
Abstract
The specific role of cadherin receptors in cytotoxicity involving Cry toxins of Bacillus thuringiensis and their interactions with cell membrane has not been defined. To elucidate the involvement of toxin-membrane and toxin-receptor interactions in cytotoxicity, we established a cell-based system utilizing High Five insect cells stably expressing BT-R1, the cadherin receptor for Cry1Ab toxin. Cry1Ab toxin is incorporated into cell membrane in both oligomeric and monomeric form. Monomeric toxin binds specifically to BT-R1 whereas incorporation of oligomeric toxin is nonspecific and lipid dependent. Toxin oligomers in the cell membrane do not produce lytic pores and do not kill insect cells. Rather, cell death correlates with binding of the Cry1Ab toxin monomer to BT-R1, which apparently activates a Mg2+-dependent cellular signaling pathway.
Similar content being viewed by others

Introduction
Protein toxins are major virulence factors for many bacterial pathogens that cause diseases in plants and animals.1, 2 Toxins affect host cells primarily in two ways. They form lytic pores in cell membrane by self-assembly of toxin oligomers or they elicit toxic reactions in cells through various events, including signal transduction, altered metabolism and inflammation.1, 2, 3 In either situation, toxin molecules may incorporate into cell membrane as oligomers like the lytic pore-forming toxins and the A-B family of toxins.4, 5, 6, 7, 8 X-ray crystallographic studies have shown that toxin oligomer complexes most likely assemble in β-barrel or umbrella-like channels in lipid membranes.9, 10, 11, 12 However, the involvement of such oligomers in cell death is not well understood because most toxin oligomer complexes themselves apparently do not punch holes in membrane and do not simply cause cell death by lysis.13, 14 Instead, the interaction of toxins with cells involves complicated pathways, the end result of which is cell death.15, 16, 17
The Cry proteins of Bacillus thuringiensis (Bt) represent more than 100 phylogenetically related toxins with varied entomopathogenic activities.18 Like many other bacterial toxins, Cry toxins incorporate into lipid bilayer rafts as well as brush border membrane vesicles (BBMV).19 Lipid bilayer- and BBMV-associated Cry toxin molecules have been reported to appear in oligomeric (dimeric and tetrameric) and monomeric forms,20, 21 and these associations have been correlated with changes in membrane permeability.22 The oligomeric forms of Cry toxin were presumed to be pores that cause cell death by osmotic lysis. In other words, Cry toxin action was believed to involve the formation of lytic pores that are the result of assembly of toxin molecules as oligomers in membrane.22, 23, 24, 25 However, there is no definitive correlation between the association of Cry toxin oligomer complexes and toxic action. Studies of mutated Cry toxin proteins have shown that neither the toxin oligomer complex nor commensurate changes in membrane vesicle permeability correlate directly with toxicity.26, 27, 28, 29 Furthermore, Cry toxin proteins can interact with lipid bilayer rafts and BBMV in the absence of a cadherin receptor, which determines specific insecticidal activity of Cry toxins.
The cadherin receptor BT-R1, identified in the tobacco hornworm Manduca sexta,30, 31, 32 represents a family of cadherins that are expressed in the midgut epithelium of various insects susceptible to Cry1A toxins. Cry toxins bind to their respective cadherin receptors with high affinity and specificity, the disruption or absence of which results in loss of susceptibility to Cry toxin.33, 34, 35 So far, the role of BT-R1 in the cytotoxic action of Cry toxin has not been defined.
In the present study, we utilized a cell-based system consisting of High Five (H5) insect cells stably expressing BT-R1 to determine whether toxicity of Cry1Ab is brought about by lytic pore formation or by specific interaction of the toxin with BT-R1. This particular cell-based system allowed us to investigate toxin-membrane and toxin-receptor interactions, along with toxin-induced cellular responses simultaneously. Results of our studies reveal that Cry1Ab toxin does form oligomeric complexes in the membranes of cells expressing BT-R1 as well as in those of cells devoid of receptor. Toxin oligomers integrated into cell membrane do not produce lytic pores and do not kill the cells whereas monomeric Cry1Ab toxin specifically binds to BT-R1, activating a Mg2+-dependent cellular signaling pathway that leads to necrotic cell death.
Results
Cry1Ab toxin induces cell death only in the presence of BT-R1
H5 cells originate from undifferentiated ovarian cells of the cabbage looper Trichoplusia ni.36 They do not express receptors for Cry toxins and Cry1Ab is not toxic to the cells. Cells expressing BT-R1 (S5) were generated from H5 cells by establishing stabilized expression of the full-length BT-R1 cDNA under the insect actin promoter with a nonlytic insect expression vector. BT-R1 was expressed as a membrane protein and expression was localized in the cytoplasmic membrane of the S5 cells as determined by immunofluorescent staining using anti-BT-R1 antibody (Figure 1a, S5). Small areas inside the cell located between the cell nucleus and cell membrane also stained positively, suggesting that expressed BT-R1 may be present in ER vesicles. Western blot experiments with protein extracts from cytoplasmic and membrane pellet fractions of the cells also showed the presence of BT-R1 in both fractions (Figure 1b, lanes S and P of S5, respectively). Even though the deduced molecular weight of BT-R1 is 190 kDa, based on amino-acid sequence of the protein ((NCBI access ID: AAG37912), expressed protein was detected as a 210-kDa band in S5 cells, indicating that post-translational modification of the protein is similar to that of natural BT-R1 isolated directly from M. sexta larvae.31 Immunoligand-binding assays using Cry1Ab toxin indicated that Cry1Ab specifically bound to BT-R1 expressed in the transfected S5 cells (Figure 1c). No BT-R1 was detected in H5 cells by either immunofluorescent staining or Western blotting (Figure 1).

Expression of BT-R1 in S5 cells. (a) S5 and H5 cells were immunostained by using BT-R1 antibody (green). Cell nuclei were stained by Hoechst 33342 (blue). The merged view of S5 cells shows the localization of BT-R1 on the cell membrane, after the twin cells completed cell division. Bar=10 μm. (b) Supernatant (S) corresponding to cytoplasmic proteins and pellet (P) corresponding to membrane associated proteins were analyzed by Western blotting using BT-R1 antibody. BT-R1 (210 kDa) was detected in protein extracts obtained from S5 cells. A nonspecific weak band (70 kDa) was referenced as internal control in the blots. (c) Binding of Cry1Ab toxin to BT-R1 (210 kDa) was intense in S5 protein extracts, as detected in immunoligand blotting assay by using Cry1Ab antibody
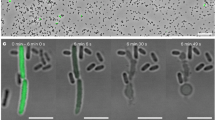
Cry1Ab was not toxic to H5 cells. H5 cells underwent normal cell growth and cell division when they were exposed to 180 nM Cry1Ab toxin in the culture medium (Figure 2a). Conversely, the vast majority of S5 cells were killed by 180 nM Cry1Ab toxin as seen by Trypan blue staining (Figure 2a). In addition, toxin-exposed S5 cells exhibited dramatic morphological changes, such as altered size, shape and overall appearance. S5 cells were sensitive to Cry1Ab in a dose-dependent manner (Figure 2b). Whereas, 180 nM toxin killed most of the S5 cells (>80% of the entire population), a lower concentration (15 nM) of the toxin killed only 7% of the cells (Figure 2b). The LC50 of Cry1Ab for S5 cells was 65 nM.

Selective cytotoxicity of Cry1Ab on S5 cells. (a) Trypan blue staining of nuclei represents dead S5 cells under Cry1Ab toxin (180 nM) treatment. The toxin had no lethal effect on H5 cells. Bar=40 μm. (b) S5 cells were sensitive to Cry1Ab in a dose-dependent manner, whereas H5 cells were insensitive to the toxin. Data are presented as the mean±S.D. of six experiments
Cry1Ab oligomers insert into cellular membrane regardless of BT-R1
Studies of artificial lipid rafts and BBMV prepared from larval midguts have shown that Cry toxins are incorporated into these lipid membranes in both oligomeric and monomeric forms.37 The toxin oligomers, usually in dimeric and tetrameric form, are SDS-resistant and could be detected by SDS-PAGE Western blotting.21, 26, 38 We were interested in determining whether Cry1Ab interacts with the cytoplasmic membrane of living cells in a similar way, and, if so, whether these interactions contribute to cytotoxicity. Therefore, we analyzed toxin–membrane interactions in S5 and H5 cells by examining protein samples prepared from membrane and cytoplasmic fractions before and after treatment of the cells with Cry1Ab. Antibody against Cry1Ab was used to detect the toxin in protein fractions corresponding to membrane pellet (P) and supernatant containing soluble proteins (S). After treatment with Cry1Ab, the toxin was detected only in the membrane pellet (Figure 3a, lanes 4 and 6), not in the cytoplasmic fraction (Figure 3a, lanes 3 and 5), indicating that Cry1Ab incorporates into the membrane of living cells. The dimeric (∼140 kDa) and tetrameric (∼240 kDa) forms of Cry1Ab were detected in the membrane fractions of both S5 and H5 cells (Figure 3a, lanes 4 and 6, black arrows), demonstrating that toxin oligomers insert into the membrane regardless of receptor. Moreover, the toxin oligomers also were SDS-resistant. Toxin oligomers remained intact irrespective of sample treatment prior to gel electrophoresis and Western blotting. Heat treatment (including boiling) in the presence of β-mercaptoethanol and SDS did not dissociate toxin oligomers (data not shown). Significantly, the monomeric form of the toxin was detected only in the membrane fraction of S5 cells (Figure 3a, lane 4, white arrow) containing BT-R1. Whether BT-R1 associated with an oligomeric form of the toxin or exclusively with the monomer in situ is not known.

Interaction between Cry1Ab and cell membrane. (a) After treatment with Cry1Ab (180 nM), the incorporation of toxin into S5 and H5 cell membrane fractions (lane P) and cytoplasmic fractions (lane S) were assessed by Western blotting using antibody against Cry1Ab. Toxin was not detected in the soluble cytoplasmic portion of cell protein extracts (lanes 3 and 5). Toxin treatment (+) and no treatment (−) are indicated. The black arrows point to Cry1Ab dimers (140 kDa) and tetramers (240 kDa). The white arrow points to the monomeric (60 kDa) form of Cry1Ab, which was unique to S5 cells. Purified Cry1Ab was used as a control (lane T). (b) Dose-dependent incorporation of Cry1Ab into the membrane of S5 and H5 cells. Lanes represent membrane fractions from cells treated with Cry1Ab at various concentrations. (c) Time-dependent association of Cry1Ab monomer with S5 cell membrane. Cells were treated with toxin (180 nM) before extracting cellular membrane proteins. Incubation times (0–20 min) are indicated on the lanes. (d) Time-dependent Cry1Ab incorporation profile for toxin oligomers and monomer. S5 and H5 cells were incubated with Cry1Ab (180 nM) before extracting cellular membrane proteins. Incubation times (0–60 min) are indicated on the lanes. Black arrows point to the oligomeric forms of Cry1Ab. White arrows point to toxin monomer observed only in the membrane fraction of S5 cells. (e) Oligomeric and monomeric forms of the toxin incorporated into the S5 cell membrane were compared quantitatively based on the results obtained from time-dependent incorporation profiles of toxin tetramer and monomer. The intensity of the monomer band associated with membrane after 15 min of incubation was set as the baseline for comparison. The amount of toxin monomer associated with cell membrane reached saturation within 15 min (black bars) whereas incorporation of toxin oligomers into the cell membrane increased according to time of exposure to the toxin (gray bars). (f) Oligomeric forms (dimer and tetramer) of Cry1Ab, similar to those observed in the membrane fractions of S5 and H5 cells, were detected in the membrane of synthetic liposomes prepared with different lipid compositions. L1, L2 and L3 represent different liposomes composed of different lipids (PC, phosphatidylcholine; PS, phosphatidylserine; Chol, cholesterol; SM, sphingomyelin). The ratios of lipids used in the liposome preparations are shown. (g) Passage of membrane-incorporated Cry1Ab oligomers from generation to generation. Membrane fractions of H5 cells treated with Cry1Ab were examined for two generations by Western blotting using Cry1Ab antibody. After toxin treatment, H5 cells remained viable and underwent normal growth and cell division. G0 represents cells before toxin treatment. Toxin oligomers were detected in the membrane fractions of cells throughout two generations after toxin treatment, albeit in decreasing amounts. G0+T represents cells after toxin treatment. G1 is the first generation of cells derived from G0+T. G2 is the second generation of cells derived from G1. G2+T represents the G2 cells after treatment with Cry1Ab. Membrane-incorporated Cry1Ab oligomers were diluted from one generation to the other
To better understand the difference in the Cry1Ab-membrane-incorporation profiles of S5 and H5 cells, we analyzed toxin–membrane interaction in a dose- and time-dependent manner. In the dose-dependent experiments, S5 and H5 cells were treated for 1 h with different concentrations of Cry1Ab toxin before measuring toxin incorporation (Figure 3b). Comparable levels of oligomeric (120-kDa dimer and 240-kDa tetramer, black arrows) and monomeric toxin (60 kDa, white arrow) were detected in the membrane of S5 cells when treated with different concentrations (60, 120 and 180 nM) of toxin (Figure 3b). This result demonstrates that incorporation of Cry1Ab into S5 cell membrane does not depend on toxin concentration over the range tested. In contrast, in H5 cells, incorporation of toxin dimers and tetramers (black arrows) into the membrane fractions occurred in a dose-dependent fashion (Figure 3b). Significantly, no monomeric form of the toxin appeared in H5 cells (Figure 3b), confirming that incorporation of toxin monomers is unique to S5 cells expressing BT-R1.
To understand the dynamics of toxin–membrane interaction, incorporation of toxin by S5 and H5 cells was measured at 15 min intervals for 60 min. Incorporation profiles for monomeric and oligomeric toxin were different (Figure 3c and d). Monomeric toxin was incorporated into the S5 cell membrane rapidly. Indeed, monomer was detected within 5 min of toxin exposure, reaching a saturation level in 15 min (Figure 3c, white arrow). Prolonged incubation (60 min) of S5 cells with Cry1Ab toxin did not show any increase in the amount of toxin monomer incorporated into membrane (Figure 3d, white arrow). No toxin monomer was detected in H5 cells (Figure 3d). Incorporation of oligomeric toxin into both S5 and H5 cells happened relatively slowly, appearing in 15 min after addition of toxin (Figure 3d, black arrows) and increasing steadily during the period of incubation with the toxin. These results indicate that the specificity of toxin interaction, which leads to cytotoxicity, correlates directly with the association of monomeric toxin with the S5 cells expressing BT-R1. The saturation kinetics of monomer incorporation into the S5 cells points out the high specificity and affinity of Cry1Ab toxin for its binding partner BT-R1 on the cell surface at a given time (Figure 3e, dark bars). In contrast, the increase in incorporation of Cry1Ab toxin oligomers into cell membrane is not receptor-dependent, but relies on the availability of lipid membrane (Figure 3e, gray bars).
To confirm that Cry1Ab oligomers assembled in the cell membrane of both S5 and H5 cells in a lipid-dependent fashion, we measured the incorporation of Cry1Ab toxin into protein-free liposomes composed of different lipids. As seen in Figure 3f, Cry1Ab was incorporated into liposomes in similar oligomeric forms (dimer and tetramer) as in cellular membrane (Figure 3b). Certain lipid components are required for bacterial toxin oligomer assembly and insertion. For example, cholesterol (Chol) and sphingomyelin (SM) are two known specific receptors for pore-forming bacterial toxins.39, 40 Therefore, we were interested in knowing whether a lipid receptor such as Chol or SM is required for toxin–membrane interaction of Cry1Ab. Cry1Ab oligomer complexes were incorporated equally into liposomes composed of different lipid molecules, with or without Chol and SM (Figure 3f). These results demonstrate that incorporation of Cry1Ab oligomer into cell membrane is a lipid-dependent property, which does not rely on a specific receptor, as was reported recently.41
To further substantiate our observations that nonspecific interaction of the Cry1Ab toxin with cell membrane does not lead to cell death and that the assembly of toxin oligomers in the membrane does not represent lytic pores, we examined the growth and division of receptor-free H5 cells after Cry1Ab treatment. In these experiments, the viability and growth of H5 cells were monitored along with the incorporation of Cry1Ab into the cell membrane for three continuous passages of toxin-treated cells. H5 cells were incubated first in the presence of Cry1Ab toxin (180 nM) for 8 h and then, in the absence of toxin. Both toxin-treated cells and untreated cells remained viable through the three passages with a doubling time of 18 h. Interestingly, decreasing amounts of Cry toxin oligomers were detected in the membrane fractions of cells at each passage (Figure 3g; initial generation (G0)+T to second generation (G2)). This finding suggests that that membrane-incorporated toxin is carried along with the cells during cell division and that toxin incorporation into the cell membrane does not harm the cells. The G2 cells contained a relatively small amount of toxin oligomers that apparently was inherited from the parent cells (first generation (G1)). The G2 cells were capable of incorporating additional toxin oligomers into their cell membrane (Figure 3g; G2+T). Obviously, Cry1Ab oligomers do not represent lytic pores because they do not affect cell growth and division when incorporated into cell membrane.
Inhibition of toxin-receptor interaction prevents Cry1Ab-induced cell death
Since Cry1Ab binding to BT-R1 on S5 cells results in cell death, we wanted to know whether inhibiting the binding of toxin to the BT-R1 receptor averts cell death. Previously, we showed that blocking the binding of Cry1Ab toxin to BT-R1 using the toxin-binding region (TBR) of BT-R1 thwarts the lethal effect of the toxin in M. sexta larvae.32, 42, 43 In this study, we used a 12 kDa fragment (Figure 4a) containing TBR linked to maltose-binding protein (MBP), which facilitated expression and purification of the fragment (Figure 4b). MBP did not interact with the toxin (Figure 4b). The Cry1Ab toxin was bound specifically to TBR as shown by immunoligand binding analysis (Figure 4b). Therefore, we used the TBR to inhibit binding of Cry1Ab to BT-R1 in living cells. Blocking the receptor active site of the toxin with equimolar TBR prevented toxin from binding to BT-R1 on the S5 cells (Figure 4c, inset). Also as seen in Figure 4c, toxicity decreased in a dose-dependent manner when S5 cells were treated with different molar ratios of TBR and Cry1Ab (0.25 : 1–2 : 1) premixed for 30 min. Toxicity was inhibited maximally when soluble TBR was present at a 1 : 1 M ratio with Cry1Ab or higher (Figure 4c).

Inhibition of Cry1Ab binding and cytotoxicity. (a) The toxin-binding region (TBR) has been defined within the extracellular membrane proximate area of BT-R1.32 (b) The specific binding of Cry1Ab to TBR is demonstrated by immunoligand binding assay. MBP-TBR is the fusion protein of TBR and maltose-binding protein (MBP), which can be digested to MBP and TBR by thermolysin protease, as shown in the SDS gel (left panel). Cry1Ab bound to both TBR and MBP-TBR, but not MBP, in immunoligand binding assays (right panel). (c) S5 cells were treated with Cry1Ab (180 nM) that was preincubated with different concentrations of TBR. Cytotoxicity (Y-axis) brought about by toxin-TBR mixtures were recorded. The X-axis indicates the molar ratios of TBR to Cry1Ab in the preincubation step. At 1 : 1 M ratio between Cry1Ab and TBR, the cytotoxic action was eliminated, indicating that binding of toxin molecules to the receptor on S5 cells was inhibited. Data are represented as the mean ±S.D. of six experiments. Inhibition of Cry1Ab binding to BT-R1 by TBR was confirmed by immunoligand blotting analysis using protein samples prepared from S5 cells. Toxin interaction with BT-R1 was eliminated when toxin was preincubated with TBR (+TBR) as compared to untreated toxin (-TBR), which bound to the receptor on blots (inset c). (d) Membrane-associated Cry1Ab monomers were specifically blocked by TBR. Cry1Ab was incubated with TBR at 1 : 1 M ratio for 30 min and then added to cell culture medium at different concentrations as indicated on the respective lanes. Membrane-incorporated Cry1Ab was detected using antibody against the toxin. Toxin dimers and tetramers (black arrows) accumulated in the membrane fractions of both S5 and H5 cells as previously observed (Figure 3) whereas association of Cry1Ab monomers with S5 cell membrane was abolished completely upon preincubation of toxin with TBR. Purified Cry1Ab (lane T) is shown as control for the monomeric form of the toxin
Importantly, when binding of toxin to the receptor was blocked by the TBR, no monomeric Cry1Ab toxin was detectable in S5 cell membrane whereas oligomers of the toxin were readily apparent (Figure 4d). Evidently, the association of monomeric, not oligomeric, toxin with cell membrane is the manifestation of the specific interaction of toxin monomer with BT-R1 on the cell surface. The incorporation of toxin oligomers into cell membrane is not only nonspecific but inconsequential as well.
Cytotoxic action of Cry1Ab involves Mg2+-dependent signaling downstream of toxin binding to BT-R1
Previously, we showed that calcium is required to maintain the structural integrity of BT-R144 and that binding of Cry1Ab toxin to BT-R1 inhibits calcium-induced adhesion of midgut epithelial vesicles derived from M. sexta.45 We were interested in knowing whether the binding of Cry1Ab toxin to BT-R1 and subsequent cell death are dependent on calcium ions. To answer this question, we tested the binding of toxin to BT-R1 of S5 cells in the presence of the divalent cation chelators ethylenediaminetetra-acetic acid (EDTA) and ethylene-glycol-bis(2-aminoethyl ether)-N,N,N,N′-tetraacetic acid (EGTA). The presence of EDTA (5 mM) and EGTA (5 mM) did not affect the binding of Cry1Ab toxin to BT-R1 as determined by immunoligand blots using Cry1Ab antibody (Figure 5a). To examine what effect Cry1Ab has on S5 cells in the absence of extracellular Ca2+, S5 cells were preincubated with 5 mM EDTA or EGTA and then treated with toxin. Neither EDTA nor EGTA itself adversely affected the S5 cells (data not shown). As can be seen in Figure 5b, toxin was readily incorporated into cell membrane as both oligomers (black arrows) and monomer (white arrows). The amount of toxin oligomer and monomer incorporated into cell membrane was similar to that for cells not treated with EDTA or EGTA. Toxin was not detected in the soluble portion (Figure 5b, S; also see Figure 3a, S) of the cell lysates, suggesting that the toxin is localized in the membrane and is not internalized across the cell membrane. Fluorescent microscopic analysis confirmed that the interaction of Cry1Ab with the S5 cells was not affected by removal of Ca2+, or any other divalent cation, by EDTA or EGTA (Figure 5c).

Effects of chelating agents on toxin–receptor binding and cytotoxic action of Cry1Ab. (a) Binding of Cry1Ab toxin to BT-R1 in the presence of EDTA and EGTA by immunoligand blotting assay. The presence of EDTA (5 mM) and EGTA (5 mM) did not affect the binding of Cry1Ab to the BT-R1 in the S5 cell lysates. (b) Cry1Ab monomer and oligomers were detected in the cellular fractions of EDTA- and EGTA-treated S5 cells by Western blotting. Toxin oligomers and monomer were detected in the membrane pellets (P) after Cry1Ab treatment (+) as compared to samples that were not treated (−). Neither EDTA nor EGTA treatment affected the membrane-association profiles for toxin oligomers and monomer. Black arrows point to Cry1Ab dimers and tetramers. White arrow points to the monomeric form of Cry1Ab. Purified Cry1Ab (lane T) is shown as control for the monomeric form of the toxin. (c) Cry1Ab toxin (T) bound to S5 cells within 10 min of toxin treatment as detected by immunofluorescent staining (green) using antibody against Cry1Ab. Cell nuclei were stained with Hoechst 33342 (blue). Treatment with EDTA or EGTA did not impair Cry1Ab toxin binding to S5 cells. No binding of toxin to H5 cells occurred. Bar=10 μm. (d) Trypan blue staining of nuclei of S5 cells pre-treated with EDTA or EGTA. EDTA-treated cells (EDTA+Cry1Ab) were protected from the cytotoxic action of Cry1Ab whereas EGTA-treated cells (EGTA+Cry1Ab) were not. Cry1Ab cytotoxicity was recovered by addition of Mg2+ to the medium of EDTA-treated S5 cells 10 min before toxin addition (EDTA+Mg2++Cry1Ab) whereas addition of Ca2+ (EDTA+Ca2++Cry1Ab) did not, demonstrating that the cytotoxic action of Cry1Ab depends strictly on the presence of Mg2+. Bar=50 μm
Although toxin–receptor and toxin–membrane interactions are not dependent on calcium, it was surprising to see that EDTA, not EGTA, completely abolished toxin-induced death of the S5 cells (Figure 5d). Cells pretreated with EGTA were fully susceptible to Cry1Ab toxin. EGTA preferentially chelates Ca2+ whereas EDTA chelates Mg2+ as well as Ca2+. To determine whether Mg2+ is required for cytotoxicity, Mg2+ (5 mM) and Ca2+ (5 mM) each were added separately to cells pretreated with EDTA. Remarkably, Mg2+ restored Cry1Ab-mediated cytotoxicity. Ca2+ had no such effect (Figure 5d). Apparently, binding of Cry1Ab to BT-R1, which is requisite for cytotoxicity, is linked to a Mg2+-dependent signaling pathway associated with cell death.
Discussion
The postulated mechanism of Cry toxin action is that oligomerized toxin inserts into the cell membrane and forms lytic pores, which brings about a drastic ion flux that disrupts and destroys the epithelial cells lining the midgut of a susceptible insect.22, 25 This assumption is based on studies of Cry toxin interaction with synthetic lipid rafts and BBMV prepared from insect midgut tissue.46 Although this mechanism, as proposed, has been generally accepted, no direct relationship between pore formation and cytotoxicity has been demonstrated because artificial membrane systems are not physiologically active. More importantly, the participation of a cadherin receptor such as BT-R1 has not been reconciled in the pore formation scenario. Since the primary determinant of cytotoxicity is binding of toxin to a specific receptor, the involvement of receptor cannot be dismissed. Certainly, correlations made between the formation of a toxin oligomer complex in membrane and cytotoxic action are not consistent. For example, (i) Cry toxin can insert into bilayer lipid rafts and BBMV irrespective of toxin receptors,20 (ii) toxin oligomer complexes can form in BBMV prepared from insects that are not susceptible to Cry toxin29, 47 and (iii) Cry toxin mutants that are not insecticidal induce changes in membrane permeability similar to wild-type toxin.27, 28
In the present study, we used an H5 insect cell-based system that allowed us to simultaneously correlate toxin–receptor and toxin–membrane interactions with cellular responses associated with cytotoxicity. An important distinction of this system is the ability to examine incorporation of Cry1Ab toxin into cell membrane and to monitor the interaction of the toxin with BT-R1 in live insect cells. We learned that Cry1Ab toxin is incorporated into cell membrane in oligomeric (dimeric and tetrameric) forms in cells stably expressing BT-R1 (S5) as well as in receptor-free (H5) cells (Figure 3a). Importantly, the Cry1Ab toxin killed S5 cells but not H5 cells. Cytotoxicity and cell death were the direct result of univalent binding of toxin monomers to BT-R1 expressed on the cell surface. The oligomeric form of Cry1Ab toxin was not involved in the cytotoxic pathway. Blocking the interaction of monomeric toxin with BT-R1 prevented toxicity and cell death but it did not interfere with incorporation of oligomeric toxin into the cell membrane (Figure 4d). H5 cells devoid of BT-R1 remained viable upon exposure to Cry1Ab (up to 800 nM) and continued to grow and divide successfully (Figure 3g). That toxin molecules were incorporated into the cell membrane of H5 cells as oligomers and were maintained there through several generations without any consequence to the cells is noteworthy. We also observed toxin oligomers in protein-free liposomes, indicating that such incorporation is lipid- and not receptor-dependent. Most significantly, incorporation of Cry1Ab oligomers into cell membrane does not result in the formation of lytic pores and, therefore, argues against the ‘pore-formation model’ postulated previously.22, 23, 24, 25
In the pore-forming model of Cry toxin action, the toxin monomers are considered precursors to oligomeric assembly.21, 25 The formation of toxin oligomer also has been postulated to be the result of the interaction of monomers that were bound to the cadherin receptor.48 Our results clearly show differential association profiles for the oligomeric and monomeric forms of Cry1Ab toxin in cell membrane, the consequence of two distinct modes of interaction with the insect cells. Both monomers and oligomers were associated with S5 cells expressing BT-R1 whereas no monomers, only oligomers, were associated with H5 cells devoid of the receptor (Figure 3a–d). Indeed, the two forms of the toxin appear to be mutually exclusive relative to toxin action because binding of monomer to BT-R1 on S5 cells, along with cytotoxicity, was precluded by the TBR without inhibiting oligomer incorporation into membrane. Moreover, binding of toxin monomers to BT-R1 on S5 cells was extremely rapid, regardless of toxin concentration, reaching saturation immediately after addition of toxin (Figure 3c–e). In contrast, the assembly of oligomers in cell membrane with or without BT-R1 was similar. Incorporation of toxin oligomers was dose- and time-dependent, with incorporation continuing over an extended period of toxin exposure (Figure 3b–e). These findings contradict the notion that membrane-associated monomers are precursors to oligomer assembly.48 Our studies are the first to demonstrate two distinct modes of interaction between Cry1Ab toxin and insect cells. One interaction is receptor independent and promotes assembly and insertion of toxin oligomers into cell membrane and the other involves univalent binding of toxin monomers to BT-R1, the latter of which leads to cell death.
The binding of toxins and microbial surface components to receptors on host cells is a crucial step in most bacterial infections that allows bacterial invasion by avoiding or subverting normal cell defense functions. Many bacteria have evolved to recruit host cell surface receptors that are linked to critical cellular mechanisms, such as signal transduction pathways, to trigger aberrant responses including inflammatory reactions, cytoskeletal rearrangement and cell death.1, 2, 3 An intriguing result of our studies is that binding of Cry1Ab toxin to the cadherin receptor BT-R1 on insect cells promotes cytotoxicity associated with magnesium-dependent cellular responses. In fact, cytotoxicity brought about by Cry1Ab appears to depend strictly on magnesium because the absence of magnesium prevented cell death even though the toxin bound to BT-R1 (Figure 5). Removal of magnesium did not interfere with the incorporation of toxin oligomers and monomer into cell membrane (Figure 5b). These findings demonstrate that the interaction of toxin with the receptor is prerequisite, but not sufficient, to induce cytotoxicity, which apparently is linked to magnesium-dependent cellular responses. Recently, Cry1Ab toxin was shown to bind equally to toxin-susceptible and toxin-resistant cells, substantiating that neither resistance of cells to the toxin nor cytotoxicity can be explained solely by toxin binding.49 Our results support the view that cytotoxicity is mediated by binding of toxin to BT-R1, which triggers magnesium-dependent cellular responses that lead to cell death.
We propose a new model for the action of Cry toxins (Figure 6). According to the model, there are two distinct modes of interaction between toxin and target cell. In one mode, Cry toxin molecules are assembled as oligomers in the cell membrane. This interaction is lipid dependent and nonspecific and does not lead to cell death. In the other mode, toxin monomer binds to BT-R and triggers a magnesium-dependent signaling pathway that ultimately leads to cell death. Our model argues that toxin oligomers formed in cell membrane represent products of nonspecific interaction between toxin and lipid membrane components and, apparently, poses no harmful effects on the host cell. The model agrees with the paradigm for many bacterial toxins that challenge host cells by targeting cell surface receptors and manipulating critical reactions associated with various cellular responses.1, 2 Obviously, cells can alter these responses and enable themselves to avoid toxin action. In fact, recent studies with another member of the Cry toxin family, Cry5B, showed that exposure of cells to Cry toxin induces changes in a mitogen-activated protein kinase pathway and stimulates cellular defenses necessary in coping with toxin attack.15

Proposed model for Cry toxin action. According to the model, there are two kinds of interaction between toxin and cell. The first is a nonspecific toxin–lipid interaction, mediating assembly of Cry toxin molecules as oligomers and their insertion into membrane. The membrane-incorporated oligomer complex does not form lytic pores in the membrane and has no toxic effect on cells. The second kind is specific interaction between Cry toxin and the cadherin receptor BT-R, which mediates cytotoxicity. Binding of toxin to receptor brings about cell death through activation of a Mg2+-dependent signaling pathway downstream of the toxin–receptor interaction. The model predicts that cytotoxicity associated with Cry toxin depends strictly on Mg2+-dependent cellular responses that are triggered upon toxin binding to BT-R, leading to necrotic cell death
The high affinity and specific binding of Cry1Ab to BT-R1 indicates that BT-R1, indeed, is the cell surface ligand recruited for targeting host cells.31, 50 BT-R1 homologs are the principal determinant for Cry1A toxin action in lepidopteran insects.30, 31, 32, 33, 34, 35 No BT-R1 homologs has been identified in vertebrates, suggesting that these particular cadherin receptors represent a unique family of proteins in invertebrates, particularly insects, and may explain why Cry toxins are not toxic to mammalian cells. Perhaps, Cry toxins once constituted virulence factors with the ability to destroy cells through oligomerization and incorporation of toxin molecules into cell membranes. Clearly, Cry toxins have gained specificity as well as efficiency in exerting cytotoxicity through selectively interacting with certain surface molecules on insect cells. Elucidation of the molecular mechanism of Cry toxin action and the cellular events involved in cytotoxicity is critical to understanding host specificity and to more intelligent use of Cry toxins to control pest insects in a safe and environmentally compatible manner.
Materials and Methods
Preparation of Cry1Ab toxin
Cry1Ab toxin was prepared as previously described31 by tryptic digestion of protoxin obtained from the parasporal crystal of Bt subsp. berliner.
Cell cultures
H5 cells (Invitrogen) were cultured as a monolayer in 25-cm2 tissue culture flasks containing 5 ml of insect-Xpress medium (Cambrex) supplemented with gentamycin (10 μg/ml). G418 (800 μg/ml) was used to select for and maintain transfected cells.
Cloning and expression of BT-R1
The cDNA encoding BT-R1 ((GenBank: AF319973) was cloned into the plasmid pXINSECT-DEST38 (Invitrogen). H5 cells were cotransfected with the recombinant plasmid and pBmA:neo (1 : 20 ratio) (Invitrogen) using the transfection reagent Cellfectin (Invitrogen). A single clone designated as S5 that was resistant to G418 was selected for subculturing and subsequent experimentation. Expression of BT-R1 in S5 cells was confirmed by SDS-PAGE and Western blotting using antibody raised against BT-R1.
Cloning and purification of TBR
The region encoding the toxin-binding site (TBR) of BT-R1 (residues: 1349–1460) was cloned into the pMal plasmid (New England Biolabs) for expression in Escherichia coli as an N-terminal MBP fusion protein. The protein was purified by anion-exchange chromatography followed by affinity chromatography on amylose resin. The MBP fusion partner was removed by digestion with thermolysin protease.
Assay for cytotoxicity
Cells were seeded in 96-well plates (1 × 104 cell/well) and allowed to grow attached to the surface of the plate bottom. Growth medium was replaced with fresh medium containing Cry1Ab at various concentrations and the cells were incubated for 4 h. Cell death was determined by Trypan blue exclusion. In all, 10 μl of Trypan blue (0.4%, wt/vol) were added directly to each well and incubated for 5 min. Stained cells were viewed immediately under a microscope (Nikon TE600) and photomicrographs were taken with an RTE/CCD-1300 camera (Roper Scientific) at × 200. Cells were not detached from the plate bottom within the time of observation and photographing. Microphotographs were analyzed by using imaging software (MetaMorph 4, Universal Imaging) to count the number of blue-stained dead cells (NB) and transparent viable cells (NT), respectively. Cytotoxicity was calculated by the ratio NB/(NB+NT).
Preparation of cell extracts
Toxin-treated and untreated cells (1 × 106) were washed in PBS (4°C) and lysed in CytoBuster protein extraction reagent (Novagen). Cell lysates were centrifuged at 13 000 × g for 10 min at 4°C and the supernatants were collected as soluble fraction. The membrane pellets were dissolved in membrane protein buffer (5 M urea, 2 M thiourea, 2% (w/v) 3[(3-cholamidopropyl)dimethylammonio]-propanesulfonic acid (CHAPS), 40 mM Tris·HCl) and collected as membrane fraction. The S5 and H5 cells were incubated with Cry1Ab at a specified concentration and time before preparation of cell extracts for measurement of toxin incorporation. All cell extracts were freshly prepared for Western and immunoligand blotting analysis.
Preparation of liposomes
Egg phosphatidylcholine (PC), brain phosphatidylserine (PS), brain SM and Chol were purchased from Avanti Polar Lipids. Liposomes with different lipid compositions were prepared according to the manufacturer's instructions. The liposomes were hydrated in PBS buffer by vortexing. For toxin-incorporation analysis, the liposomes were incubated with Cry1Ab (180 nM) for 2 h and the mixture was then incubated in SDS sample buffer for 10 min at 95°C before Western blot analysis.
Western and immunoligand blotting analysis
Western blots were carried out using equal amounts of protein (10 μg) separated on 7% SDS-PAGE and transferred to polyvinyl difluoride (PVDF) membrane (Millipore). Before loading on the gels, protein samples were mixed with equal amounts of 2 × loading buffer (2% (w/v) SDS, 5 M urea, 2 M thiourea, 4% (w/v) CHAPS, 100 mM TrisHCl, 20% glycerol, 10 mM β-ME, 0.03% (w/v) bromophenol blue) by vortexing for 1 min at room temperature with or without boiling. Detection was accomplished using primary antibodies against BT-R1 or Cry1Ab, followed by incubation with horseradish peroxidase-coupled secondary antibodies (Sigma). Proteins were visualized by ECL plus detection reagent (Amersham). Immunoligand blots were performed with Cry1Ab and protein extracts from H5 and S5 cells. After the protein samples were transferred from gel to PVDF membrane, the membrane was incubated with Cry1Ab toxin (3nM) in blocking buffer for 1 h and washed three times in PBS-T. Detection of bound toxin molecules was accomplished using antibody against Cry1Ab.
Immunofluorescent staining
After growing over night in 8-well glass chambers, toxin-treated and untreated cells were washed in PBS and fixed in 4% parafomaldehyde solution (PFA). Fixed cells were permeabilized with 0.2% Triton X-100 at room temperature. The cells were rinsed three times and blocked with 1% BSA in PBS for 30 min. The cells were incubated with antibodies against BT-R1 or Cry1Ab followed by the Alexa Fluor 488 chicken anti-rabbit IgG antibody (Molecular Probes), and then postfixed in 4% PFA containing 2 μg/ml of Hoechst 33342 stain (Molecular Probes). The stained samples on glass slides were viewed under a fluorescence microscope (Nikon TE600) and microphotographs were taken with an RTE/CCD-1300 camera (Roper Scientific) at × 400.
Accession codes
Abbreviations
- BBMV:
-
brush border membrane vesicles
- CHAPS:
-
3[(3-cholamidopropyl)dimethylammonio]-propanesulfonic acid
- Chol:
-
cholesterol
- EDTA:
-
ethylenediaminetetra-acetic acid
- EGTA:
-
ethylene-glycol-bis(2-aminoethyl ether)-N,N,N,N′-tetraacetic acid
- H5:
-
High Five
- MBP:
-
maltose-binding protein
- PC:
-
phosphatidylcholine
- PFA:
-
paraformaldehyde
- PS:
-
phosphatidylserine
- PVDF:
-
polyvinyl difluoride
- S5:
-
BT-R1-transfected High Five
- TBR:
-
toxin-binding region
- SM:
-
sphingomyelin
References
Finlay BB and Cossart P (1997) Exploitation of mammalian host cell functions by bacterial pathogens. Science 276: 718–725
Schmitt CK, Meysick KC and O'Brien AD (1999) Bacterial toxins: friends or foes? Emerg. Infect. Dis. 5: 224–234
Schiavo G and van der Goot FG (2001) The bacterial toxin toolkit. Nat. Rev. Mol. Cell Biol. 2: 530–537
Montecucco C (1998) Protein toxins and membrane transport. Curr. Opin. Cell Biol. 10: 530–536
Blaustein RO, Koehler TM, Collier RJ and Finkelstein A (1989) Anthrax toxin: channel-forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl. Acad. Sci. USA 86: 2209–2213
Knapp O, Benz R, Gibert M, Marvaud JC and Popoff MR (2002) Interaction of Clostridium perfringens iota-toxin with lipid bilayer membranes. Demonstration of channel formation by the activated binding component Ib and channel block by the enzyme component Ia. J. Biol. Chem. 277: 6143–6152
Schmid A, Benz R, Just I and Aktories K (1994) Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes. Formation of cation-selective channels and inhibition of channel function by chloroquine. J. Biol. Chem. 269: 16706–16711
Wilmsen HU, Pattus F and Buckley JT (1990) Aerolysin, a hemolysin from Aeromonas hydrophila, forms voltage-gated channels in planar lipid bilayers. J. Membr. Biol. 115: 71–81
Ascenzi P, Visca P, Ippolito G, Spallarossa A, Bolognesi M and Montecucco C (2002) Anthrax toxin: a tripartite lethal combination. FEBS Lett. 531: 384–388
Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H and Gouaux JE (1996) Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274: 1859–1866
Gilbert RJ, Jimenez JL, Chen S, Tickle IJ, Rossjohn J, Parker M, Andrew PW and Saibil HR (1999) Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae. Cell 97: 647–655
Menestrina G, Dalla SM, Comai M, Coraiola M, Viero G, Werner S, Colin DA, Monteil H and Prevost G (2003) Ion channels and bacterial infection: the case of beta-barrel pore-forming protein toxins of Staphylococcus aureus. FEBS Lett. 552: 54–60
Fivaz M, Abrami L, Tsitrin Y and van der Goot FG (2001) Not as simple as just punching a hole. Toxicon 39: 1637–1645
Valeva A, Walev I, Gerber A, Klein J, Palmer M and Bhakdi S (2000) Staphylococcal alpha-toxin: repair of a calcium-impermeable pore in the target cell membrane. Mol. Microbiol. 36: 467–476
Huffman DL, Abrami L, Sasik R, Corbeil J, van der Goot FG and Aroian RV (2004) Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 101: 10995–11000
Rose F, Dahlem G, Guthmann B, Grimminger F, Maus U, Hanze J, Duemmer N, Grandel U, Seeger W and Ghofrani HA (2002) Mediator generation and signaling events in alveolar epithelial cells attacked by S. aureus alpha-toxin.. Am. J. Physiol Lung Cell Mol. Physiol 282: L207–L214
Nelson KL, Brodsky RA and Buckley JT (1999) Channels formed by subnanomolar concentrations of the toxin aerolysin trigger apoptosis of T lymphomas. Cell Microbiol. 1: 69–74
Agaisse H and Lereclus D (1995) How does Bacillus thuringiensis produce so much insecticidal crystal protein? J. Bacteriol 177: 6027–6032
Puntheeranurak T, Uawithya P, Potvin L, Angsuthanasombat C and Schwartz JL (2004) Ion channels formed in planar lipid bilayers by the dipteran-specific Cry4B Bacillus thuringiensis toxin and its alpha1-alpha5 fragment. Mol. Membr. Biol. 21: 67–74
Schwartz JL, Lu YJ, Sohnlein P, Brousseau R, Laprade R, Masson L and Adang MJ (1997) Ion channels formed in planar lipid bilayers by Bacillus thuringiensis toxins in the presence of Manduca sexta midgut receptors. FEBS Lett. 412: 270–276
Gomez I, Sanchez J, Miranda R, Bravo A and Soberon M (2002) Cadherin-like receptor binding facilitates proteolytic cleavage of helix alpha-1 in domain I and oligomer pre-pore formation of Bacillus thuringiensis Cry1Ab toxin. FEBS Lett. 513: 242–246
Grochulski P, Masson L, Borisova S, Pusztai-Carey M, Schwartz JL, Brousseau R and Cygler M (1995) Bacillus thuringiensis CryIA(a) insecticidal toxin: crystal structure and channel formation. J. Mol. Biol. 254: 447–464
Li JD, Carroll J and Ellar DJ (1991) Crystal structure of insecticidal delta-endotoxin from Bacillus thuringiensis at 2.5 A resolution. Nature 353: 815–821
Gazit E, La Rocca P, Sansom MS and Shai Y (1998) The structure and organization within the membrane of the helices composing the pore-forming domain of Bacillus thuringiensis delta-endotoxin are consistent with an ‘umbrella-like’ structure of the pore. Proc. Natl. Acad. Sci. USA 95: 12289–12294
Rajamohan F, Lee MK and Dean DH (1998) Bacillus thuringiensis insecticidal proteins: molecular mode of action. Prog. Nucl. Acid Res. Mol. Biol. 60: 1–27
Kumar AS and Aronson AI (1999) Analysis of mutations in the pore-forming region essential for insecticidal activity of a Bacillus thuringiensis delta-endotoxin. J. Bacteriol 181: 6103–6107
Vachon V, Prefontaine G, Rang C, Coux F, Juteau M, Schwartz JL, Brousseau R, Frutos R, Laprade R and Masson L (2004) Helix 4 mutants of the Bacillus thuringiensis insecticidal toxin Cry1Aa display altered pore-forming abilities. Appl. Environ. Microbiol. 70: 6123–6130
Vachon V, Prefontaine G, Coux F, Rang C, Marceau L, Masson L, Brousseau R, Frutos R, Schwartz JL and Laprade R (2002) Role of helix 3 in pore formation by the Bacillus thuringiensis insecticidal toxin Cry1Aa. Biochemistry 41: 6178–6184
Luo K, Banks D and Adang MJ (1999) Toxicity, binding, and permeability analyses of four Bacillus thuringiensis cry1 delta-endotoxins using brush border membrane vesicles of Spodoptera exigua and Spodoptera frugiperda. Appl. Environ. Microbiol. 65: 457–464
Vadlamudi RK, Weber E, Ji I, Ji TH and Bulla Jr LA (1995) Cloning and expression of a receptor for an insecticidal toxin of Bacillus thuringiensis. J. Biol. Chem. 270: 5490–5494
Vadlamudi RK, Ji TH and Bulla Jr LA (1993) A specific binding protein from Manduca sexta for the insecticidal toxin of Bacillus thuringiensis subsp. berliner. J. Biol. Chem. 268: 12334–12340
Dorsch JA, Candas M, Griko NB, Maaty WS, Midboe EG, Vadlamudi RK and Bulla Jr LA (2002) Cry1A toxins of Bacillus thuringiensis bind specifically to a region adjacent to the membrane-proximal extracellular domain of BT-R(1) in Manduca sexta: involvement of a cadherin in the entomopathogenicity of Bacillus thuringiensis. Insect Biochem. Mol. Biol. 32: 1025–1036
Gahan LJ, Gould F and Heckel DG (2001) Identification of a gene associated with Bt resistance in Heliothis virescens. Science 293: 857–860
Nagamatsu Y, Toda S, Koike T, Miyoshi Y, Shigematsu S and Kogure M (1998) Cloning, sequencing, and expression of the Bombyx mori receptor for Bacillus thuringiensis insecticidal CryIA(a) toxin. Biosci. Biotechnol. Biochem. 62: 727–734
Morin S, Henderson S, Fabrick JA, Carriere Y, Dennehy TJ, Brown JK and Tabashnik BE (2004) DNA-based detection of Bt resistance alleles in pink bollworm. Insect Biochem. Mol. Biol. 34: 1225–1233
Davis TR, Trotter KM, Granados RR and Wood HA (1992) Baculovirus expression of alkaline phosphatase as a reporter gene for evaluation of production, glycosylation and secretion. Biotechnology (NY) 10: 1148–1150
Rausell C, Pardo-Lopez L, Sanchez J, Munoz-Garay C, Morera C, Soberon M and Bravo A (2004) Unfolding events in the water-soluble monomeric Cry1Ab toxin during transition to oligomeric pre-pore and to membrane inserted pore channel. J. Biol. Chem. 279: 55168–55175
Aronson AI, Geng C and Wu L (1999) Aggregation of Bacillus thuringiensis Cry1A toxins upon binding to target insect larval midgut vesicles. Appl. Environ. Microbiol. 65: 2503–2507
Yamaji-Hasegawa A, Makino A, Baba T, Senoh Y, Kimura-Suda H, Sato SB, Terada N, Ohno S, Kiyokawa E, Umeda M and Kobayashi T (2003) Oligomerization and pore formation of a sphingomyelin-specific toxin, lysenin. J. Biol. Chem. 278: 22762–22770
Tweten RK, Parker MW and Johnson AE (2001) The cholesterol-dependent cytolysins. Curr. Top. Microbiol. Immunol. 257: 15–33
Griffitts JS, Haslam SM, Yang T, Garczynski SF, Mulloy B, Morris H, Cremer PS, Dell A, Adang MJ and Aroian RV (2005) Glycolipids as receptors for Bacillus thuringiensis crystal toxin. Science 307: 922–925
Midboe EG, Candas M and Bulla Jr LA (2003) Expression of a midgut-specific cadherin BT-R1 during the development of Manduca sexta larva. Comp Biochem. Physiol B Biochem. Mol. Biol. 135: 125–137
Meng J, Candas M, Keeton TP and Bulla Jr LA (2001) Expression in Spodoptera frugiperda (Sf21) insect cells of BT-R(1), a cadherin-related receptor from Manduca sexta for Bacillus thuringiensis Cry1Ab toxin. Protein Expr. Purif. 22: 141–147
Candas M, Francis BR, Griko NB, Midboe EG and Bulla Jr LA (2002) Proteolytic cleavage of the developmentally important cadherin BT-R1 in the midgut epithelium of Manduca sexta. Biochemistry 41: 13717–13724
Griko N, Candas M, Zhang X, Junker M and Bulla Jr LA (2004) Selective antagonism to the cadherin BT-R1 interferes with calcium-induced adhesion of epithelial membrane vesicles. Biochemistry 43: 1393–1400
Carroll J and Ellar DJ (1997) Analysis of the large aqueous pores produced by a Bacillus thuringiensis protein insecticide in Manduca sexta midgut-brush-border-membrane vesicles. Eur. J. Biochem. 245: 797–804
Masson L, Mazza A, Brousseau R and Tabashnik B (1995) Kinetics of Bacillus thuringiensis toxin binding with brush border membrane vesicles from susceptible and resistant larvae of Plutella xylostella. J. Biol. Chem. 270: 11887–11896
de Maagd RA, Bravo A and Crickmore N (2001) How Bacillus thuringiensis has evolved specific toxins to colonize the insect world. Trends Genet. 17: 193–199
Li H, Gonzalez-Cabrera J, Oppert B, Ferre J, Higgins RA, Buschman LL, Radke GA, Zhu KY and Huang F (2004) Binding analyses of Cry1Ab and Cry1Ac with membrane vesicles from Bacillus thuringiensis-resistant and -susceptible Ostrinia nubilalis. Biochem. Biophys. Res. Commun. 323: 52–57
Franklin SE, Young L, Watson D, Cigan A, Meyer T and Bulla Jr LA (1997) Southern analysis of BT-R1, the Manduca sexta gene encoding the receptor for the Cry1Ab toxin of Bacillus thuringiensis. Mol. Gen. Genet. 256: 517–524
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by J Abrams
Rights and permissions
About this article
Cite this article
Zhang, X., Candas, M., Griko, N. et al. Cytotoxicity of Bacillus thuringiensis Cry1Ab toxin depends on specific binding of the toxin to the cadherin receptor BT-R1 expressed in insect cells. Cell Death Differ 12, 1407–1416 (2005). https://doi.org/10.1038/sj.cdd.4401675
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401675
Keywords
This article is cited by
-
Up-regulation of apoptotic- and cell survival-related gene pathways following exposures of western corn rootworm to B. thuringiensis crystalline pesticidal proteins in transgenic maize roots
BMC Genomics (2021)
-
Enhancement of insect susceptibility and larvicidal efficacy of Cry4Ba toxin by calcofluor
Parasites & Vectors (2018)
-
Assessment of a commercial spider venom peptide against spotted-wing Drosophila and interaction with adjuvants
Journal of Pest Science (2018)
-
A Proteomic Analysis Provides Novel Insights into the Stress Responses of Caenorhabditis elegans towards Nematicidal Cry6A Toxin from Bacillus thuringiensis
Scientific Reports (2017)
-
Expression of recombinant and mosaic Cry1Ac receptors from Helicoverpa armigera and their influences on the cytotoxicity of activated Cry1Ac to Spodoptera litura Sl-HP cells
Cytotechnology (2016)
