A Simple and Rapid Protocol for the Isolation of Murine Bone Marrow Suitable for the Differentiation of Dendritic Cells

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
2.2. Equipment
- Class II biological safety cabinet;
- Centrifuge;
- Water bath;
- Single channel pipettes;
- Serological pipettes;
- Inverted phase contrast microscope;
- Humidified incubator at 37 °C and 10% CO2;
- Hemocytometer;
- Dissection instruments, including MAYO Scissors 14.5 cm Straight Tungsten Carbide (Elite Medical, cat. 13-5200) to cut bone epiphyses.
3. Procedure
3.1. Bone Dissection (Sterile Work Required)
- Disinfect euthanised mice thoroughly with 80% ethanol and briefly dry the coat with paper towels, transfer mice into a biosafety cabinet;
- Cut skin from the middle of the lower abdomen along both hind legs;
- Remove excess muscle tissue to expose the leg bones (femur and tibia);
- Remove bones by dislocating the femur from the pubic bone;
- Remove excess muscle and tendons from the bones;
- Separate the femur and tibia;
- Transfer bones into ice-cold RF2 medium and keep them on ice until ready to proceed with the bone marrow isolation step (Section 3.2/Section 3.3).
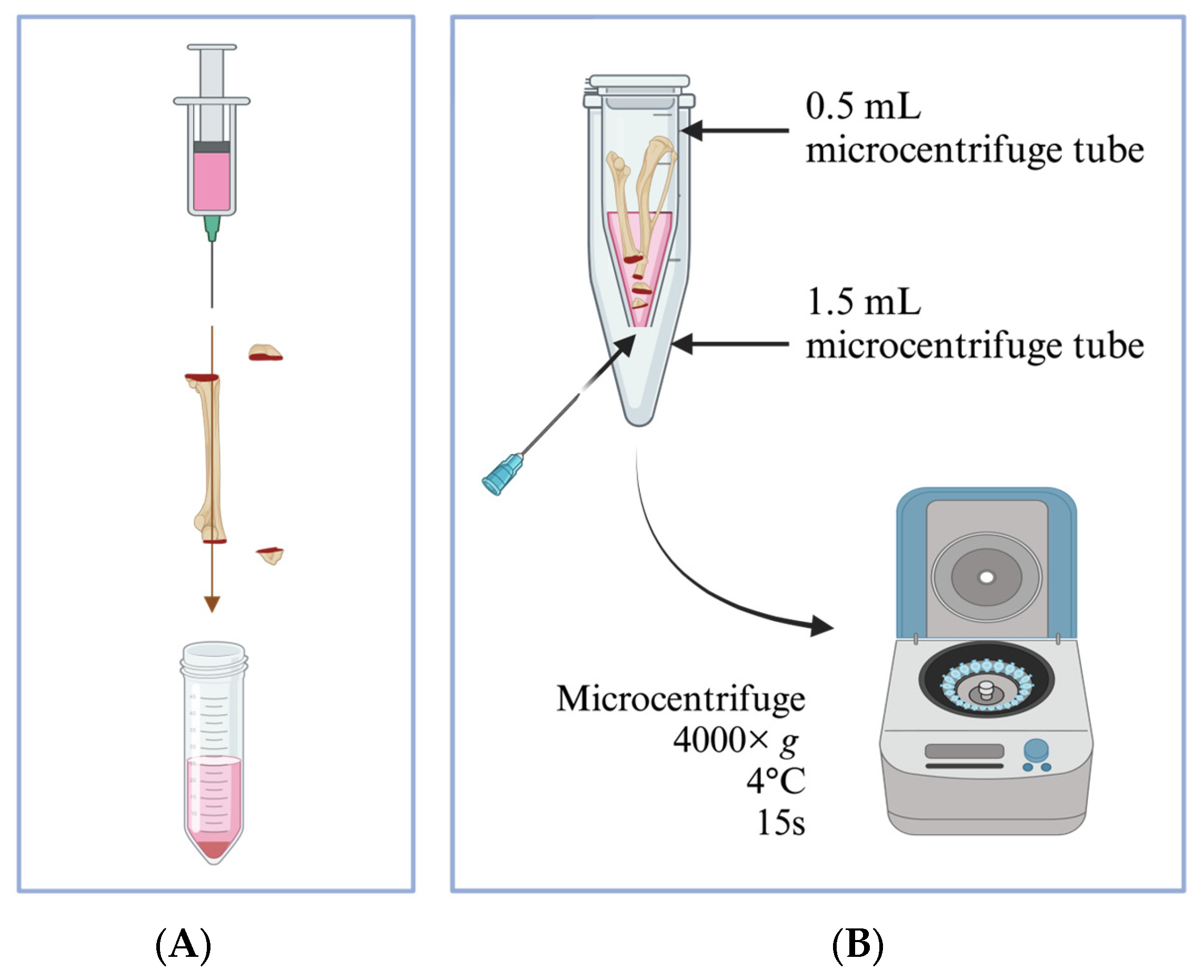
3.2. Bone Marrow Isolation Using the Flushing Method
- Cut open both ends of the bone to expose the bone marrow;
- Fill a 20 mL syringe with 20 mL ice-cold complete DC medium and attach to a 21-gauge needle;
- Flush out the bone marrow into a centrifuge tube (Figure 1A);
- Re-suspend the bone marrow aggregates by gently pipetting several times with a serological pipette.
3.3. Bone Marrow Isolation Using the Centrifugation Method
- Use a 21-gauge needle to pierce through the bottom of a sterile 0.5 mL microcentrifuge tube and place it into a sterile 1.5 mL microcentrifuge tube with the lid removed;
- Cut each bone open at the epiphysis; do not discard the epiphysis. Position the 2 bones and 2 epiphyses with the cut sides downwards inside the 0.5 mL microcentrifuge tube (Figure 1B);
- Add 100–150 µL ice-cold DC medium into the 0.5 mL tube containing bones and close its cap;
- Centrifuge at 4000× g for 15 s at 4 °C; the bone marrow will pellet in the 1.5 mL tube. Observe visually for a color change towards a cleared white bone, and spin one more time in case the bone marrow is still remaining in the bone;
- Discard the bones and 0.5 mL tubes, transfer the bone marrow from the 1.5 mL tubes into a 50 mL centrifuge tube, and add approximately 5 mL DC media, keeping the tube on ice. Each 50 mL tube can contain a maximum of bone marrow from 4 bones of 1 mouse (i.e., 2 tibiae + 2 femurs);
- Top up to 10 mL of ice-cold DC medium after the bone marrow has been collected into the tube;
- Pipette gently to make a single-cell suspension.
3.4. Bone Marrow Cell Culture and DC Generation
- Collect isolated bone marrow into a 50 mL conical centrifuge tube in DC medium and centrifuge at 400× g for 7 min at 4 °C;
- Remove supernatant and re-suspend cells in RBC lysis buffer. The volume of RBC lysis buffer depends on the number of mice: 1 mL lysis buffer for each mouse used (2 femurs, 2 tibiae);
- Mix by gentle pipetting and incubate for 30–40 s;
- Immediately top up with 9 mL ice-cold DC medium and underlay with 3–5 mL FCS.
- Centrifuge (400× g for 5 min at 4 °C) to pellet cells at the bottom. Re-suspend the bone marrow in 30 mL cold DC media;
- Pass cells through a 70 nm cell strainer, underlay with 3–5 mL FCS;
- Re-suspend cells in a defined volume of cold DC medium and count cells;
- Culture bone marrow cells at 1.5 × 106 cells/mL/3.2 cm2 in DC medium + Flt3L at 200 ng/mL. Accordingly, culturing can be performed in flasks or plates under the following conditions:96-well round bottom plate: 0.3 × 106 cells in 200 µL of medium per well;6-well plate: 4.5 × 106 cells in 3 mL of medium per well;25 cm2 flask: 15 × 106 cells in 10 mL medium;75 cm2 flask: 45 × 106 cells in 30 mL medium;10 cm dish: 25.5 × 106 cells in 17 mL medium.
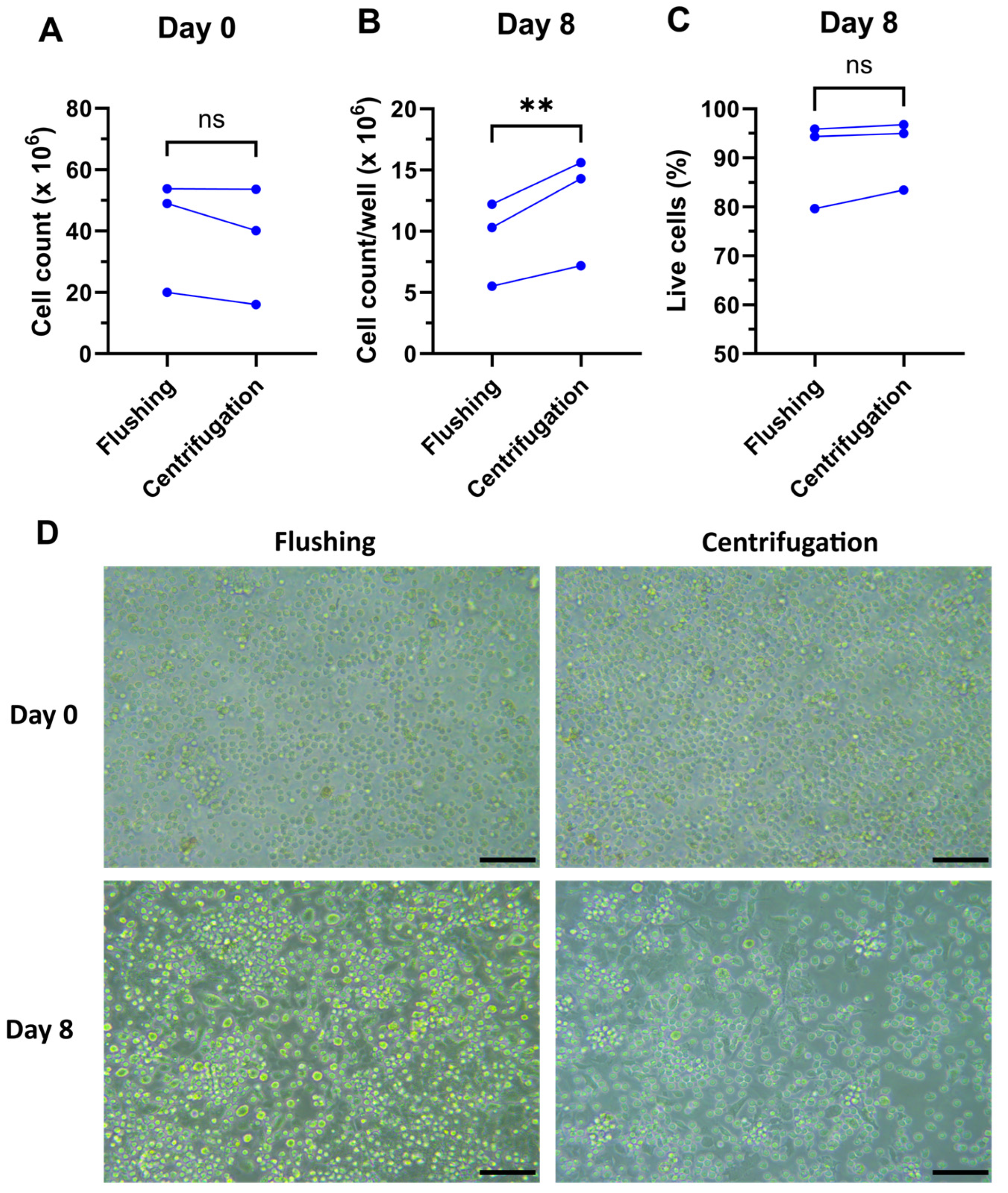
- Differentiate the bone marrow culture for 8 days. During the 8-day incubation period in a humidified incubator at 37 °C and 10% CO2, avoid mechanical forces such as shaking, moving, or otherwise handling the cells as much as possible. After 8 days of culture, the total cell yield should be similar to the cell input on Day 0 and overall approximately 40 × 106 per mouse (2 femurs, 2 tibiae).Day 8:
- After the 8-day incubation, cells are harvested. After removing all the media, cells are collected by adding 0.25 mM EDTA (e.g., 2 mL/6-well) and incubated at 37 °C for no more than 3 min. Most cells should detach from the surface and the remaining attaching cells can be gently detached by flushing with the DC medium. The differentiation into DCs can be checked using flow cytometry.
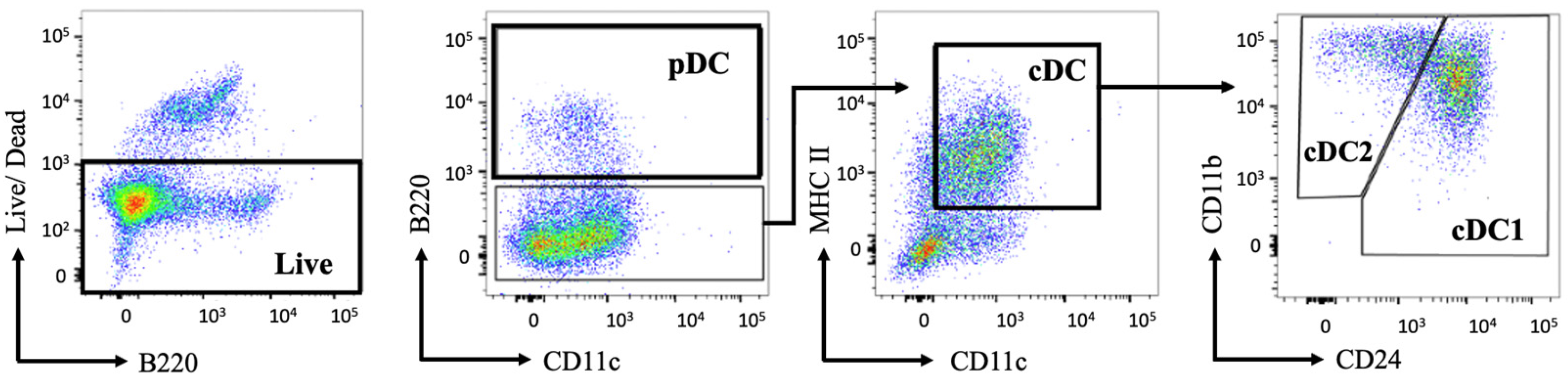
3.5. Flow Cytometry Staining
- Wash cells twice in PBS and resuspend at 5 × 105 cells in 100 μL of live/dead Aqua plus Fc Block mix in 1× PBS per stain;
- Incubate cells for 20 min in the dark on ice;
- Wash cells with 100 μL 1× PBS and centrifuge at 400× g for 5 min at 4 °C;
- Re-suspend cells in 100 μL of antibody cocktail (MHC II-AF700, CD11c-APC, CD45R/B220-FITC, CD11b-PeCy7, CD24-eF450, and CLEC9A-BV605). Single-color controls with cells or compensation beads can also be prepared at this time;
- Incubate cells for 30 min in the dark on ice;
- Wash cells with 100 μL 1× PBS and centrifuge at 400× g for 5 min at 4 °C;
- (Optional): Re-suspend cells in 100 μL of 1% PFA and incubate in the dark at room temperature for 20 min. Wash cells with 100 μL 1× PBS and centrifuge at 400× g for 5 min;
- Re-suspend cells in 100 μL 1× PBS for flow cytometric analysis.
3.6. Statistical Analysis
4. Expected Results
4.1. Total Cell Yields
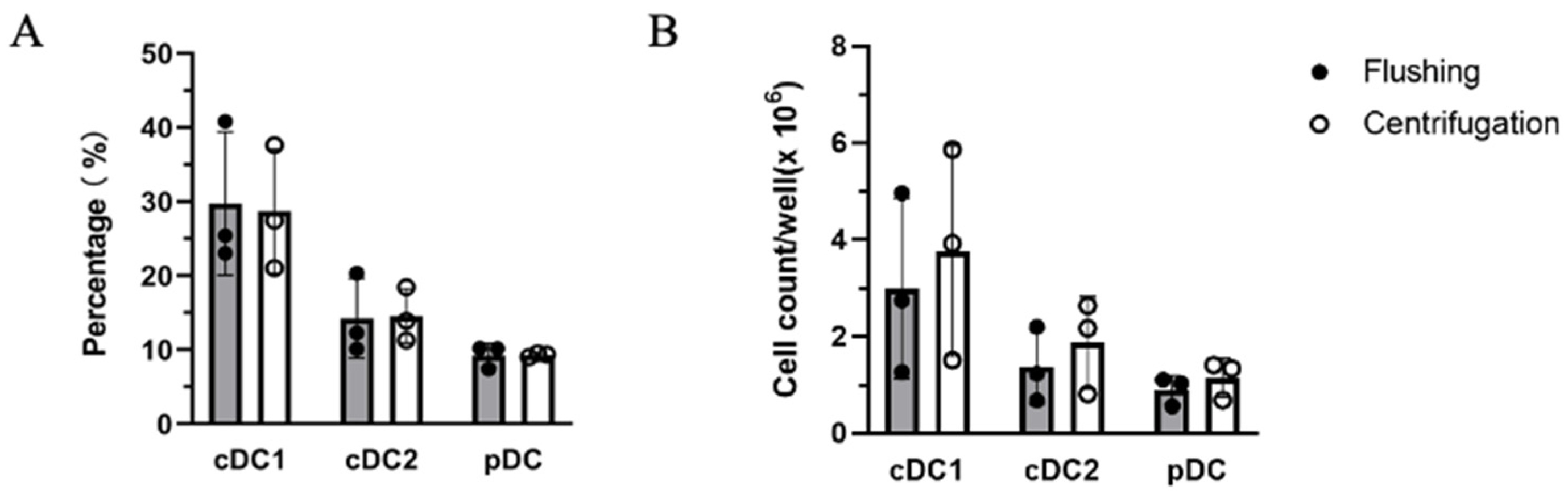
4.2. Flow Cytometric Analysis of Surface Marker Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mach, N.; Gillessen, S.; Wilson, S.B.; Sheehan, C.; Mihm, M.; Dranoff, G. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 2000, 60, 3239–3246. [Google Scholar]
- Daro, E.; Butz, E.; Smith, J.; Teepe, M.; Maliszewski, C.R.; McKenna, H.J. Comparison of the functional properties of murine dendritic cells generated in vivo with Flt3 ligand, GM-CSF and Flt3 ligand plus GM-SCF. Cytokine 2002, 17, 119–130. [Google Scholar] [CrossRef]
- Naik, S.H.; Proietto, A.I.; Wilson, N.S.; Dakic, A.; Schnorrer, P.; Fuchsberger, M.; Lahoud, M.H.; O’Keeffe, M.; Shao, Q.-x.; Chen, W.-f. Cutting edge: Generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 2005, 174, 6592–6597. [Google Scholar] [CrossRef]
- Naik, S.H.; O’Keeffe, M.; Proietto, A.; Shortman, H.H.K.; Wu, L. CD8+, CD8−, and plasmacytoid dendritic cell generation in vitro using flt3 ligand. Dendritic Cell Protoc. 2010, 595, 167–176. [Google Scholar]
- Mellman, I. Dendritic cells: Master regulators of the immune response. Cancer Immunol. Res. 2013, 1, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Macri, C.; Pang, E.S.; Patton, T.; O’Keeffe, M. Dendritic cell subsets. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2018; Volume 84, pp. 11–21. [Google Scholar]
- Heath, W.R.; Belz, G.T.; Behrens, G.M.; Smith, C.M.; Forehan, S.P.; Parish, I.A.; Davey, G.M.; Wilson, N.S.; Carbone, F.R.; Villadangos, J.A. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol. Rev. 2004, 199, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Tullett, K.M.; Tan, P.S.; Park, H.Y.; Schittenhelm, R.B.; Michael, N.; Li, R.; Policheni, A.N.; Gruber, E.; Huang, C.; Fulcher, A.J.; et al. RNF41 regulates the damage recognition receptor Clec9A and antigen cross-presentation in mouse dendritic cells. Elife 2020, 9, e63452. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Blees, H.; Henry, C.M.; Buck, M.D.; Schulz, O.; Rogers, N.C.; Childs, E.; Zelenay, S.; Rhys, H.; Domart, M.C.; et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat. Immunol. 2021, 22, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Caminschi, I.; Proietto, A.I.; Ahmet, F.; Kitsoulis, S.; Shin Teh, J.; Lo, J.C.; Rizzitelli, A.; Wu, L.; Vremec, D.; van Dommelen, S.L.; et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 2008, 112, 3264–3273. [Google Scholar] [CrossRef] [PubMed]
- Radford, K.J.; Tullett, K.M.; Lahoud, M.H. Dendritic cells and cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 26–32. [Google Scholar] [CrossRef]
- Shortman, K.; Lahoud, M.H.; Caminschi, I. Improving vaccines by targeting antigens to dendritic cells. Exp. Mol. Med. 2009, 41, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Quan, N. Immune cell isolation from mouse femur bone marrow. Bio-Protocol 2015, 5, e1631. [Google Scholar] [CrossRef] [PubMed]
- Kirkling, M.E.; Reizis, B. In Vitro Generation of Murine CD8α+ DEC205+ XCR1+ Cross-Presenting Dendritic Cells from Bone Marrow–Derived Hematopoietic Progenitors. In Dendritic Cells: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2023; pp. 109–119. [Google Scholar]
- Kirkling, M.E.; Cytlak, U.; Lau, C.M.; Lewis, K.L.; Resteu, A.; Khodadadi-Jamayran, A.; Siebel, C.W.; Salmon, H.; Merad, M.; Tsirigos, A. Notch signaling facilitates in vitro generation of cross-presenting classical dendritic cells. Cell Rep. 2018, 23, 3658–3672.e3656. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B. Guidelines for mouse and human DC generation. Immunology 2022, 53, 2249816. [Google Scholar] [CrossRef]
- Amend, S.R.; Valkenburg, K.C.; Pienta, K.J. Murine hind limb long bone dissection and bone marrow isolation. JoVE (J. Vis. Exp.) 2016, 110, e53936. [Google Scholar]
- Vining, K.H.; Mooney, D.J. Mechanical forces direct stem cell behaviour in development and regeneration. Nat. Rev. Mol. Cell Biol. 2017, 18, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Dong, Z.; Li, B.; Lu, F.; Li, Y. Mechanical Force Directs Proliferation and Differentiation of Stem Cells. Tissue Eng. Part B Rev. 2023, 29, 141–150. [Google Scholar] [CrossRef]
- Lee-Thedieck, C.; Spatz, J.P. Biophysical regulation of hematopoietic stem cells. Biomater. Sci. 2014, 2, 1548–1561. [Google Scholar] [CrossRef]
- Brasel, K.; De Smedt, T.; Smith, J.L.; Maliszewski, C.R. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood 2000, 96, 3029–3039. [Google Scholar] [CrossRef]
- Pandey, K.; Ramarathinam, S.H.; Purcell, A.W. Isolation of HLA bound peptides by immunoaffinity capture and identification by mass spectrometry. Curr. Protoc. 2021, 1, e92. [Google Scholar] [CrossRef]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry–based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [Google Scholar] [CrossRef] [PubMed]
- Nombela-Arrieta, C.; Manz, M.G. Quantification and three-dimensional microanatomical organization of the bone marrow. Blood Adv. 2017, 1, 407–416. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Source | Identifier |
|---|---|---|
| Pipette filter tips: 10 µL 20 µL | Neptune Scientific San Diego, CA, U.S.A. | Cat# BT10 Cat# BT20 |
| Stripettes: 5 mL | Corning Costar New York, NY, U.S.A. | Cat# CLS4487 |
| Conical centrifuge tubes: 50 mL | Greiner Bio-One Singapore, Singapore | Cat# 227270 |
| Microcentrifuge tubes: 0.5 mL 1.5 mL | Eppendorf Melbourne, Australia | Cat# 0030123603 Cat# 0030123611 |
| Falcon 70 µm cell strainer | Corning Costar New York, NY, U.S.A. | Cat # 352350 |
| Falcon 6-well plate-bottom plate | Corning Costar New York, NY, U.S.A. | Cat# 38016 |
| Reagents | Composition | Source | Identifier |
|---|---|---|---|
| RF2 | RPMI 1640 medium | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 21870-076 |
| 2% v/v fetal calf serum (FCS) | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# A3161001 | |
| Complete murine DC medium (308 ± 2 osmol/L) | RPMI 1640 | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 21870-076 |
| 10% v/v heat-inactivated FCS | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# A3161001 | |
| 1% v/v GlutaMax | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 35050-061 | |
| 1% v/v HEPES | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 15630-080 | |
| 1% v/v penicillin/streptomycin | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 15140-122 | |
| 100 µM β-Mercaptoethanol | Sigma-Aldrich St. Louis, MO, U.S.A. | Cat# 60-24-2 | |
| 2.5 mM EDTA | 2.5 mM EDTA | N/A | N/A |
| 1× PBS | 1× PBS | N/A | N/A |
| Erythrocyte lysis buffer | 8.3 g/L ammonium chloride in 0.01 M Tris-HCl buffer. | Sigma-Aldrich St. Louis, U.S.A. | Cat# R7757 |
| In vivo MAb recombinant Flt3L-Ig (hum/hum) | N/A | Bio X Cell Lebanon, PA, U.S.A. | Cat# BE0098 |
| Reagents and Antibodies | Source | Identifier | Working Dilution (in 100 μL) |
|---|---|---|---|
| Live/Dead Fixable Aqua Dead cell Stain Kit | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 34957 | 1:500 |
| Rat anti-mouse CD16/CD32 (mouse BD Fc block, clone 2.4G2) | BD Biosciences Franklin Lakes, NJ, U.S.A. | Cat# 553142 | 1:400 |
| Rat anti-mouse/human CD45R (B220, clone RA-6B2) | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 11-0452-85 | 1:300 |
| Rat anti-mouse MHC class II (clone M5/114.15.2) | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 56-5321-82 | 1:300 |
| Hamster anti-mouse CD11c (clone HL3) | BD Biosciences Franklin Lakes, NJ, U.S.A. | Cat# 550261 | 1:300 |
| Rat anti-mouse CD24 (clone M1/69) | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 48-0242-82 | 1:300 |
| Rat anti-CD11b (clone M1/70) | BD Biosciences Franklin Lakes, NJ, U.S.A. | Cat# 552850 | 1:200 |
| Rat anti-mouse CD370 (Clec9A, clone 10B4) | BD Biosciences Franklin Lakes, NJ, U.S.A. | Cat# 744511 | 1:100 |
| Compensation beads | Thermo Fisher Scientific Waltham, MA, U.S.A. | Cat# 01-3333-42 | 1:20 |
| PFA (Paraformaldehyde) | ProSciTech Kirwan, Australia | C004 | 1% in 1× PBS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, R.; Bafit, M.; Tullett, K.M.; Tan, P.S.; Lahoud, M.H.; O’Keeffe, M.; Purcell, A.W.; Braun, A. A Simple and Rapid Protocol for the Isolation of Murine Bone Marrow Suitable for the Differentiation of Dendritic Cells. Methods Protoc. 2024, 7, 20. https://doi.org/10.3390/mps7020020
Song R, Bafit M, Tullett KM, Tan PS, Lahoud MH, O’Keeffe M, Purcell AW, Braun A. A Simple and Rapid Protocol for the Isolation of Murine Bone Marrow Suitable for the Differentiation of Dendritic Cells. Methods and Protocols. 2024; 7(2):20. https://doi.org/10.3390/mps7020020
Chicago/Turabian StyleSong, Runqiu, Mariam Bafit, Kirsteen M. Tullett, Peck Szee Tan, Mireille H. Lahoud, Meredith O’Keeffe, Anthony W. Purcell, and Asolina Braun. 2024. "A Simple and Rapid Protocol for the Isolation of Murine Bone Marrow Suitable for the Differentiation of Dendritic Cells" Methods and Protocols 7, no. 2: 20. https://doi.org/10.3390/mps7020020