
BCL2 Protein Progressively Declines during Robust CLL Clonal Expansion: Potential Impact on Venetoclax Clinical Efficacy and Insights on Mechanism
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
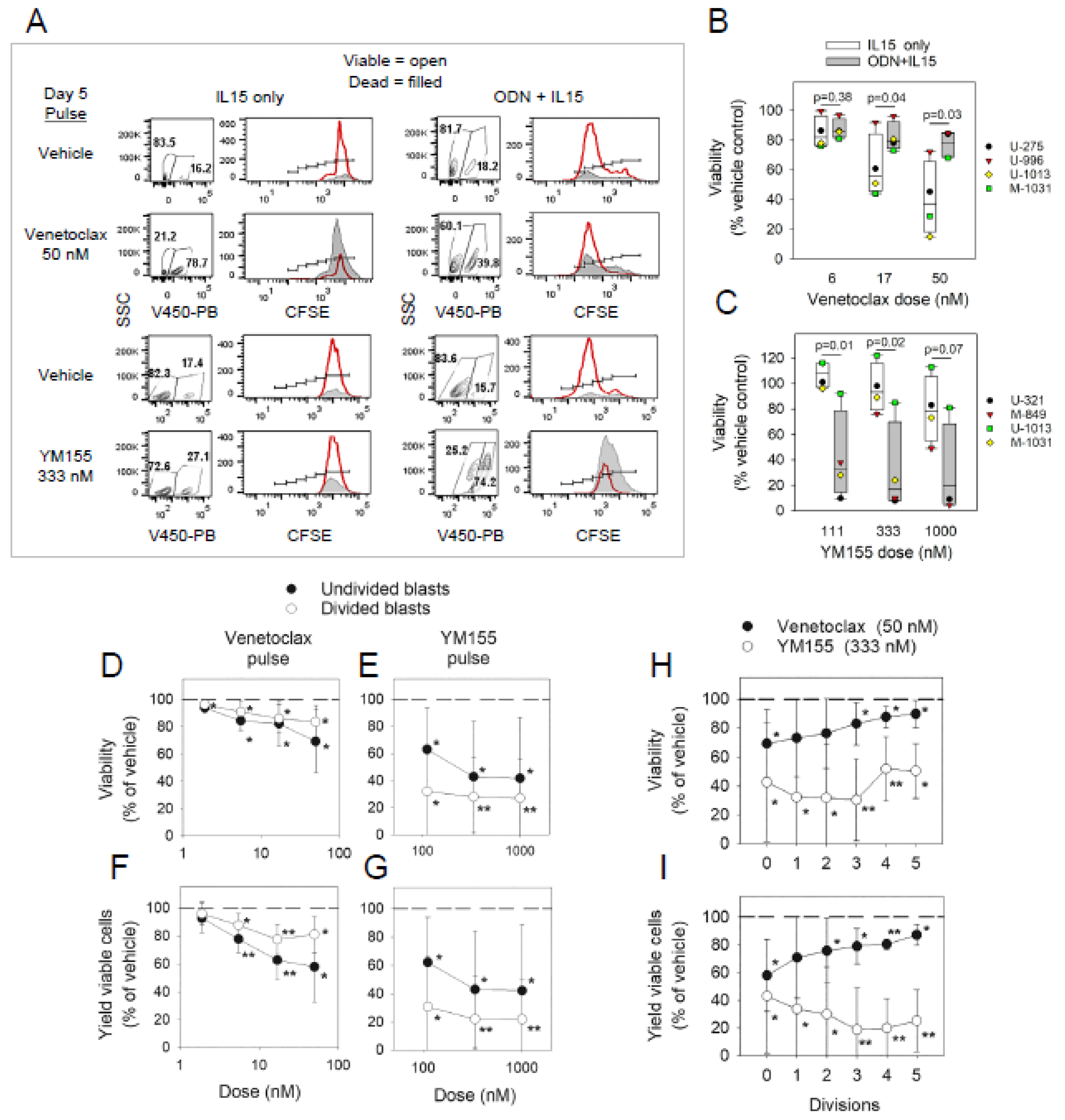
2.1. Sensitivity of non-Cycling and Cycling CLL Cells to Specific Inhibitors of BCL2 or Surviving
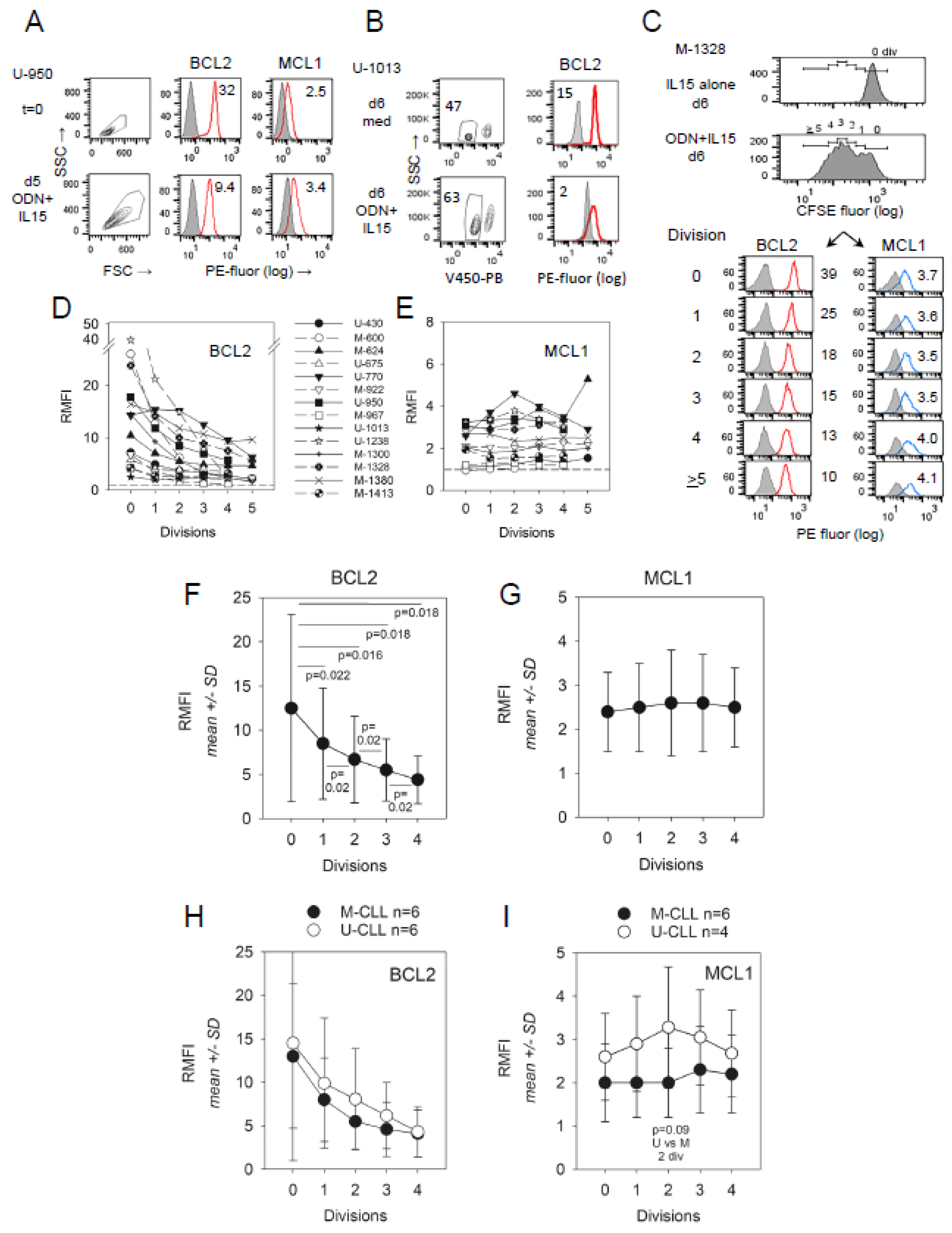
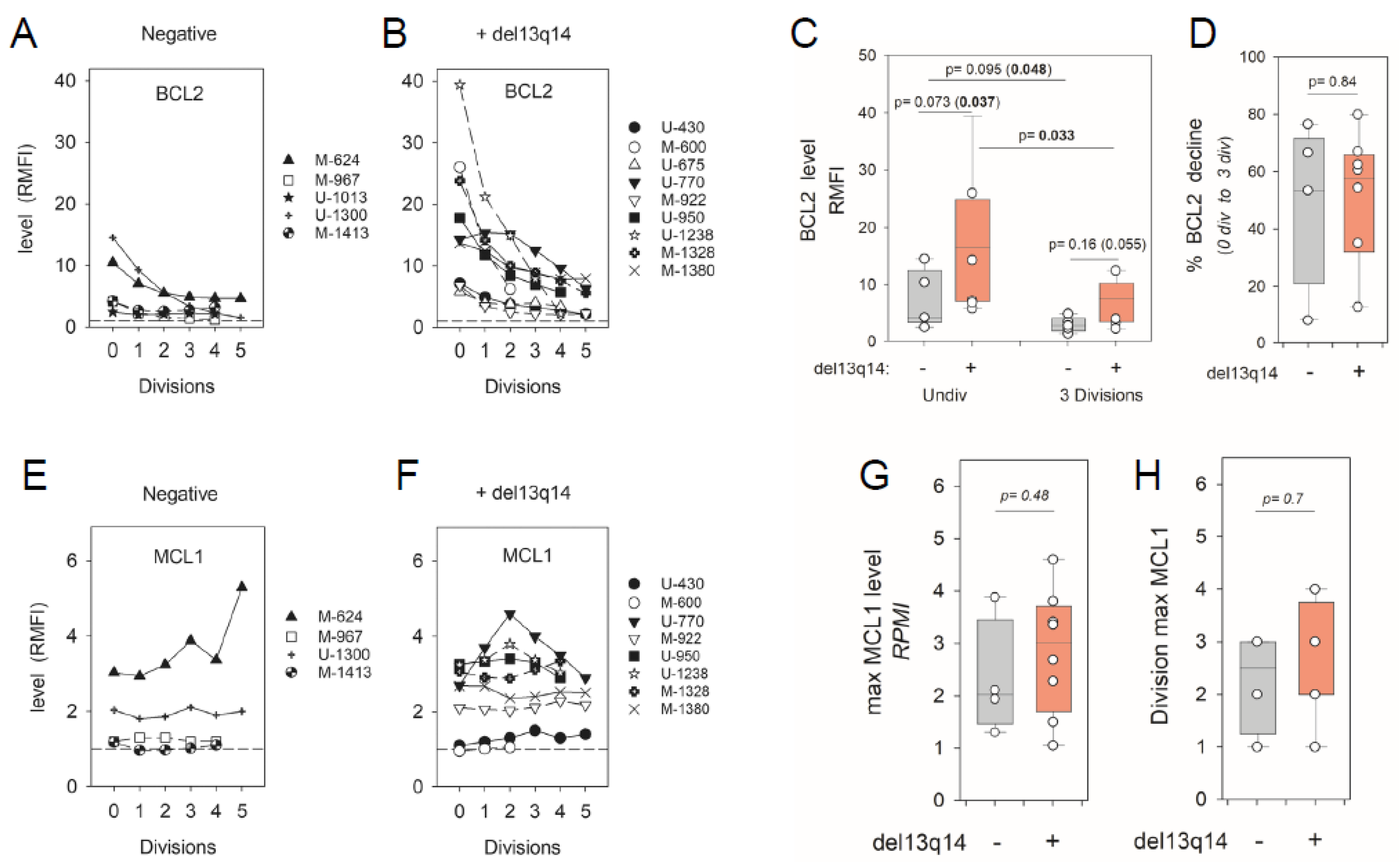
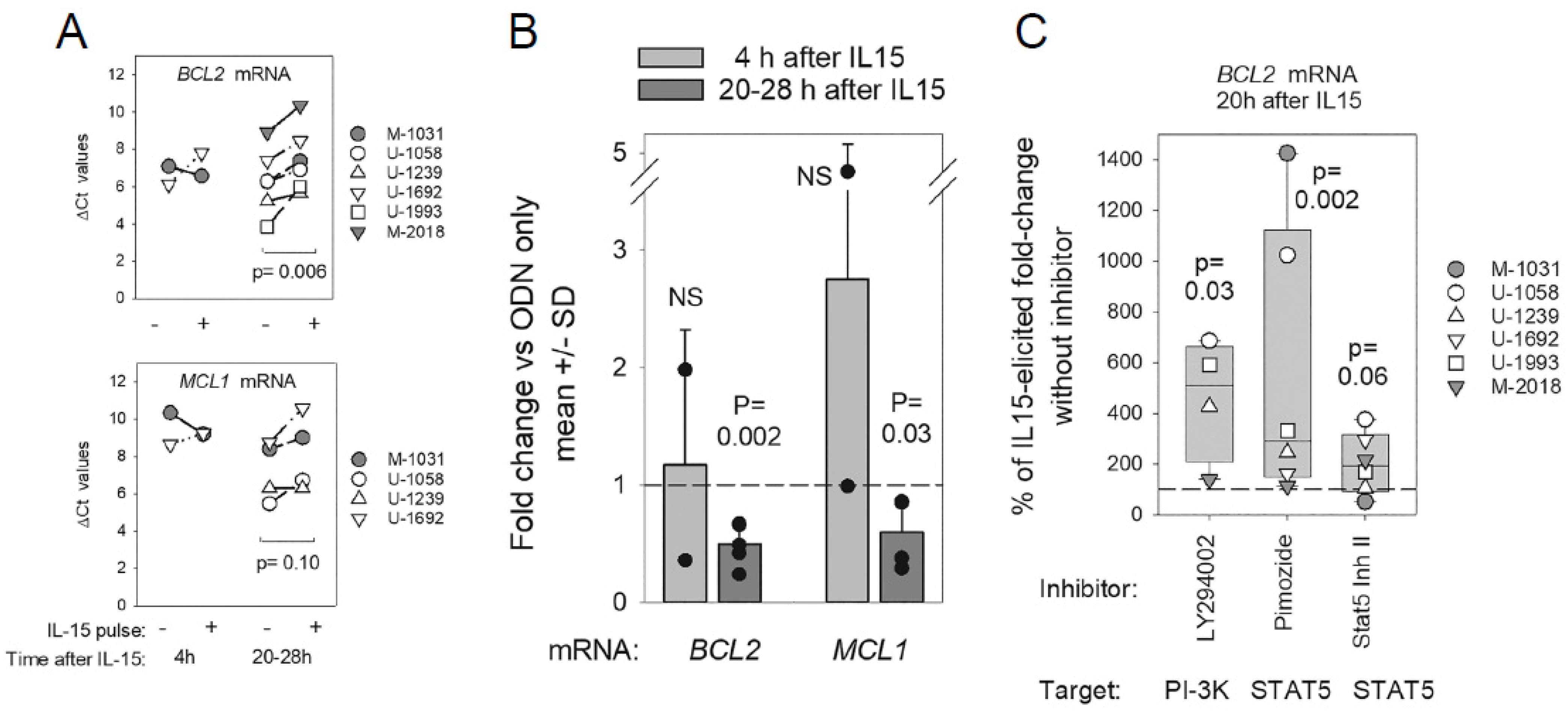
2.2. Impact of CLL del(13q) on BCL2 and MCL1 Protein Levels during ODN + IL15-Driven Cell Clonal Expansion
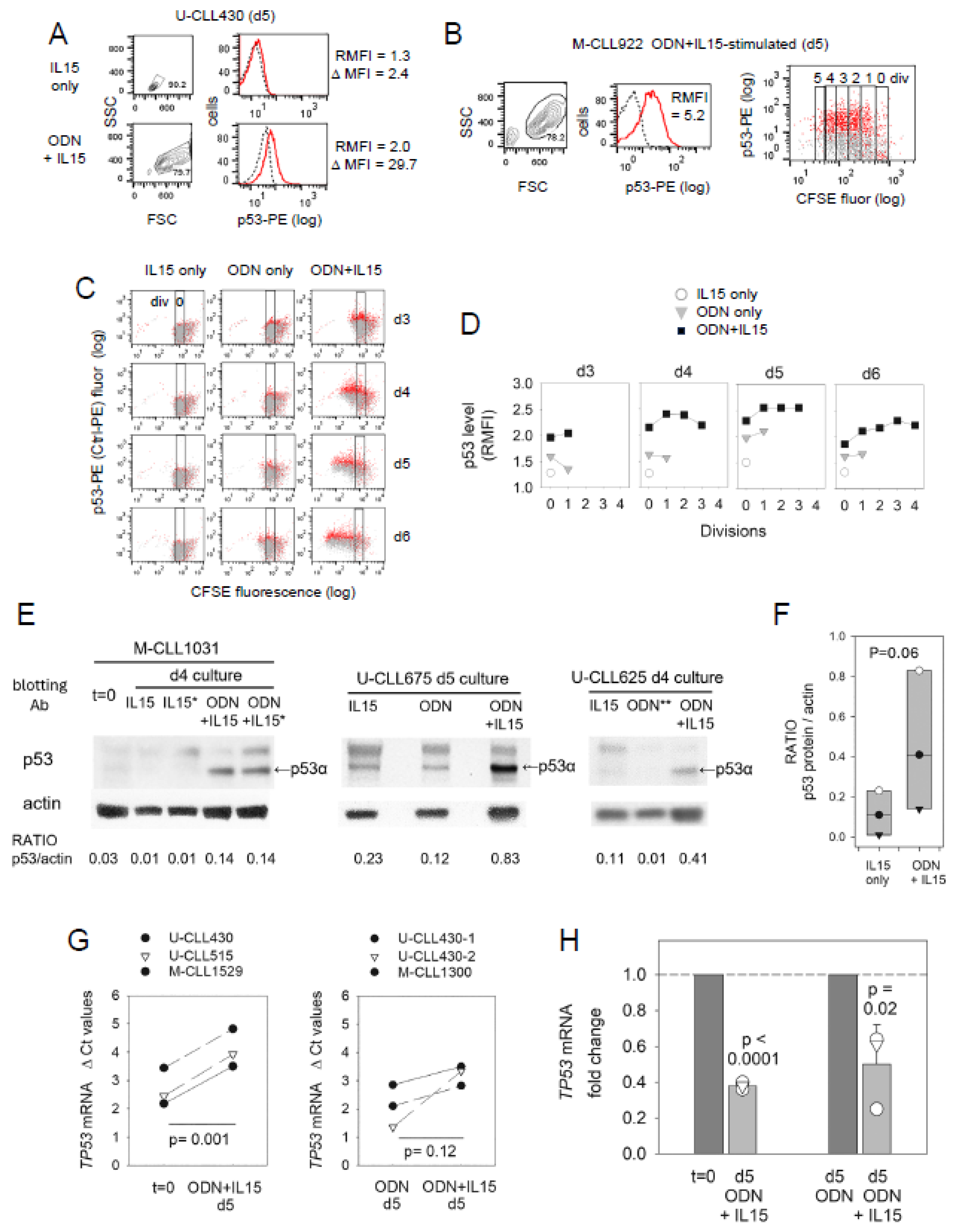
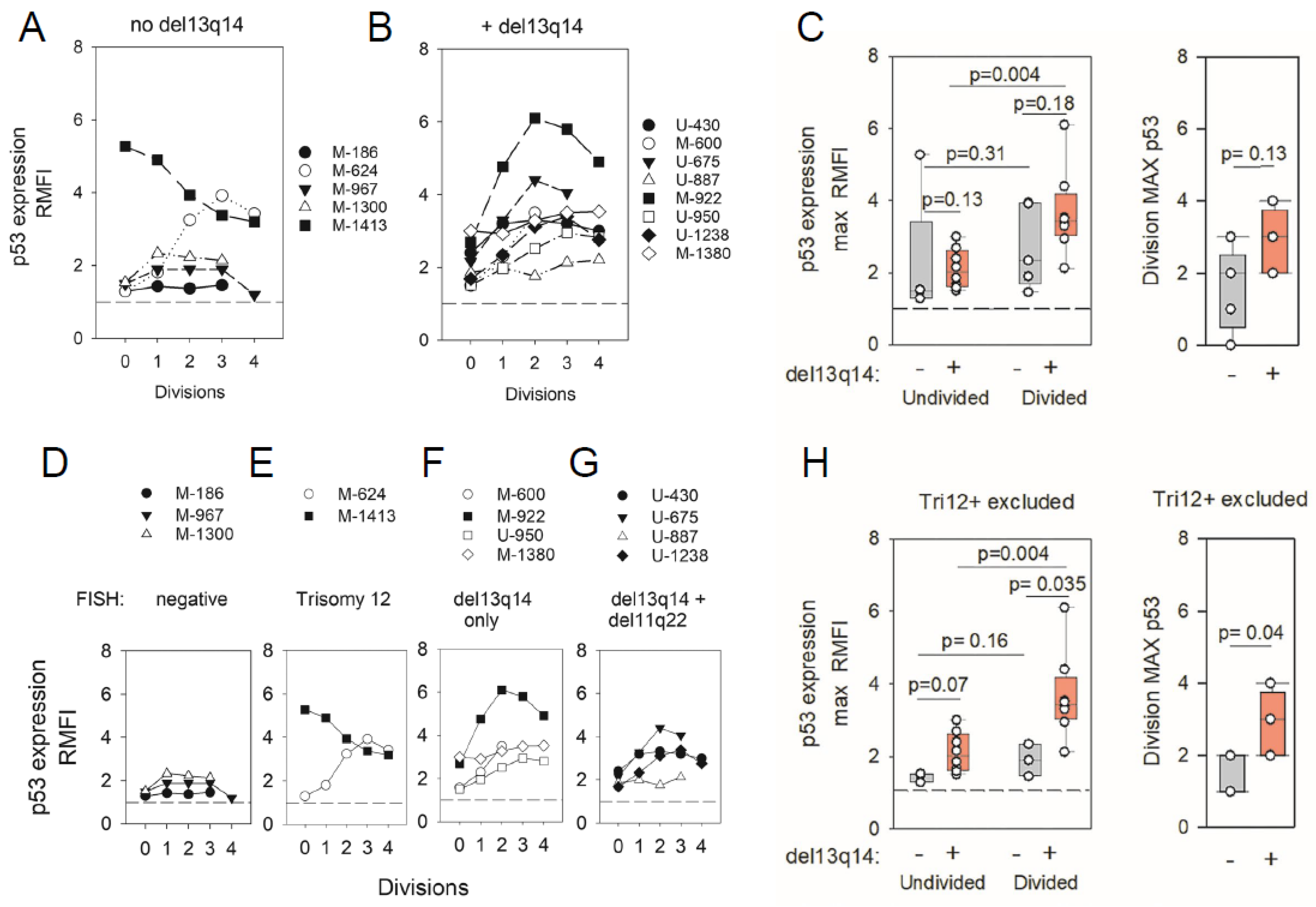
2.3. ODN + IL15-Stimulated CLL Cells Express Elevated Protein Levels of p53 Transcription Factor (TF)
2.4. Early Effects of IL15 and Downstream STAT5 and PI-3K on BCL2/MCL1 mRNA Levels


2.5. Evidence for miR15a/miR16-1 Mediated Feedback Repression of p53 TF
3. Discussion
4. Materials and Methods
4.1. CLL Patient Samples
4.2. CLL Cell Isolation from Patient Blood (n = 29 CLL) and Spleen (n = 1 CLL)
4.3. Purity of CLL Populations
4.4. Culture Conditions
4.5. Inhibitors
4.6. Intracellular Staining for Cytoplasmic BCL2, MCL1, and Survivin Proteins
4.7. Staining for Nuclear p53 Protein
4.8. Flow Cytometric (FACS) Analyses
4.9. Immunoblotting for p53 Protein
4.10. Quantitative Assessment of BCL2, MCL1, and TP53 mRNA by Reverse Transcription Real-Time PCR (q-RT-PCR)
4.11. Phase Microscopy
4.12. Statistics
4.13. Study Approval
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rai, K.R.; Jain, P. Chronic lymphocytic leukemia (CLL)-Then and now. Am. J. Hematol. 2016, 91, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Mongini, P.K.; Gupta, R.; Boyle, E.; Nieto, J.; Lee, H.; Stein, J.; Bandovic, J.; Stankovic, T.; Barrientos, J.; Kolitz, J.E.; et al. TLR-9 and IL-15 Synergy Promotes the in vitro Clonal Expansion of Chronic Lymphocytic Leukemia B Cells. J. Immunol. 2015, 195, 901–923. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Larsson, L.G.; Gottardi, D.; Gaidano, G.; Carlsson, M.; Nilsson, K.; Caligaris-Cappio, F. Growth- and differentiation-associated expression of bcl-2 in B-chronic lymphocytic leukemia cells. Blood 1992, 79, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Hanada, M.; Delia, D.; Aiello, A.; Stadtmauer, E.; Reed, J.C. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 1993, 82, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Zaja, F.; Di Loreto, C.; Amoroso, V.; Salmaso, F.; Russo, D.; Silvestri, F.; Fanin, R.; Damiani, D.; Infanti, L.; Mariuzzi, L.; et al. BCL-2 immunohistochemical evaluation in B-cell chronic lymphocytic leukemia and hairy cell leukemia before treatment with fludarabine and 2-chloro-deoxy-adenosine. Leuk. Lymphoma 1998, 28, 567–572. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.; Schneider, C.; Zucknick, M.; Bogelein, D.; Schulze, K.; Zenz, T.; Mohr, J.; Philippen, A.; Huber, H.; Buhler, A.; et al. Protein expression analysis of chronic lymphocytic leukemia defines the effect of genetic aberrations and uncovers a correlation of CDK4, P27 and P53 with hierarchical risk. Haematologica 2010, 95, 1880–1888. [Google Scholar] [CrossRef]
- Aqeilan, R.I.; Calin, G.A.; Croce, C.M. miR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 2010, 17, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Cimmino, A.; Fabbri, M.; Ferracin, M.; Wojcik, S.E.; Shimizu, M.; Taccioli, C.; Zanesi, N.; Garzon, R.; Aqeilan, R.I.; et al. miR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 5166–5171. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Itchaki, G.; Brown, J.R. The potential of venetoclax (ABT-199) in chronic lymphocytic leukemia. Ther. Adv. Hematol. 2016, 7, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Ma, S.; Kipps, T.J.; Coutre, S.E.; Davids, M.S.; Eichhorst, B.; Hallek, M.; Byrd, J.C.; Humphrey, K.; Zhou, L.; et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood 2019, 134, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Dohner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Thompson, E.R.; Nguyen, T.; Birkinshaw, R.W.; Gong, J.N.; Chen, X.; McBean, M.; Thijssen, R.; Conway, T.; Anderson, M.A.; et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood 2020, 135, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Thomalla, D.; Beckmann, L.; Grimm, C.; Oliverio, M.; Meder, L.; Herling, C.D.; Nieper, P.; Feldmann, T.; Merkel, O.; Lorsy, E.; et al. Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies. Blood 2022, 140, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, R.; Tian, L.; Anderson, M.A.; Flensburg, C.; Jarratt, A.; Garnham, A.L.; Jabbari, J.S.; Peng, H.; Lew, T.E.; Teh, C.E.; et al. Single-cell multiomics reveal the scale of multilayered adaptations enabling CLL relapse during venetoclax therapy. Blood 2022, 140, 2127–2141. [Google Scholar] [CrossRef] [PubMed]
- Khalsa, J.K.; Cha, J.; Utro, F.; Naeem, A.; Murali, I.; Kuang, Y.; Vasquez, K.; Li, L.; Tyekucheva, S.; Fernandes, S.M.; et al. Genetic events associated with venetoclax resistance in CLL identified by whole-exome sequencing of patient samples. Blood 2023, 142, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.; Thompson, E.; Tam, C. Soho state of the Art Updates and Next Questions|Mechanisms of Resistance to BCL2 Inhibitor Therapy in Chronic Lymphocytic Leukemia and Potential Future Therapeutic Directions. Clin. Lymphoma Myeloma Leuk. 2022, 22, 795–804. [Google Scholar] [CrossRef]
- Chong, S.J.F.; Zhu, F.; Dashevsky, O.; Mizuno, R.; Lai, J.X.; Hackett, L.; Ryan, C.E.; Collins, M.C.; Iorgulescu, J.B.; Guieze, R.; et al. Hyperphosphorylation of BCL-2 family proteins underlies functional resistance to venetoclax in lymphoid malignancies. J. Clin. Investig. 2023, 133, e170169. [Google Scholar] [CrossRef]
- Granziero, L.; Ghia, P.; Circosta, P.; Gottardi, D.; Strola, G.; Geuna, M.; Montagna, L.; Piccoli, P.; Chilosi, M.; Caligaris-Cappio, F. Survivin is expressed on CD40 stimulation and interfaces proliferation and apoptosis in B-cell chronic lymphocytic leukemia. Blood 2001, 97, 2777–2783. [Google Scholar] [CrossRef]
- Smit, L.A.; Hallaert, D.Y.; Spijker, R.; de Goeij, B.; Jaspers, A.; Kater, A.P.; van Oers, M.H.; van Noesel, C.J.; Eldering, E. Differential Noxa/Mcl-1 balance in peripheral versus lymph node chronic lymphocytic leukemia cells correlates with survival capacity. Blood 2007, 109, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Cojocari, D. Hematologic Tumor Cell Resistance to the BCL-2 Inhibitor Venetoclax: A Product of Its Microenvironment? Front. Oncol. 2018, 8, 458. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ye, H. Progress in understanding the mechanisms of resistance to BCL-2 inhibitors. Exp. Hematol. Oncol. 2022, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Damle, R.N.; Calissano, C.; Chiorazzi, N. Chronic lymphocytic leukaemia: A disease of activated monoclonal B cells. Best Pract. Res. Clin. Haematol. 2010, 23, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Petlickovski, A.; Laurenti, L.; Li, X.; Marietti, S.; Chiusolo, P.; Sica, S.; Leone, G.; Efremov, D.G. Sustained signaling through the B-cell receptor induces mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood 2005, 105, 4820–4827. [Google Scholar] [CrossRef] [PubMed]
- Bojarczuk, K.; Sasi, B.K.; Gobessi, S.; Innocenti, I.; Pozzato, G.; Laurenti, L.; Efremov, D.G. BCR signaling inhibitors differ in their ability to overcome mcl-1-mediated resistance of CLL B cells to ABT-199. Blood 2016, 127, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, R.; Slinger, E.; Weller, K.; Geest, C.R.; Beaumont, T.; van Oers, M.H.; Kater, A.P.; Eldering, E. Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica 2015, 100, e302–e306. [Google Scholar] [CrossRef] [PubMed]
- Jayappa, K.D.; Portell, C.A.; Gordon, V.L.; Capaldo, B.J.; Bekiranov, S.; Axelrod, M.J.; Brett, L.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Petricoin, E.F.; et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017, 1, 933–946. [Google Scholar] [CrossRef]
- Patten, P.E.; Chu, C.C.; Albesiano, E.; Damle, R.N.; Yan, X.J.; Kim, D.; Zhang, L.; Magli, A.R.; Barrientos, J.; Kolitz, J.E.; et al. IGHV-unmutated and IGHV-mutated chronic lymphocytic leukemia cells produce activation-induced deaminase protein with a full range of biologic functions. Blood 2012, 120, 4802–4811. [Google Scholar] [CrossRef]
- Saito, M.; Novak, U.; Piovan, E.; Basso, K.; Sumazin, P.; Schneider, C.; Crespo, M.; Shen, Q.; Bhagat, G.; Califano, A.; et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 11294–11299. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Haque, S.; Nieto, J.; Trott, J.; Inman, J.K.; McCormick, S.; Chiorazzi, N.; Mongini, P.K. A p53 axis regulates B cell receptor-triggered, innate immune system-driven B cell clonal expansion. J. Immunol. 2012, 188, 6093–6108. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; Isaacson, P.G. Proliferation centres in B-cell malignant lymphoma, lymphocytic (B-CLL): An immunophenotypic study. Histopathology 1994, 24, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Li, W.; Yan, X.J.; Barrientos, J.; Kolitz, J.E.; Allen, S.L.; Rai, K.; Chiorazzi, N.; Mongini, P.K.A. Mechanism for IL-15-Driven B Cell Chronic Lymphocytic Leukemia Cycling: Roles for AKT and STAT5 in Modulating Cyclin D2 and DNA Damage Response Proteins. J. Immunol. 2019, 202, 2924–2944. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Yan, X.J.; Barrientos, J.; Kolitz, J.E.; Allen, S.L.; Rai, K.; Chiorazzi, N.; Mongini, P.K.A. Mechanistic Insights Into CpG DNA and IL-15 Synergy in Promoting B cell Chronic Lymphocytic Leukemia Clonal Expansion. J. Immunol. 2018, 201, 1570–1585. [Google Scholar] [CrossRef] [PubMed]
- Kita, A.; Mitsuoka, K.; Kaneko, N.; Nakata, M.; Yamanaka, K.; Jitsuoka, M.; Miyoshi, S.; Noda, A.; Mori, M.; Nakahara, T.; et al. Sepantronium bromide (YM155) enhances response of human B-cell non-hodgkin lymphoma to rituximab. J. Pharmacol. Exp. Ther. 2012, 343, 178–183. [Google Scholar] [CrossRef]
- Mackler, B.F.; Altman, L.C.; Rosenstreich, D.L.; Oppenheim, J.J. Induction of lymphokine production by EAC and of blastogenesis by soluble mitogens during human B-cell activation. Nature 1974, 249, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.E.; Parker, D.C. Cross-linking of B lymphocyte Fc gamma receptors and membrane immunoglobulin inhibits anti-immunoglobulin-induced blastogenesis. J. Immunol. 1984, 132, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.N.; Beesetty, P.; Sulentic, C.; Kozak, J.A. Rapid Quantification of Mitogen-Induced Blastogenesis in T Lymphocytes for Identifying Immunomodulatory Drugs. J. Vis. Exp. 2016, 118, e55212. [Google Scholar]
- Austen, B.; Powell, J.E.; Alvi, A.; Edwards, I.; Hooper, L.; Starczynski, J.; Taylor, A.M.; Fegan, C.; Moss, P.; Stankovic, T. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood 2005, 106, 3175–3182. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Purroy, N.; Abrisqueta, P.; Carabia, J.; Carpio, C.; Calpe, E.; Palacio, C.; Castellvi, J.; Crespo, M.; Bosch, F. Targeting the proliferative and chemoresistant compartment in chronic lymphocytic leukemia by inhibiting survivin protein. Leukemia 2014, 28, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K.; et al. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Wei, A.H.; Huang, D.C.S. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood 2021, 138, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Peperzak, V.; Vikstrom, I.; Walker, J.; Glaser, S.P.; LePage, M.; Coquery, C.M.; Erickson, L.D.; Fairfax, K.; Mackay, F.; Strasser, A.; et al. Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 2013, 14, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Tellier, J.; Nutt, S.L. Plasma cells: The programming of an antibody-secreting machine. Eur. J. Immunol. 2019, 49, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, jcs223826. [Google Scholar] [CrossRef]
- Albadari, N.; Li, W. Survivin Small Molecules Inhibitors: Recent Advances and Challenges. Molecules 2023, 28, 1376. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Croce, C.M. Role of miR-15/16 in CLL. Cell Death Differ. 2015, 22, 6–11. [Google Scholar] [CrossRef]
- Degheidy, H.A.; Gadalla, S.M.; Farooqui, M.Z.; Abbasi, F.; Arthur, D.C.; Bauer, S.R.; Wilson, W.H.; Wiestner, A.; Stetler-Stevenson, M.A.; Marti, G.E. Bcl-2 level as a biomarker for 13q14 deletion in CLL. Cytometry B Clin. Cytom. 2013, 84, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, G.; Feng, L.; Zhang, W.; Pelicano, H.; Wang, F.; Ogasawara, M.A.; Lu, W.; Amin, H.M.; Croce, C.M.; et al. Loss of p53 and altered miR15-a/16-1short right arrowMCL-1 pathway in CLL: Insights from TCL1-Tg:p53(-/-) mouse model and primary human leukemia cells. Leukemia 2014, 28, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Carrington, E.M.; Zhan, Y.; Brady, J.L.; Zhang, J.G.; Sutherland, R.M.; Anstee, N.S.; Schenk, R.L.; Vikstrom, I.B.; Delconte, R.B.; Segal, D.; et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 2017, 24, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.P.; Koss, B.; Bathina, M.; Perciavalle, R.M.; Bisanz, K.; Opferman, J.T. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol. Cell Biol. 2010, 30, 3099–3110. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Luo, Q.; Liu, Z. Ubiquitination and deubiquitination of MCL1 in cancer: Deciphering chemoresistance mechanisms and providing potential therapeutic options. Cell Death Dis. 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Sutton, L.A.; Rosenquist, R. Deciphering the molecular landscape in chronic lymphocytic leukemia: Time frame of disease evolution. Haematologica 2015, 100, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Tausch, E.; Rossi, D.; Sutton, L.A.; Soussi, T.; Zenz, T.; Kater, A.P.; Niemann, C.U.; Gonzalez, D.; Davi, F.; et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia 2018, 32, 1070–1080. [Google Scholar] [CrossRef]
- Fabbri, M.; Bottoni, A.; Shimizu, M.; Spizzo, R.; Nicoloso, M.S.; Rossi, S.; Barbarotto, E.; Cimmino, A.; Adair, B.; Wojcik, S.E.; et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA 2011, 305, 59–67. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Balatti, V.; Croce, C.M. BCL2 and miR-15/16: From gene discovery to treatment. Cell Death Differ. 2018, 25, 21–26. [Google Scholar] [CrossRef]
- Martinez-Valdez, H.; Guret, C.; de Bouteiller, O.; Fugier, I.; Banchereau, J.; Liu, Y.J. Human germinal center B cells express the apoptosis-inducing genes Fas, c-myc, P53, and Bax but not the survival gene bcl-2. J. Exp. Med. 1996, 183, 971–977. [Google Scholar] [CrossRef]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Haque, S.; Yan, X.J.; Rosen, L.; McCormick, S.; Chiorazzi, N.; Mongini, P.K. Effects of prostaglandin E2 on p53 mRNA transcription and p53 mutagenesis during T-cell-independent human B-cell clonal expansion. FASEB J. 2014, 28, 627–643. [Google Scholar] [CrossRef]
- Lezina, L.; Spriggs, R.V.; Beck, D.; Jones, C.; Dudek, K.M.; Bzura, A.; Jones, G.D.D.; Packham, G.; Willis, A.E.; Wagner, S.D. CD40L/IL-4-stimulated cll demonstrates variation in translational regulation of DNA damage response genes including ATM. Blood Adv. 2018, 2, 1869–1881. [Google Scholar] [CrossRef]
- Saville, M.K.; Sparks, A.; Xirodimas, D.P.; Wardrop, J.; Stevenson, L.F.; Bourdon, J.C.; Woods, Y.L.; Lane, D.P. Regulation of p53 by the ubiquitin-conjugating enzymes UBCH5B/C in vivo. J. Biol. Chem. 2004, 279, 42169–42181. [Google Scholar] [CrossRef]
- Khoury, M.P.; Bourdon, J.C. p53 Isoforms: An Intracellular Microprocessor? Genes. Cancer 2011, 2, 453–465. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Investig. 2005, 115, 755–764. [Google Scholar] [CrossRef]
- van Gent, R.; Kater, A.P.; Otto, S.A.; Jaspers, A.; Borghans, J.A.; Vrisekoop, N.; Ackermans, M.A.; Ruiter, A.F.; Wittebol, S.; Eldering, E.; et al. In vivo dynamics of stable chronic lymphocytic leukemia inversely correlate with somatic hypermutation levels and suggest no major leukemic turnover in bone marrow. Cancer Res. 2008, 68, 10137–10144. [Google Scholar] [CrossRef]
- Murphy, E.J.; Neuberg, D.S.; Rassenti, L.Z.; Hayes, G.; Redd, R.; Emson, C.; Li, K.; Brown, J.R.; Wierda, W.G.; Turner, S.; et al. Leukemia-cell proliferation and disease progression in patients with early stage chronic lymphocytic leukemia. Leukemia 2017, 31, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Trott, J.S.; Haque, S.; McCormick, S.; Chiorazzi, N.; Mongini, P.K. A cyclooxygenase-2/prostaglandin E2 pathway augments activation-induced cytosine deaminase expression within replicating human B cells. J. Immunol. 2010, 185, 5300–5314. [Google Scholar] [CrossRef] [PubMed]
- Rush, J.S.; Liu, M.; Odegard, V.H.; Unniraman, S.; Schatz, D.G. Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc. Natl. Acad. Sci. USA 2005, 102, 13242–13247. [Google Scholar] [CrossRef]
- Calissano, C.; Damle, R.N.; Hayes, G.; Murphy, E.J.; Hellerstein, M.K.; Moreno, C.; Sison, C.; Kaufman, M.S.; Kolitz, J.E.; Allen, S.L.; et al. In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood 2009, 114, 4832–4842. [Google Scholar] [CrossRef]
- Cuthill, K.M.; Zhang, Y.; Pepper, A.; Boelen, L.; Coulter, E.; Asquith, B.; Devereux, S.; Macallan, D.C. Identification of proliferative and non-proliferative subpopulations of leukemic cells in CLL. Leukemia 2022, 36, 2233–2241. [Google Scholar] [CrossRef]
- Inoue, T.; Shinnakasu, R.; Kawai, C.; Ise, W.; Kawakami, E.; Sax, N.; Oki, T.; Kitamura, T.; Yamashita, K.; Fukuyama, H.; et al. Exit from germinal center to become quiescent memory B cells depends on metabolic reprograming and provision of a survival signal. J. Exp. Med. 2020, 218, e20200866. [Google Scholar] [CrossRef]
- Kotmayer, L.; Laszlo, T.; Mikala, G.; Kiss, R.; Levay, L.; Hegyi, L.L.; Grof, S.; Nagy, T.; Barna, G.; Farkas, P.; et al. Landscape of BCL2 Resistance Mutations in a Real-World Cohort of Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia Treated with Venetoclax. Int. J. Mol. Sci. 2023, 24, 5802. [Google Scholar] [CrossRef]
- Correia, C.; Maurer, M.J.; McDonough, S.J.; Schneider, P.A.; Ross, P.E.; Novak, A.J.; Feldman, A.L.; Cerhan, J.R.; Slager, S.L.; Witzig, T.E.; et al. Relationship between BCL2 mutations and follicular lymphoma outcome in the chemoimmunotherapy era. Blood Cancer J. 2023, 13, 81. [Google Scholar] [CrossRef]
- Xiang, H.; Noonan, E.J.; Wang, J.; Duan, H.; Ma, L.; Michie, S.; Boxer, L.M. The immunoglobulin heavy chain gene 3′ enhancers induce BCL2 deregulation and lymphomagenesis in murine B cells. Leukemia 2011, 25, 1484–1493. [Google Scholar] [CrossRef]
- Le Noir, S.; Laffleur, B.; Carrion, C.; Garot, A.; Lecardeur, S.; Pinaud, E.; Denizot, Y.; Skok, J.; Cogne, M. The IgH locus 3′ cis-regulatory super-enhancer co-opts AID for allelic transvection. Oncotarget 2017, 8, 12929–12940. [Google Scholar] [CrossRef]
- O’Connor, D.S.; Grossman, D.; Plescia, J.; Li, F.; Zhang, H.; Villa, A.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc. Natl. Acad. Sci. USA 2000, 97, 13103–13107. [Google Scholar] [CrossRef] [PubMed]
- Chandele, A.; Prasad, V.; Jagtap, J.C.; Shukla, R.; Shastry, P.R. Upregulation of survivin in g2/m cells and inhibition of caspase 9 activity enhances resistance in staurosporine-induced apoptosis. Neoplasia 2004, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Welm, A.; Bishop, J.M. Cell division and cell survival in the absence of survivin. Proc. Natl. Acad. Sci. USA 2004, 101, 15100–15105. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.C.; Huang, W.; Scheres, S.H.; Shi, Y. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Miletic, A.V.; Jellusova, J.; Cato, M.H.; Lee, C.R.; Baracho, G.V.; Conway, E.M.; Rickert, R.C. Essential Role for Survivin in the Proliferative Expansion of Progenitor and Mature B Cells. J. Immunol. 2016, 196, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Calissano, C.; Damle, R.N.; Marsilio, S.; Yan, X.J.; Yancopoulos, S.; Hayes, G.; Emson, C.; Murphy, E.J.; Hellerstein, M.K.; Sison, C.; et al. Intraclonal complexity in chronic lymphocytic leukemia: Fractions enriched in recently born/divided and older/quiescent cells. Mol. Med. 2011, 17, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Calissano, C.; Damle, R.N.; Marsilio, S.; Yan, X.J.; Yancopoulos, S.; Hayes, G.; Emson, C.; Murphy, E.J.; Hellerstein, M.K.; Sison, C.; et al. Correction to: Intraclonal Complexity in Chronic Lymphocytic Leukemia: Fractions Enriched in Recently Born/Divided and Older/Quiescent Cells. Mol. Med. 2022, 28, 35. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Spitzer, M.H.; Joy, J.P.; Kaileh, M.; Qiu, X.; Nolan, G.P.; Sen, R. Postmitotic G1 phase survivin drives mitogen-independent cell division of B lymphocytes. Proc. Natl. Acad. Sci. USA 2022, 119, e2115567119. [Google Scholar] [CrossRef] [PubMed]
- Shadman, M. Diagnosis and Treatment of Chronic Lymphocytic Leukemia: A Review. JAMA 2023, 329, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Quartermaine, C.; Ghazi, S.M.; Yasin, A.; Awan, F.T.; Fradley, M.; Wiczer, T.; Kalathoor, S.; Ferdousi, M.; Krishan, S.; Habib, A.; et al. Cardiovascular Toxicities of BTK Inhibitors in Chronic Lymphocytic Leukemia: JACC: CardioOncology State-of-the-Art Review. JACC Cardio Oncol. 2023, 5, 570–590. [Google Scholar] [CrossRef]
- Kaneko, N.; Kita, A.; Yamanaka, K.; Mori, M. Combination of YM155, a survivin suppressant with a STAT3 inhibitor: A new strategy to treat diffuse large B-cell lymphoma. Leuk. Res. 2013, 37, 1156–1161. [Google Scholar] [CrossRef]
- Kaneko, N.; Mitsuoka, K.; Amino, N.; Yamanaka, K.; Kita, A.; Mori, M.; Miyoshi, S.; Kuromitsu, S. Combination of YM155, a survivin suppressant, with bendamustine and rituximab: A new combination therapy to treat relapsed/refractory diffuse large B-cell lymphoma. Clin. Cancer Res. 2014, 20, 1814–1822. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Lopez-Jimenez, J.; Smith, S.E.; Steinberg, J.; Keating, A.; Sasse, C.; Jie, F.; Thyss, A. A multicenter phase ii study of sepantronium bromide (YM155) plus rituximab in patients with relapsed aggressive B-cell Non-Hodgkin lymphoma. Leuk. Lymphoma 2016, 57, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Willimott, S.; Baou, M.; Naresh, K.; Wagner, S.D. CD154 induces a switch in pro-survival Bcl-2 family members in chronic lymphocytic leukaemia. Br. J. Haematol. 2007, 138, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Mongini, P.K.; Inman, J.K.; Han, H.; Kalled, S.L.; Fattah, R.J.; McCormick, S. Innate immunity and human B cell clonal expansion: Effects on the recirculating B2 subpopulation. J. Immunol. 2005, 175, 6143–6154. [Google Scholar] [CrossRef] [PubMed]
- Sewastianik, T.; Straubhaar, J.R.; Zhao, J.J.; Samur, M.K.; Adler, K.; Tanton, H.E.; Shanmugam, V.; Nadeem, O.; Dennis, P.S.; Pillai, V.; et al. miR-15a/16-1 deletion in activated B cells promotes plasma cell and mature B-cell neoplasms. Blood 2021, 137, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Budhram-Mahadeo, V.; Morris, P.J.; Smith, M.D.; Midgley, C.A.; Boxer, L.M.; Latchman, D.S. p53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J. Biol. Chem. 1999, 274, 15237–15244. [Google Scholar] [CrossRef] [PubMed]
- Willimott, S.; Wagner, S.D. miR-125b and miR-155 contribute to BCL2 repression and proliferation in response to CD40 ligand (CD154) in human leukemic B-cells. J. Biol. Chem. 2012, 287, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Fizzotti, M.; Pileri, S.; Liso, A.; Pasqualucci, L.; Flenghi, L. Bcl-6 protein expression in normal and neoplastic lymphoid tissues. Ann. Oncol. 1997, 8 (Suppl. S2), 101–104. [Google Scholar] [CrossRef] [PubMed]
- Patten, P.E.; Ferrer, G.; Chen, S.S.; Simone, R.; Marsilio, S.; Yan, X.J.; Gitto, Z.; Yuan, C.; Kolitz, J.E.; Barrientos, J.; et al. Chronic lymphocytic leukemia cells diversify and differentiate in vivo via a nonclassical Th1-dependent, Bcl-6-deficient process. JCI Insight 2016, 1, e86288. [Google Scholar] [CrossRef]
- Eischen, C.M.; Packham, G.; Nip, J.; Fee, B.E.; Hiebert, S.W.; Zambetti, G.P.; Cleveland, J.L. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene 2001, 20, 6983–6993. [Google Scholar] [CrossRef]
- Huang, D.C.; O’Reilly, L.A.; Strasser, A.; Cory, S. The anti-apoptosis function of BCL-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 1997, 16, 4628–4638. [Google Scholar] [CrossRef]
- O’Reilly, L.A.; Harris, A.W.; Tarlinton, D.M.; Corcoran, L.M.; Strasser, A. Expression of a Bcl-2 transgene reduces proliferation and slows turnover of developing B lymphocytes in vivo. J. Immunol. 1997, 159, 2301–2311. [Google Scholar] [CrossRef]
- Zinkel, S.; Gross, A.; Yang, E. Bcl2 family in DNA damage and cell cycle control. Cell Death Differ. 2006, 13, 1351–1359. [Google Scholar] [CrossRef]
- Masir, N.; Jones, M.; Lee, A.M.; Goff, L.K.; Clear, A.J.; Lister, A.; Marafioti, T.; Mason, D.Y. The expression of Bcl-2 by proliferating cells varies in different categories of B-cell lymphoma. Histopathology 2010, 56, 617–626. [Google Scholar] [CrossRef]
- Iqbal, J.; Sanger, W.G.; Horsman, D.E.; Rosenwald, A.; Pickering, D.L.; Dave, B.; Dave, S.; Xiao, L.; Cao, K.; Zhu, Q.; et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004, 165, 159–166. [Google Scholar] [CrossRef]
- Correia, C.; Schneider, P.A.; Dai, H.; Dogan, A.; Maurer, M.J.; Church, A.K.; Novak, A.J.; Feldman, A.L.; Wu, X.; Ding, H.; et al. BCL2 mutations are associated with increased risk of transformation and shortened survival in follicular lymphoma. Blood 2015, 125, 658–667. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef]
- Schuetz, J.M.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Tan, K.; Ben-Nierah, S.; Boyle, M.; Slack, G.W.; Marra, M.A.; Connors, J.M.; et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 2012, 26, 1383–1390. [Google Scholar] [CrossRef]
- Bouamar, H.; Jiang, D.; Wang, L.; Lin, A.P.; Ortega, M.; Aguiar, R.C. MicroRNA 155 control of p53 activity is context dependent and mediated by Aicda and Socs1. Mol. Cell Biol. 2015, 35, 1329–1340. [Google Scholar] [CrossRef]
- Arbore, G.; Henley, T.; Biggins, L.; Andrews, S.; Vigorito, E.; Turner, M.; Leyland, R. MicroRNA-155 is essential for the optimal proliferation and survival of plasmablast B cells. Life Sci. Alliance 2019, 2, e201800244. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef]
- Sutton, L.A.; Young, E.; Baliakas, P.; Hadzidimitriou, A.; Moysiadis, T.; Plevova, K.; Rossi, D.; Kminkova, J.; Stalika, E.; Pedersen, L.B.; et al. Different spectra of recurrent gene mutations in subsets of chronic lymphocytic leukemia harboring stereotyped B-cell receptors. Haematologica 2016, 101, 959–967. [Google Scholar] [CrossRef]
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Habe, S.; Winkler, D.; Buhler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G.; et al. TP53 mutation and survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2010, 28, 4473–4479. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef]
- Autore, F.; Strati, P.; Laurenti, L.; Ferrajoli, A. Morphological, immunophenotypic, and genetic features of chronic lymphocytic leukemia with trisomy 12: A comprehensive review. Haematologica 2018, 103, 931–938. [Google Scholar] [CrossRef]
- Boucher, J.; Charalambous, M.; Zarse, K.; Mori, M.A.; Kleinridders, A.; Ristow, M.; Ferguson-Smith, A.C.; Kahn, C.R. Insulin and insulin-like growth factor 1 receptors are required for normal expression of imprinted genes. Proc. Natl. Acad. Sci. USA 2014, 111, 14512–14517. [Google Scholar] [CrossRef]
- Yaktapour, N.; Ubelhart, R.; Schuler, J.; Aumann, K.; Dierks, C.; Burger, M.; Pfeifer, D.; Jumaa, H.; Veelken, H.; Brummer, T.; et al. Insulin-like growth factor-1 receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood 2013, 122, 1621–1633. [Google Scholar] [CrossRef]
- Maura, F.; Mosca, L.; Fabris, S.; Cutrona, G.; Matis, S.; Lionetti, M.; Agnelli, L.; Barbieri, M.; D’Anca, M.; Manzoni, M.; et al. Insulin growth factor 1 receptor expression is associated with NOTCH1 mutation, trisomy 12 and aggressive clinical course in chronic lymphocytic leukaemia. PLoS ONE 2015, 10, e0118801. [Google Scholar] [CrossRef]
- Rudich, S.M.; Winchester, R.; Mongini, P.K. Human B cell activation. Evidence for diverse signals provided by various monoclonal anti-IgM antibodies. J. Exp. Med. 1985, 162, 1236–1255. [Google Scholar] [CrossRef]
- Boucher, J.; Tseng, Y.H.; Kahn, C.R. Insulin and insulin-like growth factor-1 receptors act as ligand-specific amplitude modulators of a common pathway regulating gene transcription. J. Biol. Chem. 2010, 285, 17235–17245. [Google Scholar] [CrossRef]
- Winkler, D.; Schneider, C.; Krober, A.; Pasqualucci, L.; Lichter, P.; Dohner, H.; Stilgenbauer, S. Protein expression analysis of chromosome 12 candidate genes in chronic lymphocytic leukemia (CLL). Leukemia 2005, 19, 1211–1215. [Google Scholar] [CrossRef]
- Malek, S.N. The biology and clinical significance of acquired genomic copy number aberrations and recurrent gene mutations in chronic lymphocytic leukemia. Oncogene 2013, 32, 2805–2817. [Google Scholar] [CrossRef]
- Strati, P.; Shanafelt, T.D. Monoclonal B-cell lymphocytosis and early-stage chronic lymphocytic leukemia: Diagnosis, natural history, and risk stratification. Blood 2015, 126, 454–462. [Google Scholar] [CrossRef]
- Kolijn, P.M.; Hosnijeh, F.S.; Spath, F.; Hengeveld, P.J.; Agathangelidis, A.; Saleh, M.; Casabonne, D.; Benavente, Y.; Jerkeman, M.; Agudo, A.; et al. High-risk subtypes of chronic lymphocytic leukemia are detectable as early as 16 years prior to diagnosis. Blood 2022, 139, 1557–1563. [Google Scholar] [CrossRef]
- Ojha, J.; Secreto, C.; Rabe, K.; Ayres-Silva, J.; Tschumper, R.; Dyke, D.V.; Slager, S.; Fonseca, R.; Shanafelt, T.; Kay, N.; et al. Monoclonal B-cell lymphocytosis is characterized by mutations in CLL putative driver genes and clonal heterogeneity many years before disease progression. Leukemia 2014, 28, 2395–2398. [Google Scholar] [CrossRef]
- Fazi, C.; Scarfo, L.; Pecciarini, L.; Cottini, F.; Dagklis, A.; Janus, A.; Talarico, A.; Scielzo, C.; Sala, C.; Toniolo, D.; et al. General population low-count CLL-like MBL persists over time without clinical progression, although carrying the same cytogenetic abnormalities of CLL. Blood 2011, 118, 6618–6625. [Google Scholar] [CrossRef]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Bottcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef]
- Lin, W.Y.; Fordham, S.E.; Sunter, N.; Elstob, C.; Rahman, T.; Willmore, E.; Shepherd, C.; Strathdee, G.; Mainou-Fowler, T.; Piddock, R.; et al. Genome-wide association study identifies risk loci for progressive chronic lymphocytic leukemia. Nat. Commun. 2021, 12, 665. [Google Scholar] [CrossRef]
- Kwok, M.; Wu, C.J. Clonal Evolution of High-Risk Chronic Lymphocytic Leukemia: A Contemporary Perspective. Front. Oncol. 2021, 11, 790004. [Google Scholar] [CrossRef]
- Kolijn, P.M.; Spath, F.; Khouja, M.; Hengeveld, P.J.; van der Straten, L.; Darzentas, N.; Hultdin, M.; McKay, J.D.; Pott, C.; Vermeulen, R.C.H.; et al. Genetic drivers in the natural history of chronic lymphocytic leukemia development as early as 16 years before diagnosis. Blood 2023, 142, 1399–1403. [Google Scholar] [CrossRef]
- Kim, S.B.; Chae, G.W.; Lee, J.; Park, J.; Tak, H.; Chung, J.H.; Park, T.G.; Ahn, J.K.; Joe, C.O. Activated Notch1 interacts with p53 to inhibit its phosphorylation and transactivation. Cell Death Differ. 2007, 14, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Rosati, E.; Baldoni, S.; De Falco, F.; Del Papa, B.; Dorillo, E.; Rompietti, C.; Albi, E.; Falzetti, F.; Di Ianni, M.; Sportoletti, P. NOTCH1 aberrations in chronic lymphocytic leukemia. Front. Oncol. 2018, 8, 229. [Google Scholar] [CrossRef]
- Wang, L.; Brooks, A.N.; Fan, J.; Wan, Y.; Gambe, R.; Li, S.; Hergert, S.; Yin, S.; Freeman, S.S.; Levin, J.Z.; et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell 2016, 30, 750–763. [Google Scholar] [CrossRef]
- Aptullahoglu, E.; Wallis, J.P.; Marr, H.; Marshall, S.; Bown, N.; Willmore, E.; Lunec, J. SF3B1 Mutations Are Associated with Resistance to Non-Genotoxic MDM2 Inhibition in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2023, 24, 11335. [Google Scholar] [CrossRef]
- Ntoufa, S.; Gerousi, M.; Laidou, S.; Psomopoulos, F.; Tsiolas, G.; Moysiadis, T.; Papakonstantinou, N.; Mansouri, L.; Anagnostopoulos, A.; Stavrogianni, N.; et al. RPS15 mutations rewire RNA translation in chronic lymphocytic leukemia. Blood Adv. 2021, 5, 2788–2792. [Google Scholar] [CrossRef]
- Diop, F.; Moia, R.; Favini, C.; Spaccarotella, E.; De Paoli, L.; Bruscaggin, A.; Spina, V.; Terzi-di-Bergamo, L.; Arruga, F.; Tarantelli, C.; et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica 2020, 105, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Mongini, P.K.; Blessinger, C.A.; Dalton, J.P. Affinity requirements for induction of sequential phases of human B cell activation by membrane IgM-cross-linking ligands. J. Immunol. 1991, 146, 1791–1800. [Google Scholar] [CrossRef]
- Judge, A.D.; Zhang, X.; Fujii, H.; Surh, C.D.; Sprent, J. Interleukin 15 controls both proliferation and survival of a subset of memory-phenotype CD8(+) T cells. J. Exp. Med. 2002, 196, 935–946. [Google Scholar] [CrossRef]
- Carson, W.E.; Giri, J.G.; Lindemann, M.J.; Linett, M.L.; Ahdieh, M.; Paxton, R.; Anderson, D.; Eisenmann, J.; Grabstein, K.; Caligiuri, M.A. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J. Exp. Med. 1994, 180, 1395–1403. [Google Scholar] [CrossRef]
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002, 298, 2199–2202. [Google Scholar] [CrossRef]
- Criado, I.; Blanco, E.; Rodriguez-Caballero, A.; Alcoceba, M.; Contreras, T.; Gutierrez, M.L.; Romero, A.; Fernandez-Navarro, P.; Gonzalez, M.; Solano, F.; et al. Residual normal B-cell profiles in monoclonal B-cell lymphocytosis versus chronic lymphocytic leukemia. Leukemia 2018, 32, 2701–2705. [Google Scholar] [CrossRef]
- Blanco, E.; Perez-Andres, M.; Arriba-Mendez, S.; Contreras-Sanfeliciano, T.; Criado, I.; Pelak, O.; Serra-Caetano, A.; Romero, A.; Puig, N.; Remesal, A.; et al. Age-associated distribution of normal B-cell and plasma cell subsets in peripheral blood. J. Allergy Clin. Immunol. 2018, 141, 2208–2219.e2216. [Google Scholar] [CrossRef]
- Ghamlouch, H.; Ouled-Haddou, H.; Guyart, A.; Regnier, A.; Trudel, S.; Claisse, J.F.; Fuentes, V.; Royer, B.; Marolleau, J.P.; Gubler, B. TLR9 Ligand (CpG Oligodeoxynucleotide) Induces CLL B-Cells to Differentiate into CD20(+) Antibody-Secreting Cells. Front. Immunol. 2014, 5, 292. [Google Scholar] [CrossRef]
- Budeus, B.; Dampmann, M.; Kibler, A.; Brauser, M.; Bronischewski, K.; Homp, E.; Taudien, S.; Johansson, P.; Bloehdorn, J.; Stilgenbauer, S.; et al. Chronic lymphocytic leukemia includes a tumor subset resembling memory B cells that develop early and persist. bioRxiv 2022. [Google Scholar] [CrossRef]
- Krutzik, P.O.; Clutter, M.R.; Nolan, G.P. Coordinate analysis of murine immune cell surface markers and intracellular phosphoproteins by flow cytometry. J. Immunol. 2005, 175, 2357–2365. [Google Scholar] [CrossRef]
- Radler, P.D.; Wehde, B.L.; Wagner, K.U. Crosstalk between STAT5 activation and PI3K/AKT functions in normal and transformed mammary epithelial cells. Mol. Cell Endocrinol. 2017, 451, 31–39. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef]
- Muller, J.; Sperl, B.; Reindl, W.; Kiessling, A.; Berg, T. Discovery of chromone-based inhibitors of the transcription factor stat5. Chembiochem 2008, 9, 723–727. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Yamaguchi, S.; Hasegawa, M.; Nemoto, T.; Inoue, M.; Suzuki, K.; Hirokawa, K.; Kitagawa, M. Differential expression of survivin in bone marrow cells from patients with acute lymphocytic leukemia and chronic lymphocytic leukemia. Leuk. Res. 2004, 28, 487–494. [Google Scholar] [CrossRef]
- Li, F.; Yang, J.; Ramnath, N.; Javle, M.M.; Tan, D. Nuclear or cytoplasmic expression of survivin: What is the significance? Int. J. Cancer 2005, 114, 509–512. [Google Scholar] [CrossRef]
- Khan, S.; Aspe, J.R.; Asumen, M.G.; Almaguel, F.; Odumosu, O.; Acevedo-Martinez, S.; De Leon, M.; Langridge, W.H.; Wall, N.R. Extracellular, cell-permeable survivin inhibits apoptosis while promoting proliferative and metastatic potential. Br. J. Cancer 2009, 100, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Figel, S.; Birkemeier, M.; Dharma, S.S.; Barone, T.; Steinmetz, E.; Ciesielski, M.; Fenstermaker, R. Wild type, dEX3 and 2B survivin isoforms localize to the tumor cell plasma membrane, are secreted in exosomes, and interact with extracellular tubulin. Biochem. Biophys. Rep. 2021, 28, 101174. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLL A | IGHV Status | IGHV Gene | del13q14 B | Tri12+ B,C | del11q22 B | del17p B | ATM D Mut | Therapy (Mo Prior) |
|---|---|---|---|---|---|---|---|---|
| 275 | M | 3-30 | +(16% ho; 6% het) | neg | neg | neg | nd | |
| 321 | U | 4-34 | neg | +(53%) | neg | neg | nd | |
| 430 | U | 1-69*01 | +(97% het) | neg | +(10% het) | neg | + | 276 mo |
| 515 | U | 4-39*01 | +(6.5%) | nd | nd | nd | nd | |
| 600 | M | 4-34*01 | +(99% ho) | neg | neg | neg | nd | |
| 624 | M | 3-7*01 | neg | +(10%) | neg | neg | WT | |
| 625 | U | 1-69 | neg | neg | +(80% het) | neg | nd | |
| 631 | U | 3-20 | neg | neg | +(90% het) | neg | nd | |
| 675 | U | 3-23*01 | +(80% het) | neg | +(19% het) | neg | nd | 39 mo |
| 770 | U | 3-15*01 | +(89% het) | neg | neg | neg | + | |
| 849 | M | 3-72 | +(68% het) | +(96%) | neg | neg | nd | 72 mo |
| 887 | U | 3-30-3*01 | +(30% het) | neg | +(27%) | neg | WT | |
| 922 | M | 4-34*07 | +(45% het) | neg | neg | neg | nd | |
| 950 | U | 2-5*10 | +(89% het) | neg | neg | net | nd | 8 mo |
| 967 | M | 3-7*01 | neg | neg | neg | neg | nd | |
| 996 | U | 1-69 | neg | neg | neg | neg | nd | |
| 1013 | U | 3-33 | neg | +(52%) | neg | neg | nd | |
| 1031 | M | 4-39 | neg | neg | neg | neg | nd | |
| 1158 | U | 3-15*01 | +(97%) | nd | +(48%) | nd | nd | |
| 1238 | U | 3-30-3*01 | +(55% het) | neg | +(69% het) | neg | nd | |
| 1239 | U | 3-30*03 | neg | neg | neg | neg | WT | |
| 1300 | M | 3-7*02 | neg | neg | neg | neg | WT | |
| 1328 | M | 4-61*01 | +(86%) | neg | neg | neg | nd | |
| 1380 | M | 3-7*01 | +(50% ho; 22% het) | neg | neg | neg | nd | |
| 1413 | M | 4-39*01 | neg | +(20%) | neg | neg | nd | |
| 1529 | M | 4-59*01 | +(30% ho; 60% het) | neg | neg | neg | nd | |
| 1692 | U | 2-7 | neg | +(85%) | neg | neg | nd | |
| 1993 | U | 3-11*01 | +(90%) | nd | nd | +(90%) | nd | |
| 2018 | M | 3-84*01 | +(61%) | +(61%) | nd | nd | nd |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Haque, S.; Gupta, R.; Kolitz, J.E.; Allen, S.L.; Rai, K.; Chiorazzi, N.; Mongini, P.K.A. BCL2 Protein Progressively Declines during Robust CLL Clonal Expansion: Potential Impact on Venetoclax Clinical Efficacy and Insights on Mechanism. Lymphatics 2024, 2, 50-78. https://doi.org/10.3390/lymphatics2020005
Lee H, Haque S, Gupta R, Kolitz JE, Allen SL, Rai K, Chiorazzi N, Mongini PKA. BCL2 Protein Progressively Declines during Robust CLL Clonal Expansion: Potential Impact on Venetoclax Clinical Efficacy and Insights on Mechanism. Lymphatics. 2024; 2(2):50-78. https://doi.org/10.3390/lymphatics2020005
Chicago/Turabian StyleLee, Hyunjoo, Shabirul Haque, Rashmi Gupta, Jonathan E. Kolitz, Steven L. Allen, Kanti Rai, Nicholas Chiorazzi, and Patricia K. A. Mongini. 2024. "BCL2 Protein Progressively Declines during Robust CLL Clonal Expansion: Potential Impact on Venetoclax Clinical Efficacy and Insights on Mechanism" Lymphatics 2, no. 2: 50-78. https://doi.org/10.3390/lymphatics2020005