
Structured Framework and Genome Analysis of Magnaporthe grisea Inciting Pearl Millet Blast Disease Reveals Versatile Metabolic Pathways, Protein Families, and Virulence Factors


Abstract
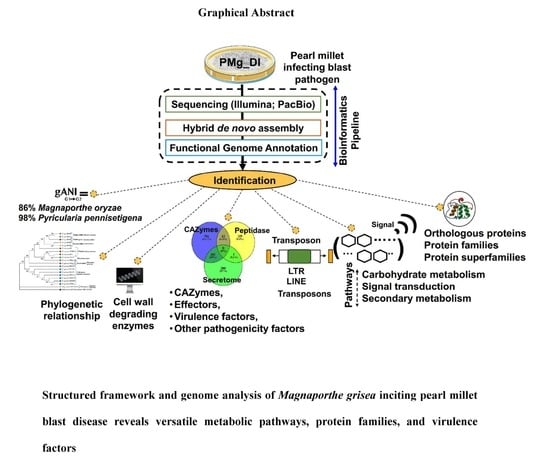
:
1. Introduction
2. Materials and Methods
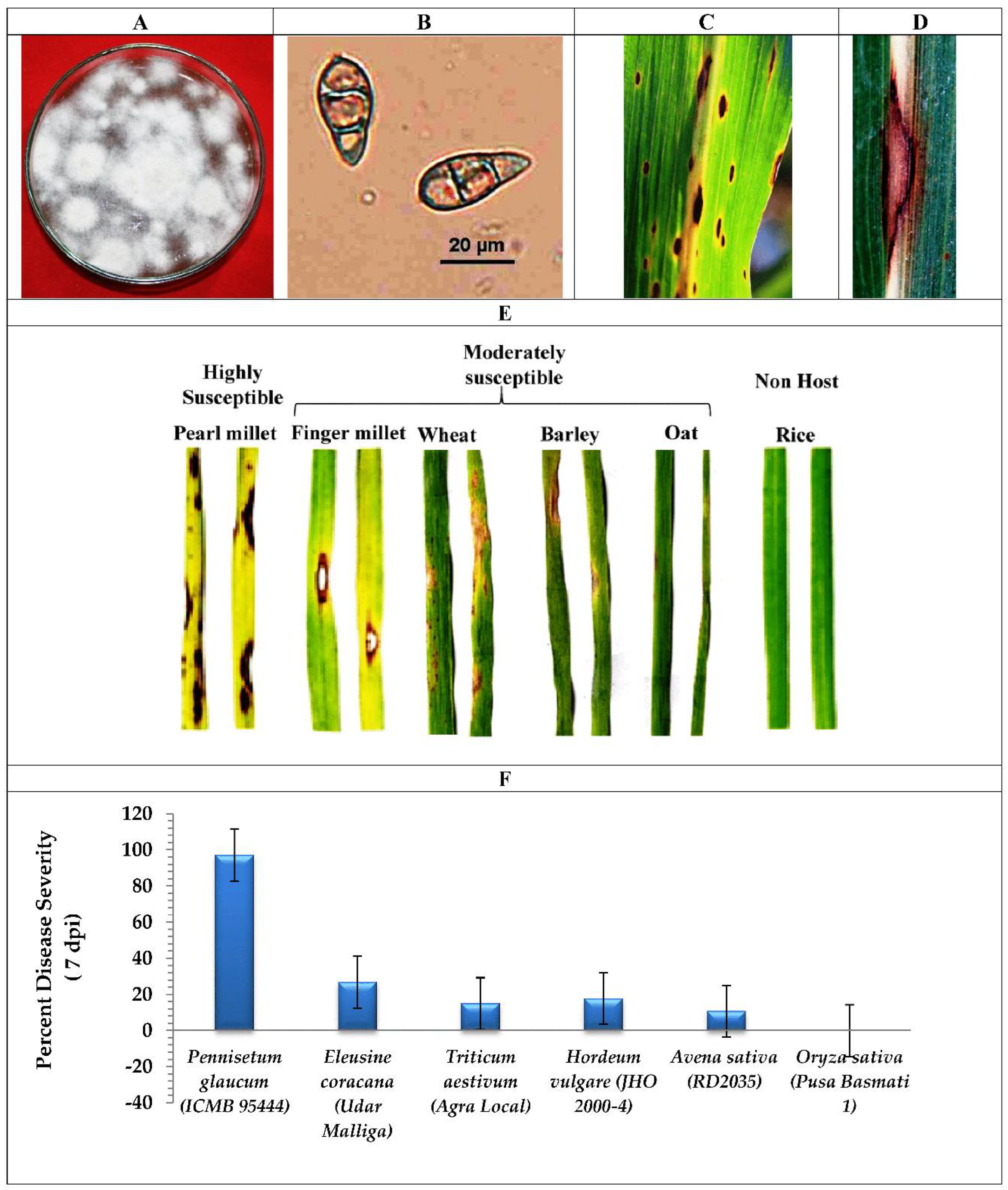
2.1. Fungus Isolation and Host Range Susceptibility Study
2.2. Experimental Materials, Data Collection, Sequencing, and Assembly
2.3. Functional Genome Annotation of M. grisea PMg_Dl
2.4. Phylogenetic Tree Analysis
3. Results
3.1. Genome Assembly, Assessment, and Genes Identification of M. grisea PMg_Dl
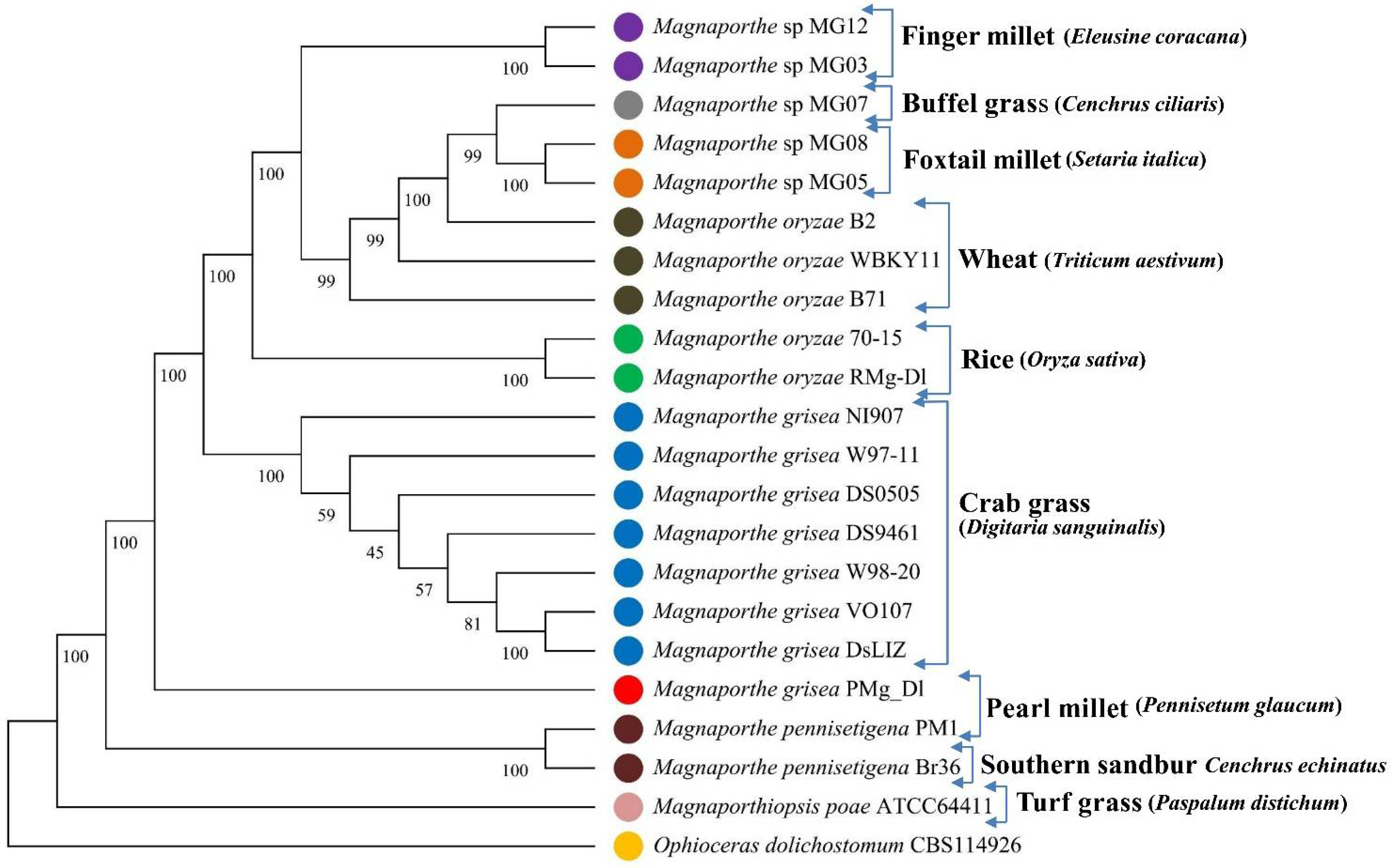
3.2. Phylogenetic Tree
3.3. Determination of Transposon and SSRs
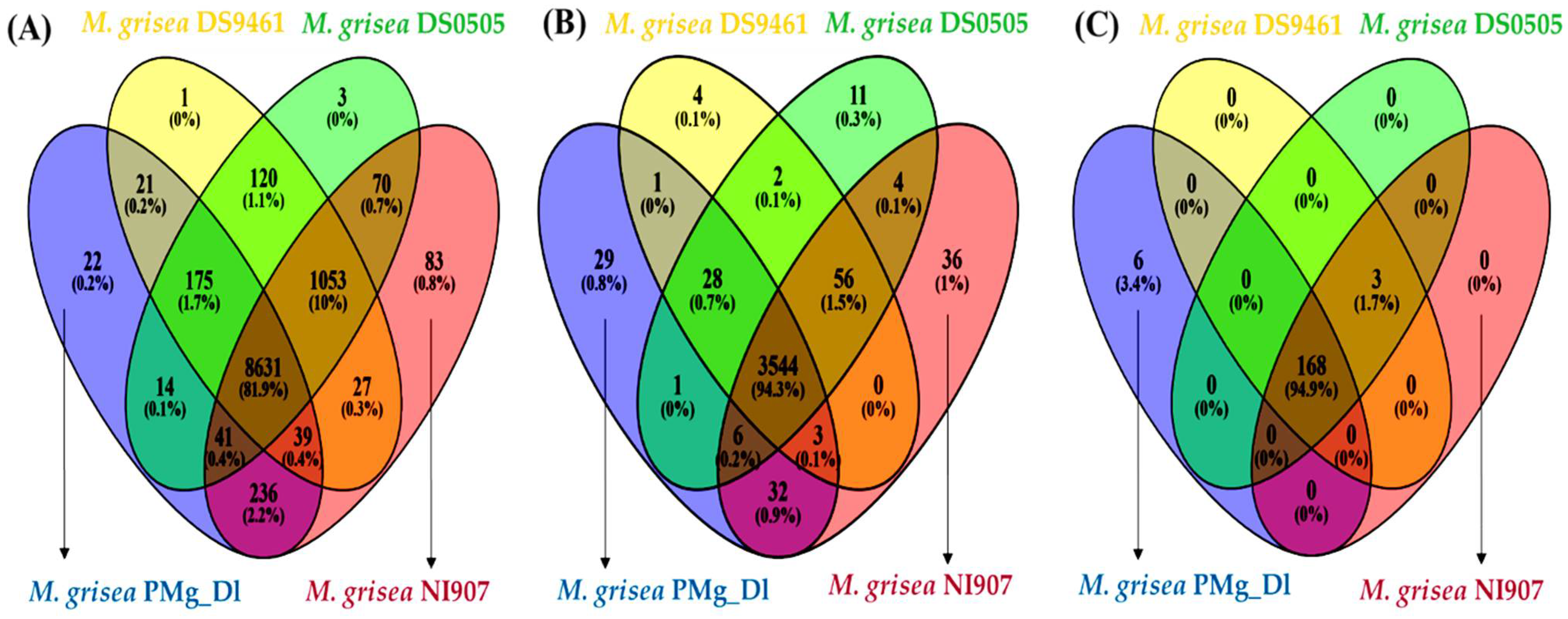
3.4. Analysis of Orthologous Genes, Protein Family, and CAZymes in Assembled Genomes
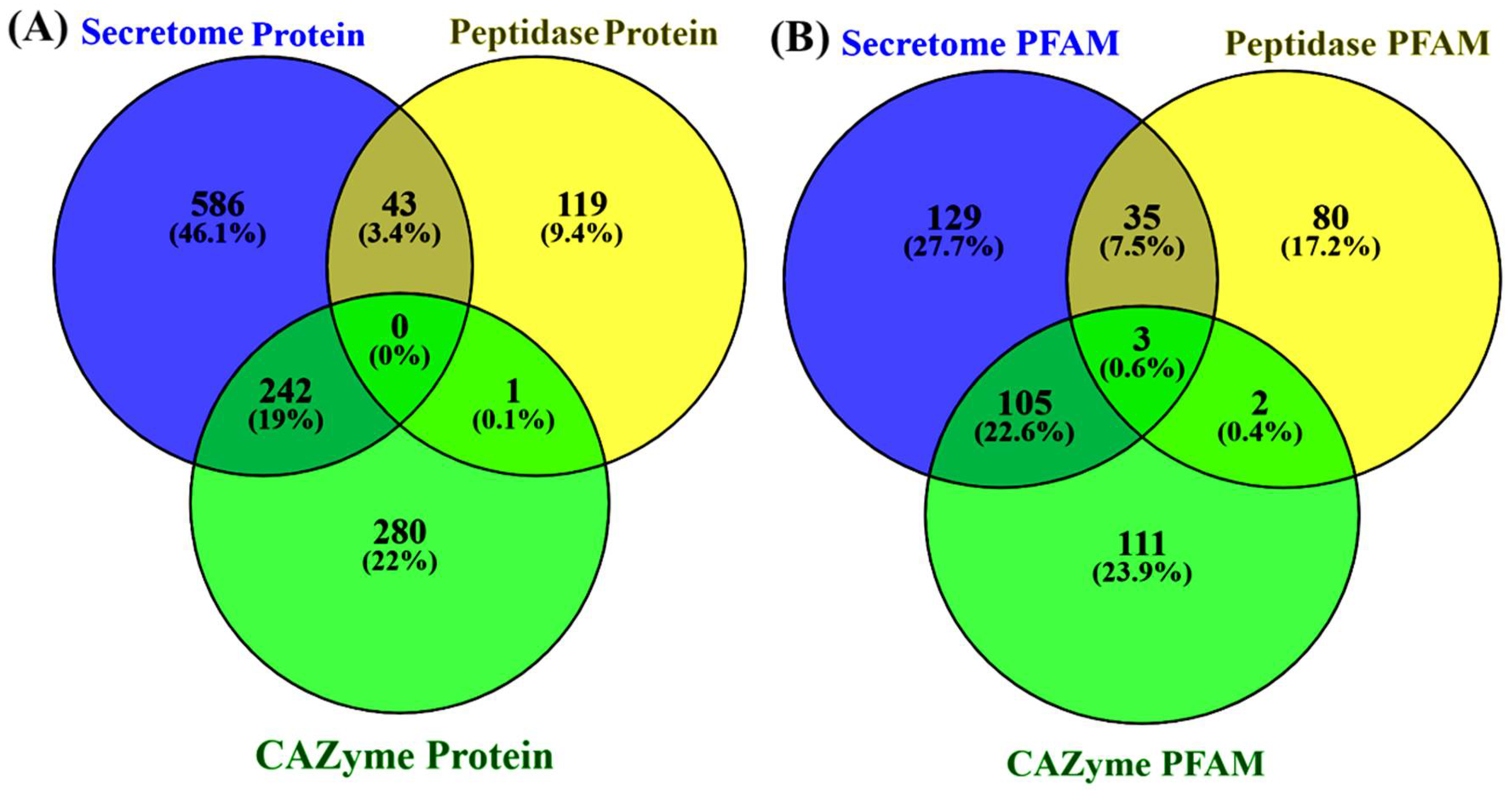
3.5. Determination of Secretome and Peptidase Proteins
3.6. Genome Functional Annotation Using Gene Ontology
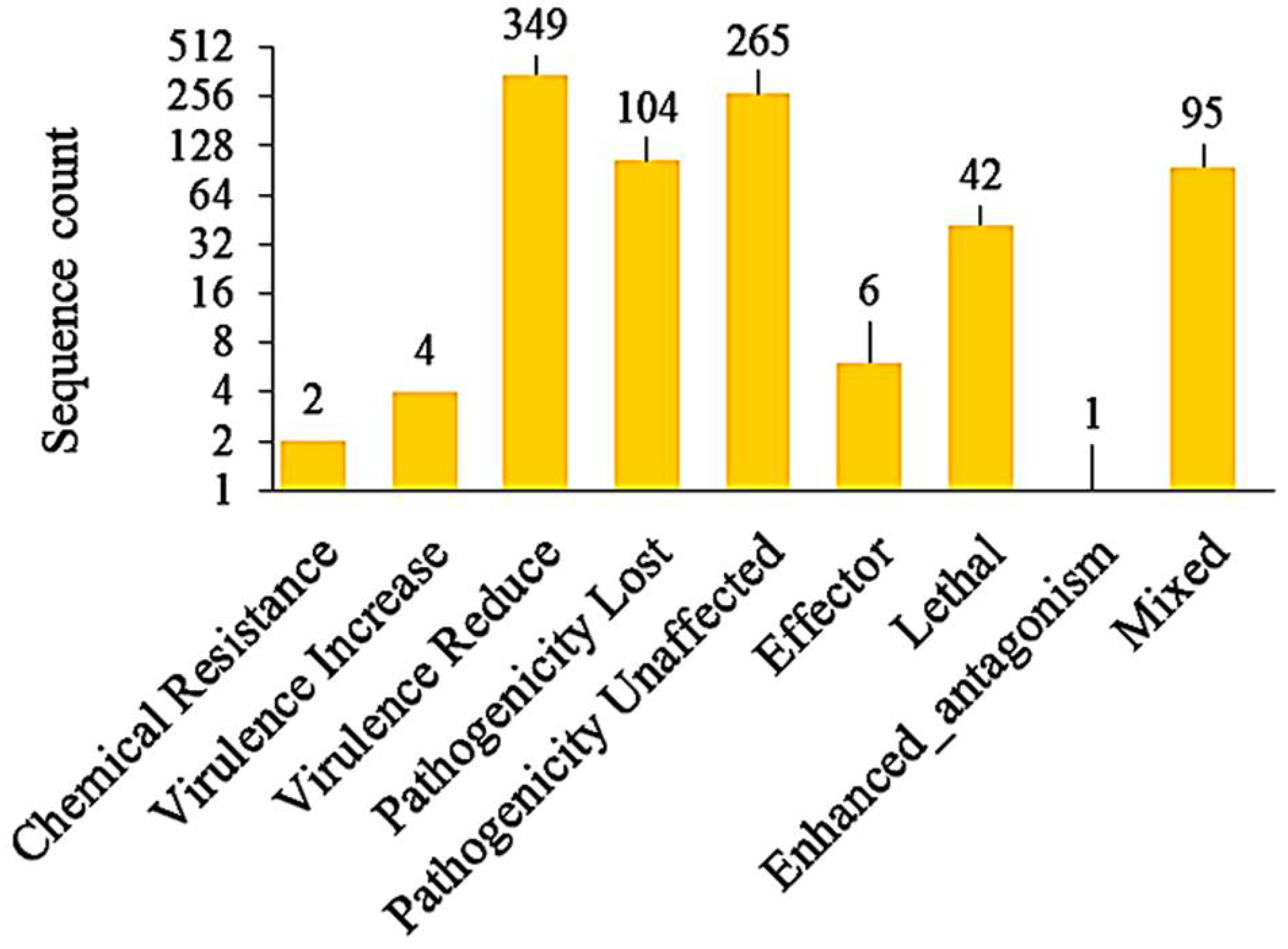
3.7. Identification of Pathogenicity Genes, VFs, and Effectors
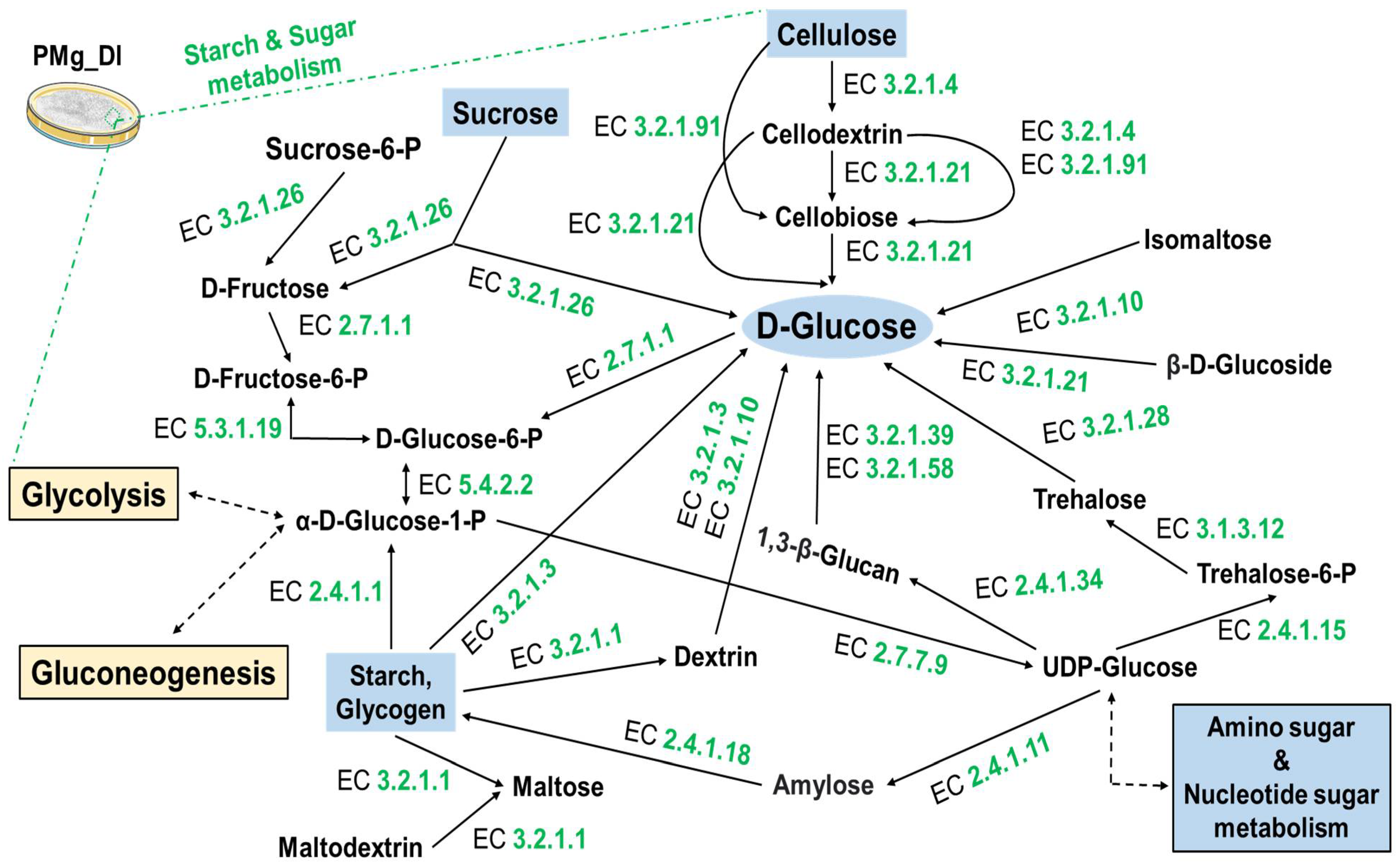
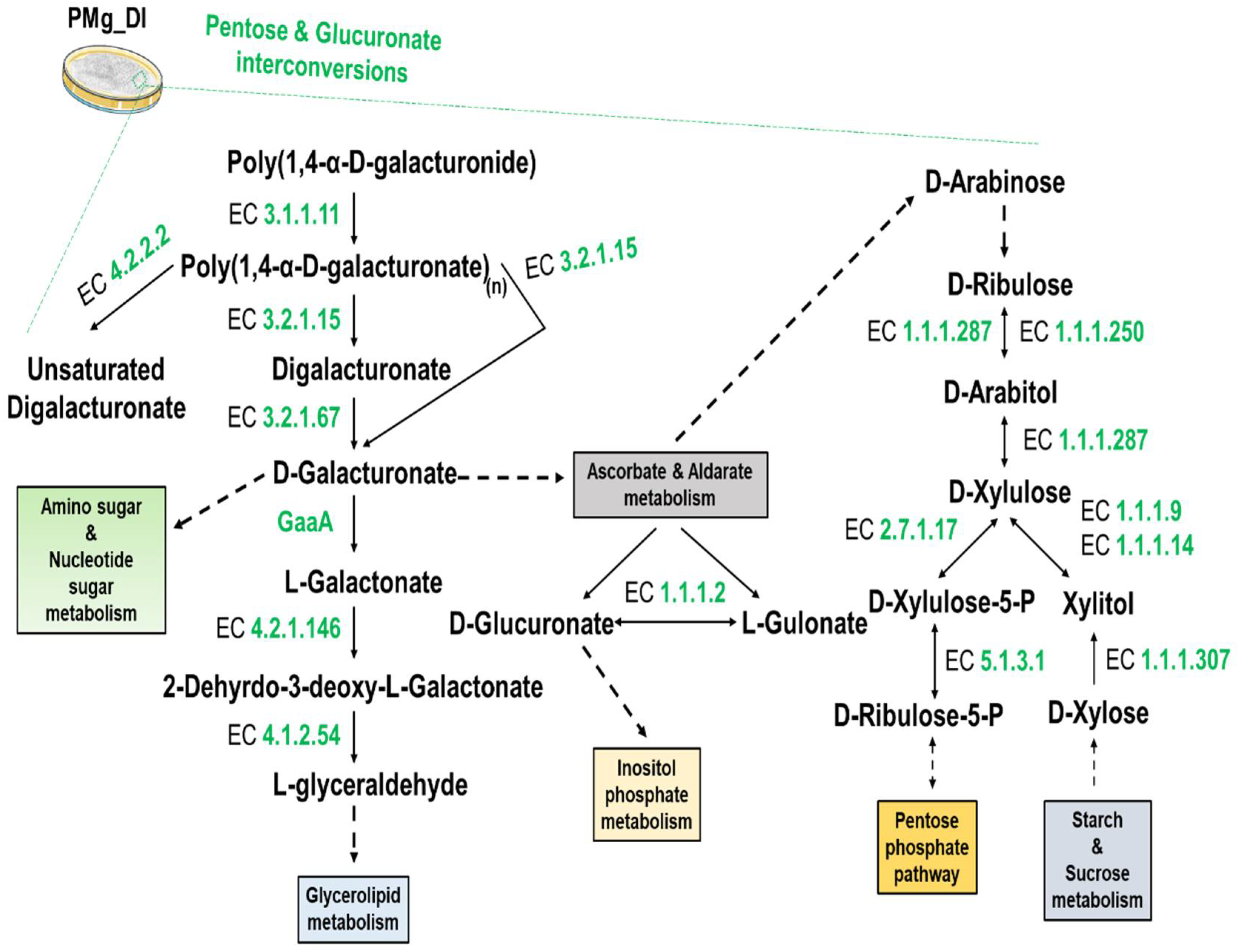
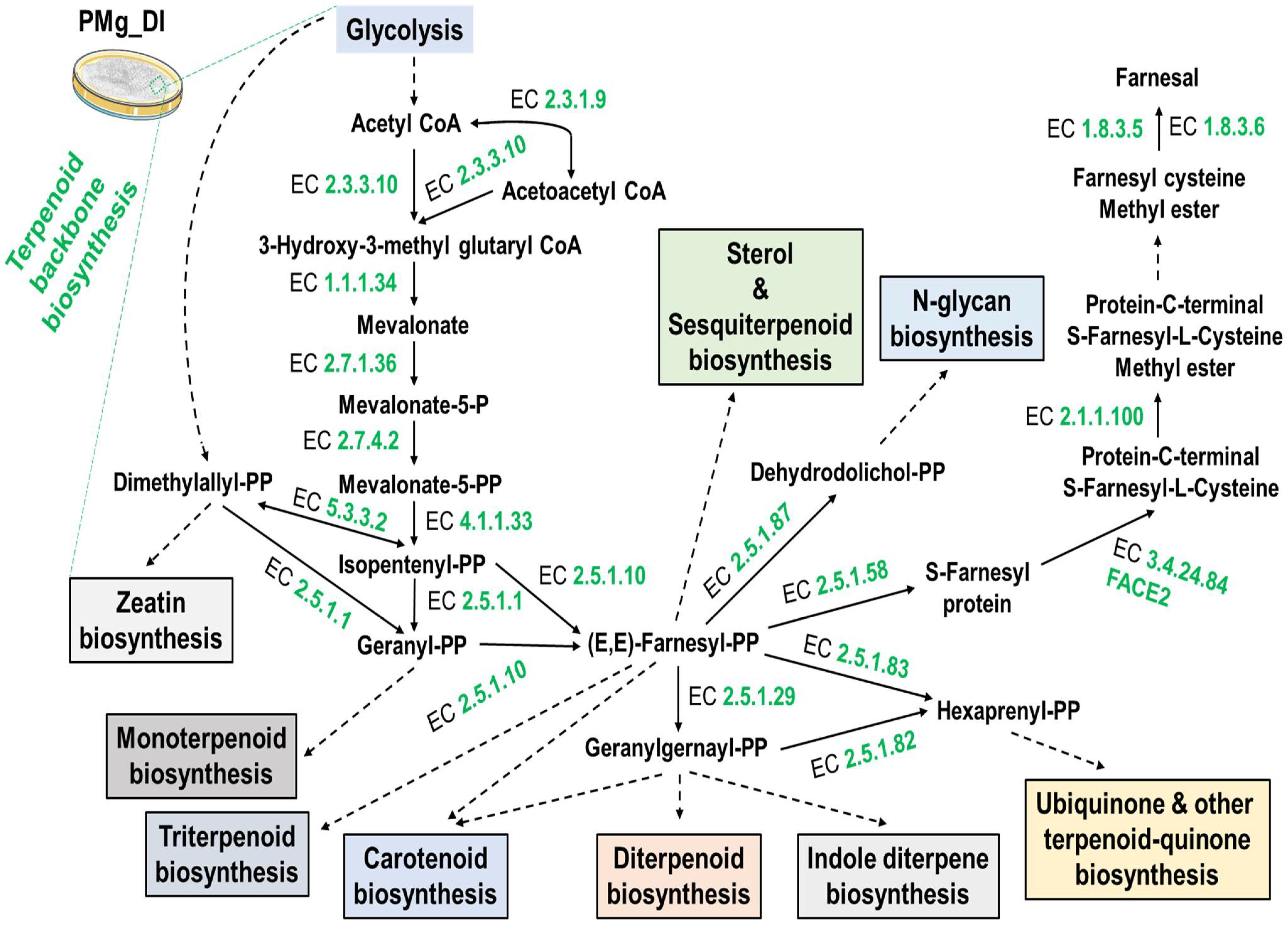
3.8. Identification of Metabolic Pathways in M. grisea PMg_Dl
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sood, S.; Singh, M. (Eds.) Introduction. In Millets and Pseudo Cereals; Woodhead Publishing: Sawston, UK, 2021; Chapter 1; pp. 1–6. [Google Scholar] [CrossRef]
- Ronald, P. Plant genetics, sustainable agriculture, and global food security. Genetics 2011, 188, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, O.P.; Gupta, S.K.; Govindaraj, M.; Sharma, R.; Varshney, R.K.; Srivastava, R.K.; Rathore, A.; Mahala, R.S. Genetic gains in pearl millet in India: Insights into historic breeding strategies and future perspective. Front. Plant Sci. 2021, 12, 396. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.D.; Sharma, B.; Saini, H.K. Processing, value addition, and health benefits. In Millets and Pseudo Cereals; Singh, M., Sood, S., Eds.; Woodhead Publishing: Sawston, UK, 2021. [Google Scholar] [CrossRef]
- Das, I.K. Millet Diseases: Current Status and Their Management. In Millets and Sorghum: Biology and Genetic Improvement; Wiley: Hoboken, NJ, USA, 2017; pp. 291–322. [Google Scholar] [CrossRef]
- Pordel, A.; Tharreau, D.; Cros-Arteil, S.; Shams, E.; Moumeni, A.; Mirzadi Gohari, A.; Javan-Nikkhah, M. Pyricularia oryzae causing blast on foxtail millet in Iran. Plant Dis. 2018, 102, 1853. [Google Scholar] [CrossRef]
- Nagaraja, A.; Chethana, B.S.; Jain, A.K. Biotic stresses and their management. In Millets and Pseudo Cereals; Singh, M., Sood, S., Eds.; Woodhead Publishing: Sawston, UK, 2021; Chapter 7; pp. 119–142. [Google Scholar] [CrossRef]
- Anil Kumar, B.M.; Hosahatti, R.; Tara Satyavathi, C.; Prakash, G.; Sharma, R.; Narasimhulu, R.; Chandra Nayaka, S. Pearl millet blast resistance: Current status and recent advancements in genomic selection and genome editing approaches. In Blast Disease of Cereal Crops: Evolution and Adaptation in Context of Climate Change; Anil Kumar, B.M., Hosahatti, R., Satyavathi, C.T., Prakash, G., Sharma, R., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 183–200. [Google Scholar] [CrossRef]
- Sharma, T.R.; Rai, A.K.; Gupta, S.K.; Vijayan, J.; Devanna, B.N.; Ray, S. Rice blast management through host-plant resistance: Retrospect and prospects. Agric. Res. 2012, 1, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.; Singh, B.; Dhakate, P.; Rahman, M.; Islam, M.A. Rice, Marker-assisted breeding, and disease resistance. In Disease Resistance in Crop Plants: Molecular, Genetic and Genomic Perspectives; Wani, S.H., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 83–111. [Google Scholar] [CrossRef]
- Jukanti, A.K.; Gowda, C.L.L.; Rai, K.N.; Manga, V.K.; Bhatt, R.K. Crops that feed the world 11. Pearl millet (Pennisetum glaucum L.): An important source of food security, nutrition, and health in the arid and semi-arid tropics. Food Secur. 2016, 8, 307–329. [Google Scholar] [CrossRef]
- Mbinda, W.; Masaki, H. Breeding strategies and challenges in the improvement of blast disease resistance in finger millet. A Current Review. Front. Plant Sci. 2021, 11, 2151. [Google Scholar] [CrossRef]
- Chandra, N.S.; Srivastava, R.K.; Udayashankar, A.C.; Lavanya, S.N.; Prakash, G.; Bishnoi, H.R.; Kadvani, D.L.; Vir, S.O.; Niranjana, S.R.; Prakash, H.S.; et al. Magnaporthe Blast of Pearl Millet in India Present Status and Prospects; Indian Council of Agricultural Research: New Delhi, India, 2017; pp. 1–33. [Google Scholar]
- Singh, S.; Sharma, R.; Chandra Nayaka, S.; Satyavathi, C.T.; Raj, C. Understanding pearl millet blast caused by Magnaporthe grisea and strategies for its management. In Blast Disease of Cereal Crops: Evolution and Adaptation in Context of Climate Change; Nayaka, S.C., Hosahatti, R., Prakash, G., Satyavathi, C.T., Sharma, R., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 151–172. [Google Scholar] [CrossRef]
- Sahu, K.P.; Patel, A.; Kumar, M.; Sheoran, N.; Mehta, S.; Reddy, B.; Eke, P.; Prabhakaran, N.; Kumar, A. Integrated metabarcoding and culturomic-based microbiome profiling of rice phyllosphere reveal diverse and functional bacterial communities for blast disease suppression. Front. Microbiol. 2021, 12, 780458. [Google Scholar] [CrossRef]
- Mehta, S.; Kumar, A.; Achary, V.M.M.; Ganesan, P.; Rathi, N.; Singh, A.; Sahu, K.P.; Lal, S.K.; Das, T.K.; Reddy, M.K. Antifungal activity of glyphosate against fungal blast disease on glyphosate-tolerant OsmEPSPS transgenic rice. Plant Sci. 2021, 311, 111009. [Google Scholar] [CrossRef]
- Prakash, G.; Kumar, A.; Sheoran, N.; Aggarwal, R.; Satyavathi, C.T.; Chikara, S.K.; Ghosh, A.; Jain, R.K. First draft genome sequence of a pearl millet blast pathogen, Magnaporthe grisea strain PMg_Dl, obtained using pacbio single-molecule real-time and Illumina next seq 500 sequencing. Microbiol. Resour. Announc. 2019, 8, e01499-18. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, S.; Joshi, S.M.; Athoni, B.K.; Patil, P.V.; Jogaiah, S. Elucidation of genetic relatedness of Magnaporthe grisea, an incitant of pearl millet blast disease by molecular markers associated with virulence of host differential cultivars. Microb. Pathog. 2020, 149, 104533. [Google Scholar] [CrossRef]
- Reddy, B.; Kumar, A.; Mehta, S.; Sahu, K.P. Methods of assessments of microbial diversity and their functional role in soil fertility and crop productivity. In Plant, Soil and Microbes in Tropical Ecosystems; Dubey, S.K., Verma, S.K., Eds.; Springer: Singapore, 2021; pp. 293–314. [Google Scholar] [CrossRef]
- Kumar, A.; Sheoran, N.; Prakash, G.; Ghosh, A.; Chikara, S.K.; Rajashekara, H.; Singh, U.D.; Aggarwal, R.; Jain, R.K. Genome sequence of a unique Magnaporthe oryzae RMg-Dl isolate from India that causes blast disease in diverse cereal crops, obtained using PacBio single-molecule and Illumina HiSeq2500 sequencing. Genome Announc. 2017, 5, e01570-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Dilokpimol, A.; Mäkelä, M.R.; Hildén, K.; Bervoets, S.; Riley, R.; Grigoriev, I.V.; Hainaut, M.; Henrissat, B.; de Vries, R.P.; et al. The draft genome sequence of the ascomycete fungus Penicillium subrubescens reveals a highly enriched content of plant biomass related CAZymes compared to related fungi. J. Biotechnol. 2017, 246, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.; Kumar, A.; Mehta, S.; Sheoran, N.; Chinnusamy, V.; Prakash, G. Hybrid de novo genome-reassembly reveals new insights on pathways and pathogenicity determinants in rice blast pathogen Magnaporthe oryzae RMg_Dl. Sci. Rep. 2021, 11, 22922. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Leggett, R.M.; Clavijo, B.J.; Clissold, L.; Clark, M.D.; Caccamo, M. NextClip: An analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 2014, 30, 566–568. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Warren, R.L.; Sutton, G.G.; Jones, S.J.M.; Holt, R.A. Assembling millions of short DNA sequences using SSAKE. Bioinformatics 2006, 23, 500–501. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Humann, J.L.; Lee, T.; Ficklin, S.; Main, D. Structural and functional annotation of eukaryotic genomes with GenSAS. Methods Mol. Biol. 2019, 1962, 29–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalili, V.; Afgan, E.; Gu, Q.; Clements, D.; Blankenberg, D.; Goecks, J.; Taylor, J.; Nekrutenko, A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res. 2020, 48, W395–W402. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, H.; Wu, P.; Entwistle, S.; Li, X.; Yohe, T.; Yi, H.; Yang, Z.; Yin, Y. dbCAN-seq: A database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2018, 46, D516–D521. [Google Scholar] [CrossRef]
- Lum, G.; Min, X.J. FunSecKB: The fungal secretome knowledgebase. Database 2011, 2011, bar001. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Bateman, A. How to use the MEROPS database and website to help understand peptidase specificity. Protein Sci. 2021, 30, 83–92. [Google Scholar] [CrossRef]
- Kall, L.; Krogh, A.; Sonnhammer, E.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Dodds, P. EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant Microbe Interact. 2021, 35, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, A.; Minio, A.; Griggio, F.; Delledonne, M.; Infantino, A.; Aragona, M. The genome assembly of the fungal pathogen Pyrenochaeta lycopersici from single-molecule real-time sequencing sheds new light on its biological complexity. PLoS ONE 2018, 13, e0200217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Yao, B.; Zhang, C. DFVF: Database of fungal virulence factors. Database 2012, 2012, bas032. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, X.; Zhang, Z. The Magnaporthe grisea species complex and plant pathogenesis. Mol. Plant Pathol. 2016, 17, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, L.; Vermani, M.; Kaur Ahluwalia, S.; Vijayaraghavan, P. Molecular virulence determinants of Magnaporthe oryzae: Disease pathogenesis and recent interventions for disease management in rice plant. Mycology 2021, 12, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Cools, H.J.; Hammond-Kosack, K.E. Exploitation of genomics in fungicide research: Current status and future perspectives. Mol. Plant Pathol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Mat Razali, N.; Cheah, B.H.; Nadarajah, K. Transposable Elements Adaptive role in genome plasticity, pathogenicity and evolution in fungal phytopathogens. Int. J. Mol. Sci. 2019, 20, 3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, P.H. Diverse transposable element landscapes in pathogenic and nonpathogenic yeast models: The value of a comparative perspective. Mobile DNA 2020, 11, 16. [Google Scholar] [CrossRef]
- Canfora, L.; Malusa, E.; Tkaczuk, C.; Tartanus, M.; Labanowska, B.H.; Pinzari, F. Development of a method for detection and quantification of B. brongniartii and B. bassiana in soil. Sci. Rep. 2016, 6, 22933. [Google Scholar] [CrossRef]
- Moges, A.D.; Admassu, B.; Belew, D.; Yesuf, M.; Njuguna, J.; Kyalo, M.; Ghimire, S.R. Development of microsatellite markers and analysis of genetic diversity and population structure of Colletotrichum gloeosporioides from Ethiopia. PLoS ONE 2016, 11, e0151257. [Google Scholar] [CrossRef]
- Sheoran, N.; Ganesan, P.; Mughal, N.M.; Yadav, I.S.; Kumar, A. Genome assisted molecular typing and pathotyping of rice blast pathogen, Magnaporthe oryzae, reveals a genetically homogenous population with high virulence diversity. Fungal Biol. 2021, 125, 733–747. [Google Scholar] [CrossRef]
- Peterson, D.; Li, T.; Calvo, A.M.; Yin, Y. Categorization of Orthologous Gene Clusters in 92 Ascomycota Genomes Reveals Functions Important for Phytopathogenicity. J. Fungi 2021, 7, 337. [Google Scholar] [CrossRef]
- Iquebal, M.A.; Tomar, R.S.; Parakhia, M.V.; Singla, D.; Jaiswal, S.; Rathod, V.M.; Padhiyar, S.M.; Kumar, N.; Rai, A.; Kumar, D. Draft whole genome sequence of groundnut stem rot fungus Athelia rolfsii revealing genetic architect of its pathogenicity and virulence. Sci. Rep. 2017, 7, 5299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervé, C.; Rogowski, A.; Blake, A.W.; Marcus, S.E.; Gilbert, H.J.; Knox, J.P. Carbohydrate-binding modules promote the enzymatic deconstruction of intact plant cell walls by targeting and proximity effects. Proc. Natl. Acad. Sci. USA 2010, 107, 15293–15298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.R. Correction: Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2014, 15, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.T.; Jeon, J.; Choi, J.; Cheong, K.; Song, H.; Choi, G.; Kang, S.; Lee, Y.H. Kingdom-Wide Analysis of Fungal Small Secreted Proteins (SSPs) Reveals their Potential Role in Host Association. Front. Plant Sci. 2016, 7, 186. [Google Scholar] [CrossRef] [Green Version]
- Yike, I. Fungal proteases and their pathophysiological effects. Mycopathologia 2011, 171, 299–323. [Google Scholar] [CrossRef]
- Erickson, R.H.; Kim, Y.S. Digestion and absorption of dietary protein. Annu. Rev. Med. 1990, 41, 133–139. [Google Scholar] [CrossRef]
- Hu, G.; Leger, R.J. A phylogenomic approach to reconstructing the diversification of serine proteases in fungi. J. Evol. Biol. 2004, 17, 1204–1214. [Google Scholar] [CrossRef]
- Xia, Y. Proteases in pathogenesis and plant defence. Cell. Microbiol. 2004, 6, 905–913. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, J.R. Effectors and effector delivery in Magnaporthe oryzae. PLoS Pathog. 2014, 10, e1003826. [Google Scholar] [CrossRef] [Green Version]
- Hogenhout, S.A.; Van der Hoorn, R.A.; Terauchi, R.; Kamoun, S. Emerging concepts in effector biology of plant-associated organisms. Mol. Plant Microbe Interact. 2009, 22, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Penselin, D.; Munsterkotter, M.; Kirsten, S.; Felder, M.; Taudien, S.; Platzer, M.; Ashelford, K.; Paskiewicz, K.H.; Harrison, R.J.; Hughes, D.J.; et al. Comparative genomics to explore phylogenetic relationship, cryptic sexual potential and host specificity of Rhynchosporium species on grasses. BMC Genom. 2016, 17, 953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cornelissen, B.; Rep, M. Host-specificity factors in plant pathogenic fungi. Fungal Genet. Biol. 2020, 144, 103447. [Google Scholar] [CrossRef] [PubMed]
- Bentham, A.R.; Petit-Houdenot, Y.; Win, J.; Chuma, I.; Terauchi, R.; Banfield, M.J.; Kamoun, S.; Langner, T. A single amino acid polymorphism in a conserved effector of the multihost blast fungus pathogen expands host-target binding spectrum. PLoS Pathog. 2021, 17, e1009957. [Google Scholar] [CrossRef] [PubMed]
- Borah, N.; Albarouki, E.; Schirawski, J. Comparative Methods for Molecular Determination of Host-Specificity Factors in Plant-Pathogenic Fungi. Int. J. Mol. Sci. 2018, 19, 863. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Reddy, B.; Dubey, S.K. De novo genome assembly and comparative annotation reveals metabolic versatility in cellulolytic bacteria from cropland and forest soils. Funct. Integr. Genom. 2020, 20, 89–101. [Google Scholar] [CrossRef]
- Lee, D.K.; Ahn, S.; Cho, H.Y.; Yun, H.Y.; Park, J.H.; Lim, J.; Lee, J.; Kwon, S.W. Metabolic response induced by parasitic plant-fungus interactions hinder amino sugar and nucleotide sugar metabolism in the host. Sci. Rep. 2016, 6, 37434. [Google Scholar] [CrossRef] [Green Version]
- Lees, J.G.; Dawson, N.L.; Sillitoe, I.; Orengo, C.A. Functional innovation from changes in protein domains and their combinations. Curr. Opin. Struct. Biol. 2016, 38, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Nandineni, M.R. Genome sequencing and comparative genomics reveal a repertoire of putative pathogenicity genes in chilli anthracnose fungus Colletotrichum truncatum. PLoS ONE 2017, 12, e0183567. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Tong, X.; Han, M.; Hu, H.; Dai, F. Genome-wide identification and characterization of WD40 protein genes in the Silkworm, Bombyx mori. Int. J. Mol. Sci. 2018, 19, 527. [Google Scholar] [CrossRef] [Green Version]
- Jain, B.P. Genome wide analysis of WD40 proteins in Saccharomyces cerevisiae and their orthologs in Candida albicans. Protein J. 2019, 38, 58–75. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; Zwiers, L.-H.; De Waard, M.A. Secretion of natural and synthetic toxic compounds from filamentous fungi by membrane transporters of the ATP-binding Cassette and major facilitator superfamily. Eur. J. Plant Pathol. 2002, 108, 719–734. [Google Scholar] [CrossRef]
- Kumar, S.; Lekshmi, M. Functional and structural roles of the major facilitator superfamily bacterial multidrug efflux pumps. Microorganisms 2020, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Mehrabi, R.; Xu, J.R. Mitogen-activated protein kinase pathways and fungal pathogenesis. Eukaryot. Cell 2007, 6, 1701–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, Y.; Zhong, S. The role of mitogen-activated protein (MAP) kinase signaling components in the fungal development, stress response and virulence of the fungal cereal pathogen Bipolaris sorokiniana. PLoS ONE 2015, 10, e0128291. [Google Scholar] [CrossRef] [Green Version]
- Roohparvar, R.; Huser, A.; Zwiers, L.H.; De Waard, M.A. Control of Mycosphaerella graminicola on wheat seedlings by medical drugs known to modulate the activity of ATP-binding cassette transporters. Appl. Environ. Microbiol. 2007, 73, 5011–5019. [Google Scholar] [CrossRef] [Green Version]
- Hamel, L.P.; Nicole, M.C.; Duplessis, S.; Ellis, B.E. Mitogen-activated protein kinase signaling in plant-interacting fungi: Distinct messages from conserved messengers. Plant Cell 2012, 24, 1327–1351. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Kovalchuk, A.; Kerio, S.; Asiegbu, F.O. Distribution and bioinformatic analysis of the cerato-platanin protein family in Dikarya. Mycologia 2013, 105, 1479–1488. [Google Scholar] [CrossRef]
- Shor, E.; Chauhan, N. A case for two-component signaling systems as antifungal drug targets. PLoS Pathol. 2015, 11, e1004632. [Google Scholar] [CrossRef] [Green Version]
- Adachi, K.; Hamer, J.E. Divergent cAMP signaling pathways regulate growth and pathogenesis in the rice blast fungus Magnaporthe grisea. Plant Cell 1998, 10, 1361–1374. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.Z.; Wang, Z.F.; Liu, Y.D.; Zhang, G.Z.; Li, G. The whole-genome sequencing and analysis of a Ganoderma lucidum strain provide insights into the genetic basis of its high triterpene content. Genomics 2021, 113, 840–849. [Google Scholar] [CrossRef]
- Moktali, V.; Park, J.; Fedorova-Abrams, N.D.; Park, B.; Choi, J.; Lee, Y.H.; Kang, S. Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genom. 2012, 13, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Kim, J.E.; Lee, Y.W.; Son, H. Fungal Cytochrome P450s and the P450 Complement (CYPome) of Fusarium graminearum. Toxins 2018, 10, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attributes | M. grisea PMg_Dl |
|---|---|
| Illumina NextSeq 500 PE | 43,962,401 (PE reads); 13.1 Gb |
| Illumina NextSeq 500 MP | 17,160,010 (MP reads); 3.4 Gb |
| PacBio RS II | 148,768 single end; 1.1 Gb |
| Genome size | 47,897,363 |
| Number of scaffolds | 341 |
| Scaffold N50 | 765,468 |
| Largest scaffold | 2,246,534 |
| GC% | 47.3 |
| No. of genes | 10,218 |
| Proteins | 10,184 |
| rRNA | 38 |
| tRNA | 209 |
| SSR | 79,317 |
| SSR bases | 243,245 |
| Virulence factors | 51 |
| CAZymes | 539 |
| Peptidase | 163 |
| Secretome proteins | 871 |
| Effector proteins | 594 |
| Family | Copy Number (M. grisea PMg_Dl) |
|---|---|
| DNA | 326 |
| DNA/TcMar-Fot1 | 471 |
| DNA/TcMar-Mariner | 38 |
| DNA/TcMar-Tc1 | 20 |
| DNA/hAT-Ac | 54 |
| DNA/hAT-Restless | 54 |
| Type: DNA | 971 |
| LINE/Tad1 | 147 |
| Type: LINE | 173 |
| LTR/Copia | 1672 |
| LTR/Gypsy | 837 |
| Type: LTR | 2509 |
| Type: EVERYTHING_TE | 3653 |
| Type: Simple_repeat | 28 |
| Type: Unknown | 7033 |
| LINE/CRE-Cnl1 | 26 |
| DNA/TcMar-Ant1 | 8 |
| Total | 18,020 |
| Family | M. grisea PMg_Dl * | M. grisea DS9461 * | M. grisea DS0505 * | M. grisea NI907 * |
|---|---|---|---|---|
| AA | 127 | 123 | 121 | 117 |
| CBM | 11 | 15 | 15 | 15 |
| CE | 50 | 48 | 50 | 51 |
| GH | 253 | 256 | 254 | 252 |
| GT | 91 | 95 | 94 | 96 |
| PL | 7 | 6 | 6 | 6 |
| Total | 539 | 543 | 540 | 537 |
| Carbohydrate Metabolism Pathways | M. grisea PMg_Dl (Gene Count) |
|---|---|
| KO:00010 Glycolysis/Gluconeogenesis | 25 |
| KO:00020 Citrate cycle | 20 |
| KO:00030 Pentose phosphate pathway | 18 |
| KO:00040 Pentose and glucuronate inter conversions | 18 |
| KO:00051 Fructose and mannose metabolism | 21 |
| KO:00052 Galactose metabolism | 14 |
| KO:00053 Ascorbate and aldarate metabolism | 6 |
| KO:00500 Starch and sucrose metabolism | 26 |
| KO:00520 Amino sugar and nucleotide sugar metabolism | 26 |
| KO:00620 Pyruvate metabolism | 28 |
| KO:00630 Glyoxylate and dicarboxylate metabolism | 21 |
| KO:00650 Butanoate metabolism | 12 |
| KO:00640 Propanoate metabolism | 18 |
| KO:00660 C5-Branched dibasic acid metabolism | 3 |
| KO:00562 Inositol phosphate metabolism | 21 |
| Total | 277 |
| Signal Transduction Pathways | M. grisea PMg_Dl (Gene Count) |
|---|---|
| KO:02020 Two-component system | 19 |
| KO:04014 Ras signaling pathway | 19 |
| KO:04015 Rap1 signaling pathway | 10 |
| KO:04010 MAPK signaling pathway | 17 |
| KO:04013 MAPK signaling pathway—fly | 14 |
| KO:04016 MAPK signaling pathway—plant | 4 |
| KO:04011 MAPK signaling pathway—yeast | 56 |
| KO:04012 ErbB signaling pathway | 6 |
| KO:04310 Wnt signaling pathway | 13 |
| KO:04330 Notch signaling pathway | 4 |
| KO:04340 Hedgehog signaling pathway | 5 |
| KO:04341 Hedgehog signaling pathway—fly | 7 |
| KO:04350 TGF-beta signaling pathway | 7 |
| KO:04390 Hippo signaling pathway | 9 |
| KO:04391 Hippo signaling pathway—fly | 7 |
| KO:04392 Hippo signaling pathway—multiple species | 6 |
| KO:04370 VEGF signaling pathway | 9 |
| KO:04371 Apelin signaling pathway | 14 |
| KO:04630 Jak-STAT signaling pathway | 4 |
| KO:04064 NF-kappa B signaling pathway | 5 |
| KO:04668 TNF signaling pathway | 3 |
| KO:04066 HIF-1 signaling pathway | 15 |
| KO:04068 FoxO signaling pathway | 14 |
| KO:04020 Calcium signaling pathway | 11 |
| KO:04070 Phosphatidylinositol signaling system | 18 |
| KO:04072 Phospholipase D signaling pathway | 13 |
| KO:04071 Sphingolipid signaling pathway | 20 |
| KO:04024 cAMP signaling pathway | 12 |
| KO:04022 cGMP-PKG signaling pathway | 11 |
| KO:04151 PI3K-Akt signaling pathway | 24 |
| KO:04152 AMPK signaling pathway | 23 |
| KO:04150 mTOR signaling pathway | 38 |
| Total | 437 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, B.; Mehta, S.; Prakash, G.; Sheoran, N.; Kumar, A. Structured Framework and Genome Analysis of Magnaporthe grisea Inciting Pearl Millet Blast Disease Reveals Versatile Metabolic Pathways, Protein Families, and Virulence Factors. J. Fungi 2022, 8, 614. https://doi.org/10.3390/jof8060614
Reddy B, Mehta S, Prakash G, Sheoran N, Kumar A. Structured Framework and Genome Analysis of Magnaporthe grisea Inciting Pearl Millet Blast Disease Reveals Versatile Metabolic Pathways, Protein Families, and Virulence Factors. Journal of Fungi. 2022; 8(6):614. https://doi.org/10.3390/jof8060614
Chicago/Turabian StyleReddy, Bhaskar, Sahil Mehta, Ganesan Prakash, Neelam Sheoran, and Aundy Kumar. 2022. "Structured Framework and Genome Analysis of Magnaporthe grisea Inciting Pearl Millet Blast Disease Reveals Versatile Metabolic Pathways, Protein Families, and Virulence Factors" Journal of Fungi 8, no. 6: 614. https://doi.org/10.3390/jof8060614