
Syntheses and Applications of Symmetrical Dinuclear Half-Sandwich Ruthenium(II)–Dipicolinamide Complexes as Catalysts in the Transfer Hydrogenation of Ketones

Abstract
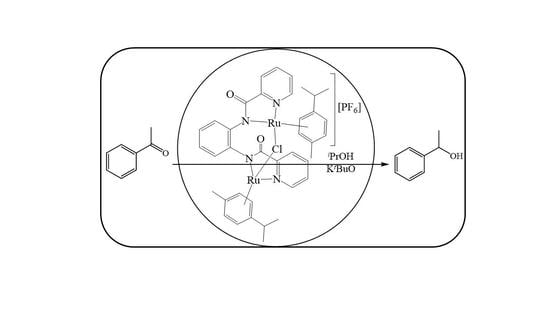
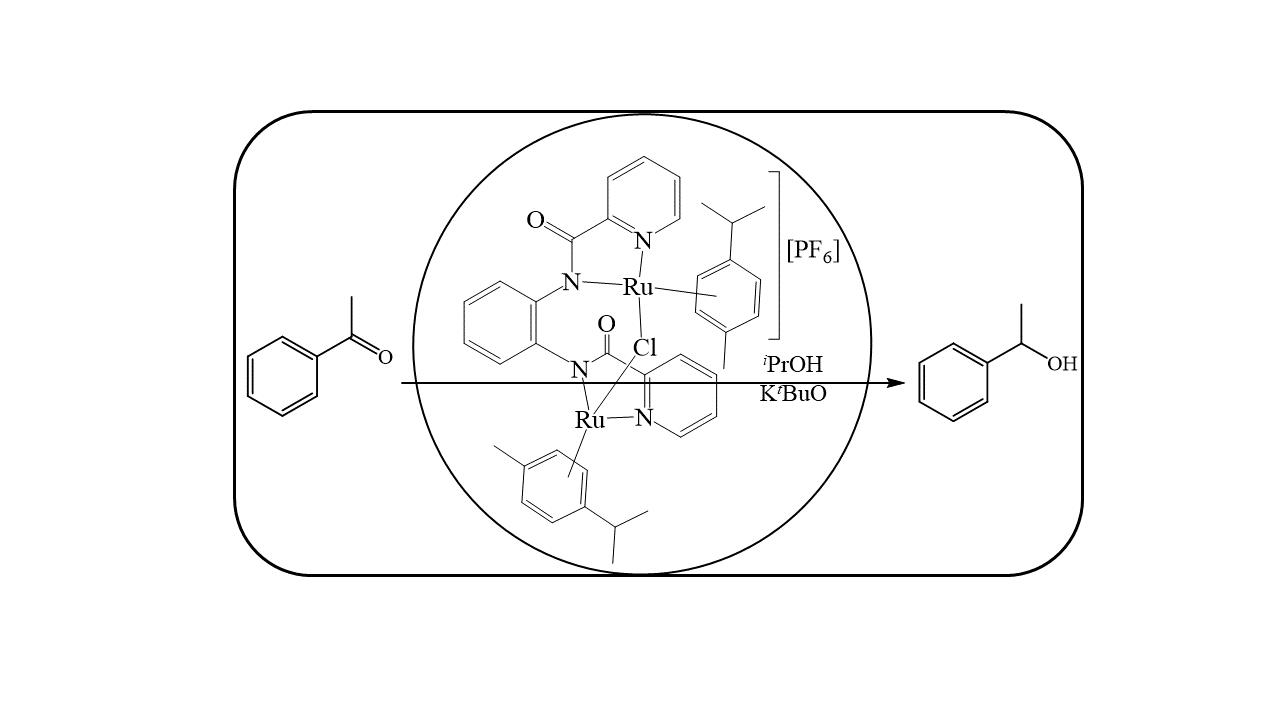
:
1. Introduction
2. Results and Discussion
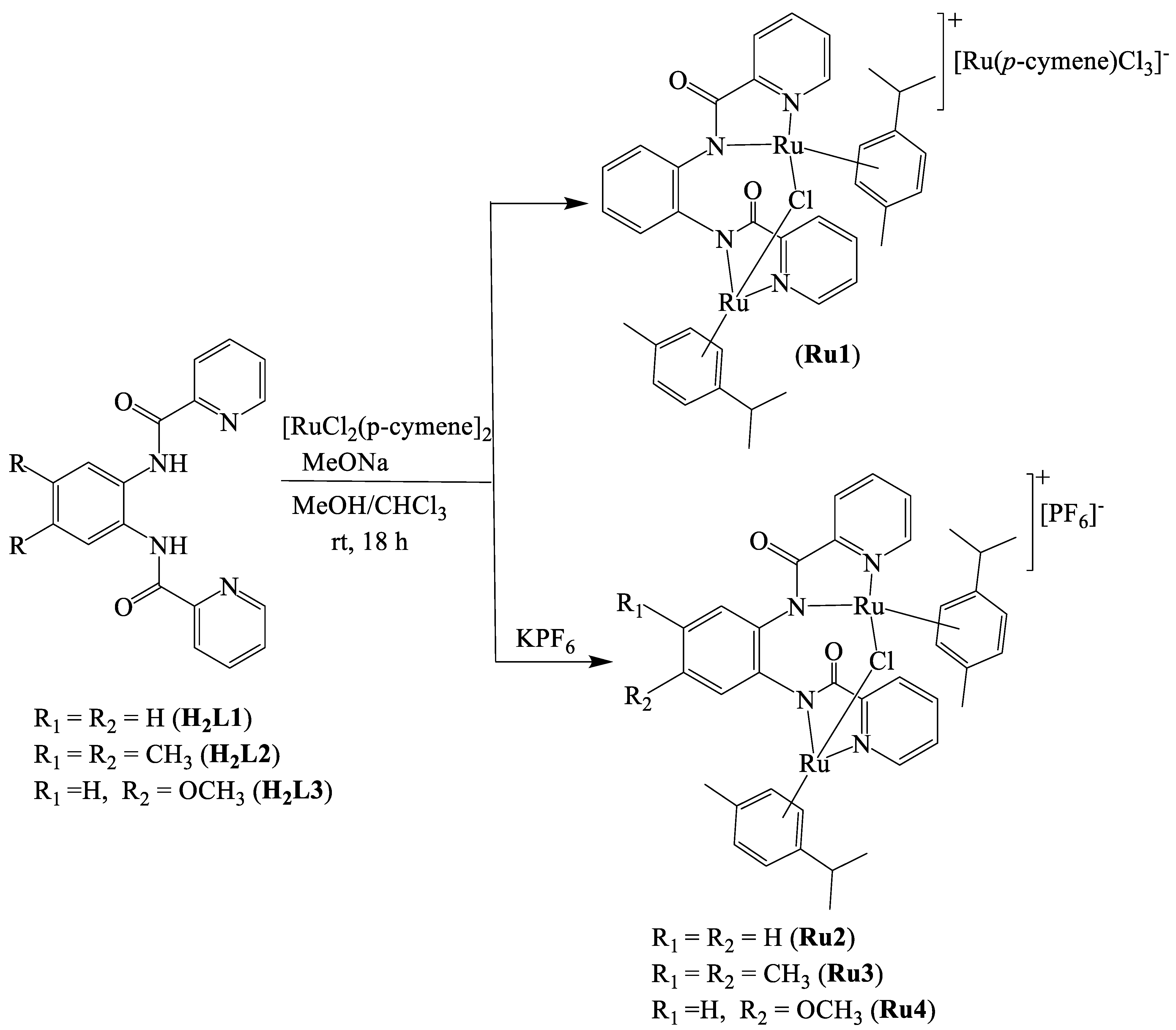
2.1. Synthesis and Characterisation of Ligands and Complexes
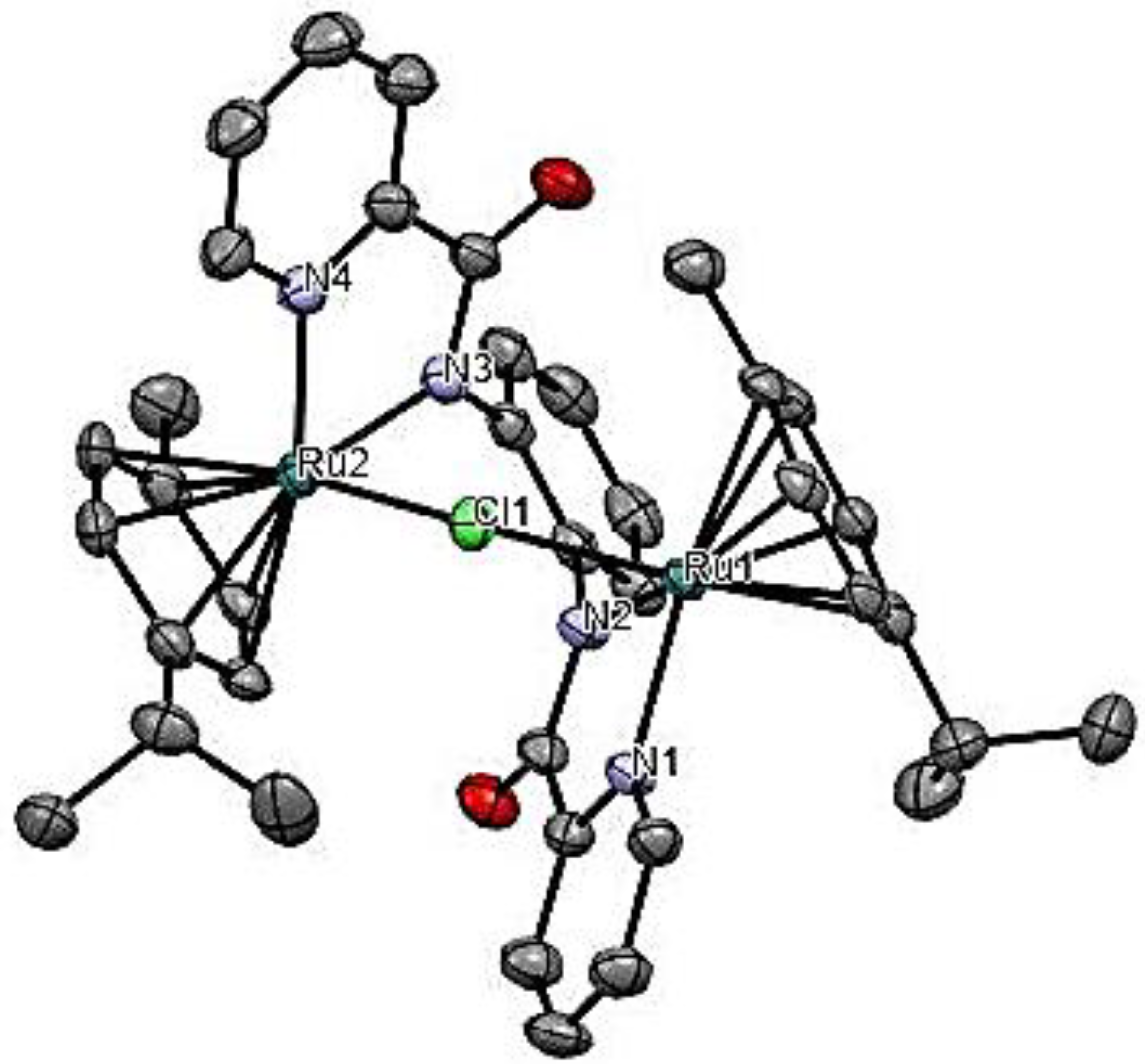
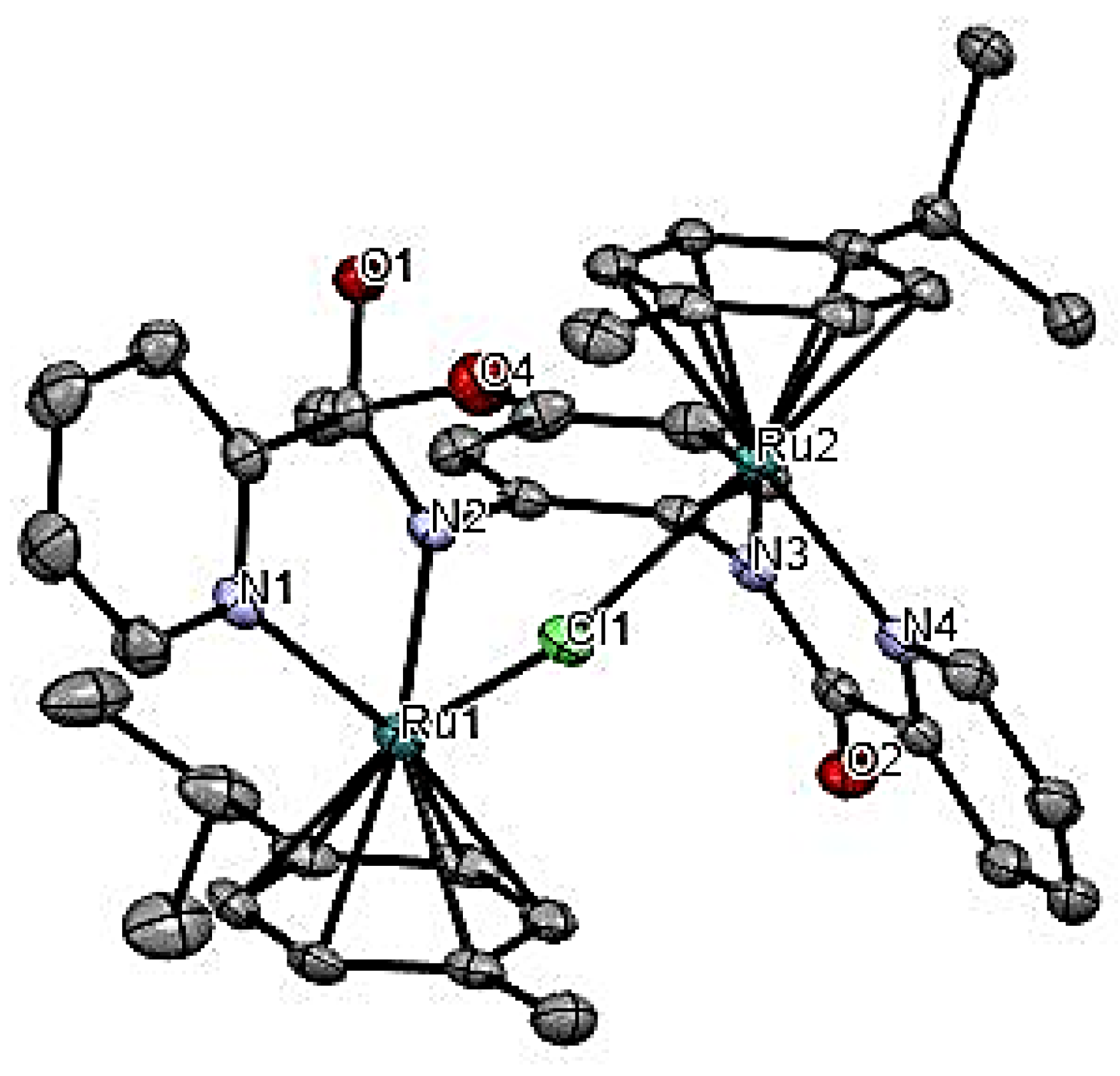
2.2. Molecular Structures of Ruthenium(II) Complexes Ru1 and Ru3
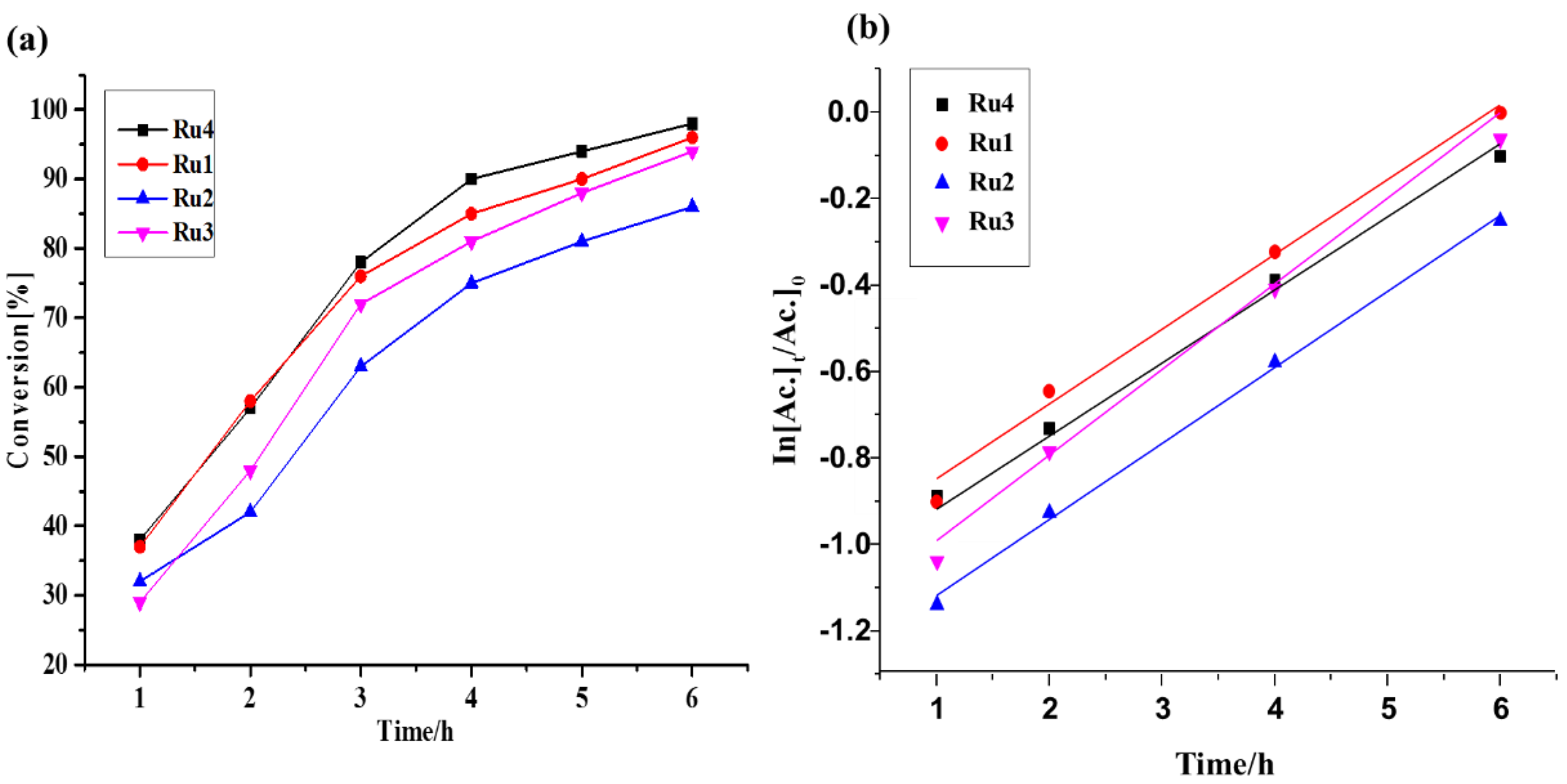
2.3. Transfer Hydrogenation of Ketones
2.3.1. Influence of Catalyst Structure on the TH of Acetophenone
2.3.2. Investigation of the Ketone Substrate Scope
3. Conclusions
4. Experimental Section
4.1. Materials and Instrumentations
4.2. X-ray Data Collection, Structure, and Refinement
4.3. Synthesis of Dinuclear Ruthenium(II) Carboxamide Complexes
4.3.1. [{Ru(η6-p-cymene)2-μ-Cl}2L1][Ru(η6-p-cymene)Cl3] (Ru1)
4.3.2. [{Ru(η6-p-cymene)2-μ-Cl}2L1][PF6] (Ru2)
4.3.3. [{Ru(η6-p-cymene)2-μ-Cl}2L2][PF6] (Ru3)
4.3.4. [{Ru(η6-p-cymene)2-μ-Cl}2L3][PF6] (Ru4)
4.3.5. Transfer Hydrogenation of Experiments
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornils, B.; Herrmann, W.A. Concepts in homogeneous catalysis: The industrial view. J. Catal. 2003, 216, 23–31. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Q.; Chen, H.; Li, R.-X. A novel cationic dinuclear ruthenium complex: Synthesis, characterization and catalytic activity in the transfer hydrogenation of ketones. Inorg. Chem. Commun. 2011, 14, 1884–1888. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Foubelo, F.; Najera, C.; Yus, M. Catalytic asymmetric transfer hydrogenation of ketones: Recent advances. Tetrahedron Asymmetry 2015, 26, 769–790. [Google Scholar] [CrossRef] [Green Version]
- Aydemir, M.; Durap, F.; Baysal, A.; Meric, N.; Buldağ, A.; Gümgüm, B.; Özkar, S.; Yıldırım, L.T. Novel neutral phosphinite bridged dinuclear ruthenium (II) arene complexes and their catalytic use in transfer hydrogenation of aromatic ketones: X-ray structure of a new Schiff base, N3, N3′-di-2-hydroxybenzylidene-[2, 2′] bipyridinyl-3, 3′-diamine. J. Mol. Catal. A Chem. 2010, 326, 75–81. [Google Scholar] [CrossRef]
- Pye, D.R.; Mankad, N.P. Bimetallic catalysis for C–C and C–X coupling reactions. Chem. Sci. 2017, 8, 1705–1718. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Chai, H.; Wang, L.; Yu, Z. Exceptionally Active Assembled Dinuclear Ruthenium (II)-NNN Complex Catalysts for Transfer Hydrogenation of Ketones. Organometallics 2017, 36, 2914–2921. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The golden age of transfer hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Chai, H.; Wang, Q.; Liu, T.; Yu, Z. Diruthenium (II)–NNN pincer complex catalysts for transfer hydrogenation of ketones. Dalton Trans. 2016, 45, 17843–17849. [Google Scholar] [CrossRef]
- Yoshida, K.; Kamimura, T.; Kuwabara, H.; Yanagisawa, A. Chiral bicyclic NHC/Ir complexes for catalytic asymmetric transfer hydrogenation of ketones. Chem. Commun. 2015, 51, 15442–15445. [Google Scholar] [CrossRef]
- Volpe, A.; Baldino, S.; Tubaro, C.; Baratta, W.; Basato, M.; Graiff, C. Dinuclear Di (N-heterocyclic carbene) Iridium (III) Complexes as Catalysts in Transfer Hydrogenation. Eur. J. Inorg. Chem. 2016, 2016, 247–251. [Google Scholar] [CrossRef]
- Ashley, J.M.; Farnaby, J.H.; Hazari, N.; Kim, K.E.; Luzik, E.D., Jr.; Meehan, R.E.; Meyer, E.B.; Schley, N.D.; Schmeier, T.J.; Tailor, A.N. Axially chiral dimeric Ir and Rh complexes bridged by flexible NHC ligands. Inorg. Chim. Acta 2012, 380, 399–410. [Google Scholar] [CrossRef]
- Madern, N.; Talbi, B.; Salmain, M. Aqueous phase transfer hydrogenation of aryl ketones catalysed by achiral ruthenium (II) and rhodium (III) complexes and their papain conjugates. Appl. Organomet. Chem. 2013, 27, 6–12. [Google Scholar] [CrossRef]
- Dayan, O.; Çetinkaya, B. Mono-and binuclear ruthenium (II) complexes containing pyridine-2, 6-diimine (Pydim) ligands: Synthesis, characterization and catalytic activity in the transfer hydrogenation of acetophenone. J. Mol. Catal. A: Chem. 2007, 271, 134–141. [Google Scholar] [CrossRef]
- Nath, B.D.; Takaishi, K.; Ema, T. Macrocyclic multinuclear metal complexes acting as catalysts for organic synthesis. Catal. Sci. Technol. 2020, 10, 12–34. [Google Scholar] [CrossRef]
- Maity, A.; Sil, A.; Patra, S.K. Ruthenium (II) Complexes of 4′-(Aryl)-2, 2′: 6′, 2″-terpyridyl Ligands as Simple Catalysts for the Transfer Hydrogenation of Ketones. Eur. J. Inorg. Chem. 2018, 2018, 4063–4073. [Google Scholar] [CrossRef]
- Bratko, I.; Gomez, M. Polymetallic complexes linked to a single-frame ligand: Cooperative effects in catalysis. Dalton Trans. 2013, 42, 10664–10681. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wu, K.; Wang, L.; Fan, H.; Zhou, Y.-G.; Yu, Z. Assembled multinuclear ruthenium (II)–NNNN complexes: Synthesis, catalytic properties, and DFT calculations. Organometallics 2019, 39, 93–104. [Google Scholar] [CrossRef]
- Kumah, R.T.; Vijayan, P.; Ojwach, S.O. Carboxamide carbonyl-ruthenium (ii) complexes: Detailed structural and mechanistic studies in the transfer hydrogenation of ketones. New J. Chem. 2022. [Google Scholar] [CrossRef]
- Yadav, S.; Vijayan, P.; Yadav, S.; Gupta, R. Ruthenium complexes of phosphine–amide based ligands as efficient catalysts for transfer hydrogenation reactions. Dalton Trans. 2021, 50, 3269–3279. [Google Scholar] [CrossRef]
- Arafa, W.A. Sonochemical Preparation of Dipicolinamide Mn-complexes and Their Application as Catalysts Towards Sono-synthesis of Ketones. J. Heterocycl. Chem. 2019, 56, 1403–1412. [Google Scholar] [CrossRef]
- Meghdadi, S.; Mereiter, K.; Amirnasr, M.; Karimi, F.; Amiri, A. Synthesis, crystal structure and electrochemistry of cobalt (III) carboxamide complexes with amine and azide ancillary ligands. Polyhedron 2014, 68, 60–69. [Google Scholar] [CrossRef]
- Meghdadi, S.; Mereiter, K.; Shams Mohammadi, N.; Amiri, A. Synthesis, characterization and X-ray crystal structure of a cobalt (III) complex with 2-bis (pyridine-2-carboxamido)-4, 5-dimethylbenzene ligand. Inorg. Chem. Res. 2017, 1, 69–78. [Google Scholar]
- Aradhyula, B.P.R.; Kaminsky, W.; Kollipara, M.R. Half-sandwich d6 metal complexes with bis (pyridine carboxamide) benzene ligand: Synthesis and spectral analysis. J. Mol. Struct. 2017, 1149, 162–170. [Google Scholar] [CrossRef]
- Almodares, Z.; Lucas, S.J.; Crossley, B.D.; Basri, A.M.; Pask, C.M.; Hebden, A.J.; Phillips, R.M.; McGowan, P.C. Rhodium, iridium, and ruthenium half-sandwich picolinamide complexes as anticancer agents. Inorg. Chem. 2014, 53, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Meghdadi, S.; Amirnasr, M.; Kiani, M.; Fadaei Tirani, F.; Bagheri, M.; Schenk, K.J. Benign synthesis of quinolinecarboxamide ligands, H2bqbenzo and H2bqb and their Pd (II) complexes: X-ray crystal structure, electrochemical and antibacterial studies. J. Coord. Chem. 2017, 70, 2409–2424. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Botubol-Ares, J.M.; Cordón-Ouahhabi, S.; Moutaoukil, Z.; Collado, I.G.; Jiménez-Tenorio, M.; Puerta, M.C.; Valerga, P. Methylene-Linked Bis-NHC Half-Sandwich Ruthenium Complexes: Binding of Small Molecules and Catalysis toward Ketone Transfer Hydrogenation. Organometallics 2021, 40, 792–803. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Henderson, M.A.; Koivisto, B.D.; Osthoff, H.D.; Berlinguette, C.P. Electronic modification of the [RuII (tpy)(bpy)(OH2)] 2+ scaffold: Effects on catalytic water oxidation. J. Am. Chem. Soc. 2010, 132, 16094–16106. [Google Scholar] [CrossRef]
- Pospech, J.; Fleischer, I.; Franke, R.; Buchholz, S.; Beller, M. Alternative metals for homogeneous catalyzed hydroformylation reactions. Angew. Chem. Int. Ed. 2013, 52, 2852–2872. [Google Scholar] [CrossRef]
- Pachisia, S.; Kishan, R.; Yadav, S.; Gupta, R. Half-Sandwich Ruthenium Complexes of Amide-Phosphine Based Ligands: H-Bonding Cavity Assisted Binding and Reduction of Nitro-substrates. Inorg. Chem. 2021, 60, 2009–2022. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Vijayan, P.; Gupta, R. Ruthenium complexes of N/O/S based multidentate ligands: Structural diversities and catalysis perspectives. J. Organomet. Chem. 2021, 954, 122081. [Google Scholar] [CrossRef]
- Govindaswamy, P.; Canivet, J.m.; Therrien, B.; Süss-Fink, G.; Štěpnička, P.; Ludvík, J. Mono and dinuclear rhodium, iridium and ruthenium complexes containing chelating 2, 2′-bipyrimidine ligands: Synthesis, molecular structure, electrochemistry and catalytic properties. J. Organomet. Chem. 2007, 692, 3664–3675. [Google Scholar] [CrossRef]
- Viji, M.; Tyagi, N.; Naithani, N.; Ramaiah, D. Aryl appended neutral and cationic half-sandwich ruthenium (ii)–NHC complexes: Synthesis, characterisation and catalytic applications. New J. Chem. 2017, 41, 12736–12745. [Google Scholar] [CrossRef]
- Meriç, N.; Durap, F.; Aydemir, M.; Baysal, A. The application of tunable tridendate P-based ligands for the Ru (II)-catalysed transfer hydrogenation of various ketones. Appl. Organomet. Chem. 2014, 28, 803–808. [Google Scholar] [CrossRef]
- Raja, N.; Ramesh, R. Binuclear ruthenium (II) pyridazine complex catalyzed transfer hydrogenation of ketones. Tetrahedron Lett. 2012, 53, 4770–4774. [Google Scholar] [CrossRef]
- Martins, J.E.; Clarkson, G.J.; Wills, M. Ru (II) complexes of N-alkylated TsDPEN ligands in asymmetric transfer hydrogenation of ketones and imines. Org. Lett. 2009, 11, 847–850. [Google Scholar] [CrossRef]
- Wang, Q.; Chai, H.; Yu, Z. Dimeric ruthenium (II)-NNN complex catalysts bearing a pyrazolyl-pyridylamino-pyridine ligand for transfer hydrogenation of ketones and acceptorless dehydrogenation of alcohols. Organometallics 2017, 36, 3638–3644. [Google Scholar] [CrossRef]
- Armarego, W.L. Purification of Laboratory Chemicals. Butterworth-Heinemann: Oxford, UK, 2017. [Google Scholar]
- Fulmer, G.R.; Miller, A.J.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. Cs Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Karaca, E.Ö. The catalytic activity of new iridium(I) N-heterocyclic carbene complexes for hydrogen transfer reaction of ketones. Mon. Für Chem. Chem. Mon. 2021, 152, 287–293. [Google Scholar] [CrossRef]
- Bernier, C.M.; Merola, J.S. Design of Iridium N-Heterocyclic Carbene Amino Acid Catalysts for Asymmetric Transfer Hydrogenation of Aryl Ketones. Catalysts 2021, 11, 671. [Google Scholar] [CrossRef]
- Yoshida, M.; Hirahata, R.; Inoue, T.; Shimbayashi, T.; Fujita, K.-i. Iridium-catalyzed transfer hydrogenation of ketones and aldehydes using glucose as a sustainable hydrogen donor. Catalysts 2019, 9, 503. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Han, X.; Xu, J.; Liu, P.; Li, F. Transfer Hydrogenation of Ketones and Imines with Methanol under Base-Free Conditions Catalyzed by an Anionic Metal–Ligand Bifunctional Iridium Catalyst. J. Org. Chem. 2020, 85, 2242–2249. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Ru1 | Ru4 |
|---|---|---|
| Empirical formula | C48H54Cl4N4O2Ru3+[CH2Cl2] | C39H42ClF6N4O3PRu2 |
| Formula weight | 1418.74 | 997.32 |
| Temperature/K | 100 (2) | 100 (2) |
| Wavelength(Å) | 1.54178 | 1.54170 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P-1 | P21/c |
| Unit cell dimensions; | ||
| a | 13.121 (2)Å | 15.3868 (4) |
| b | 15.820 (3)Å | 19.3230 (5) |
| c | 16.506 (4)Å | 13.7638 (4) |
| α | 61.430 (6)◦ | 90 |
| β | 71.530 (11)◦ | 101.771 (1) |
| ɣ | 72.142 (7)◦ | 90 |
| Volume | 2803.9 (10)Å3 | 4006.18 (19) |
| Z | 2 | 4 |
| Density (calculated) / Mg/m3 | 1.680 | 1.654 |
| Absorption coefficient/ mm−1 | 11.186 | 7.713 |
| F(000) | 1424.0 | 200080 |
| Crystal Size (mm3) | 0.14 × 0.095 × 0.07 | 0.15 × 0.15 × 0.15 |
| Theta range for data collection | 71.961 | 68.225 |
| Reflections collected | 11036 | 7297 |
| Completeness | 97.6% | 99.4% |
| Refinement method | Full-matrix least-square on F2 | Full-matrix least-square on F2 |
| Goodness-of-fit on F2 | 1.076 | 1.057 |
| Final R indices [I>2sigma(I)] | R1 = 0.0511, wR2 = 0.1447 | R1 = 0.0278, wR2 = 0.0782 |
| R indices (all data) | R1 = 0.0567, wR2 = 0.1509 | R1 = 0.0282, wR2 = 0.0785 |
| Largest diff. peak and hole/eA−3 | 1.55/−2.57 | 1.42/−0.50 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst Loading/× 10−3 mol% (ppm) | Base | b Conversion[%] | Yield% | TON × 104 | TOF × 103/h−1 |
| 1 | - | KtBuO | 2 | 1 | - | - |
| 2 | 5.00 (50) | - | 32 | 30 | 0.64 | 1.07 |
| 3 | 2.50 (25) | KtBuO | 41 | 39 | 1.56 | 2.60 |
| 4 | 5.00 (50) | KtBuO | 86 | 85 | 1.72 | 2.87 |
| 5 | 15.00 (150) | KtBuO | 95 | 94 | 0.63 | 1.05 |
| 6 | 25.00 (250) | KtBuO | 96 | 95 | 0.26 | 0.43 |
| 7 | 55.00 (550) | KtBuO | 99 | 98 | 0.13 | 0.22 |
| 8 | 5.00 (50) | KOH | 76 | 76 | 1.71 | 2.85 |
| 9 | 5.00 (50) | K2CO3 | 29 | 28 | 0.58 | 0.97 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | b Conv. [%] | b Yield [%] | TON × 104 | TOF/h−1 x 103 | kobs × 10−1/h−1 |
| 1 | [RuCl2(p-cymene)2] | 12 | 11 | 0.02 | 0.03 | -- |
| 2 | Ru1 | 99 | 98 | 1.96 | 3.27 | 1.97 |
| 3 | Ru2 | 86 | 85 | 1.72 | 2.87 | 1.69 |
| 4 | Ru3 | 96 | 94 | 1.88 | 3.13 | 1.76 |
| 5 | Ru4 | 92 | 92 | 1.84 | 3.07 | 1.73 |
| Entry | Ketone | b Yield (%) | Entry | Ketone | b Yield% |
|---|---|---|---|---|---|
| 1 |  | 86 | 8 |  | 75 |
| 2 * |  | 99 | 9 |  | 78 |
| 3 * |  | 99 | 10 |  | 88 |
| 4 |  | 70 | 11 |  | 84 |
| 5 |  | 81 | 12 |  | 76 |
| 6 |  | 79 | 13 |  | 73 |
| 7 |  | 78 | 14 |  | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumah, R.T.; Mvelase, S.T.; Ojwach, S.O. Syntheses and Applications of Symmetrical Dinuclear Half-Sandwich Ruthenium(II)–Dipicolinamide Complexes as Catalysts in the Transfer Hydrogenation of Ketones. Inorganics 2022, 10, 190. https://doi.org/10.3390/inorganics10110190
Kumah RT, Mvelase ST, Ojwach SO. Syntheses and Applications of Symmetrical Dinuclear Half-Sandwich Ruthenium(II)–Dipicolinamide Complexes as Catalysts in the Transfer Hydrogenation of Ketones. Inorganics. 2022; 10(11):190. https://doi.org/10.3390/inorganics10110190
Chicago/Turabian StyleKumah, Robert Tettey, Sabathile Thandeka Mvelase, and Stephen Otieno Ojwach. 2022. "Syntheses and Applications of Symmetrical Dinuclear Half-Sandwich Ruthenium(II)–Dipicolinamide Complexes as Catalysts in the Transfer Hydrogenation of Ketones" Inorganics 10, no. 11: 190. https://doi.org/10.3390/inorganics10110190