz, P. A. et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol. Metab. 20, 51–62 (2019).
Holst, J. J. & Rosenkilde, M. M. GIP as a therapeutic target in diabetes and obesity: insight from incretin co-agonists. J. Clin. Endocrinol. Metabol. 105, e2710–e2716 (2020).
Kuhne, S. G. & Stengel, A. Alteration of peptidergic gut-brain signaling under conditions of obesity. J. Physiol. Pharmacol. https://doi.org/10.26402/jpp.2019.5.01 (2019).
Liou, A. P. et al. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140, 903–912 (2011).
Liou, A. P. et al. The extracellular calcium-sensing receptor is required for cholecystokinin secretion in response to L-phenylalanine in acutely isolated intestinal I cells. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G538–G546 (2011).
Raybould, H. E., Meyer, J. H., Tabrizi, Y., Liddle, R. A. & Tso, P. Inhibition of gastric emptying in response to intestinal lipid is dependent on chylomicron formation. Am. J. Physiol. 274, R1834–R1838 (1998).
Burton-Freeman, B., Davis, P. A. & Schneeman, B. O. Interaction of fat availability and sex on postprandial satiety and cholecystokinin after mixed-food meals. Am. J. Clin. Nutr. 80, 1207–1214 (2004).
French, S. J., Murray, B., Rumsey, R. D., Sepple, C. P. & Read, N. W. Is cholecystokinin a satiety hormone? Correlations of plasma cholecystokinin with hunger, satiety and gastric emptying in normal volunteers. Appetite 21, 95–104 (1993).
Sayegh, A. I. The role of cholecystokinin receptors in the short-term control of food intake. Prog. Mol. Biol. Transl. Sci. 114, 277–316 (2013).
Reeve, J. R., Eysselein, V., Walsh, J. H., Ben-Avram, C. M. & Shively, J. E. New molecular forms of cholecystokinin. Microsequence analysis of forms previously characterized by chromatographic methods. J. Biol. Chem. 261, 16392–16397 (1986).
Sayegh, A. I., Washington, M. C., Raboin, S. J., Aglan, A. H. & Reeve, J. R. Jr. CCK-58 prolongs the intermeal interval, whereas CCK-8 reduces this interval: not all forms of cholecystokinin have equal bioactivity. Peptides 55, 120–125 (2014).
Washington, M. C., Williams, K. & Sayegh, A. I. The feeding responses evoked by endogenous cholecystokinin are regulated by different gastrointestinal sites. Horm. Behav. 78, 79–85 (2016).
Washington, M. C., Mhalhal, T. R. & Sayegh, A. I. Cholecystokinin-33, but not cholecystokinin-8 shows gastrointestinal site specificity in regulating feeding behaviors in male rats. Horm. Behav. 85, 36–42 (2016).
Sayegh, A. I. et al. Celiac and the cranial mesenteric arteries supply gastrointestinal sites that regulate meal size and intermeal interval length via cholecystokinin-58 in male rats. Horm. Behav. 67, 48–53 (2015).
Chandra, R. & Liddle, R. A. Cholecystokinin. Curr. Opin. Endocrinol. Diabetes Obes. 14, 63–67 (2007).
Degen, L. et al. Effect of CCK-1 receptor blockade on ghrelin and PYY secretion in men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1391–R1399 (2007).
Feinle, C., Meier, O., Otto, B., D’Amato, M. & Fried, M. Role of duodenal lipid and cholecystokinin A receptors in the pathophysiology of functional dyspepsia. Gut 48, 347–355 (2001).
Feinle, C. et al. Effects of fat digestion on appetite, APD motility, and gut hormones in response to duodenal fat infusion in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 284, G798–G807 (2003).
Matzinger, D. et al. The role of long chain fatty acids in regulating food intake and cholecystokinin release in humans. Gut 46, 688–693 (2000).
Ritter, R. C., Covasa, M. & Matson, C. A. Cholecystokinin: proofs and prospects for involvement in control of food intake and body weight. Neuropeptides 33, 387–399 (1999).
Schwartz, G. J., McHugh, P. R. & Moran, T. H. Gastric loads and cholecystokinin synergistically stimulate rat gastric vagal afferents. Am. J. Physiol. 265, R872–R876 (1993).
Rogers, R. C. & Hermann, G. E. Mechanisms of action of CCK to activate central vagal afferent terminals. Peptides 29, 1716–1725 (2008).
Broberger, C., Holmberg, K., Shi, T. J., Dockray, G. & Hökfelt, T. Expression and regulation of cholecystokinin and cholecystokinin receptors in rat nodose and dorsal root ganglia. Brain Res. 903, 128–140 (2001).
Moran, T. H., Ameglio, P. J., Schwartz, G. J. & McHugh, P. R. Blockade of type A, not type B, CCK receptors attenuates satiety actions of exogenous and endogenous CCK. Am. J. Physiol. 262, R46–R50 (1992).
Pi-Sunyer, X., Kissileff, H. R., Thornton, J. & Smith, G. P. C-terminal octapeptide of cholecystokinin decreases food intake in obese men. Physiol. Behav. 29, 627–630 (1982).
Gibbs, J., Young, R. C. & Smith, G. P. Cholecystokinin decreases food intake in rats. J. Comp. Physiol. Psychol. 84, 488–495 (1973).
Ritter, R. C. & Ladenheim, E. E. Capsaicin pretreatment attenuates suppression of food intake by cholecystokinin. Am. J. Physiol. 248, R501–R504 (1985).
Smith, G. P., Jerome, C. & Norgren, R. Afferent axons in abdominal vagus mediate satiety effect of cholecystokinin in rats. Am. J. Physiol. 249, R638–R641 (1985).
Diepenbroek, C. et al. Validation and characterization of a novel method for selective vagal deafferentation of the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 313, G342–G352 (2017).
Duca, F. A. & Lam, T. K. T. Gut microbiota, nutrient sensing and energy balance. Diabetes Obes. Metab. 16, 68–76 (2014).
Ritter, R. C. Increased food intake and CCK receptor antagonists: beyond abdominal vagal afferents. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R991–R993 (2004).
Yox, D. P., Brenner, L. & Ritter, R. C. CCK-receptor antagonists attenuate suppression of sham feeding by intestinal nutrients. Am. J. Physiol. 262, R554–R561 (1992).
Blevins, J. E., Stanley, B. G. & Reidelberger, R. D. Brain regions where cholecystokinin suppresses feeding in rats. Brain Res. 860, 1–10 (2000).
Hildebrand, P. & Beglinger, C. The role of cholecystokinin as a regulator of gastrointestinal functions. Praxis 87, 1821–1824 (1998).
Beglinger, S. et al. Role of fat hydrolysis in regulating glucagon-like Peptide-1 secretion. J. Clin. Endocrinol. Metab. 95, 879–886 (2010).
Sakata, Y. et al. Postabsorptive factors are important for satiation in rats after a lipid meal. Am. J. Physiol. 271, G438–G442 (1996).
Bensaı̈d, A. et al. Protein is more potent than carbohydrate for reducing appetite in rats. Physiol. Behav. 75, 577–582 (2002).
Caron, J., Domenger, D., Dhulster, P., Ravallec, R. & Cudennec, B. Protein digestion-derived peptides and the peripheral regulation of food intake. Front. Endocrinol. 8, 85–85 (2017).
Ryan, A. T. et al. Intraduodenal protein modulates antropyloroduodenal motility, hormone release, glycemia, appetite, and energy intake in lean men. Am. J. Clin. Nutr. 96, 474–482 (2012).
Covasa, M., Marcuson, J. K. & Ritter, R. C. Diminished satiation in rats exposed to elevated levels of endogenous or exogenous cholecystokinin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R331–R337 (2001).
Reidelberger, R. D., Castellanos, D. A. & Hulce, M. Effects of peripheral CCK receptor blockade on food intake in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R429–R437 (2003).
Darcel, N. P., Liou, A. P., Tomé, D. & Raybould, H. E. Activation of vagal afferents in the rat duodenum by protein digests requires PepT1. J. Nutr. 135, 1491–1495 (2005).
Yox, D. P., Stokesberry, H. & Ritter, R. C. Vagotomy attenuates suppression of sham feeding induced by intestinal nutrients. Am. J. Physiol. 260, R503–R508 (1991).
Cheung, G. W., Kokorovic, A., Lam, C. K., Chari, M. & Lam, T. K. Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab. 10, 99–109 (2009).
Breen, D. M. et al. Duodenal PKC-δ and cholecystokinin signaling axis regulates glucose production. Diabetes 60, 3148–3153 (2011).
Rasmussen, B. A. et al. Duodenal activation of cAMP-dependent protein kinase induces vagal afferent firing and lowers glucose production in rats. Gastroenterology 142, 834–843.e3 (2012).
Marino, C. R., Leach, S. D., Schaefer, J. F., Miller, L. J. & Gorelick, F. S. Characterization of cAMP-dependent protein kinase activation by CCK in rat pancreas. FEBS Lett. 316, 48–52 (1993).
Batterham, R. L. et al. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab. 4, 223–233 (2006).
Karra, E., Chandarana, K. & Batterham, R. L. The role of peptide YY in appetite regulation and obesity. J. Physiol. 587, 19–25 (2009).
Batterham, R. L. et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature 418, 650–654 (2002).
Konturek, S. J., Konturek, J. W., Pawlik, T. & Brzozowski, T. Brain-gut axis and its role in the control of food intake. J. Physiol. Pharmacol. 55, 137–154 (2004).
Vrang, N., Madsen, A. N., Tang-Christensen, M., Hansen, G. & Larsen, P. J. PYY(3-36) reduces food intake and body weight and improves insulin sensitivity in rodent models of diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R367–R375 (2006).
Chelikani, P. K., Haver, A. C. & Reidelberger, R. D. Comparison of the inhibitory effects of PYY(3-36) and PYY(1-36) on gastric emptying in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R1064–R1070 (2004).
Savage, A. P., Adrian, T. E., Carolan, G., Chatterjee, V. K. & Bloom, S. R. Effects of peptide YY (PYY) on mouth to caecum intestinal transit time and on the rate of gastric emptying in healthy volunteers. Gut 28, 166–170 (1987).
Holst, J. J. Enteroglucagon. Annu. Rev. Physiol. 59, 257–271 (1997).
Schepp, W. et al. Oxyntomodulin: a cAMP-dependent stimulus of rat parietal cell function via the receptor for glucagon-like peptide-1 (7-36)NH2. Digestion 57, 398–405 (1996).
Baggio, L. L., Huang, Q., Brown, T. J. & Drucker, D. J. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology 127, 546–558 (2004).
Dakin, C. L. et al. Oxyntomodulin inhibits food intake in the rat. Endocrinology 142, 4244–4250 (2001).
Du, X. et al. Differential effects of oxyntomodulin and GLP-1 on glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 303, E265–E271 (2012).
Parlevliet, E. T. et al. Oxyntomodulin ameliorates glucose intolerance in mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 294, E142–E147 (2008).
Pocai, A. Action and therapeutic potential of oxyntomodulin. Mol. Metab. 3, 241–251 (2013).
Date, Y. et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 141, 4255–4261 (2000).
Ariyasu, H. et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J. Clin. Endocrinol. Metab. 86, 4753–4758 (2001).
Stengel, A. et al. Identification and characterization of nesfatin-1 immunoreactivity in endocrine cell types of the rat gastric oxyntic mucosa. Endocrinology 150, 232–238 (2009).
Wren, A. M. et al. Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86, 5992 (2001).
Tschöp, M., Smiley, D. L. & Heiman, M. L. Ghrelin induces adiposity in rodents. Nature 407, 908–913 (2000).
Wren, A. M. et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 141, 4325–4328 (2000).
Tschöp, M. et al. Post-prandial decrease of circulating human ghrelin levels. J. Endocrinol. Invest. 24, RC19–RC21 (2001).
Nakazato, M. et al. A role for ghrelin in the central regulation of feeding. Nature 409, 194–198 (2001).
Date, Y. et al. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 123, 1120–1128 (2002).
Okada, T. et al. Analysis of peripheral ghrelin signaling via the vagus nerve in ghrelin receptor–restored GHSR-null mice. Neurosci. Lett. 681, 50–55 (2018).
Guan, X.-M. et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Mol. Brain Res. 48, 23–29 (1997).
Banks, W. A., Tschöp, M., Robinson, S. M. & Heiman, M. L. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J. Pharmacol. Exp. Ther. 302, 822 (2002).
Cowley, M. A. et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649–661 (2003).
Stengel, A. et al. Ghrelin and NUCB2/nesfatin-1 are expressed in the same gastric cell and differentially correlated with body mass index in obese subjects. Histochem. Cell Biol. 139, 909–918 (2013).
Oh-I, S. et al. Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature 443, 709–712 (2006).
Li, Z. et al. Peripheral effects of nesfatin-1 on glucose homeostasis. PLoS ONE 8, e71513 (2013).
Shimizu, H. et al. Peripheral administration of nesfatin-1 reduces food intake in mice: the leptin-independent mechanism. Endocrinology 150, 662–671 (2009).
Pan, W., Hsuchou, H. & Kastin, A. J. Nesfatin-1 crosses the blood-brain barrier without saturation. Peptides 28, 2223–2228 (2007).
Price, T. O., Samson, W. K., Niehoff, M. L. & Banks, W. A. Permeability of the blood-brain barrier to a novel satiety molecule nesfatin-1. Peptides 28, 2372–2381 (2007).
Goebel, M., Stengel, A., Wang, L. & Taché, Y. Central nesfatin-1 reduces the nocturnal food intake in mice by reducing meal size and increasing inter-meal intervals. Peptides 32, 36–43 (2011).
Stengel, A. et al. Central nesfatin-1 reduces dark-phase food intake and gastric emptying in rats: differential role of corticotropin-releasing factor2 receptor. Endocrinology 150, 4911–4919 (2009).
Gonzalez, R. et al. Nesfatin-1 exerts a direct, glucose-dependent insulinotropic action on mouse islet β- and MIN6 cells. J. Endocrinol. 208, R9–R16 (2011).
Nakata, M., Manaka, K., Yamamoto, S., Mori, M. & Yada, T. Nesfatin-1 enhances glucose-induced insulin secretion by promoting Ca(2+) influx through L-type channels in mouse islet β-cells. Endocr. J. 58, 305–313 (2011).
Grosse, J. et al. Insulin-like peptide 5 is an orexigenic gastrointestinal hormone. Proc. Natl Acad. Sci. USA 111, 11133–11138 (2014).
Lee, Y. S. et al. Insulin-like peptide 5 is a microbially regulated peptide that promotes hepatic glucose production. Mol. Metab. 5, 263–270 (2016).
Schalla, M. A., Taché, Y. & Stengel, A. Neuroendocrine peptides of the gut and their role in the regulation of food intake. Compr. Physiol. 11, 1679–1730 (2021).
Cani, P. D. et al. Microbial regulation of organismal energy homeostasis. Nat. Metab. 1, 34–46 (2019).
Nicholson, J. K. et al. Host-gut microbiota metabolic interactions. Science 336, 1262 (2012).
Bäckhed, F. et al. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl Acad. Sci. USA 101, 15718–15723 (2004).
Bäckhed, F., Manchester, J. K., Semenkovich, C. F. & Gordon, J. I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl Acad. Sci. USA 104, 979–984 (2007).
Duca, F. A. & Covasa, M. Current and emerging concepts on the role of peripheral signals in the control of food intake and development of obesity. Br. J. Nutr. 108, 778–793 (2012).
Arora, T. et al. Microbial regulation of the L cell transcriptome. Sci. Rep. 8, 1207 (2018).
Fredborg, M., Theil, P. K., Jensen, B. B. & Purup, S. G protein-coupled receptor120 (GPR120) transcription in intestinal epithelial cells is significantly affected by bacteria belonging to the Bacteroides, Proteobacteria, and Firmicutes phyla. J. Anim. Sci. 90(Suppl 4), 10–12 (2012).
McVey Neufeld, K. A., Mao, Y. K., Bienenstock, J., Foster, J. A. & Kunze, W. A. The microbiome is essential for normal gut intrinsic primary afferent neuron excitability in the mouse. Neurogastroenterol. Motil. 25, 183–e88 (2013).
McVey Neufeld, K. A., Perez-Burgos, A., Mao, Y. K., Bienenstock, J. & Kunze, W. A. The gut microbiome restores intrinsic and extrinsic nerve function in germ-free mice accompanied by changes in calbindin. Neurogastroenterol. Motil. 27, 627–636 (2015).
Anitha, M., Vijay-Kumar, M., Sitaraman, S. V., Gewirtz, A. T. & Srinivasan, S. Gut microbial products regulate murine gastrointestinal motility via Toll-like receptor 4 signaling. Gastroenterology 143, 1006–1016.e4 (2012).
Barajon, I. et al. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J. Histochem Cytochem. 57, 1013–1023 (2009).
De Vadder, F. et al. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc. Natl Acad. Sci. USA 115, 6458–6463 (2018).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014).
Ley, R. E. et al. Obesity alters gut microbial ecology. Proc. Natl Acad. Sci. USA 102, 11070–11075 (2005).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Ridaura, V. K. et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214 (2013).
Vrieze, A. et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143, 913–916.e7 (2012).
Cani, P. D. et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58, 1091–1103 (2009).
Everard, A. et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60, 2775–2786 (2011).
Cani, P. D. et al. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes 55, 1484–1490 (2006).
Delzenne, N. M., Cani, P. D., Daubioul, C. & Neyrinck, A. M. Impact of inulin and oligofructose on gastrointestinal peptides. Br. J. Nutr. 93(Suppl 1), S157–S161 (2005).
Cani, P. D., Dewever, C. & Delzenne, N. M. Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br. J. Nutr. 92, 521–526 (2004).
Cani, P. D., Joly, E., Horsmans, Y. & Delzenne, N. M. Oligofructose promotes satiety in healthy human: a pilot study. Eur. J. Clin. Nutr. 60, 567–572 (2006).
Singh, A., Zapata, R. C., Pezeshki, A., Reidelberger, R. D. & Chelikani, P. K. Inulin fiber dose-dependently modulates energy balance, glucose tolerance, gut microbiota, hormones and diet preference in high-fat-fed male rats. J. Nutr. Biochem. 59, 142–152 (2018).
Cani, P. D. et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50, 2374–2383 (2007).
Hosoi, T., Okuma, Y., Matsuda, T. & Nomura, Y. Novel pathway for LPS-induced afferent vagus nerve activation: Possible role of nodose ganglion. Auton. Neurosci. 120, 104–107 (2005).
de La Serre, C. B., de Lartigue, G. & Raybould, H. E. Chronic exposure to low dose bacterial lipopolysaccharide inhibits leptin signaling in vagal afferent neurons. Physiol. Behav. 139, 188–194 (2015).
de Lartigue, G., Barbier de la Serre, C., Espero, E., Lee, J. & Raybould, H. E. Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am. J. Physiol. Endocrinol. Metab. 301, E187–E195 (2011).
Peters, J. H., Karpiel, A. B., Ritter, R. C. & Simasko, S. M. Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology 145, 3652–3657 (2004).
Peters, J. H., Ritter, R. C. & Simasko, S. M. Leptin and CCK selectively activate vagal afferent neurons innervating the stomach and duodenum. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1544–R1549 (2006).
de Lartigue, G., Barbier de la Serre, C., Espero, E., Lee, J. & Raybould, H. E. Leptin resistance in vagal afferent neurons inhibits cholecystokinin signaling and satiation in diet induced obese rats. PLoS ONE 7, e32967 (2012).
Thomas, C., Pellicciari, R., Pruzanski, M., Auwerx, J. & Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 7, 678–693 (2008).
Trabelsi, M.-S., Lestavel, S., Staels, B. & Collet, X. Intestinal bile acid receptors are key regulators of glucose homeostasis. Proc. Nutr. Soc. 76, 192–202 (2017).
Trabelsi, M.-S. et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 6, 7629–7629 (2015).
Zhang, S. Y. et al. FXR in the dorsal vagal complex is sufficient and necessary for upper small intestinal microbiome-mediated changes of TCDCA to alter insulin action in rats. Gut 70, 1675–1683 (2021).
Waise, T. M. Z., Lim, Y. M., Danaei, Z., Zhang, S. Y. & Lam, T. K. T. Small intestinal taurochenodeoxycholic acid-FXR axis alters local nutrient-sensing glucoregulatory pathways in rats. Mol. Metab. 44, 101132 (2021).
Fang, S. et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 21, 159–165 (2015).
Pathak, P. et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 68, 1574–1588 (2018).
Pathak, P. et al. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J. Biol. Chem. 292, 11055–11069 (2017).
Katsuma, S., Hirasawa, A. & Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 329, 386–390 (2005).
den Besten, G. et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a PPARγ-dependent switch from lipogenesis to fat oxidation. Diabetes 64, 2398–2408 (2015).
Lu, Y. et al. Short chain fatty acids prevent high-fat-diet-induced obesity in mice by regulating G protein-coupled receptors and gut microbiota. Sci. Rep. 6, 37589 (2016).
Li, Z. et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 67, 1269–1279 (2018).
Tolhurst, G. et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61, 364–371 (2012).
Brooks, L. et al. Fermentable carbohydrate stimulates FFAR2-dependent colonic PYY cell expansion to increase satiety. Mol. Metab. 6, 48–60 (2017).
Larraufie, P. et al. SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci. Rep. 8, 74–74 (2018).
Petersen, N. et al. Generation of L cells in mouse and human small intestine organoids. Diabetes 63, 410–420 (2014).
Nøhr, M. K. et al. Expression of the short chain fatty acid receptor GPR41/FFAR3 in autonomic and somatic sensory ganglia. Neuroscience 290, 126–137 (2015).
Frost, G. et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 5, 3611 (2014).
Wikoff, W. R. et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl Acad. Sci. USA 106, 3698–3703 (2009).
Côté, F. et al. Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc. Natl Acad. Sci. USA 100, 13525–13530 (2003).
Blackshaw, L. A., Brookes, S. J., Grundy, D. & Schemann, M. Sensory transmission in the gastrointestinal tract. Neurogastroenterol. Motil. 19, 1–19 (2007).
Hillsley, K. & Grundy, D. Serotonin and cholecystokinin activate different populations of rat mesenteric vagal afferents. Neurosci. Lett. 255, 63–66 (1998).
Zhu, J. X., Zhu, X. Y., Owyang, C. & Li, Y. Intestinal serotonin acts as a paracrine substance to mediate vagal signal transmission evoked by luminal factors in the rat. J. Physiol. 530, 431–442 (2001).
Kirchgessner, A. L., Tamir, H. & Gershon, M. D. Identification and stimulation by serotonin of intrinsic sensory neurons of the submucosal plexus of the guinea pig gut: activity-induced expression of Fos immunoreactivity. J. Neurosci. 12, 235 (1992).
Michel, K., Sann, H., Schaaf, C. & Schemann, M. Subpopulations of gastric myenteric neurons are differentially activated via distinct serotonin receptors: projection, neurochemical coding, and functional implications. J. Neurosci. 17, 8009 (1997).
Mazda, T., Yamamoto, H., Fujimura, M. & Fujimiya, M. Gastric distension-induced release of 5-HT stimulates c-fos expression in specific brain nuclei via 5-HT3 receptors in conscious rats. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G228–G235 (2004).
Bellono, N. W. et al. Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 170, 185–198.e16 (2017).
Barrett, E., Ross, R. P., O’Toole, P. W., Fitzgerald, G. F. & Stanton, C. γ-Aminobutyric acid production by culturable bacteria from the human intestine. J. Appl. Microbiol. 113, 411–417 (2012).
Meng, F. et al. New inducible genetic method reveals critical roles of GABA in the control of feeding and metabolism. Proc. Natl Acad. Sci. USA 113, 3645–3650 (2016).
Tian, J. et al. Oral treatment with γ-aminobutyric acid improves glucose tolerance and insulin sensitivity by inhibiting inflammation in high fat diet-fed mice. PLoS ONE 6, e25338 (2011).
Yunes, R. A. et al. GABA production and structure of gadB/gadC genes in Lactobacillus and Bifidobacterium strains from human microbiota. Anaerobe 42, 197–204 (2016).
Kootte, R. S. et al. Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab. 26, 611–619.e6 (2017).
Marques, T. M. et al. Influence of GABA and GABA-producing Lactobacillus brevis DPC 6108 on the development of diabetes in a streptozotocin rat model. Benef. Microbes 7, 409–420 (2016).
Mojibian, M., Glavas, M. M. & Kieffer, T. J. Engineering the gut for insulin replacement to treat diabetes. J. Diabetes Investig. 7(Suppl 1), 87–93 (2016).
Bouchi, R. et al. FOXO1 inhibition yields functional insulin-producing cells in human gut organoid cultures. Nat. Commun. 5, 4242 (2014).
Talchai, C., Xuan, S., Kitamura, T., DePinho, R. A. & Accili, D. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nat. Genet. 44, 406–412, S1 (2012).
Chen, Z. et al. Incorporation of therapeutically modified bacteria into gut microbiota inhibits obesity. J. Clin. Invest. 124, 3391–3406 (2014).
Ramachandran, G. & Bikard, D. Editing the microbiome the CRISPR way. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 374, 20180103 (2019).
Wang, L. et al. Engineered bacteria of MG1363-pMG36e-GLP-1 attenuated obesity-induced by high fat diet in mice. Front. Cell. Infect. Microbiol. 11, 595575 (2021).
Li, B. et al. Microbiota depletion impairs thermogenesis of brown adipose tissue and browning of white adipose tissue. Cell Rep. 26, 2720–2737.e5 (2019).
Wen, J., Bo, T., Zhang, X., Wang, Z. & Wang, D. Thermo-TRPs and gut microbiota are involved in thermogenesis and energy metabolism during low temperature exposure of obese mice. J. Exp. Biol. 223, jeb218974 (2020).
Blouet, C. & Schwartz, G. J. Duodenal lipid sensing activates vagal afferents to regulate non-shivering brown fat thermogenesis in rats. PLoS ONE 7, e51898 (2012).
Grasset, E. & Burcelin, R. The gut microbiota to the brain axis in the metabolic control. Rev. Endocr. Metab. Disord. 20, 427–438 (2019).
Acknowledgements
This work was supported by grants from National Institute of Diabetes and Digestive and Kidney Diseases (R01DK121804) and National Institute of Food and Agriculture (67017-29252)
Author information
Authors and Affiliations
Contributions
HRW drafted the manuscript, SNW designed the figure, and HRW, SNW, and FAD revised and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wachsmuth, H.R., Weninger, S.N. & Duca, F.A. Role of the gut–brain axis in energy and glucose metabolism. Exp Mol Med 54, 377–392 (2022). https://doi.org/10.1038/s12276-021-00677-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00677-w
This article is cited by
-
Effects of dietary fibre on metabolic health and obesity
Nature Reviews Gastroenterology & Hepatology (2024)
-
The Novel Insight of Gut Microbiota from Mouse Model to Clinical Patients and the Role of NF-κB Pathway in Polycystic Ovary Syndrome
Reproductive Sciences (2024)
-
Gut microbiota and type 2 diabetes mellitus: a focus on the gut-brain axis
Endocrine (2024)
-
Diabetic Encephalopathy: Role of Oxidative and Nitrosative Factors in Type 2 Diabetes
Indian Journal of Clinical Biochemistry (2024)
-
Comprehensive insights: unraveling the mechanisms of gut commensals in glucose metabolism regulation
Science China Life Sciences (2024)
