


Organoplatinum Chemistry Related to Alkane Oxidation: The Effect of a Nitro Substituent in Ligands Having an Appended Phenol Group

Abstract
: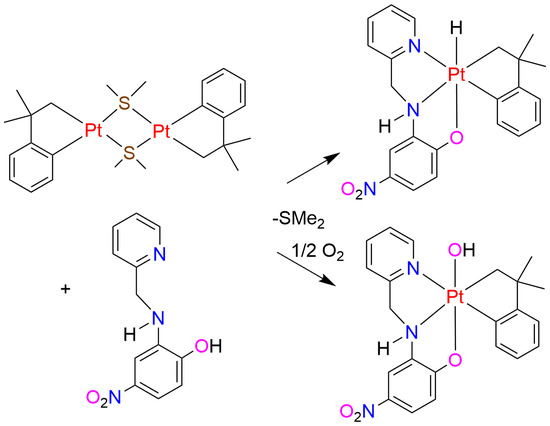
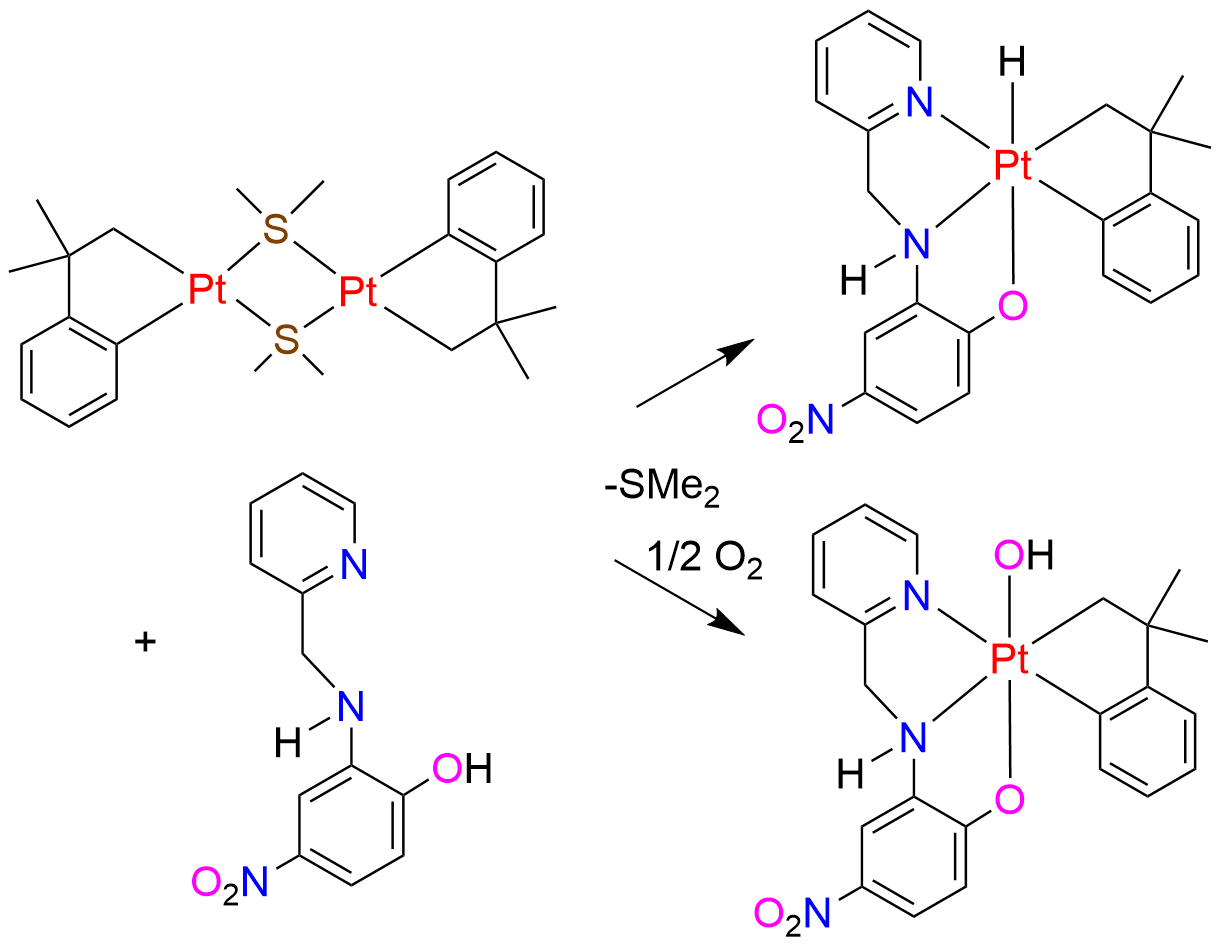{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
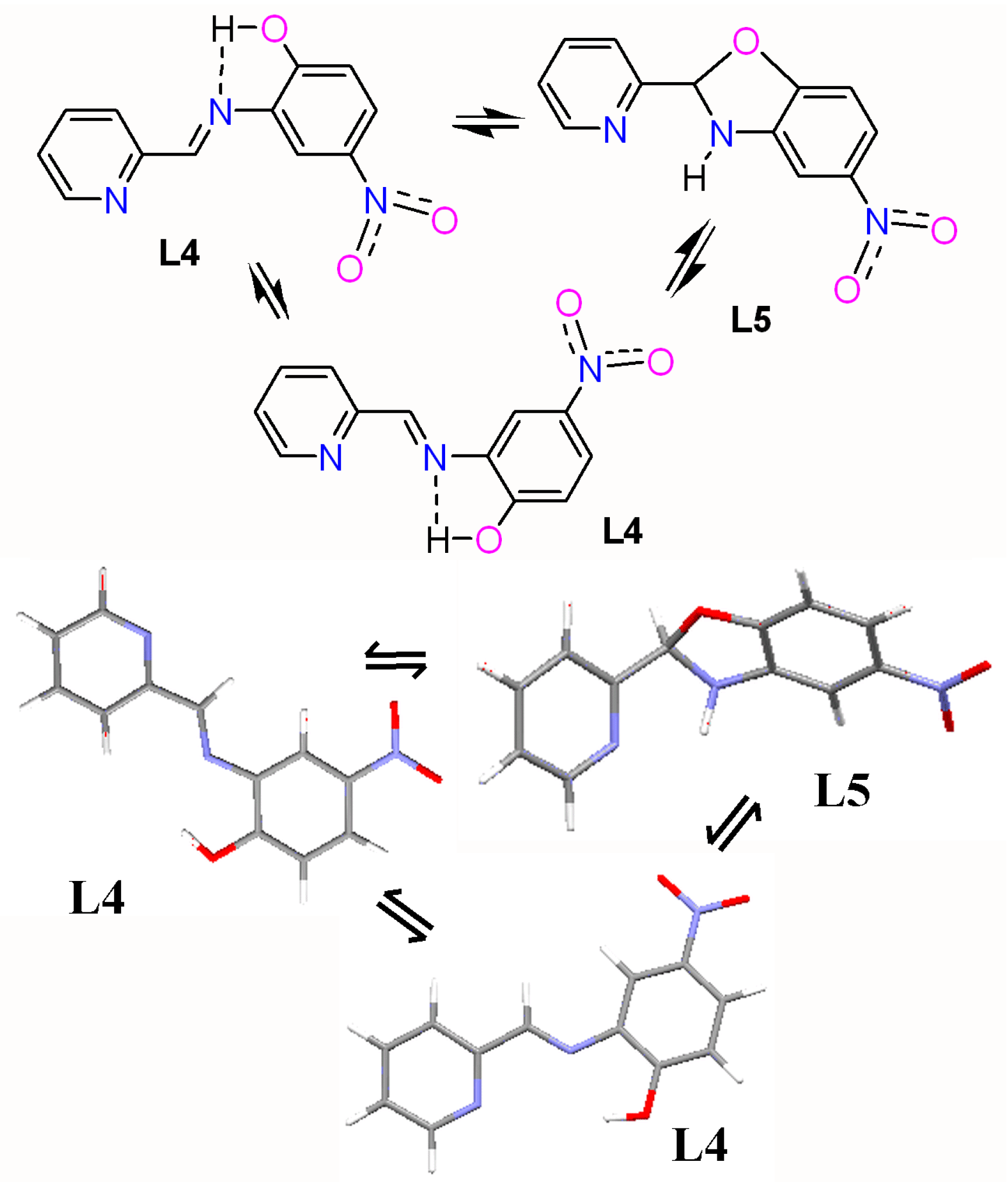
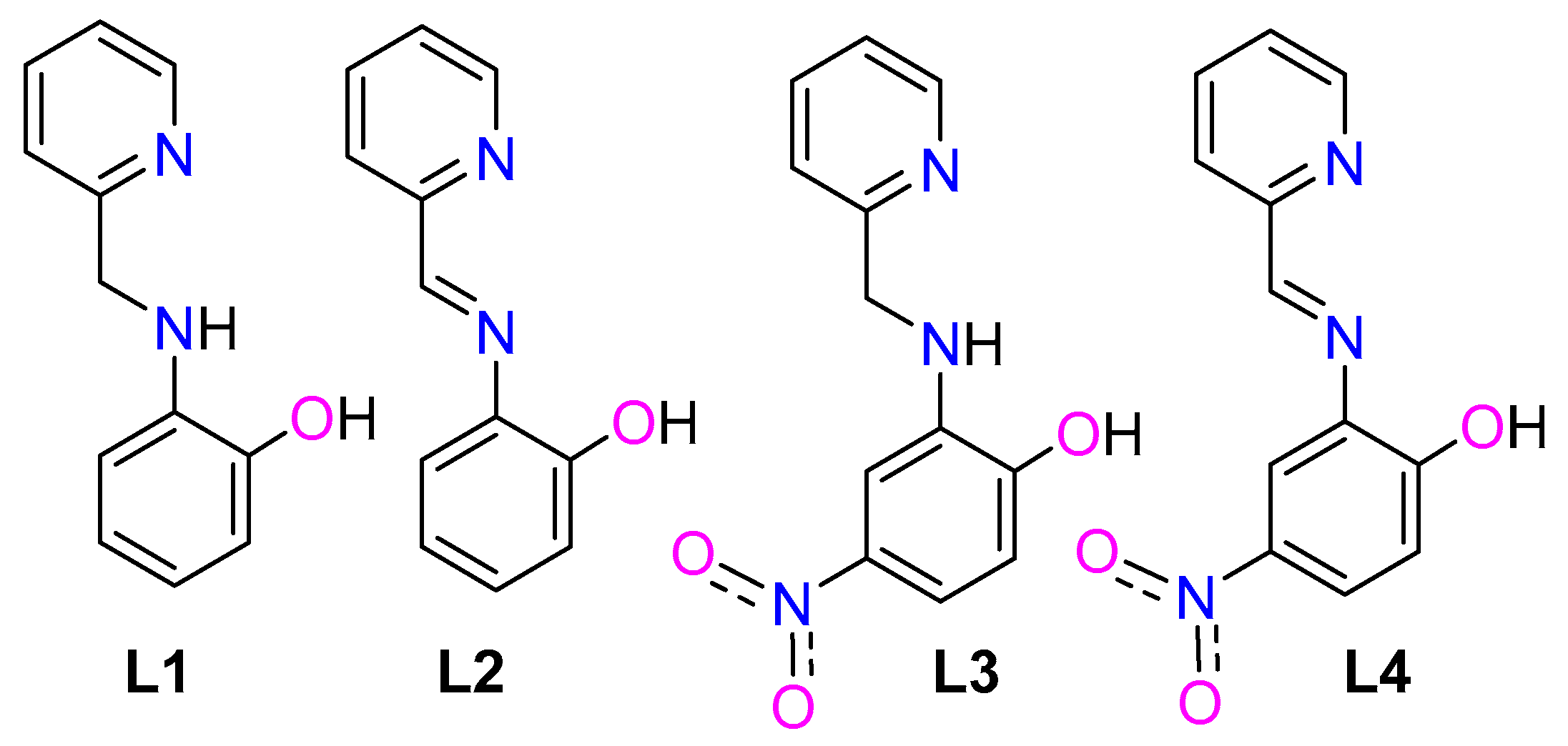
2.1. The Ligand L4
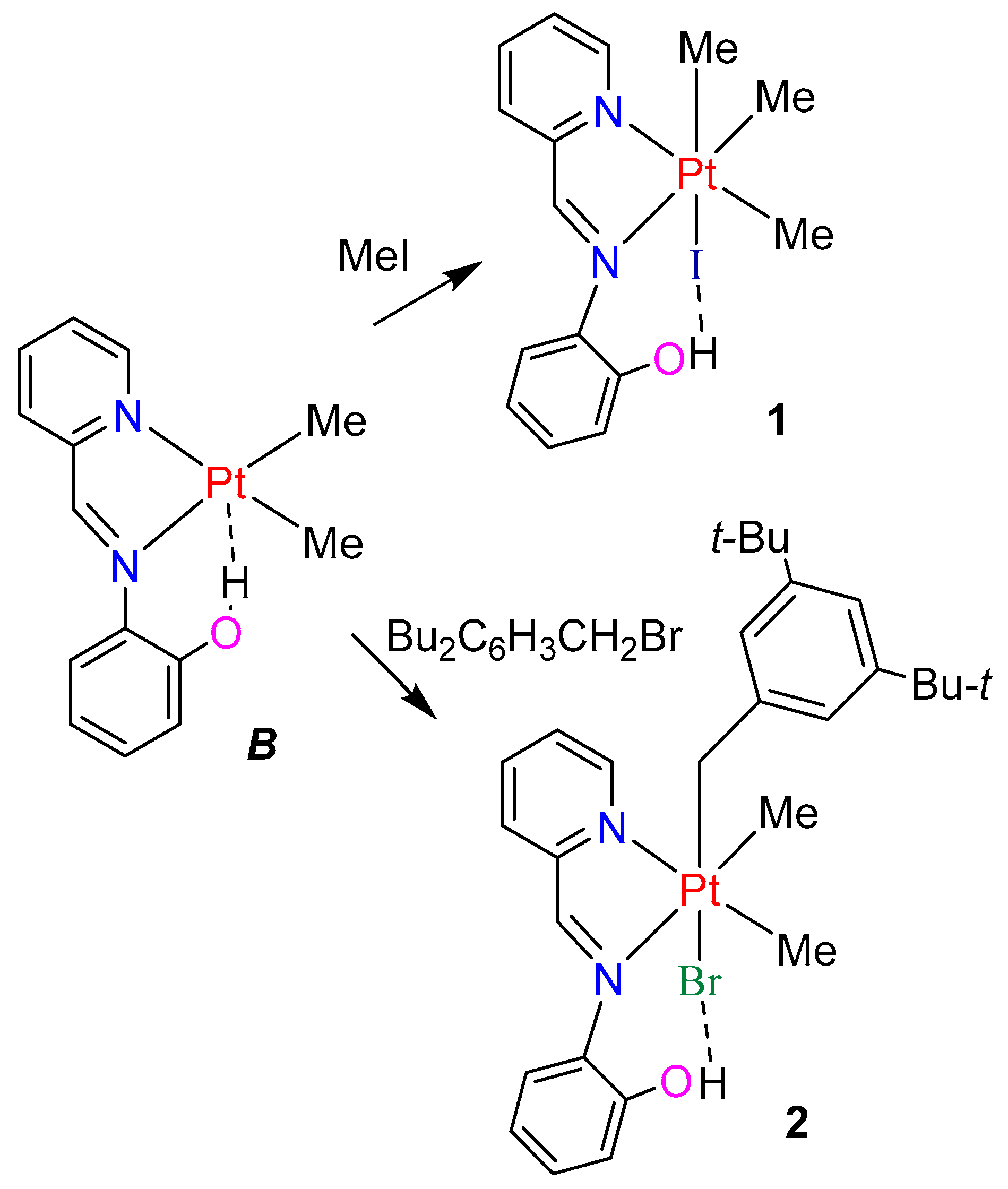
2.2. Model Platinum(IV) Complexes with Imine Ligand L2
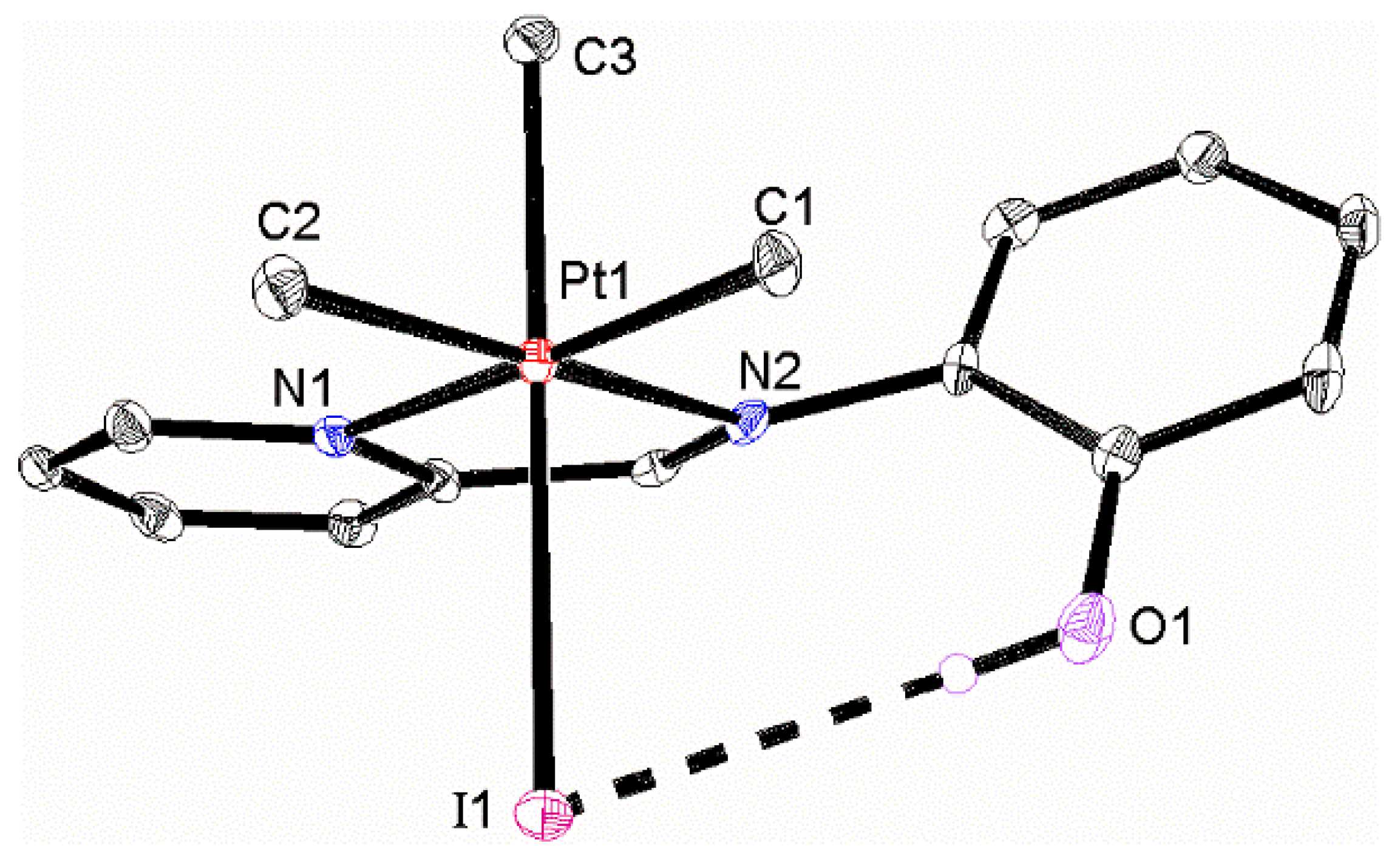
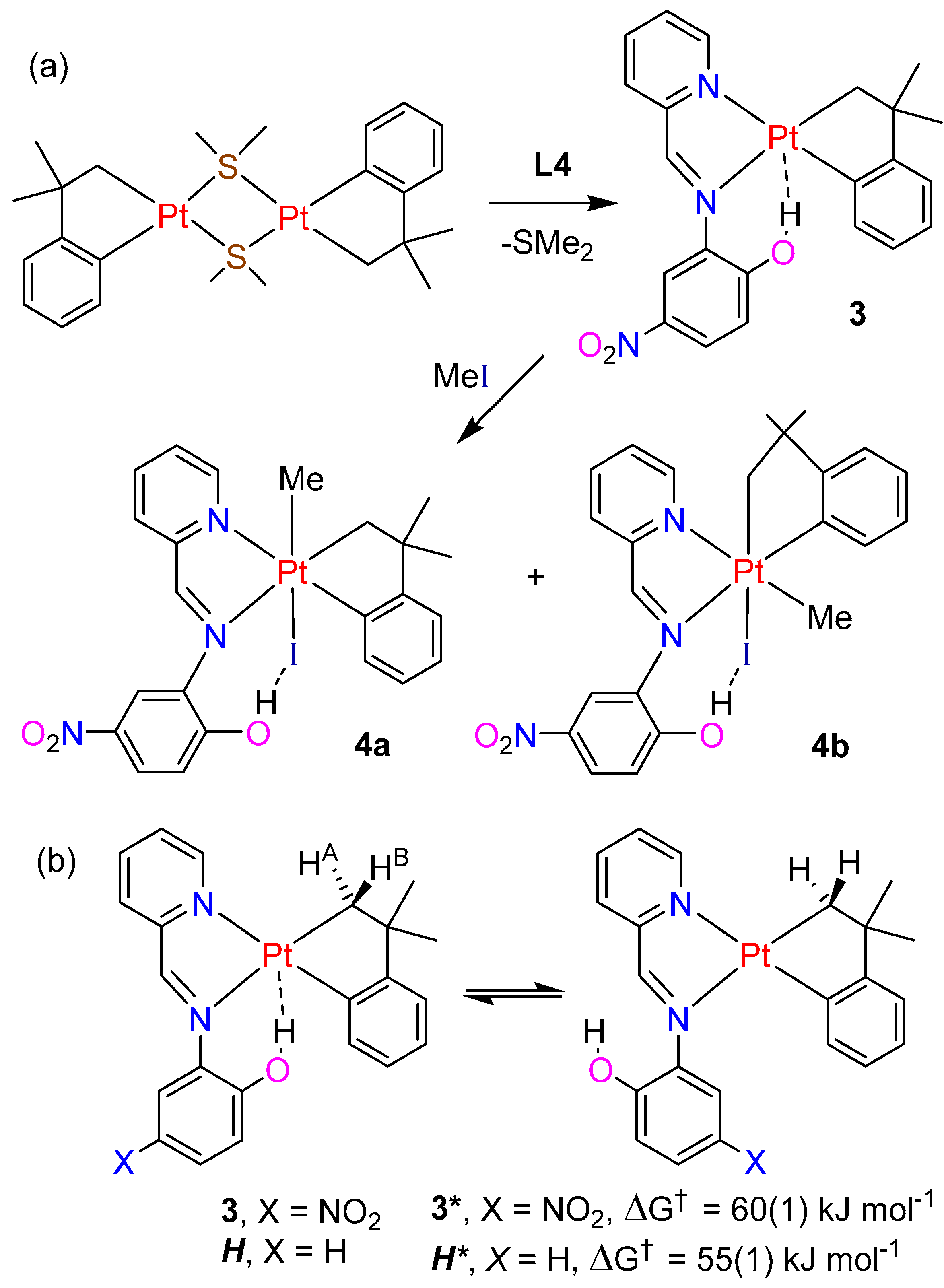
2.3. Cycloneophyl Complexes with Imine Ligand L4
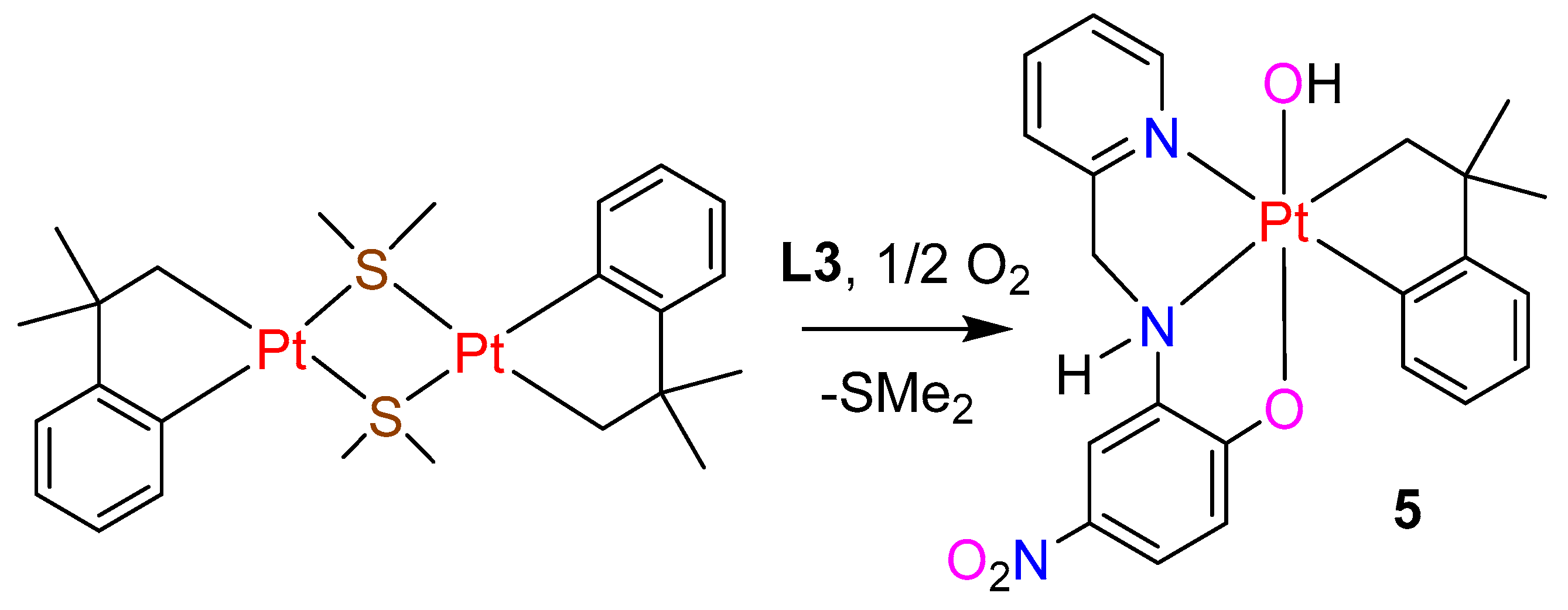
2.4. Cycloneophyl Complexes with Ligand L3
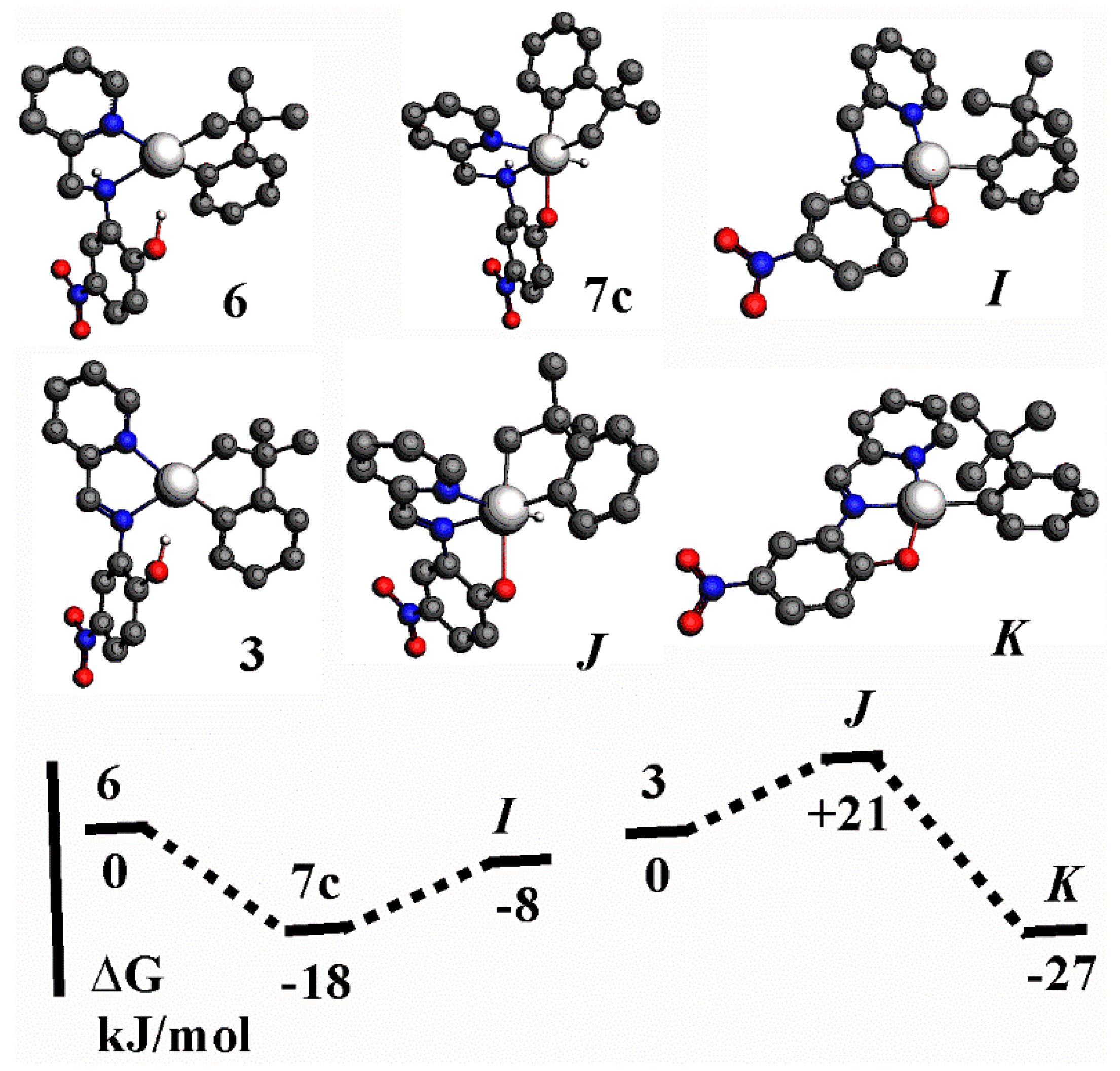
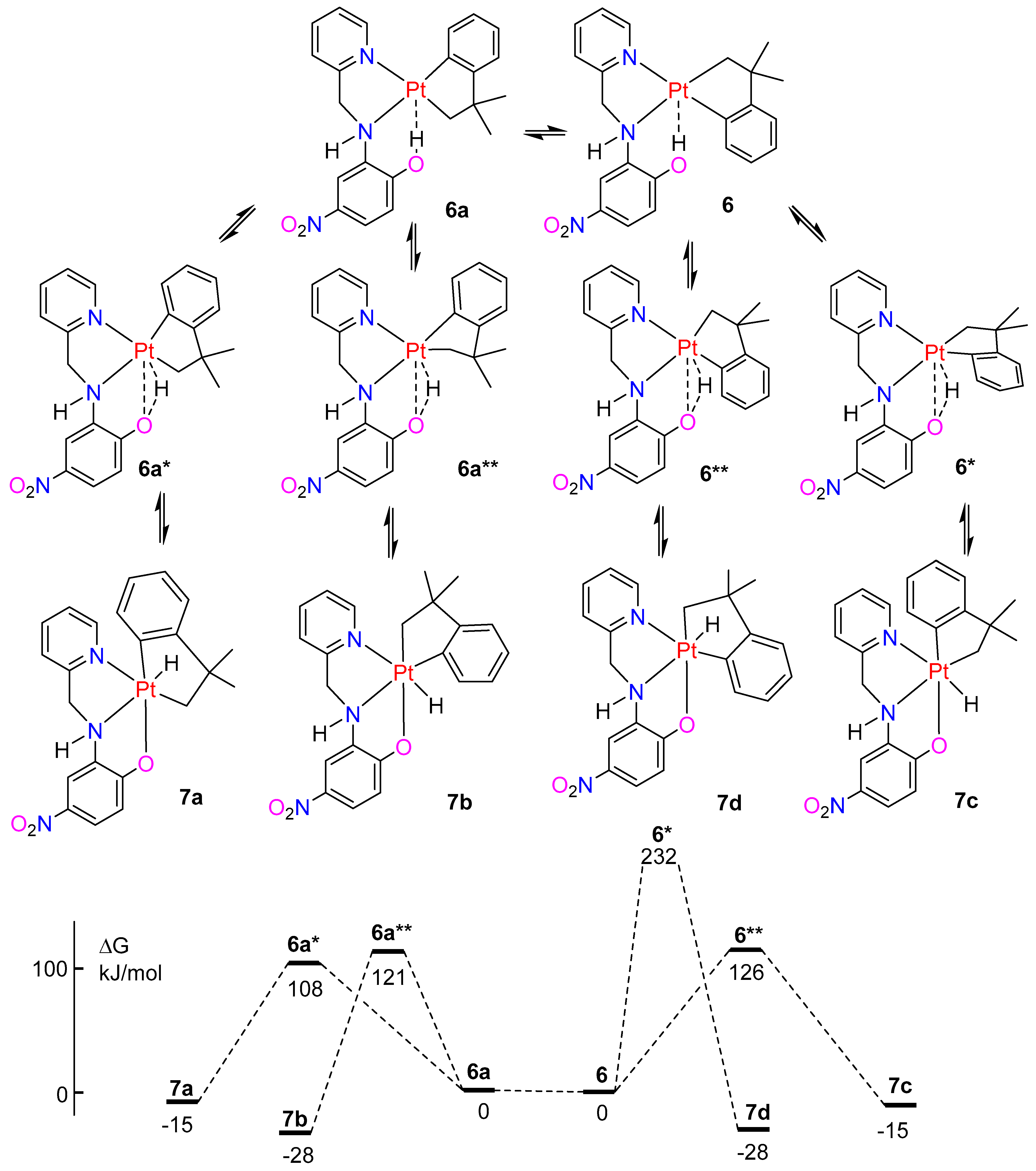
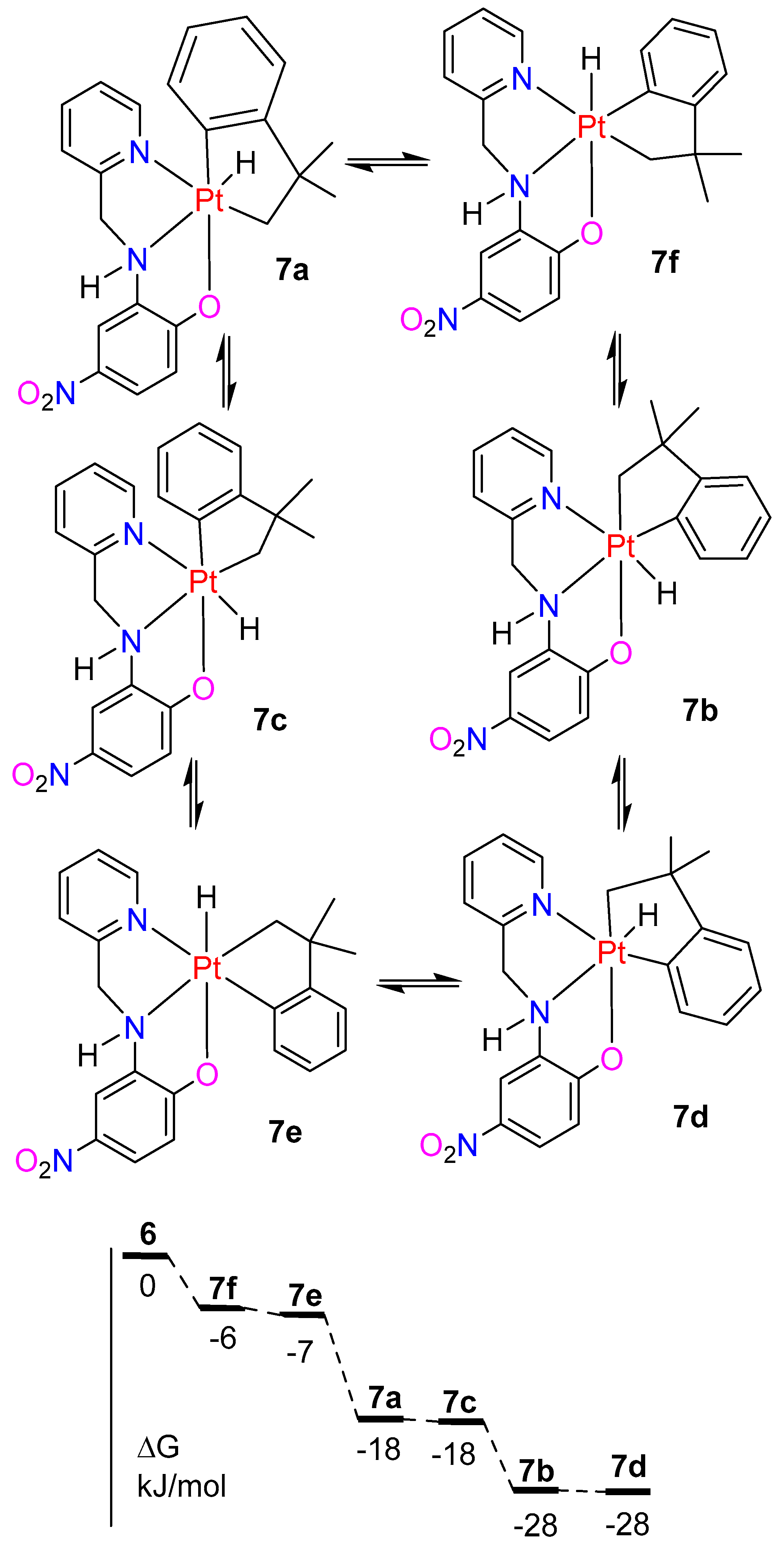
2.5. A Comparison of Complexes with L3 and L4
3. Materials and Methods
4. Conclusions
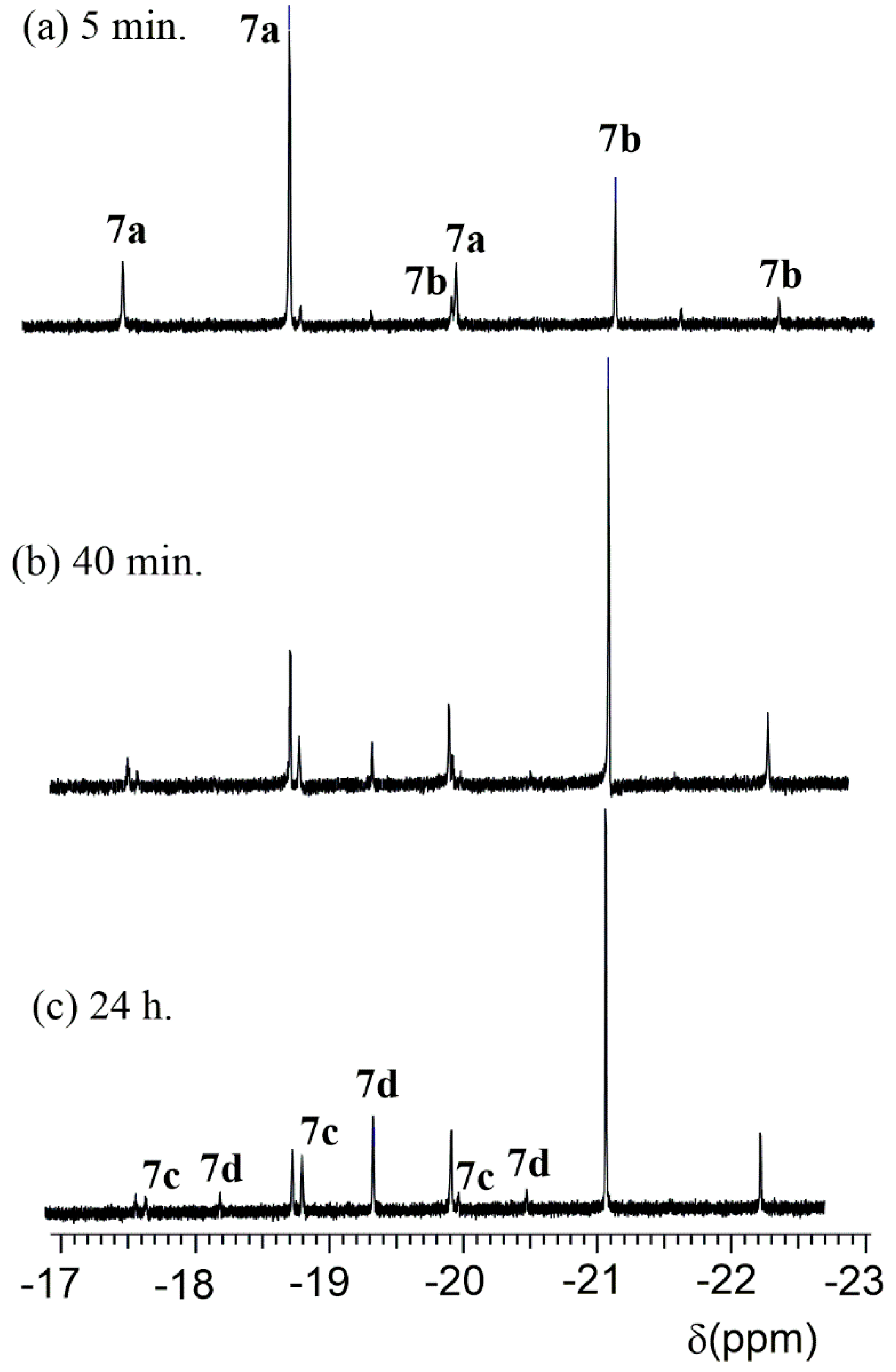
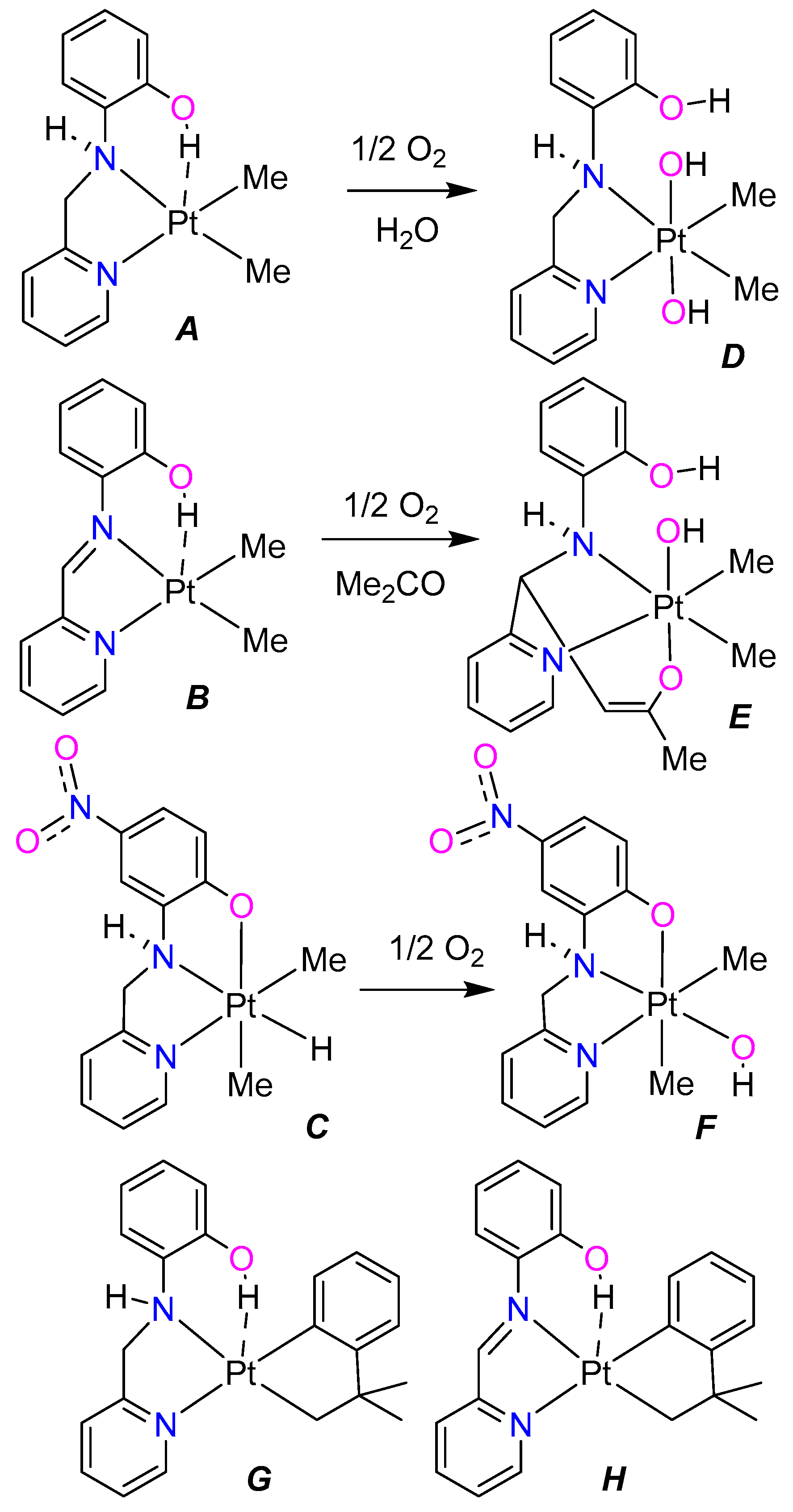
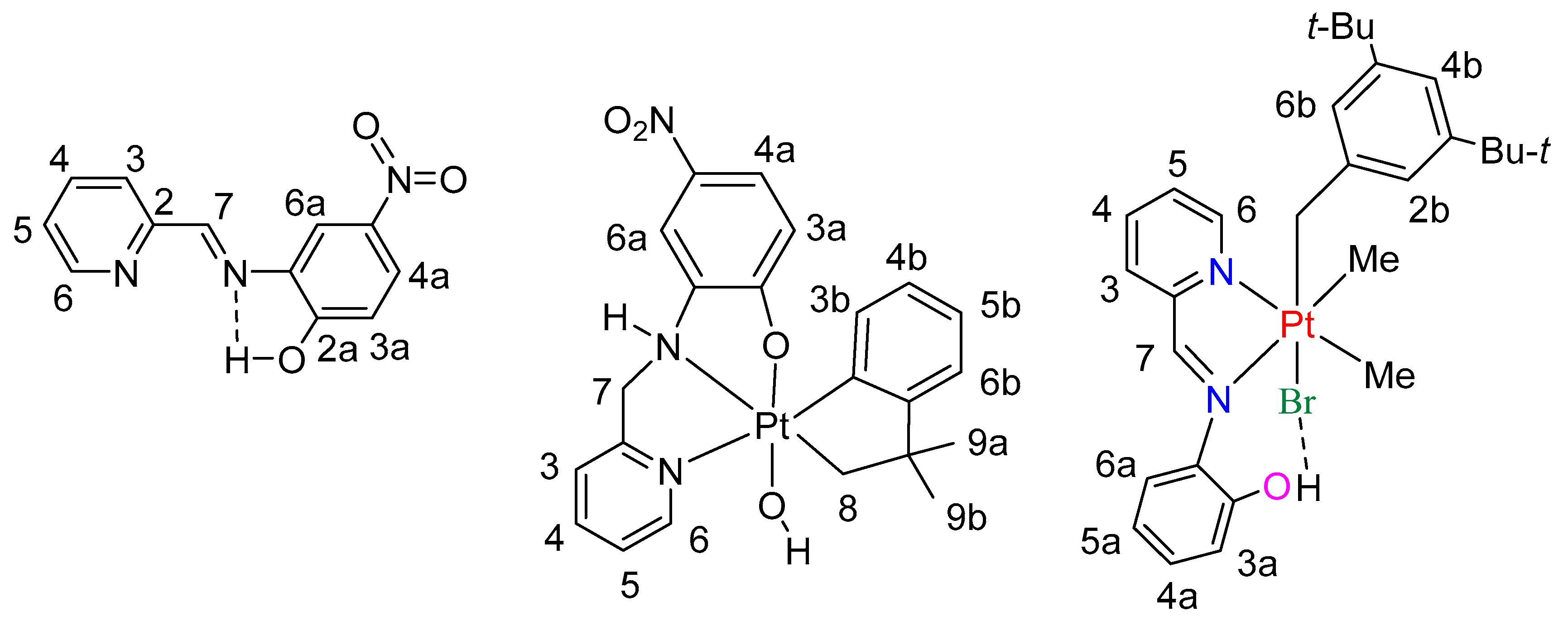
- The nitro group enhances the acidity of the phenol unit in ligands L3 and L4. This is most apparent by comparing the reactions of L1 and L3 with [Pt2(CH2CMe2C6H4)2(μ-SMe2)2] to give either the platinum(II) complex, G, (Scheme 2) or the hydridoplatinum(IV) complex 7 (as a mixture of isomers, Scheme 6 and Scheme 7). The difference is less marked in the reactions of L2 and L4 with [Pt2(CH2CMe2C6H4)2(μ-SMe2)2]. These both give the platinum(II) complex, H (Scheme 2) or 3 (Scheme 4), but there is evidence that the OH··Pt hydrogen bond is stronger in 3 than in H.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Manna, S.; Kong, W.J.; Backvall, J.E. Metal-catalyzed biomimetic aerobic oxidation of organic substrates. Adv. Catal. 2021, 69, 1–57. [Google Scholar]
- Chakrabarty, S.; Wang, Y.; Perkins, J.C.; Narayan, A.R.H. Scalable biocatalytic C-H oxyfunctionalization reactions. Chem. Soc. Rev. 2020, 49, 8137–8155. [Google Scholar] [CrossRef] [PubMed]
- Guillemard, L.; Kaplaneris, N.; Ackermann, L.; Johansson, M.J. Late-stage C-H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem. 2021, 5, 522–545. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wei, J.; Qiu, X.; Jiao, N. Homogeneous Oxygenase Catalysis. Chem. Rev. 2018, 118, 4912–4945. [Google Scholar] [CrossRef]
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef]
- Groves, J.T.; Feng, L.; Austin, R.N. Structure and Function of Alkane Monooxygenase (AlkB). Acc. Chem. Res. 2023, 56, 3665–3675. [Google Scholar] [CrossRef]
- Liu, Y.G.; You, T.; Wang, H.-X.; Tang, Z.; Zhou, C.-Y.; Che, C.-M. Iron- and cobalt-catalyzed C(sp3)-H bond functionalization reactions and their application in organic synthesis. Chem. Soc. Rev. 2020, 49, 5310–5358. [Google Scholar] [CrossRef]
- Tang, C.; Qiu, X.; Cheng, Z.; Jiao, N. Molecular oxygen-mediated oxygenation reactions involving radicals. Chem. Soc. Rev. 2021, 50, 8067–8101. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Cho, K.-B.; Lee, Y.-M.; Hong, S.; Nam, W. Mechanistic dichotomies in redox reactions of mononuclear metal-oxygen intermediates. Chem. Soc. Rev. 2020, 49, 8988–9027. [Google Scholar] [CrossRef]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef]
- Munz, D.; Strassner, T. Alkane C-H Functionalization and Oxidation with Molecular Oxygen. Inorg. Chem. 2015, 54, 5043–5052. [Google Scholar] [CrossRef] [PubMed]
- Etim, U.J.; Bai, P.; Gazit, O.M.; Zhong, Z. Low-Temperature Heterogeneous Oxidation Catalysis and Molecular Oxygen Activation. Catal. Rev. Sci. Eng. 2023, 65, 239–425. [Google Scholar] [CrossRef]
- Machan, C.W. Advances in the Molecular Catalysis of Dioxygen Reduction. ACS Catal. 2020, 10, 2640–2655. [Google Scholar] [CrossRef]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef]
- Schultz, J.W.; Rath, N.P.; Mirica, L.M. Improved Oxidative C-C Bond Formation Reactivity of High-Valent Pd Complexes Supported by a Pseudo-Tridentate Ligand. Inorg. Chem. 2020, 59, 11782–11792. [Google Scholar] [CrossRef]
- Cha, J.M.; Lee, E.S.; Yandulov, D.V. Mechanistic Studies for Pd(II)(O2) Reduction Generating Pd(0) and H2O: Formation of Pd(OH)2 as a Key Intermediate. Inorg. Chem. 2022, 61, 14544–14552. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Weinstein, A.B.; White, P.B.; Stahl, S.S. Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions. Chem. Rev. 2018, 118, 2636–2679. [Google Scholar] [CrossRef] [PubMed]
- Sberegaeva, A.V.; Zavalij, P.Y.; Vedernikov, A.N. Oxidation of a Monomethylpalladium(II) Complex with O2 in Water: Tuning Reaction Selectivity to Form Ethane, Methanol, or Methylhydroperoxide. J. Am. Chem. Soc. 2016, 138, 1446–1455. [Google Scholar] [CrossRef]
- Desnoyer, A.N.; Love, J.A. Recent advances in well-defined, late transition metal complexes that make and/or break C-N, C-O and C-S bonds. Chem. Soc. Rev. 2017, 46, 197–238. [Google Scholar] [CrossRef]
- Boisvert, L.; Goldberg, K.I. Reactions of Late Transition Metal Complexes with Molecular Oxygen. Acc. Chem. Res. 2012, 45, 899–910. [Google Scholar] [CrossRef]
- Labinger, J.A. Platinum-Catalyzed C-H Functionalization. Chem. Rev. 2017, 117, 8483–8496. [Google Scholar] [CrossRef] [PubMed]
- Prantner, J.D.; Kaminsky, W.; Goldberg, K.I. Methylplatinum(II) and Molecular Oxygen: Oxidation to Methylplatinum(IV) in Competition with Methyl Group Transfer to Form Dimethylplatinum(IV). Organometallics 2014, 33, 3227–3230. [Google Scholar] [CrossRef]
- Sberegaeva, A.V.; Watts, D.; Vedernikov, A.N. Oxidative Functionalization of Late Transition Metal-Carbon Bonds. Adv. Organomet. Chem. 2017, 67, 221–297. [Google Scholar]
- Scheuermann, M.L.; Goldberg, K.I. Reactions of Pd and Pt Complexes with Molecular Oxygen. Chem. Eur. J. 2014, 20, 14556–14568. [Google Scholar] [CrossRef] [PubMed]
- Rudakov, E.S.; Shul’pin, G.B. Stable organoplatinum complexes as intermediates and models in hydrocarbon functionalization. J. Organomet. Chem. 2015, 793, 4–16. [Google Scholar] [CrossRef]
- Watts, D.; Zavalij, P.Y.; Vedernikov, A.N. Consecutive C-H and O2 Activation at a Pt(II) Center to Produce Pt(IV) Aryls. Organometallics 2018, 37, 4177–4180. [Google Scholar] [CrossRef]
- Miller, B.; Altman, J.; Beck, W. Platinum(IV) complexes of vicinal-1,2-diamines and bis(vicinal-1,2-diamines) with an acylamino function. Evidence for a platinum hydroperoxide intermediate upon oxidation. Inorg. Chim. Acta 1997, 264, 101–108. [Google Scholar] [CrossRef]
- Look, J.L.; Wick, D.D.; Mayer, J.M.; Goldberg, K.I. Autoxidation of Platinum(IV) Hydrocarbyl Hydride Complexes to Form Platinum(IV) Hydrocarbyl Hydroperoxide Complexes. Inorg. Chem. 2009, 48, 1356–1369. [Google Scholar] [CrossRef]
- Khusnutdinova, J.R.; Zavalij, P.Y.; Vedernikov, A.N. Study of aerobic oxidation of phenyl PtII complexes (dpms)PtIIPh(L) (dpms = di(2-pyridyl)methanesulfonate; L = water, methanol, or aniline). Can. J. Chem. 2009, 87, 110–120. [Google Scholar] [CrossRef]
- Liu, W.G.; Sberegaeva, A.V.; Nielsen, R.J.; Goddard, W.A.; Vedernikov, A.N. Mechanism of O2 Activation and Methanol Production by (Di(2-pyridyl)methanesulfonate)PtIIMe(OHn)(2−n)− Complex from Theory with Validation from Experiment. J. Am. Chem. Soc. 2014, 136, 2335–2341. [Google Scholar] [CrossRef]
- Lippert, B.; Miguel, P.J.S. More of a misunderstanding than a real mismatch? Platinum and its affinity for aqua, hydroxido, and oxido ligands. Coord. Chem. Rev. 2016, 327, 333–348. [Google Scholar] [CrossRef]
- Prokopchuk, E.M.; Jenkins, H.A.; Puddephatt, R.J. Stable cationic dimethyl(hydrido)platinum(IV) complex. Organometallics 1999, 18, 2861–2866. [Google Scholar] [CrossRef]
- Moustafa, M.E.; Boyle, P.D.; Puddephatt, R.J. A biomimetic phenol substituent effect on the reaction of a dimethylplatinum(II) complex with oxygen: Proton coupled electron transfer and multiple proton relay. Chem. Commun. 2015, 51, 10334–10336. [Google Scholar] [CrossRef] [PubMed]
- Fard, M.A.; Behnia, A.; Puddephatt, R.J. Activation of Dioxygen by Dimethylplatinum(II) Complexes. Organometallics 2017, 36, 4169–4178. [Google Scholar] [CrossRef]
- Abo-Amer, A.; Boyle, P.D.; Puddephatt, R.J. Push-pull ligands to enhance the oxygen activation step in catalytic oxidation with platinum complexes. Inorg. Chim. Acta 2018, 473, 51–59. [Google Scholar] [CrossRef]
- Abo-Amer, A.; Boyle, P.D.; Puddephatt, R.J. Push-pull ligands and the oxidation of monomethylplatinum(II) complexes with oxygen or hydrogen peroxide. Inorg. Chim. Acta 2020, 507, 119580. [Google Scholar] [CrossRef]
- Abo-Amer, A.; Boyle, P.D.; Puddephatt, R.J. The remarkable effects of a ligand nitro substituent in organoplatinum chemistry related to activation of dioxygen or reductive elimination of methane. Polyhedron 2022, 217, 115722. [Google Scholar] [CrossRef]
- Behnia, A.; Fard, M.A.; Puddephatt, R.J. Complexes containing a phenol-platinum(II) hydrogen bond: Synthons for supramolecular self-assembly and precursors for hydridoplatinum(IV) complexes. Eur. J. Inorg. Chem. 2019, 2899–2906. [Google Scholar] [CrossRef]
- Fard, M.A.; Behnia, A.; Puddephatt, R.J. Platinum(II) complexes of pyridine–amine ligands with phenol substituents: Isotactic supramolecular polymers. Can. J. Chem. 2018, 97, 238–243. [Google Scholar] [CrossRef]
- Fard, M.A.; Behnia, A.; Puddephatt, R.J. Models for cooperative catalysis: Oxidative addition reactions of dimethylplatinum(II) complexes with ligands having both NH and OH functionality. ACS Omega 2019, 4, 257–268. [Google Scholar] [CrossRef]
- Behnia, A.; Fard, M.A.; Puddephatt, R.J. Stereochemistry of oxidative addition of methyl iodide and hydrogen peroxide to organoplatinum(II) complexes having an appended phenol group and the supramolecular chemistry of the platinum(IV) products. J. Organomet. Chem. 2019, 902, 120962. [Google Scholar] [CrossRef]
- Fard, M.A.; Behnia, A.; Puddephatt, R.J. Supramolecular polymer and sheet and a double cubane structure in platinum(IV) iodide chemistry: Solution of a longstanding puzzle. ACS Omega 2018, 3, 10267–10272. [Google Scholar] [CrossRef] [PubMed]
- Fard, M.A.; Puddephatt, R.J. Oxidative addition of halogens to a cycloneophylplatinum(II) complex and evidence for C-H bond activation at platinum(IV). J. Organomet. Chem. 2020, 910, 121139. [Google Scholar] [CrossRef]
- Fard, M.A.; Behnia, A.; Puddephatt, R.J. Cycloneophylplatinum Chemistry: A New Route to Platinum(II) Complexes and the Mechanism and Selectivity of Protonolysis of Platinum-Carbon Bonds. Organometallics 2018, 37, 3368–3377. [Google Scholar] [CrossRef]
- Neugebauer, M.; Schmitz, S.; Brunink, D.; Doltsinis, N.L.; Klein, A. Dynamics of the efficient cyclometalation of the undercoordinated organoplatinum complex [Pt(COD)(neoPh)]+ (neoPh=2-methyl-2-phenylpropyl). New J. Chem. 2020, 44, 19238–19249. [Google Scholar] [CrossRef]
- Campora, J.; Palma, P.; Carmona, E. The chemistry of group 10 metalacycles. Coord. Chem. Rev. 1999, 193, 207–281. [Google Scholar] [CrossRef]
- Griffiths, D.C.; Young, G.B. Mechanisms of thermolytic rearrangement of dineophylplatinum(II) complexes via intramolecular C-H activation. Organometallics 1989, 8, 875–886. [Google Scholar] [CrossRef]
- Ankianiec, B.C.; Hardy, D.T.; Thomson, S.K.; Watkins, W.N.; Young, G.B. Mechanistic studies of the thermolytic and photolytic rearrangement of [bis(diphenylphosphino)ethane]bis(neophyl)platinum(II). Organometallics 1992, 11, 2591–2598. [Google Scholar] [CrossRef]
- SHo, K.Y.; Lam, F.Y.T.; de Aguirre, A.; Maseras, F.; White, A.J.P.; Britovsek, G.J.P. Photolytic Activation of Late-Transition-Metal-Carbon Bonds and Their Reactivity toward Oxygen. Organometallics 2021, 40, 4077–4091. [Google Scholar]
- Griffiths, D.C.; Joy, L.G.; Skapski, A.C.; Wilkes, D.J.; Young, G.B. Rearrangements of neophylplatinum(II) and related derivatives via intramolecular aromatic and aliphatic delta-carbon-hydrogen bond activation—The crystal and molecular-structure of cis-(Et3P)2Pt(CH2CMe2Ph)(2-C6H4CMe3). Organometallics 1986, 5, 1744–1745. [Google Scholar] [CrossRef]
- Chappell, S.D.; Cole-Hamilton, D.J. The preparation and properties of metallacyclic compounds of the transition-elements. Polyhedron 1982, 1, 739–777. [Google Scholar] [CrossRef]
- Cámpora, J.; López, J.A.; del Rio, D.; Carmona, E.; Valerga, P.; Graiff, C.; Tiripicchio, A. Synthesis and insertion reactions of the cyclometalated palladium-alkyl complexes Pd(CH2CMe2-o-C6H4)L2.: Observation of a pentacoordinated intermediate in the insertion of SO2. Inorg. Chem. 2001, 40, 4116–4126. [Google Scholar] [CrossRef]
- Albrecht, M. Cyclometalation Using d-Block Transition Metals: Fundamental Aspects and Recent Trends. Chem. Rev. 2010, 110, 576–623. [Google Scholar] [CrossRef]
- Moustafa, M.E.; Boyle, P.D.; Puddephatt, R.J. Reactivity and mechanism in reactions of methylene halides with cycloneophylplatinum(II) complexes: Oxidative addition and methylene insertion. J. Organomet. Chem. 2021, 941, 121803. [Google Scholar] [CrossRef]
- Ramirez-Jimenez, A.; Gomez, E.; Hernandez, S. Penta- and heptacoordinated tin(IV) compounds derived from pyridine Schiff bases and 2-pyridine carboxylate: Synthesis and structural characterization. J. Organomet. Chem. 2009, 694, 2965–2975. [Google Scholar] [CrossRef]
- Chans, G.M.; Nieto-Camacho, A.; Ramirez-Apan, T.; Hernandez-Ortega, S.; Alvarez-Toledano, C.; Gomez, E. Synthetic, spectroscopic, crystallographic, and biological studies of seven-coordinated diorganotin(IV) complexes derived from Schiff bases and pyridinic carboxylic acids. Aust. J. Chem. 2016, 69, 279–290. [Google Scholar] [CrossRef]
- Iasco, O.; Riviere, E.; Guillot, R.; Buron-LeCointe, M.; Meunier, J.F.; Bousseksou, A.; Boillot, M.-L. FeII(pap-5NO2)2 and FeII(qsal-5NO2)2 Schiff-base spin-crossover complexes: A rare example with photomagnetism and room-temperature bistability. Inorg. Chem. 2015, 54, 1791–1799. [Google Scholar] [CrossRef]
- Sadimenko, A.P. Organometallic complexes of pyridyl Schiff bases. Adv. Heterocycl. Chem. 2012, 107, 133–218. [Google Scholar]
- Liles, D.C.; McPartlin, M.; Tasker, P.A.; Lip, H.C.; Lindoy, L.F. Novel polynuclear cadmium complexes resulting from ring-opening reactions of 2,6-bis(2-R-2-benzoxazolinyl)pyridine (R = Me or H): X-ray structures of [Cd4C21H17N3O2)2·(MeCO2)4]·Me2NCHO·H2O and [Cd3(C19H13N3C2)2·(MeCO2)2·(Me2NCHO)2]. Chem. Commun. 1976, 549–551. [Google Scholar] [CrossRef]
- Gurriero, P.; Bullita, E.; Vigato, P.A.; Pelli, B.; Traldi, P. Synthesis, characterization and electron impact mass spectrometry of Schiff bases rearranging to oxazoline, thiazoline and thiazole derivatives. J. Heterocycl. Chem. 1988, 25, 145–154. [Google Scholar] [CrossRef]
- Luo, Y.; Utecht, M.; Dokic, J.; Korchak, S.; Vieth, H.-M.; Haag, R.; Saalfrank, P. Cis-trans isomerisation of substituted aromatic imines: A comparative experimental and theoretical study. ChemPhysChem 2011, 12, 2311–2321. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Henling, L.M.; Labinger, J.A.; Bercaw, J.E. Structural and mechanistic investigations of the oxidation of dimethylplatinum(II) complexes by dioxygen. Inorg. Chem. 2002, 41, 3608–3619. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Wang, D.; Kramer, M.J.; Zavalij, P.Y.; Vedernikov, A.N. Oxidation of Methylplatinum(II) Complexes K[(L)PtIIMe] with O2 and C(sp3)-X (X = O and C) Reductive Elimination Reactivity of Methylplatinum(IV) Products (L)PtIVMe(OH): The Effect of Structure of Sulfonated CNN-Pincer Ligands L. Organometallics 2022, 41, 2764–2783. [Google Scholar]
- Vedernikov, A.N. Direct Functionalization of M-C (M = Pt-II, Pd-II) Bonds Using Environmentally Benign Oxidants, O2 and H2O2. Acc. Chem. Res. 2012, 45, 803–813. [Google Scholar] [CrossRef]
- Hill, G.S.; Irwin, M.J.; Levy, C.J.; Rendina, L.M.; Puddephatt, R.J.; Andersen, R.A.; McLean, L. Platinum(II) complexes of dimethylsulfide. Inorg. Synth. 1998, 32, 149–153. [Google Scholar]
- Bruker-AXS. SAINT, version 2013.8; Bruker-AXS: Madison, WI, USA, 2013. [Google Scholar]
- Bruker-AXS. SADABS, version 2012.1; Bruker-AXS: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- GSheldrick, M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jonsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Andzelm, J.; Kolmel, C.; Klamt, A. Incorporation of solvent effects into density-functional calculations of molecular energies and geometries. J. Chem. Phys. 1995, 103, 9312–9320. [Google Scholar] [CrossRef]
- ADF 2020, SCM, Vrije Universiteit, Amsterdam, The Netherlands. Available online: https://www.scm.com (accessed on 1 January 2021).















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abo-Amer, A.; Moustafa, M.E.; Boyle, P.D.; Puddephatt, R.J. Organoplatinum Chemistry Related to Alkane Oxidation: The Effect of a Nitro Substituent in Ligands Having an Appended Phenol Group. Inorganics 2024, 12, 32. https://doi.org/10.3390/inorganics12010032
Abo-Amer A, Moustafa ME, Boyle PD, Puddephatt RJ. Organoplatinum Chemistry Related to Alkane Oxidation: The Effect of a Nitro Substituent in Ligands Having an Appended Phenol Group. Inorganics. 2024; 12(1):32. https://doi.org/10.3390/inorganics12010032
Chicago/Turabian StyleAbo-Amer, Anwar, Mohamed E. Moustafa, Paul D. Boyle, and Richard J. Puddephatt. 2024. "Organoplatinum Chemistry Related to Alkane Oxidation: The Effect of a Nitro Substituent in Ligands Having an Appended Phenol Group" Inorganics 12, no. 1: 32. https://doi.org/10.3390/inorganics12010032