The Mesenchymal Cap of the Atrial Septum and Atrial and Atrioventricular Septation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Early Heart Development
3. Cardiac Mesenchyme and Its Contribution to Cardiac Septation
3.1. Endocardially Derived Mesenchyme
3.2. Cardiac Neural Crest-Derived Mesenchyme
3.3. Epicardially Derived Mesenchyme
3.4. Second Heart Field-Derived Cells
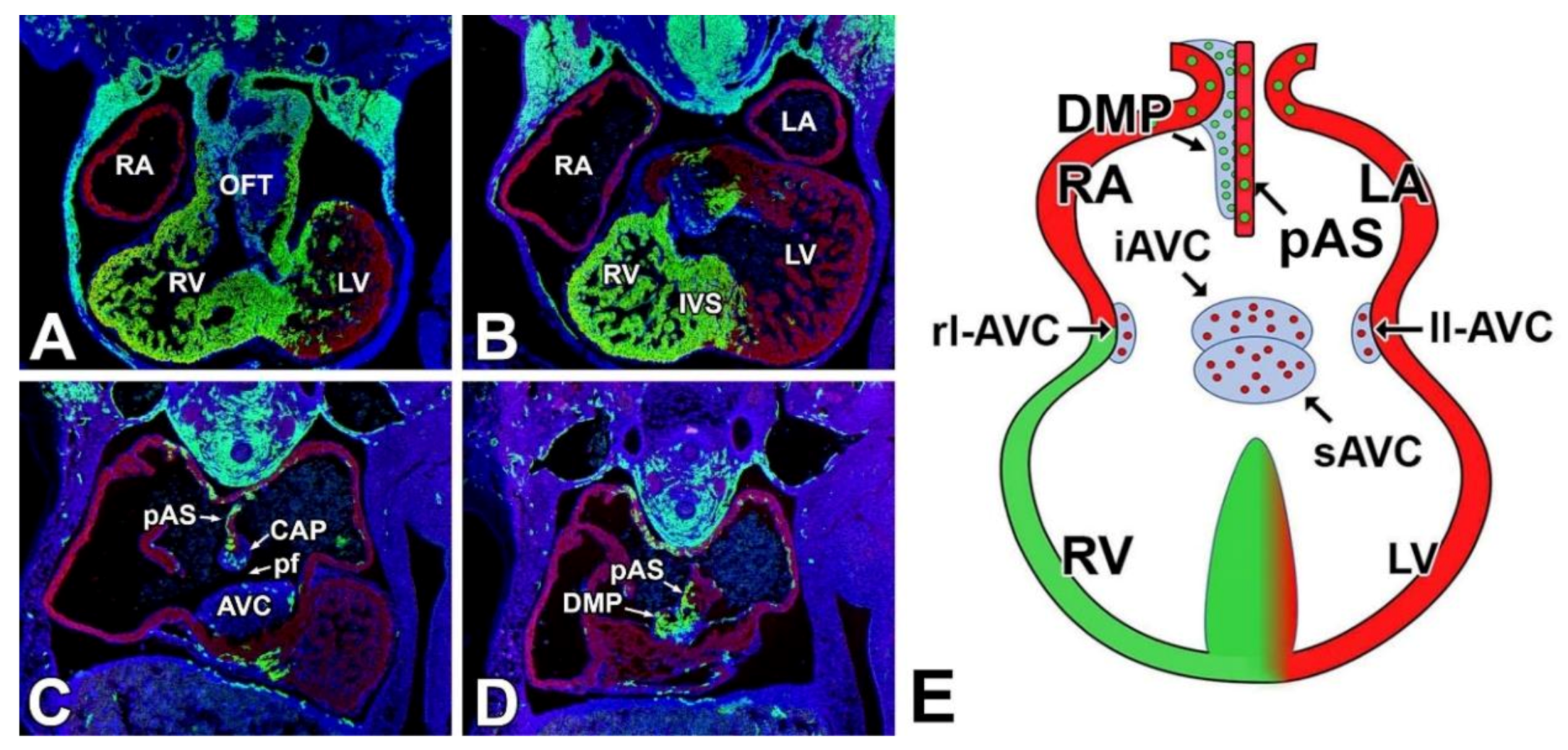
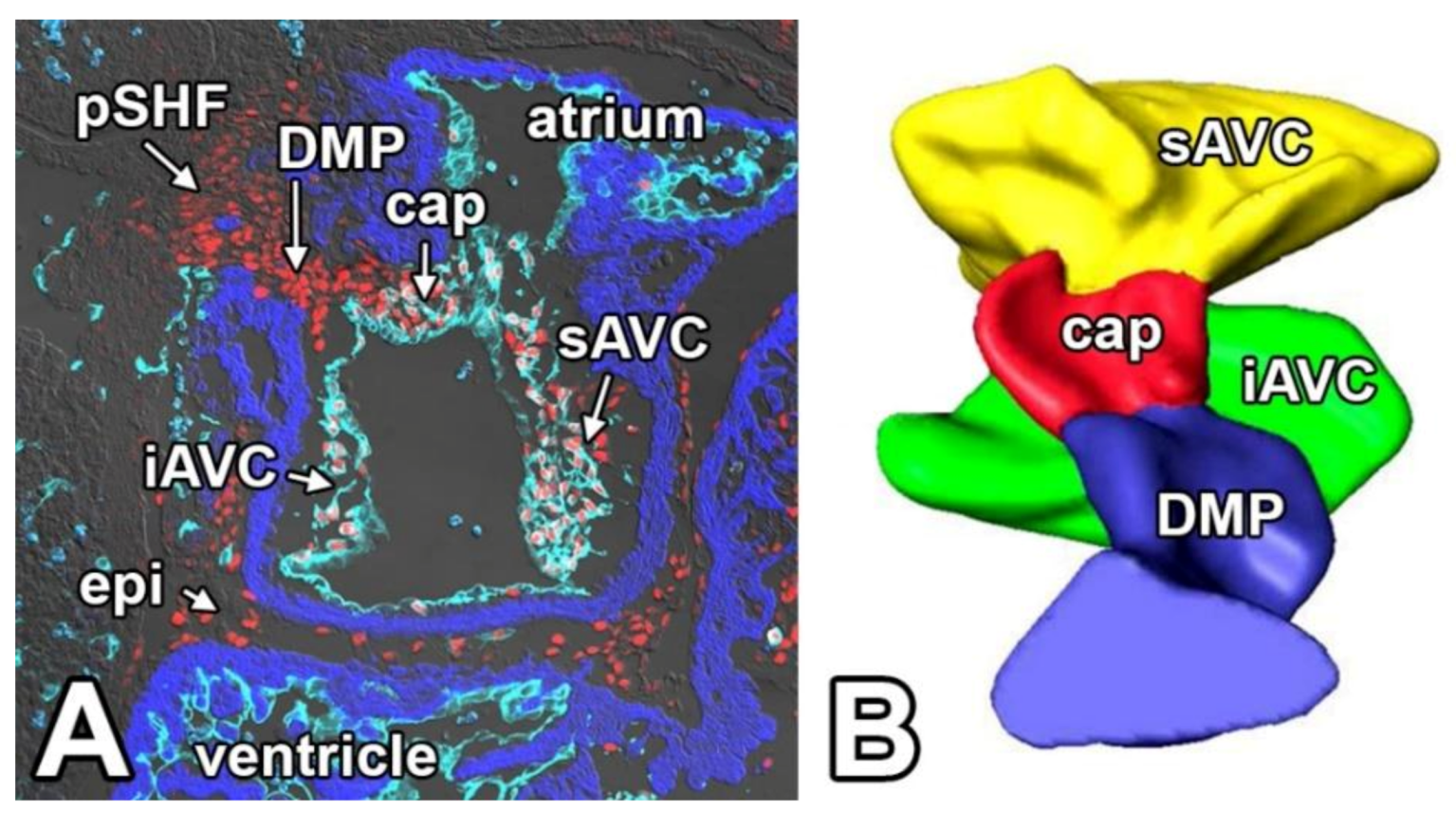
4. Atrioventricular Septal Defects—The AV Cushions and the DMP
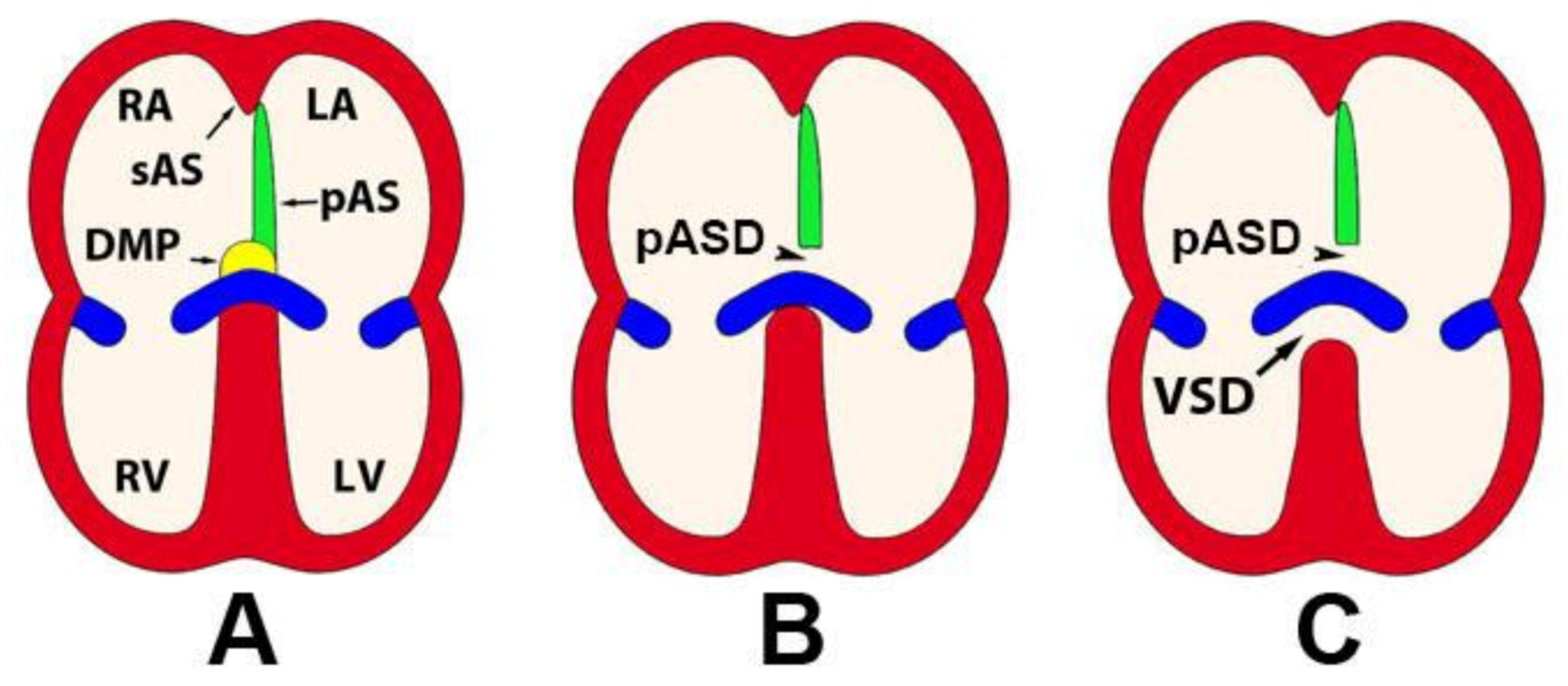
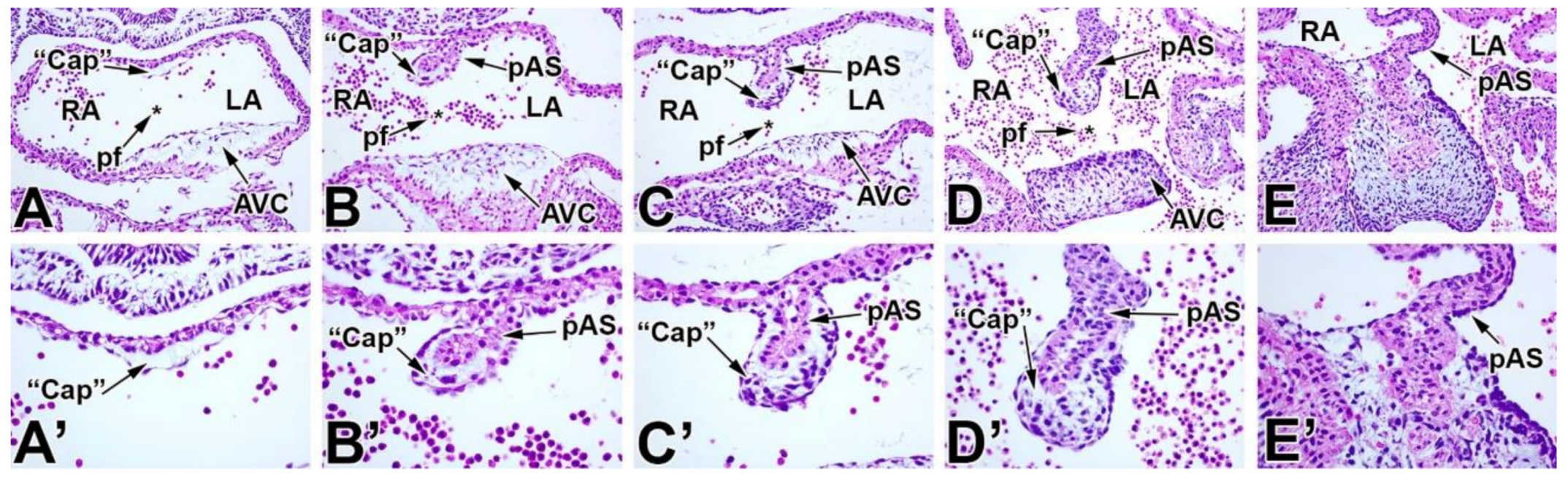
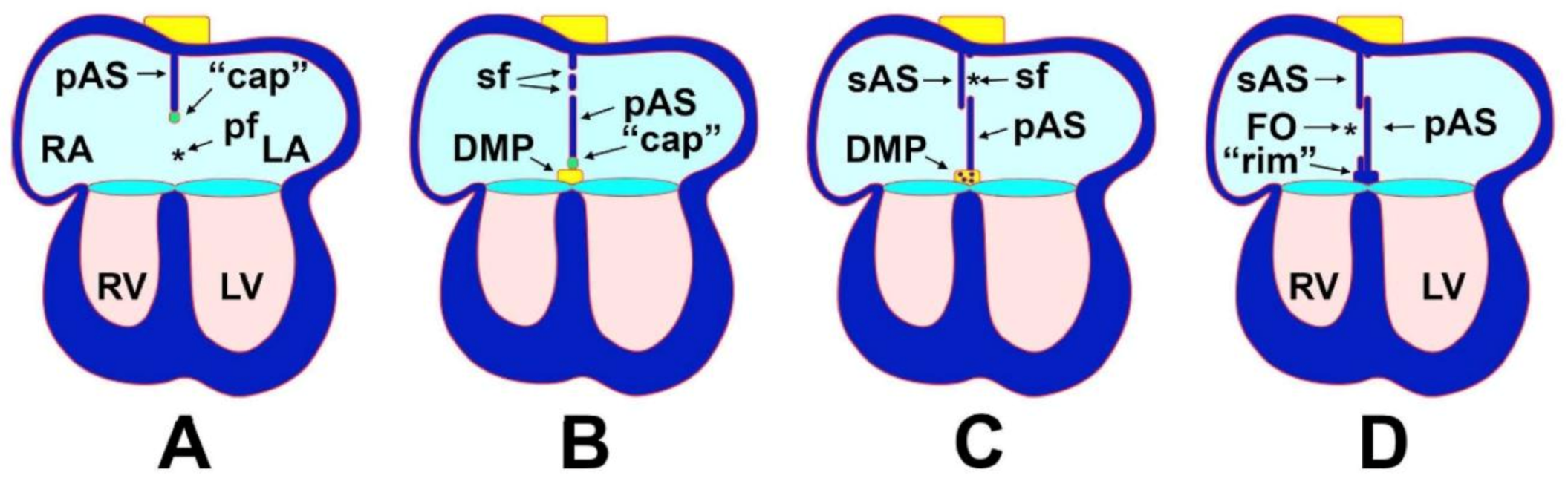
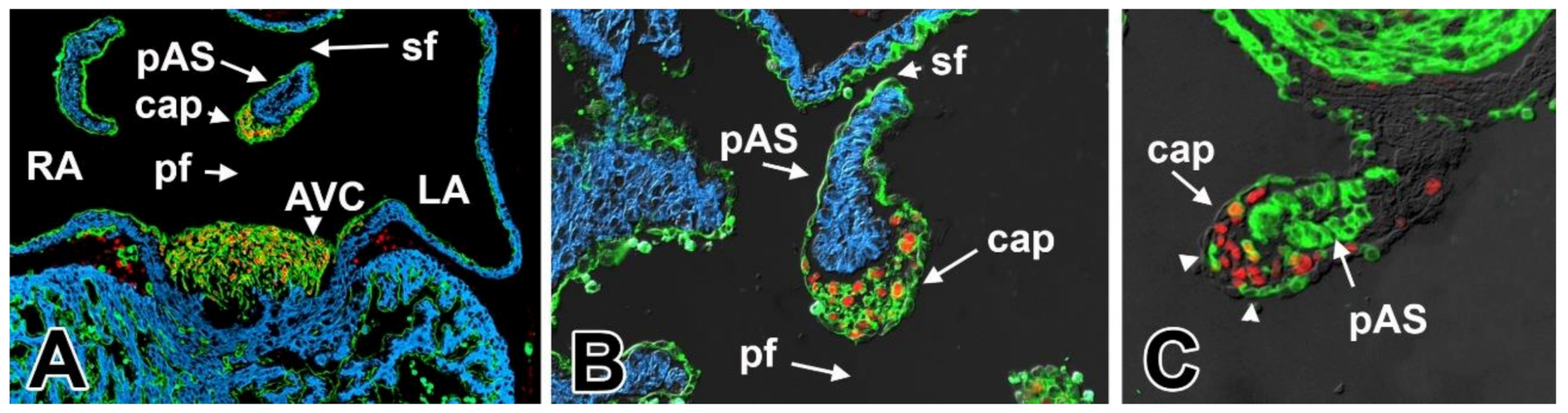
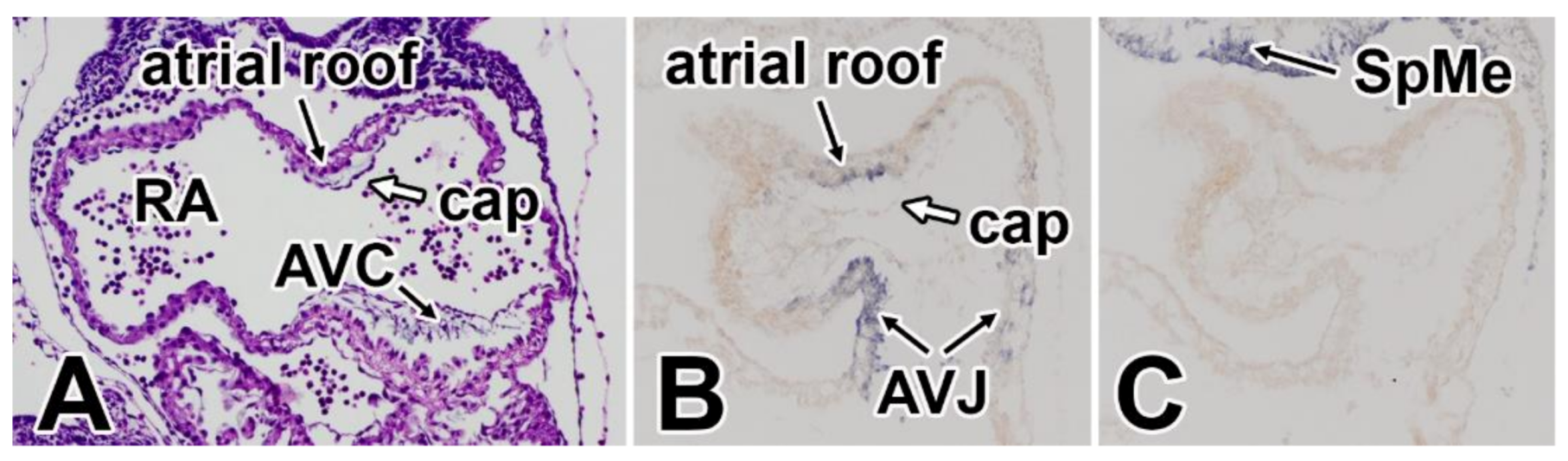
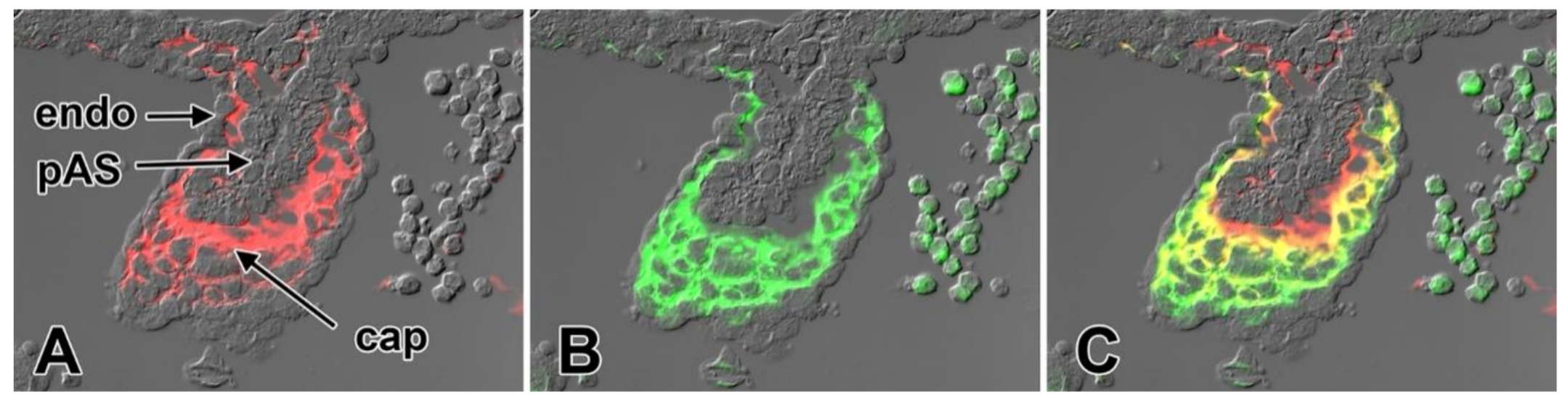
5. The Mesenchymal Cap
6. Atrial Septation
7. Development of the Mesenchymal Cap and Primary Atrial Septum
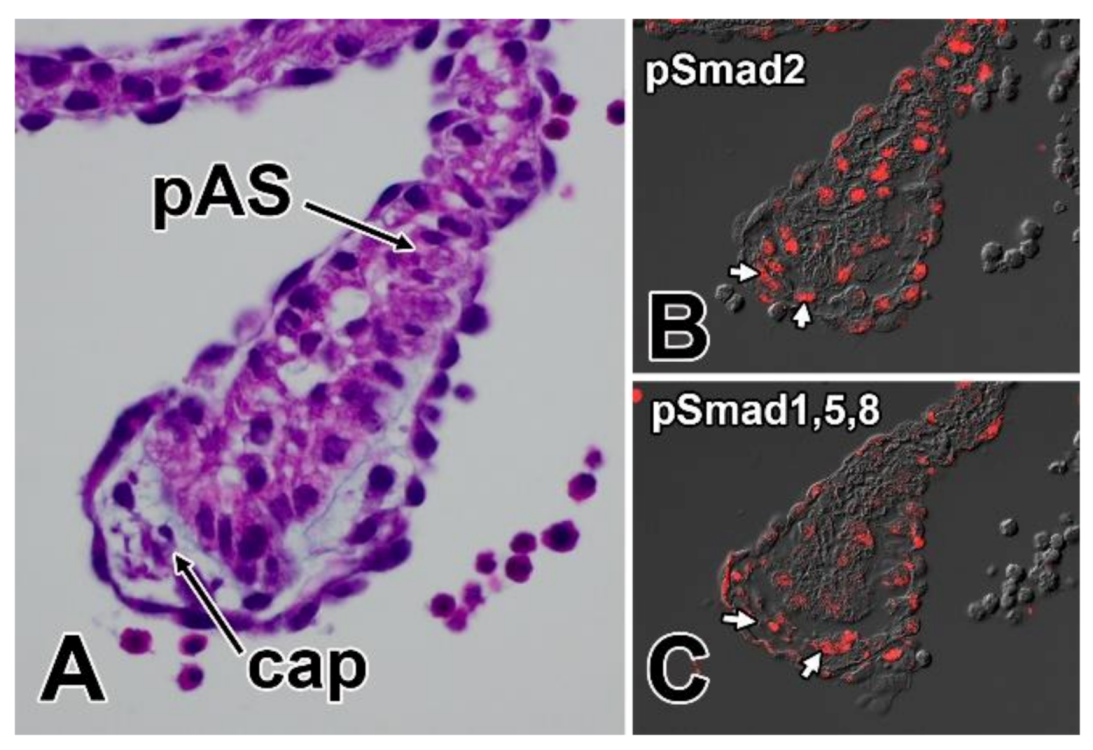
8. Regulatory Mechanisms Associated with the Development of the Mesenchymal Cap
8.1. Growth Factors and the Development of the Mesenchymal Cap—TGFbeta and BMP Signaling
8.2. Extracellular Matrix and the Mesenchymal Cap—Versican, Link Protein, and Hyaluronan
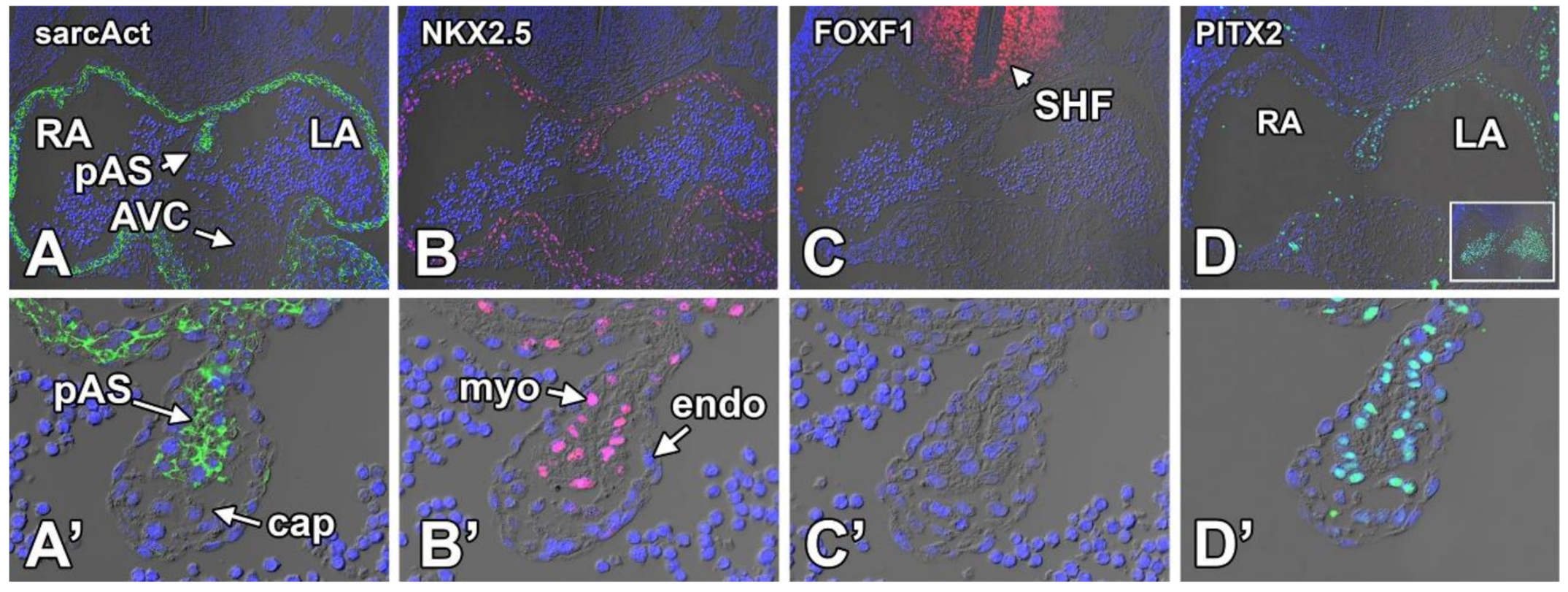
8.3. Transcription Factors and the Development of the Primary Atrial Septum and the Mesenchymal Cap
8.3.1. Sarcomeric Actin
8.3.2. NKX2.5
8.3.3. FOXF1
8.3.4. PITX2
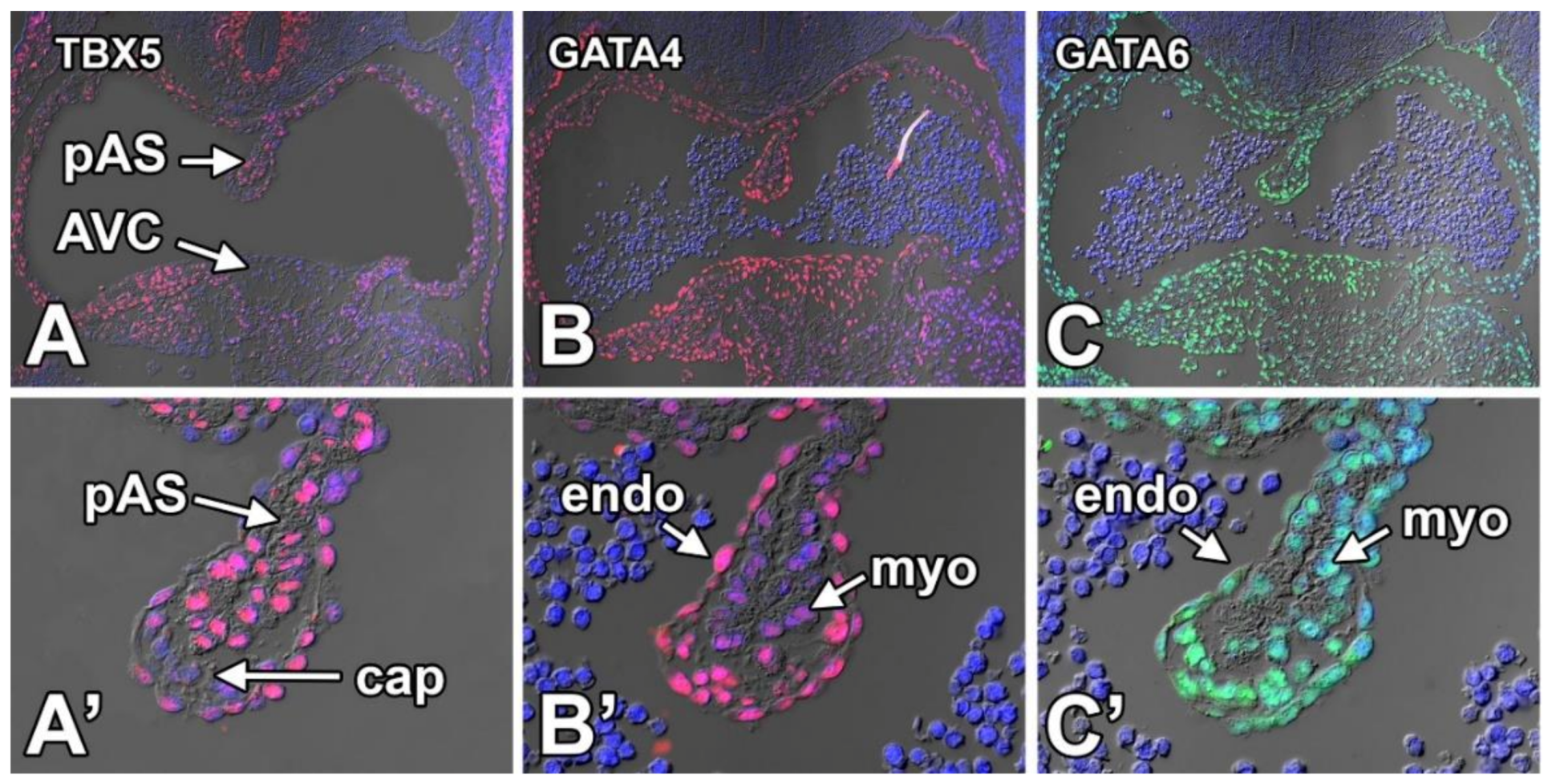
8.3.5. TBX5
8.3.6. GATA4
8.3.7. GATA6
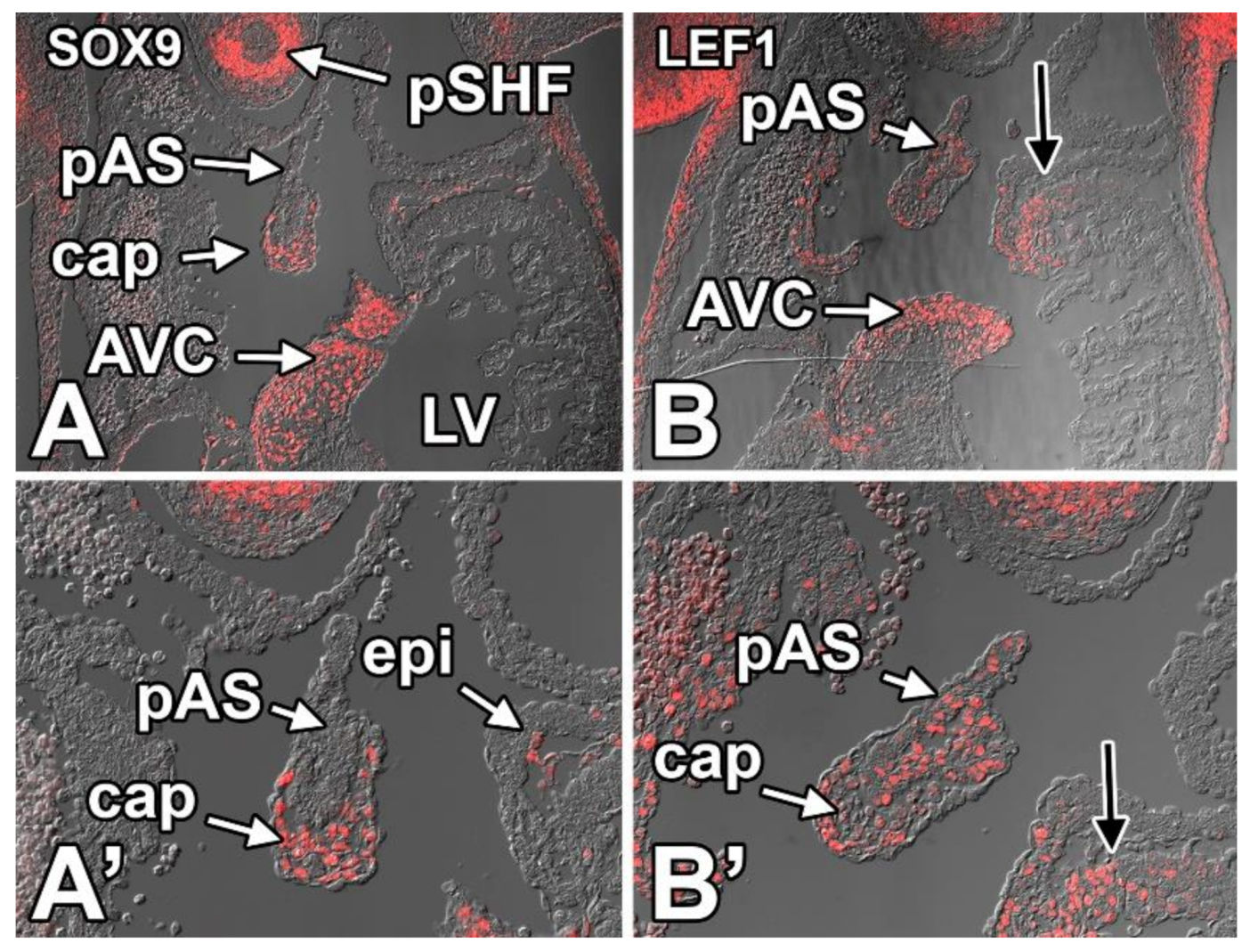
8.3.8. SOX9
8.3.9. LEF1
8.4. Conclusions
9. Discussion and Future Directions
10. Methods
10.1. Mice
10.2. Immunolabeling
Author Contributions
Funding
Conflicts of Interest
References
- Briggs, L.E.; Burns, T.A.; Lockhart, M.M.; Phelps, A.L.; Van den Hoff, M.J.; Wessels, A. Wnt/beta-catenin and sonic hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion. Dev. Dyn. 2016, 245, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Briggs, L.E.; Kakarla, J.; Wessels, A. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation 2012, 84, 117–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, L.E.; Phelps, A.L.; Brown, E.; Kakarla, J.; Anderson, R.H.; van den Hoff, M.J.; Wessels, A. Expression of the bmp receptor alk3 in the second heart field is essential for development of the dorsal mesenchymal protrusion and atrioventricular septation. Circ. Res. 2013, 112, 1420–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snarr, B.S.; Kern, C.B.; Wessels, A. Origin and fate of cardiac mesenchyme. Dev. Dyn. 2008, 237, 2804–2819. [Google Scholar] [CrossRef] [PubMed]
- Snarr, B.S.; Wirrig, E.E.; Phelps, A.L.; Trusk, T.C.; Wessels, A. A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Dev. Dyn. 2007, 236, 1287–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawles, M.E. The heart-forming areas of the early chick blastoderm. Physiol. Zool. 1943, 16, 22–44. [Google Scholar] [CrossRef]
- Rosenquist, G.C.; DeHaan, R.L. Migration of precardiac cells in the chick embryo: A radiographic study. Carnegie Inst. Wash. Contrib. Embryol. 1966, 38, 111–121. [Google Scholar]
- Dyer, L.A.; Kirby, M.L. The role of secondary heart field in cardiac development. Dev. Biol. 2009, 336, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Mjaatvedt, C.H.; Nakaoka, T.; Moreno-Rodriguez, R.; Norris, R.A.; Kern, M.J.; Eisenberg, C.A.; Turner, D.; Markwald, R.R. The outflow tract of the heart is recruited from a novel heart-forming field. Dev. Biol. 2001, 238, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.G.; Buckingham, M.E. The anterior heart-forming field: Voyage to the arterial pole of the heart. Trends Genet. 2002, 18, 210–216. [Google Scholar] [CrossRef]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldo, K.L.; Kumiski, D.H.; Wallis, K.T.; Stadt, H.A.; Hutson, M.R.; Platt, D.H.; Kirby, M.L. Conotruncal myocardium arises from a secondary heart field. Development 2001, 128, 3179–3188. [Google Scholar] [PubMed]
- Snarr, J. Risk, benefits and complications of epidural steroid injections: A case report. Aana J. 2007, 75, 183–188. [Google Scholar] [PubMed]
- Hoffmann, A.D.; Yang, X.H.; Burnicka-Turek, O.; Bosman, J.D.; Ren, X.; Steimle, J.D.; Vokes, S.A.; McMahon, A.P.; Kalinichenko, V.V.; Moskowitz, I.P. Foxf genes integrate tbx5 and hedgehog pathways in the second heart field for cardiac septation. PLoS Genet. 2014, 10, e1004604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovic, S.; Laforest, B.; Desvignes, J.P.; Lescroart, F.; Argiro, L.; Maurel-Zaffran, C.; Salgado, D.; Plaindoux, E.; De Bono, C.; Pazur, K.; et al. Hox-dependent coordination of mouse cardiac progenitor cell patterning and differentiation. Elife 2020, 9, e55124. [Google Scholar] [CrossRef] [PubMed]
- Manner, J. Cardiac looping in the chick embryo: A morphological review with special reference to terminological and biomechanical aspects of the looping process. Anat. Rec. 2000, 259, 248–262. [Google Scholar] [CrossRef]
- Manner, J. The anatomy of cardiac looping: A step towards the understanding of the morphogenesis of several forms of congenital cardiac malformations. Clin. Anat. 2009, 22, 21–35. [Google Scholar] [CrossRef]
- Wessels, A.; Sedmera, D. Developmental anatomy of the heart: A tale of mice and man. Physiol. Genom. 2003, 15, 165–176. [Google Scholar] [CrossRef]
- Bolender, D.L.; Markwald, R.R. Epithelial-mesenchymal transformation in chick atrioventricular cushion morphogenesis. Scan. Electron Microsc. 1979, 313–321. [Google Scholar]
- Markwald, R.; Eisenberg, C.; Eisenberg, L.; Trusk, T.; Sugi, Y. Epithelial-mesenchymal transformations in early avian heart development. Acta Anat. 1996, 156, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; von Gise, A.; Liu, Q.; Hu, T.; Tian, X.; He, L.; Pu, W.; Huang, X.; He, L.; Cai, C.L.; et al. Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J. Biol. Chem. 2014, 289, 18681–18692. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.B.; Boyer, A.S.; Runyan, R.B.; Barnett, J.V. Requirement of type iii tgf-beta receptor for endocardial cell transformation in the heart. Science 1999, 283, 2080–2082. [Google Scholar] [CrossRef] [PubMed]
- Sugi, Y.; Yamamura, H.; Okagawa, H.; Markwald, R.R. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev. Biol. 2004, 269, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertran, E.; Perez-Pomares, J.M.; Diez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisua-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna-Zurita, L.; Prados, B.; Grego-Bessa, J.; Luxan, G.; del Monte, G.; Benguria, A.; Adams, R.H.; Perez-Pomares, J.M.; de la Pompa, J.L. Integration of a notch-dependent mesenchymal gene program and bmp2-driven cell invasiveness regulates murine cardiac valve formation. J. Clin. Investig. 2010, 120, 3493–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessels, A.; Markman, M.W.; Vermeulen, J.L.; Anderson, R.H.; Moorman, A.F.; Lamers, W.H. The development of the atrioventricular junction in the human heart. Circ. Res. 1996, 78, 110–117. [Google Scholar] [CrossRef]
- Wessels, A.; Anderson, R.H.; Markwald, R.R.; Webb, S.; Brown, N.A.; Viragh, S.; Moorman, A.F.; Lamers, W.H. Atrial development in the human heart: An immunohistochemical study with emphasis on the role of mesenchymal tissues. Anat. Rec. 2000, 259, 288–300. [Google Scholar] [CrossRef]
- Anderson, R.H.; Brown, N.A.; Webb, S. Development and structure of the atrial septum. Heart 2002, 88, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Jensen, B.; Wang, T.; Moorman, A.F.M. Evolution and development of the atrial septum. Anat. Rec. (Hoboken) 2019, 302, 32–48. [Google Scholar] [CrossRef] [Green Version]
- Le Douarin, N.M.; Dupin, E. The “beginnings” of the neural crest. Dev. Biol. 2018, 444, S3–S13. [Google Scholar] [CrossRef]
- Hutson, M.R.; Kirby, M.L. Neural crest and cardiovascular development: A 20-year perspective. Birth Defects Res. C Embryo Today 2003, 69, 2–13. [Google Scholar] [CrossRef]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061. [Google Scholar] [CrossRef]
- Kirby, M.L.; Hutson, M.R. Factors controlling cardiac neural crest cell migration. Cell Adh. Migr. 2010, 4, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.T.; Kirby, M.L.; Forbes, G. Analysis of cranial neural crest distribution in the developing heart using quail-chick chimeras. Circ. Res. 1987, 60, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Waldo, K.; Zdanowicz, M.; Burch, J.; Kumiski, D.H.; Stadt, H.A.; Godt, R.E.; Creazzo, T.L.; Kirby, M.L. A novel role for cardiac neural crest in heart development. J. Clin. Investig. 1999, 103, 1499–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutson, M.R.; Kirby, M.L. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin. Cell Dev. Biol. 2007, 18, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.L.; Waldo, K.L. Neural crest and cardiovascular patterning. Circ. Res. 1995, 77, 211–215. [Google Scholar] [CrossRef]
- Hiriart, E.; Deepe, R.; Wessels, A. Mesothelium and malignant mesothelioma. J. Dev. Biol. 2019, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pomares, J.M.; Phelps, A.; Sedmerova, M.; Carmona, R.; Gonzalez-Iriarte, M.; Munoz-Chapuli, R.; Wessels, A. Experimental studies on the spatiotemporal expression of wt1 and raldh2 in the embryonic avian heart: A model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (epdcs). Dev. Biol. 2002, 247, 307–326. [Google Scholar] [CrossRef] [Green Version]
- Wessels, A.; Perez-Pomares, J.M. The epicardium and epicardially derived cells (epdcs) as cardiac stem cells. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2004, 276, 43–57. [Google Scholar] [CrossRef]
- Viragh, S.; Challice, C.E. The origin of the epicardium and the embryonic myocardial circulation in the mouse. Anat. Rec. 1981, 201, 157–168. [Google Scholar] [CrossRef]
- Vrancken Peeters, M.P.; Mentink, M.M.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Cytokeratins as a marker for epicardial formation in the quail embryo. Anat. Embryol. 1995, 191, 503–508. [Google Scholar]
- Gittenberger-de Groot, A.; Vrancken Peeters, M.; Mentink, M.; Gourdie, R.; Poelmann, R. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Perez-Pomares, J.M.; Macias, D.; Garcia-Garrido, L.; Munoz-Chapuli, R. Contribution of the primitive epicardium to the subepicardial mesenchyme in hamster and chick embryos. Dev. Dyn. 1997, 210, 96–105. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; Macias, D.; Garcia-Garrido, L.; Munoz-Chapuli, R. The origin of the subepicardial mesenchyme in the avian embryo: An immunohistochemical and quail-chick chimera study. Dev. Biol. 1998, 200, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Dettman, R.W.; Denetclaw, W., Jr.; Ordahl, C.P.; Bristow, J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 1998, 193, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Wessels, A.; van den Hoff, M.J.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manner, J. Does the subepicardial mesenchyme contribute myocardioblasts to the myocardium of the chick embryo heart? A quail-chick chimera study tracing the fate of the epicardial primordium. Anat. Rec. 1999, 255, 212–226. [Google Scholar] [CrossRef]
- Zhou, B.; von Gise, A.; Ma, Q.; Hu, Y.; Pu, W. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev. Bio. 2010, 338, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, M.M.; Phelps, A.L.; van den Hoff, M.J.; Wessels, A. The epicardium and the development of the atrioventricular junction in the murine heart. J. Dev. Bio. 2014, 2, 1–17. [Google Scholar] [CrossRef]
- Anderson, R.H.; Mohun, T.J.; Brown, N.A. Clarifying the morphology of the ostium primum defect. J. Anat. 2015, 226, 244–257. [Google Scholar] [CrossRef] [Green Version]
- His, W. Die area interposita, die eustachi’sche klappe und die spina vestibuli. Anat. Menschl. Embryonen 1880, 149, 52. [Google Scholar]
- Kim, J.S.; Viragh, S.; Moorman, A.F.; Anderson, R.H.; Lamers, W.H. Development of the myocardium of the atrioventricular canal and the vestibular spine in the human heart. Circ. Res. 2001, 88, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Mommersteeg, M.T.; Soufan, A.T.; de Lange, F.J.; van den Hoff, M.J.; Anderson, R.H.; Christoffels, V.M.; Moorman, A.F. Two distinct pools of mesenchyme contribute to the development of the atrial septum. Circ. Res. 2006, 99, 351–353. [Google Scholar] [CrossRef]
- Blom, N.A.; Ottenkamp, J.; Wenink, A.G.; Gittenberger-de Groot, A.C. Deficiency of the vestibular spine in atrioventricular septal defects in human fetuses with down syndrome. Am. J. Cardiol. 2003, 91, 180–184. [Google Scholar] [CrossRef]
- Sharratt, G.P.; Webb, S.; Anderson, R.H. The vestibular defect: An interatrial communication due to a deficiency in the atrial septal component derived from the vestibular spine. Cardiol. Young 2003, 13, 184–190. [Google Scholar] [CrossRef]
- De Bono, C.; Thellier, C.; Bertrand, N.; Sturny, R.; Jullian, E.; Cortes, C.; Stefanovic, S.; Zaffran, S.; Theveniau-Ruissy, M.; Kelly, R.G. T-box genes and retinoic acid signaling regulate the segregation of arterial and venous pole progenitor cells in the murine second heart field. Hum. Mol. Genet. 2018, 27, 3747–3760. [Google Scholar] [CrossRef] [PubMed]
- Al-Hay, A.A.; MacNeill, S.J.; Yacoub, M.; Shore, D.F.; Shinebourne, E.A. Complete atrioventricular septal defect, down syndrome, and surgical outcome: Risk factors. Ann. Thorac. Surg. 2003, 75, 412–421. [Google Scholar] [CrossRef]
- Dickinson, D.F.; Arnold, R.; Wilkinson, J.L. Congenital heart disease among 160 480 liveborn children in liverpool 1960 to 1969. Implications for surgical treatment. Br. Heart J. 1981, 46, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Corsten-Janssen, N.; Kerstjens-Frederikse, W.S.; du Marchie Sarvaas, G.J.; Baardman, M.E.; Bakker, M.K.; Bergman, J.E.; Hove, H.D.; Heimdal, K.R.; Rustad, C.F.; Hennekam, R.C.; et al. The cardiac phenotype in patients with a chd7 mutation. Circ. Cardiovasc. Genet. 2013, 6, 248–254. [Google Scholar] [CrossRef] [Green Version]
- McCullough, A. Further examples of endocardial cushion defect in production of a cardiac anomaly complex; a presentation of three cases from autopsy reports. J. Pediatr. 1953, 43, 429–433. [Google Scholar] [CrossRef]
- Campbell, M.; Missen, G.A. Endocardial cushion defects; common atrio-ventricular canal and ostium primum. Br. Heart J. 1957, 19, 403–418. [Google Scholar] [CrossRef] [Green Version]
- Van Mierop, L.H.S.; Alley, R.D.; Kausel, H.W.; Stranahan, A. The anatomy and embryology of endocardial cushion defects. J. Thorac. Cardiov. Surg. 1962, 43, 71. [Google Scholar] [CrossRef]
- Carmi, R.; Boughman, J.A.; Ferencz, C. Endocardial cushion defect: Further studies of “isolated” versus “syndromic” occurrence. Am. J. Med. Genet. 1992, 43, 569–575. [Google Scholar] [CrossRef]
- Anderson, R.H.; Wessels, A.; Vettukattil, J.J. Morphology and morphogenesis of atrioventricular septal defect with common atrioventricular junction. World J. Pediatr. Congenit. Heart Surg. 2010, 1, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Kulessa, H.; Tompkins, K.; Zhou, Y.; Batts, L.; Baldwin, H.S.; Hogan, B.L. An essential role of bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003, 17, 2362–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goddeeris, M.M.; Rho, S.; Petiet, A.; Davenport, C.L.; Johnson, G.A.; Meyers, E.N.; Klingensmith, J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development 2008, 135, 1887–1895. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.D.; Peterson, M.A.; Friedland-Little, J.M.; Anderson, S.A.; Moskowitz, I.P. Sonic hedgehog is required in pulmonary endoderm for atrial septation. Development 2009, 136, 1761–1770. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Hoffmann, A.D.; Burnicka-Turek, O.; Friedland-Little, J.M.; Zhang, K.; Moskowitz, I.P. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell 2012, 23, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Yuan, L.; Goss, A.M.; Wang, T.; Yang, J.; Lepore, J.J.; Zhou, D.; Schwartz, R.J.; Patel, V.; Cohen, E.D.; et al. Characterization and in vivo pharmacological rescue of a wnt2-gata6 pathway required for cardiac inflow tract development. Dev. Cell 2010, 18, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Burns, T.; Yang, Y.; Hiriart, E.; Wessels, A. The dorsal mesenchymal protrusion and the pathogenesis of atrioventricular septal defects. J. Cardiovasc. Dev. Dis. 2016, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Terada, R.; Warren, S.; Lu, J.T.; Chien, K.R.; Wessels, A.; Kasahara, H. Ablation of nkx2-5 at mid-embryonic stage results in premature lethality and cardiac malformation. Cardiovasc. Res. 2011, 91, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole-Jeffrey, C.T.; Terada, R.; Neth, M.R.; Wessels, A.; Kasahara, H. Progressive anatomical closure of foramen ovale in normal neonatal mouse hearts. Anat. Rec. (Hoboken) 2012, 295, 764–768. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.B.; Boyer, A.S.; Runyan, R.B.; Barnett, J.V. Antibodies to the type ii tgfbeta receptor block cell activation and migration during atrioventricular cushion transformation in the heart. Dev. Biol. 1996, 174, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Camenisch, T.D.; Molin, D.G.; Person, A.; Runyan, R.B.; Gittenberger-de Groot, A.C.; McDonald, J.A.; Klewer, S.E. Temporal and distinct tgfbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev. Biol. 2002, 248, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Inai, K.; Norris, R.A.; Hoffman, S.; Markwald, R.R.; Sugi, Y. Bmp-2 induces cell migration and periostin expression during atrioventricular valvulogenesis. Dev. Bio. 2008, 315, 383–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxon, J.G.; Baer, D.R.; Barton, J.A.; Hawkins, T.; Wu, B.; Trusk, T.C.; Harris, S.E.; Zhou, B.; Mishina, Y.; Sugi, Y. Bmp2 expression in the endocardial lineage is required for av endocardial cushion maturation and remodeling. Dev. Biol. 2017, 430, 113–128. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Runyan, R.B.; Markwald, R.R. Molecular Regulation of Cushion Morphogenesis; Elsevier: Cambridge, MA, USA, 2010. [Google Scholar]
- Camenisch, T.D.; Schroeder, J.A.; Bradley, J.; Klewer, S.E.; McDonald, J.A. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of erbb2-erbb3 receptors. Nat. Med. 2002, 8, 850–855. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A., Jr.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Investig. 2000, 106, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Mjaatvedt, C.H.; Yamamura, H.; Capehart, A.A.; Turner, D.; Markwald, R.R. The cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev. Biol. 1998, 202, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirrig, E.E.; Snarr, B.S.; Chintalapudi, M.R.; O’Neal, J.L.; Phelps, A.L.; Barth, J.L.; Fresco, V.M.; Kern, C.B.; Mjaatvedt, C.H.; Toole, B.P.; et al. Cartilage link protein 1 (crtl1), an extracellular matrix component playing an important role in heart development. Dev. Bio. 2007, 310, 291–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoln, J.; Kist, R.; Scherer, G.; Yutzey, K. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev. Bio. 2007, 305, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Tao, G.; Levay, A.K.; Gridley, T.; Lincoln, J. Mmp15 is a direct target of snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev. Bio. 2011, 359, 209–221. [Google Scholar] [CrossRef] [Green Version]
- de la Pompa, J.L.; Timmerman, L.A.; Takimoto, H.; Yoshida, H.; Elia, A.J.; Samper, E.; Potter, J.; Wakeham, A.; Marengere, L.; Langille, B.L.; et al. Role of the nf-atc transcription factor in morphogenesis of cardiac valves and septum. Nature 1998, 392, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Torregrosa-Carrion, R.; Luna-Zurita, L.; Garcia-Marques, F.; D’Amato, G.; Pineiro-Sabaris, R.; Bonzon-Kulichenko, E.; Vazquez, J.; de la Pompa, J.L. Notch activation promotes valve formation by regulating the endocardial secretome. Mol. Cell Proteom. 2019, 18, 1782–1795. [Google Scholar] [CrossRef]
- Bernanke, D.H.; Markwald, R.R. Effects of hyaluronic acid on cardiac cushion tissue cells in collagen matrix cultures. Tex. Rep. Biol. Med. 1979, 39, 271–285. [Google Scholar]
- Bernanke, D.H.; Markwald, R.R. Migratory behavior of cardiac cushion tissue cells in a collagen-lattice culture system. Dev. Biol. 1982, 91, 235–245. [Google Scholar] [CrossRef]
- Runyan, R.B.; Markwald, R.R. Invasion of mesenchyme into three-dimensional collagen gels: A regional and temporal analysis of interaction in embryonic heart tissue. Dev. Biol. 1983, 95, 108–114. [Google Scholar] [CrossRef]
- Gaussin, V.; Van de Putte, T.; Mishina, Y.; Hanks, M.C.; Zwijsen, A.; Huylebroeck, D.; Behringer, R.R.; Schneider, M.D. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor alk3. Proc. Natl. Acad. Sci. USA 2002, 99, 2878–2883. [Google Scholar] [CrossRef] [Green Version]
- Bartram, U.; Molin, D.G.; Wisse, L.J.; Mohamad, A.; Sanford, L.P.; Doetschman, T.; Speer, C.P.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in tgf-beta(2)-knockout mice. Circulation 2001, 103, 2745–2752. [Google Scholar] [CrossRef] [Green Version]
- Krug, E.L.; Mjaatvedt, C.H.; Markwald, R.R. Extracellular matrix from embryonic myocardium elicits an early morphogenetic event in cardiac endothelial differentiation. Dev. Biol. 1987, 120, 348–355. [Google Scholar] [CrossRef]
- Bouchey, D.; Argraves, W.S.; Little, C.D. Fibulin-1, vitronectin, and fibronectin expression during avian cardiac valve and septa development. Anat. Rec. 1996, 244, 540–551. [Google Scholar] [CrossRef]
- Kern, C.B.; Twal, W.O.; Mjaatvedt, C.H.; Fairey, S.E.; Toole, B.P.; Iruela-Arispe, M.L.; Argraves, W.S. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev. Dyn. 2006, 235, 2238–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, D.J.; Copp, A.J. Versican expression is associated with chamber specification, septation, and valvulogenesis in the developing mouse heart. Circ. Res. 1998, 83, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Posch, M.G.; Waldmuller, S.; Muller, M.; Scheffold, T.; Fournier, D.; Andrade-Navarro, M.A.; De Geeter, B.; Guillaumont, S.; Dauphin, C.; Yousseff, D.; et al. Cardiac alpha-myosin (myh6) is the predominant sarcomeric disease gene for familial atrial septal defects. PLoS ONE 2011, 6, e28872. [Google Scholar] [CrossRef]
- Basson, C.T.; Bachinsky, D.R.; Lin, R.C.; Levi, T.; Elkins, J.A.; Soults, J.; Grayzel, D.; Kroumpouzou, E.; Traill, T.A.; Leblanc-Straceski, J.; et al. Mutations in human cause limb and cardiac malformation in holt-oram syndrome. Nat. Genet. 1997, 15, 30–35. [Google Scholar] [CrossRef]
- Bruneau, B.G.; Logan, M.; Davis, N.; Levi, T.; Tabin, C.J.; Seidman, J.G.; Seidman, C.E. Chamber-specific cardiac expression of tbx5 and heart defects in holt-oram syndrome. Dev. Biol. 1999, 211, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posch, M.G.; Gramlich, M.; Sunde, M.; Schmitt, K.R.; Lee, S.H.; Richter, S.; Kersten, A.; Perrot, A.; Panek, A.N.; Al Khatib, I.H.; et al. A gain-of-function tbx20 mutation causes congenital atrial septal defects, patent foramen ovale and cardiac valve defects. J. Med. Genet. 2010, 47, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoon, C.H.; Kim, G.H.; Yoo, H.W.; Lee, B.S.; Kim, K.S.; Kim, E.A. A case of campomelic dysplasia without sex reversal. J. Korean Med. Sci. 2011, 26, 143–145. [Google Scholar] [CrossRef]
- Posch, M.G.; Perrot, A.; Schmitt, K.; Mittelhaus, S.; Esenwein, E.M.; Stiller, B.; Geier, C.; Dietz, R.; Gessner, R.; Ozcelik, C.; et al. Mutations in gata4, nkx2.5, creld1, and bmp4 are infrequently found in patients with congenital cardiac septal defects. Am. J. Med. Genet. A 2008, 146A, 251–253. [Google Scholar] [CrossRef]
- Hirayama-Yamada, K.; Kamisago, M.; Akimoto, K.; Aotsuka, H.; Nakamura, Y.; Tomita, H.; Furutani, M.; Imamura, S.; Takao, A.; Nakazawa, M.; et al. Phenotypes with gata4 or nkx2.5 mutations in familial atrial septal defect. Am. J. Med. Genet. A 2005, 135, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. Gata4 mutations cause human congenital heart defects and reveal an interaction with tbx5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef]
- Tomita-Mitchell, A.; Maslen, C.L.; Morris, C.D.; Garg, V.; Goldmuntz, E. Gata4 sequence variants in patients with congenital heart disease. J. Med. Genet. 2007, 44, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Huo, Z.; Liu, X.; Zhang, Y.; Li, L.; Zhao, H.; Yan, B.; Liu, Y.; Yang, Y.; Chen, Y.H. A novel gata6 mutation in patients with tetralogy of fallot or atrial septal defect. J. Hum. Genet. 2010, 55, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Chaboissier, M.C.; Behringer, R.R.; Rowitch, D.H.; Schedl, A.; Epstein, J.A.; de Crombrugghe, B. Essential role of sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 2004, 101, 6502–6507. [Google Scholar] [CrossRef] [Green Version]
- Garside, V.C.; Cullum, R.; Alder, O.; Lu, D.Y.; Vander Werff, R.; Bilenky, M.; Zhao, Y.; Jones, S.J.; Marra, M.A.; Underhill, T.M.; et al. Sox9 modulates the expression of key transcription factors required for heart valve development. Development 2015, 142, 4340–4350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prall, O.W.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An nkx2-5/bmp2/smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Reamon-Buettner, S.M.; Borlak, J. Nkx2-5: An update on this hypermutable homeodomain protein and its role in human congenital heart disease (chd). Hum. Mutat. 2010, 31, 1185–1194. [Google Scholar] [CrossRef] [Green Version]
- Furtado, M.B.; Wilmanns, J.C.; Chandran, A.; Perera, J.; Hon, O.; Biben, C.; Willow, T.J.; Nim, H.T.; Kaur, G.; Simonds, S.; et al. Point mutations in murine nkx2-5 phenocopy human congenital heart disease and induce pathogenic wnt signaling. JCI Insight 2017, 2, e88271. [Google Scholar] [CrossRef] [Green Version]
- Nadeau, M.; Georges, R.O.; Laforest, B.; Yamak, A.; Lefebvre, C.; Beauregard, J.; Paradis, P.; Bruneau, B.G.; Andelfinger, G.; Nemer, M. An endocardial pathway involving tbx5, gata4, and nos3 required for atrial septum formation. Proc. Natl. Acad. Sci. USA 2010, 107, 19356–19361. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.W.; Silberbach, G.M.; Kavanaugh-McHugh, A.; Cottrill, C.; Zhang, Y.; Riggs, S.; Smalls, O.; Johnson, M.C.; Watson, M.S.; Seidman, J.G.; et al. Mutations in the cardiac transcription factor nkx2.5 affect diverse cardiac developmental pathways. J. Clin. Investig. 1999, 104, 1567–1573. [Google Scholar] [CrossRef] [Green Version]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor nkx2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Snarr, B.S.; O’Neal, J.L.; Chintalapudi, M.R.; Wirrig, E.E.; Phelps, A.L.; Kubalak, S.W.; Wessels, A. Isl1 expression at the venous pole identifies a novel role for the second heart field in cardiac development. Circ. Res. 2007, 101, 971–974. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Ustiyan, V.; Pradhan, A.; Cai, Y.; Havrilak, J.A.; Bolte, C.S.; Shannon, J.M.; Kalin, T.V.; Kalinichenko, V.V. Foxf1 transcription factor is required for formation of embryonic vasculature by regulating vegf signaling in endothelial cells. Circ. Res. 2014, 115, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Franco, D.; Campione, M. The role of pitx2 during cardiac development. Linking left-right signaling and congenital heart diseases. Trends Cardiovasc. Med. 2003, 13, 157–163. [Google Scholar] [CrossRef]
- Li, Q.Y.; Newbury-Ecob, R.A.; Terrett, J.A.; Wilson, D.I.; Curtis, A.R.; Yi, C.H.; Gebuhr, T.; Bullen, P.J.; Robson, S.C.; Strachan, T.; et al. Holt-oram syndrome is caused by mutations in tbx5, a member of the brachyury (t) gene family. Nat. Genet. 1997, 15, 21–29. [Google Scholar] [CrossRef]
- Basson, C.T.; Cowley, G.S.; Solomon, S.D.; Weissman, B.; Poznanski, A.K.; Traill, T.A.; Seidman, J.G.; Seidman, C.E. The clinical and genetic spectrum of the holt-oram syndrome (heart-hand syndrome). N. Engl. J. Med. 1994, 330, 885–891. [Google Scholar] [CrossRef]
- Kisanuki, Y.Y.; Hammer, R.E.; Miyazaki, J.; Williams, S.C.; Richardson, J.A.; Yanagisawa, M. Tie2-cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Bio. 2001, 230, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Wu, B.; Tompkins, K.L.; Boyer, K.L.; Grindley, J.C.; Baldwin, H.S. Characterization of nfatc1 regulation identifies an enhancer required for gene expression that is specific to pro-valve endocardial cells in the developing heart. Development 2005, 132, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Waller, B.R., 3rd; Wessels, A. Cardiac morphogenesis and dysmorphogenesis. An immunohistochemical approach. Methods Mol. Biol. 2000, 135, 151–161. [Google Scholar] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deepe, R.; Fitzgerald, E.; Wolters, R.; Drummond, J.; Guzman, K.D.; Hoff, M.J.B.v.d.; Wessels, A. The Mesenchymal Cap of the Atrial Septum and Atrial and Atrioventricular Septation. J. Cardiovasc. Dev. Dis. 2020, 7, 50. https://doi.org/10.3390/jcdd7040050
Deepe R, Fitzgerald E, Wolters R, Drummond J, Guzman KD, Hoff MJBvd, Wessels A. The Mesenchymal Cap of the Atrial Septum and Atrial and Atrioventricular Septation. Journal of Cardiovascular Development and Disease. 2020; 7(4):50. https://doi.org/10.3390/jcdd7040050
Chicago/Turabian StyleDeepe, Ray, Emily Fitzgerald, Renélyn Wolters, Jenna Drummond, Karen De Guzman, Maurice J.B. van den Hoff, and Andy Wessels. 2020. "The Mesenchymal Cap of the Atrial Septum and Atrial and Atrioventricular Septation" Journal of Cardiovascular Development and Disease 7, no. 4: 50. https://doi.org/10.3390/jcdd7040050