
Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’s Disease


Abstract
:1. Introduction
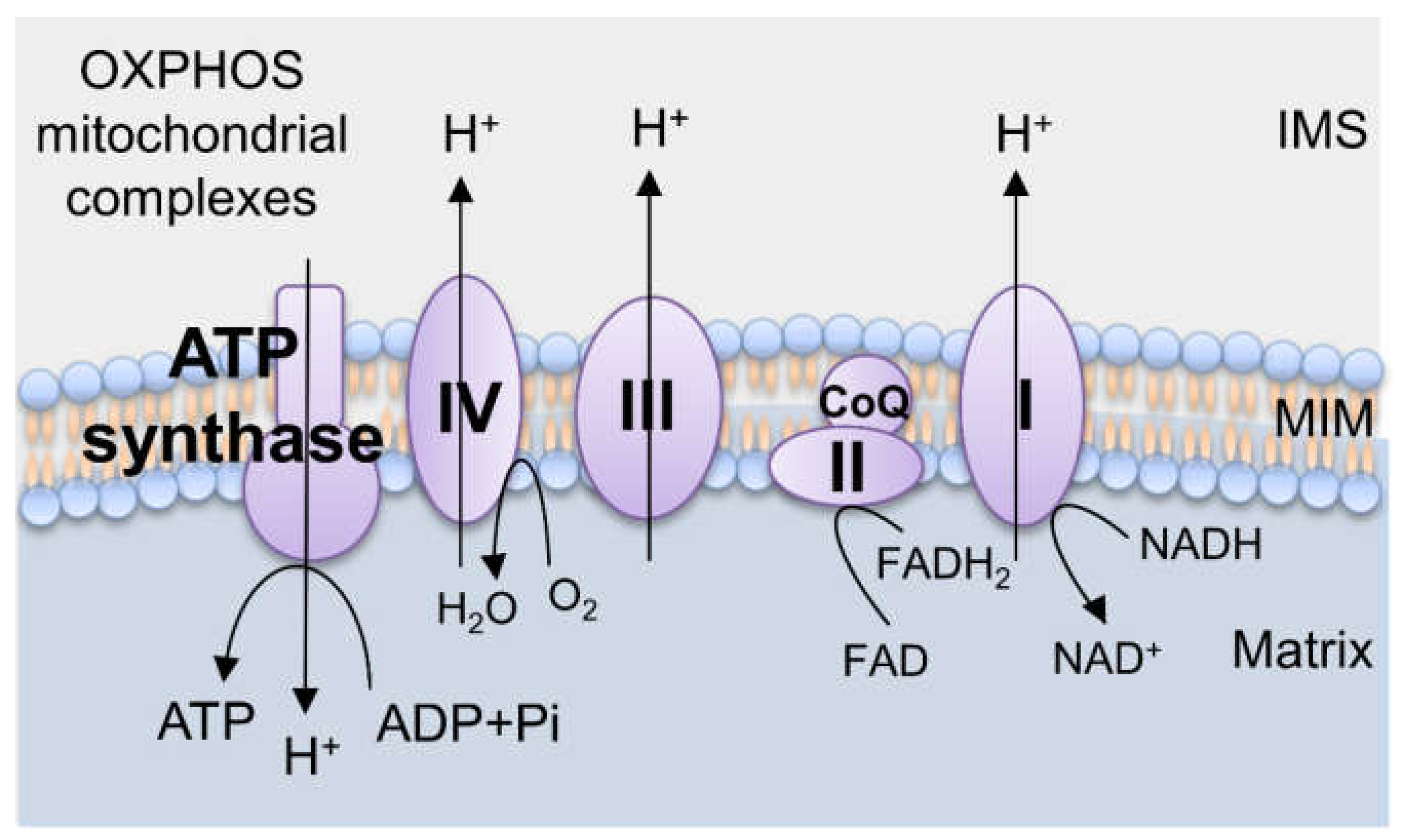
2. Mitochondrial Biology
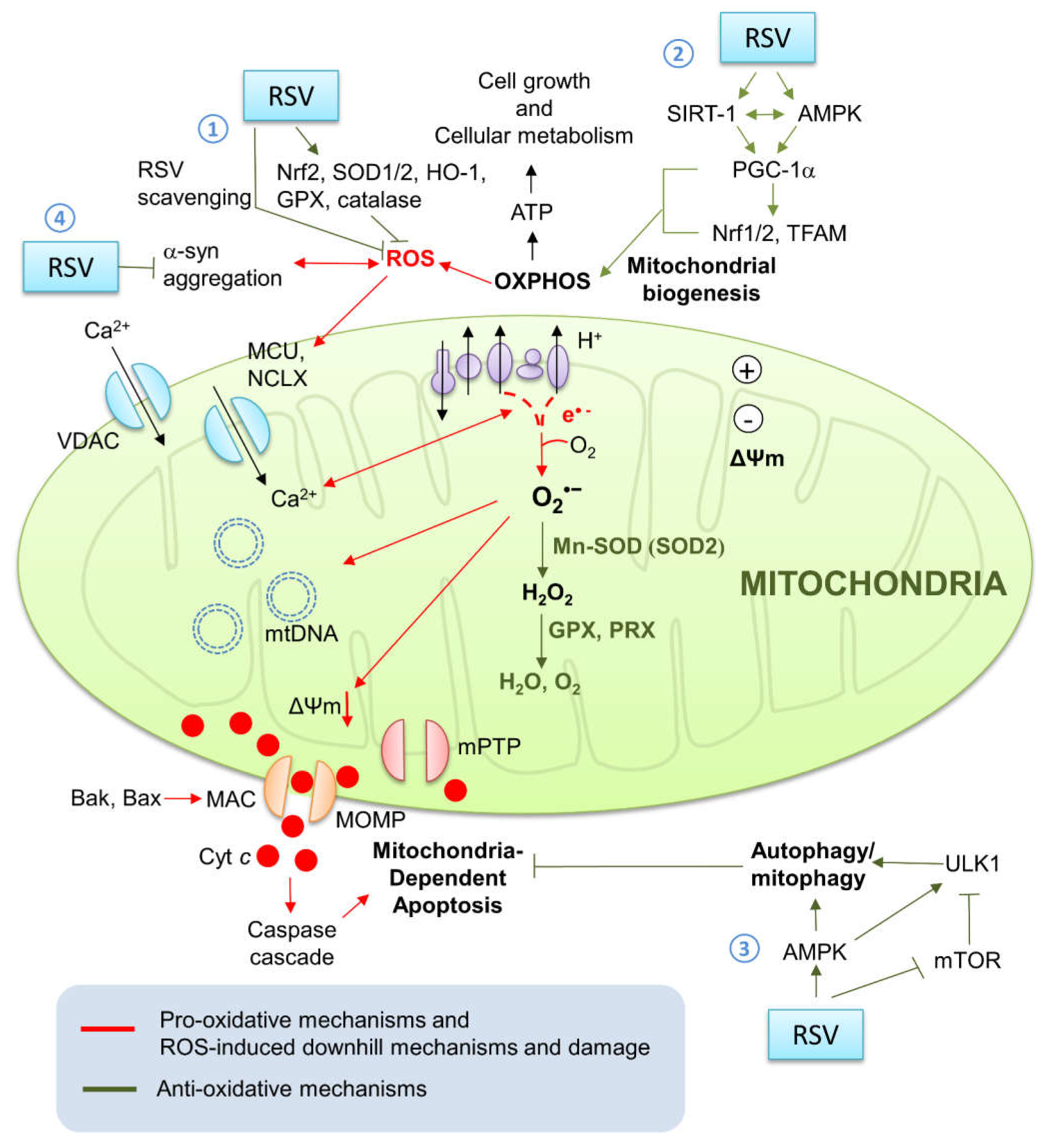
2.1. Mitochondria and ROS Generation
2.2. Mitochondrial Oxidative Stress and Antioxidative Systems
2.3. Oxidative Stress and Mitochondrial Dysfunction
2.4. Oxidative Stress and Cell Death
3. Parkinson’s Disease, Oxidative Stress, and Mitochondrial Dysfunction
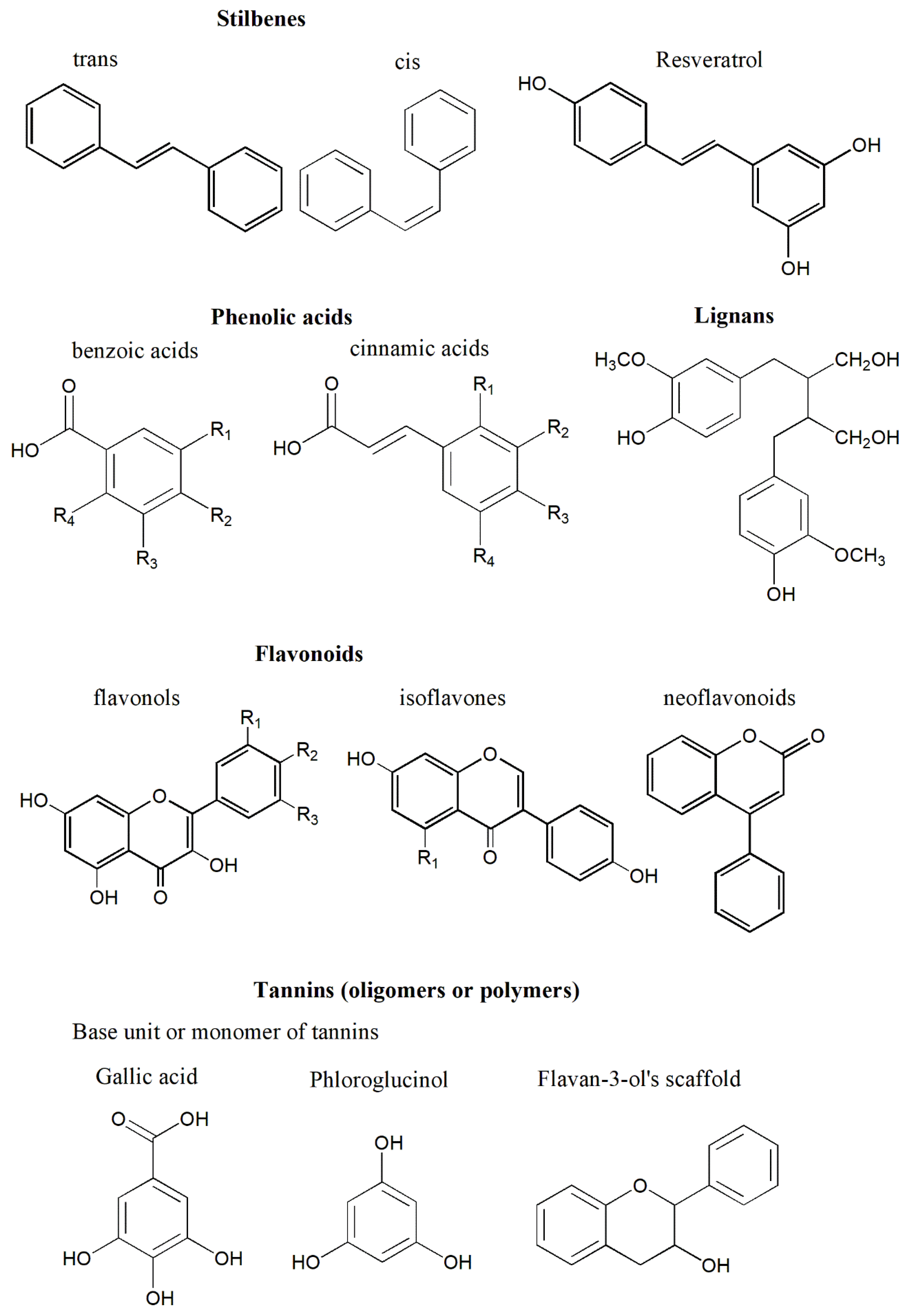
4. Polyphenols and Their Properties
4.1. Resveratrol’s Neuroprotective Effects against Parkinson’s Disease
4.2. Clinical Trials of Resveratrol on Neurodegenerative Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary
| AD | Alzheimer’s disease | MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| ADP | Adenosine diphosphate | mtDNA | Mitochondrial DNA |
| AMPK | 5′-adenosine monophosphate (AMP)-activated protein kinase | mTOR/mTORC1 | Mammalian target of rapamycin/ mammalian target of rapamycin complex I |
| APAF1 | Apoptotic peptidase-activating factor 1 | nDNA | Nucleus DNA |
| ATP | Adenosine triphosphate | NCLX | Na+/Ca2+ exchanger |
| ATP13A2 | Atpase type13a2 | NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| BAK | Bcl2-antagonist/killer | NOX | Nicotinamide adenine dinucleotide phosphate oxidase |
| BAX | Bcl-2-associated x protein | NRF | Nuclear respiratory factor |
| Ca2+ | Calcium | 6-OHDA | 6-hydroxydopamine |
| CoQ | Co-enzyme q | OM | Outer membrane |
| CR | Caloric restriction | OXPHOS | Oxidative phosphorylation |
| CVD | Cardiovascular diseases | PD | Parkinson’s disease |
| DJ-1 | Daisuke-junko-1 | PGC | Peroxisome proliferator-activated receptors (PPAR) γ coactivator |
| ER | Endoplasmic reticulum | PINK1 | Phosphatase and tensin homologue (PTEN)-induced putative kinase 1 |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 | POLG1 | Polymerase gamma 1 |
| ETC | Electric transport chain | PPAR | Peroxisome proliferator-activated receptor |
| FBXO7 | F-box only protein 7 | PRX | Peroxiredoxin |
| GPX | Gsh peroxidase | Redox | Reduction-oxidization |
| GR | Glutathione reductase | ROS | Reactive oxygen species |
| GSSG | Glutathione disulfide | rRNA | Ribosomal rna |
| GSH | Glutathione | SOD | Superoxide dismutase |
| IM | Inner membrane | (mt)SSB | (Mitochondrial) single-stranded binding protein |
| IMS | Intermembrane space | SIRT-1 | Sirtuin-1 |
| LRRK2 | Leucine rich repeat kinase 2 | SNpc | Substantia nigra pars compacta |
| MCU | Mitochondrial Ca2+ uniporter | TCA | Tricarboxylic acid |
| ΔΨm | Mitochondrial membrane potential | TFAM | Mitochondrial transcription factor A |
| MOA | Monoamine oxidase | TRX | Thioredoxin |
| MOMP | Mitochondrial outer membrane permeabilization | ULK1 | Unc-51 like autophagy activating kinase 1 |
| mPTP | Mitochondrial permeability transition pore | VPS35 | Vacuolar protein sorting 35 |
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Goetz, C.G. The history of Parkinson’s disease: Early clinical descriptions and neurological therapies. Cold Spring Harb. Perspect. Med. 2011, 1, a008862. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues e Silva, A.M.; Geldsetzer, F.; Holdorff, B.; Kielhorn, F.W.; Balzer-Geldsetzer, M.; Oertel, W.H.; Dodel, R. Who was the man who discovered the “Lewy bodies”? Mov. Disord. 2010, 25, 1765–1773. [Google Scholar] [CrossRef]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease with the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef]
- Nagatsua, T.; Sawadab, M. L-dopa therapy for Parkinson’s disease: Past, present, and future. Parkinsonism Relat. Disord. 2009, 15 (Suppl. S1), S3–S8. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 5, 229–238. [Google Scholar]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.J.; Bonifati, V. The genetics of Parkinson’s disease: Progress and therapeutic implications. Mov. Disord. 2013, 28, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Tan, L.; Yu, J.-T. The link between the SNCA gene and parkinsonism. Neurobiol. Aging 2015, 36, 1505–1518. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. S1), S32–S39. [Google Scholar] [CrossRef]
- Lin, K.-J.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Lin, H.-Y.; Lin, T.-K. The Overcrowded Crossroads: Mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [Green Version]
- Shadrina, M.I.; Slominsky, P.A.; Limborska, S.A. Molecular mechanisms of pathogenesis of Parkinson’s disease. Int. Rev. Cell Mol. Biol. 2010, 281, 229–266. [Google Scholar]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Mamelak, M. Parkinson’s Disease, the Dopaminergic Neuron and Gammahydroxybutyrate. Neurol. Ther. 2018, 7, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-K.; Cheng, C.-H.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C. Mitochondrial Dysfunction and Oxidative Stress Promote Apoptotic Cell Death in the Striatum via Cytochrome c/ Caspase-3 Signaling Cascade Following Chronic Rotenone Intoxication in Rats. Int. J. Mol. Sci. 2012, 13, 8722–8739. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Murray, C.J. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- de Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Koller, W.C. Environmental Risk Factors in Parkinson’s Disease. Neurology 1990, 40, 1218–1221. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Frey, T.G.; Mannella, C.A. The internal structure of mitochondria. Trends Biochem. Sci. 2000, 25, 319–324. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biochim. Biophys. Acta 2011, 1807, 1507–1538. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Pagadala, V.; Mueller, D.M. Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microb. Cell 2015, 2, 105–125. [Google Scholar] [CrossRef]
- Lippe, G.; Coluccino, G.; Zancani, M.; Baratta, W.; Crusiz, P. Mitochondrial F-ATP Synthase and Its Transition into an Energy-Dissipating Molecular Machine. Oxid. Med. Cell. Longev. 2019, 2019, 8743257. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2011, 35, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Boengler, K.; Heusch, G.; Schulz, R. Nuclear-encoded mitochondrial proteins and their role in cardioprotection. Biochim. Biophys. Acta 2011, 1813, 1286–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, Y.; Gao, K.; Yang, Q.; Shi, B.; Hou, P.; Ji, M. High copy number of mitochondrial DNA (mtDNA) predicts good prognosis in glioma patients. Am. J. Cancer Res. 2015, 5, 1207–1216. [Google Scholar]
- Fazzini, F.; Schöpf, B.; Blatzer, M.; Coassin, S.; Hicks, A.A.; Kronenberg, F.; Fendt, L. Plasmid-normalized quantification of relative mitochondrial DNA copy number. Sci. Rep. 2018, 8, 15347. [Google Scholar] [CrossRef]
- Hollensworth, S.B.; Shen, C.-C.; Sim, J.; Spitz, D.; Wilson, G.L.; LeDoux, S.P. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuča, K.; Musilek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef]
- De Lazzari, F.; Bubacco, L.; Whitworth, A.J.; Bisaglia, M. Superoxide Radical Dismutation as New Therapeutic Strategy in Parkinson’s Disease. Aging Dis. 2018, 9, 716–728. [Google Scholar] [CrossRef] [Green Version]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent Anion Channels Control the Release of the Superoxide Anion from Mitochondria to Cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef] [Green Version]
- Rindler, P.M.; Cacciola, A.; Kinter, M.; Szweda, L.I. Catalase-dependent H2O2consumption by cardiac mitochondria and redox-mediated loss in insulin signaling. Am. J. Physiol. Circ. Physiol. 2016, 311, H1091–H1096. [Google Scholar] [CrossRef]
- Slade, L.; Chalker, J.; Kuksal, N.; Young, A.; Gardiner, D.; Mailloux, R.J. Examination of the superoxide/hydrogen peroxide forming and quenching potential of mouse liver mitochondria. Biochim. Biophys. Acta 2017, 1861, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxid. Med. Cell. Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Seefeldt, T.; Chen, W.; Wang, X.; Matthees, D.; Hu, Y.; Guan, X. Effects of glutathione reductase inhibition on cellular thiol redox state and related systems. Arch. Biochem. Biophys. 2009, 485, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, Z.A.; Schröder, E.; Harris, J.R.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Hensley, K.; Robinson, K.; Gabbita, S.; Salsman, S.; Floyd, R. Reactive oxygen species, cell signaling, and cell injury. Free. Radic. Biol. Med. 2000, 28, 1456–1462. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- O’Dea, E.; Hoffmann, A. NF-κB signaling. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 107–115. [Google Scholar] [CrossRef]
- Schmidt, K.N.; Amstad, P.; Cerutti, P.; Baeuerle, P.A. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-κ B. Chem. Biol. 1995, 2, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 2019, 8, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, H.; Samuels, D.; Eden, J.A.; Relton, C.; Chinnery, P.F. Pathogenic Mitochondrial DNA Mutations Are Common in the General Population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefano, G.B.; Kream, R.M. Mitochondrial DNA heteroplasmy in human health and disease. Biomed. Rep. 2016, 4, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c Association with the Inner Mitochondrial Membrane Is Impaired in the CNS of G93A-SOD1 Mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Demirel, Y.A.; Gerbaud, V. Nonequilibrium Thermodynamics: Transport and Rate Processes in Physical, Chemical and Biological Systems, 4th ed.; Elsevier: Cambridge, MA, USA, 2019; Volume XXVI, p. 854. [Google Scholar]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Stewart, V.C.; Heales, S.J. Nitric oxide-induced mitochondrial dysfunction: Implications for neurodegeneration. Free Radic Biol. Med. 2003, 34, 287–303. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Castilho, R.; Vercesi, A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001, 495, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Pinton, P. The Mitochondrial Permeability Transition Pore and Cancer: Molecular Mechanisms Involved in Cell Death. Front. Oncol. 2014, 4, 302. [Google Scholar] [CrossRef] [Green Version]
- Šileikytė, J.; Forte, M. The Mitochondrial Permeability Transition in Mitochondrial Disorders. Oxid. Med. Cell. Longev. 2019, 2019, 3403075. [Google Scholar] [CrossRef] [Green Version]
- Hoek, J.; Rydström, J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem. J. 1988, 254, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, C.F.; Meyer-Fernandes, J.; Bassères, D.; Castilho, R.; Vercesi, A.E. Ca2+-dependent permeabilization of the inner mitochondrial membrane by 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS). Biochim. Biophys. Acta 1994, 1188, 93–100. [Google Scholar] [CrossRef]
- Lenartowicz, E.; Bernardi, P.; Azzone, G.F. Phenylarsine oxide induces the cyclosporin A-sensitive membrane permeability transition in rat liver mitochondria. J. Bioenerg. Biomembr. 1991, 23, 679–688. [Google Scholar] [CrossRef]
- Vercesi, A.; Ferraz, V.; Macedo, D.; Fiskum, G. Ca2+-dependent NAD(P)+-induced alterations of rat liver and hepatoma mitochondrial membrane permeability. Biochem. Biophys. Res. Commun. 1988, 154, 934–941. [Google Scholar] [CrossRef]
- Valle, V.; Fagian, M.; Parentoni, L.; Meinicke, A.; Vercesi, A. The Participation of Reactive Oxygen Species and Protein Thiols in the Mechanism of Mitochondrial Inner Membrane Permeabilization by Calcium plus Prooxidants. Arch. Biochem. Biophys. 1993, 307, 1–7. [Google Scholar] [CrossRef]
- Beal, M. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000, 23, 298–304. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Karlstad, J.; Sun, Y.; Singh, B.B. Ca2+ Signaling: An Outlook on the Characterization of Ca2+ Channels and Their Importance in Cellular Functions. Adv. Exp. Med. Biol. 2012, 740, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Hansford, R.G. Physiological role of mitochondrial Ca2+ transport. J. Bioenerg. Biomembr. 1994, 26, 495–508. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Várnai, P.; Golenár, T.; Sheu, S.-S.; Hajnóczky, G. Calcium transport across the inner mitochondrial membrane: Molecular mechanisms and pharmacology. Mol. Cell. Endocrinol. 2012, 353, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, D.; Sancak, Y.; Mootha, V.K.; Clapham, D. MCU encodes the pore conducting mitochondrial calcium currents. eLife 2013, 2, e00704. [Google Scholar] [CrossRef]
- Bernardi, P. Mitochondrial Transport of Cations: Channels, Exchangers, and Permeability Transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 2014, 56, 437–445. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2019. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Kögel, D. Autophagy in Cancer Cell Death. Biology 2019, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Syntichaki, P.; Tavernarakis, N. Death by necrosis. Uncontrollable catastrophe, or is there order behind the chaos? EMBO Rep. 2002, 3, 604–609. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Dewson, G. Mitochondria and apoptosis: Emerging concepts. F1000Prime Rep. 2015, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Pathak, T.; Trebak, M. Mitochondrial Ca(2+) signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef]
- Contreras, L.; Drago, I.; Zampese, E.; Pozzan, T. Mitochondria: The calcium connection. Biochim. Biophys. Acta 2010, 1797, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Roy, S.S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.; Zhivotovsky, B.; Orrenius, S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007, 14, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, E.K.; Wallace, C.J.; Kinnunen, P.K. Phospholipid-cytochrome c interaction: Evidence for the extended lipid anchorage. J. Biol. Chem. 2002, 277, 8822–8826. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.K.; Liou, C.W.; Chen, S.D.; Chuang, Y.C.; Tiao, M.M.; Wang, P.W.; Chuang, J.H. Mitochondrial dysfunction and biogenesis in the pathogenesis of Parkinson’s disease. Chang Gung Med. J. 2009, 32, 589–599. [Google Scholar] [PubMed]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, L.; Silva, M.V.; Falcão, A.; Soares, E.; Costa, R.; Loureiro, A.I.; Fernandes-Lopes, C.; Rocha, J.-F.; Nunes, T.; Wright, L.; et al. Pharmacokinetic and safety profile of trans-resveratrol in a rising multiple-dose study in healthy volunteers. Mol. Nutr. Food Res. 2009, 53, S7–S15. [Google Scholar] [CrossRef]
- Kopin, I.J. Features of the Dopaminergic Neurotoxin MPTP. Ann. N. Y. Acad. Sci. 1992, 648, 96–104. [Google Scholar] [CrossRef]
- Ransom, B.R.; Kunis, D.M.; Irwin, I.; Langston, J. Astrocytes convert the parkinsonism inducing neurotoxin, MPTP, to its active metabolite, MPP+. Neurosci. Lett. 1987, 75, 323–328. [Google Scholar] [CrossRef]
- Sriram, K.; Pai, K.S.; Boyd, M.R.; Ravindranath, V. Evidence for generation of oxidative stress in brain by MPTP: In vitro and in vivo studies in mice. Brain Res. 1997, 749, 44–52. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Marsden, C.D. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherer, T.B.; Betarbet, R.; Greenamyre, J.T. Environment, Mitochondria, and Parkinson’s Disease. Neuroscience 2002, 8, 192–197. [Google Scholar] [CrossRef]
- Chen, T.; Tan, J.; Wan, Z.; Zou, Y.; Afewerky, H.K.; Zhang, Z.; Zhang, T. Effects of Commonly Used Pesticides in China on the Mitochondria and Ubiquitin-Proteasome System in Parkinson’s Disease. Int. J. Mol. Sci. 2017, 18, 2507. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.; Casson, R.J.; Chidlow, G.; Wood, J.P. The mitochondrial complex I inhibitor rotenone induces endoplasmic reticulum stress and activation of GSK-3beta in cultured rat retinal cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5616–5628. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Nalls, M.A. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeve, A.; Krishnan, K.J.; Turnbull, D. Mitochondrial DNA Mutations in Disease, Aging, and Neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jimenez, R.; Lurette, O.; Hebert-Chatelain, E. Damage in Mitochondrial DNA Associated with Parkinson’s Disease. DNA Cell Biol. 2020, 39, 1421–1430. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct Membrane Association Drives Mitochondrial Fission by the Parkinson Disease-associated Protein α-Synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullin, S.; Schapira, A. α-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Kamienieva, I.; Duszyński, J.; Szczepanowska, J. Multitasking guardian of mitochondrial quality: Parkin function and Parkinson’s disease. Transl. Neurodegener. 2021, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contu, V.R.; Kotake, Y.; Toyama, T.; Okuda, K.; Miyara, M.; Sakamoto, S.; Ohta, S. Endogenous neurotoxic dopamine derivative covalently binds to Parkinson’s disease-associated ubiquitin C-terminal hydrolase L1 and alters its structure and function. J. Neurochem. 2014, 130, 826–838. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fallon, L.; Lashuel, H.A.; Liu, Z.; Lansbury, P.T., Jr. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 2002, 111, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Park. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and mitochondria: Recent advances and current views. Brain Res. 2019, 1702, 96–104. [Google Scholar] [CrossRef]
- Singh, F.; Ganley, I.G. Parkinson’s disease and mitophagy: An emerging role for LRRK2. Biochem. Soc. Trans. 2021, 49, 551–562. [Google Scholar] [CrossRef]
- Tsunemi, T.; Krainc, D. Zn²⁺ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 2013, 23, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Koentjoro, B.; Davis, R.L.; Sue, C.M. Loss of ATP13A2 impairs glycolytic function in Kufor-Rakeb syndrome patient-derived cell models. Park. Relat. Disord. 2016, 27, 67–73. [Google Scholar] [CrossRef]
- Park, J.S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Münchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, 1843.e1–1843.e7. [Google Scholar] [CrossRef]
- Park, J.S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef]
- Li, B.; Hu, Q.; Wang, H.; Man, N.; Ren, H.; Wen, L.; Nukina, N.; Fei, E.; Wang, G. Omi/HtrA2 is a positive regulator of autophagy that facilitates the degradation of mutant proteins involved in neurodegenerative diseases. Cell Death Differ. 2010, 17, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Krüger, R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef] [Green Version]
- Martins, L.M.; Morrison, A.; Klupsch, K.; Fedele, V.; Moisoi, N.; Teismann, P.; Abuin, A.; Grau, E.; Geppert, M.; Livi, G.P.; et al. Neuroprotective Role of the Reaper-Related Serine Protease HtrA2/Omi Revealed by Targeted Deletion in Mice. Mol. Cell. Biol. 2004, 24, 9848–9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N.; Kirber, M.T.; Mostoslavsky, G.; Ikezu, T.; Wolozin, B.; et al. Impairment of PARK14-dependent Ca2+ signalling is a novel determinant of Parkinson’s disease. Nat. Commun. 2016, 7, 10332. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, V.; Palermo, G.; Del Prete, E.; Mancuso, M.; Ceravolo, R. Understanding the Multiple Role of Mitochondria in Parkinson’s Disease and Related Disorders: Lesson from Genetics and Protein–Interaction Network. Front. Cell Dev. Biol. 2021, 9, 636506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Lee, J.C.T.; Tan, E.K. Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat. Res. Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.-L.; Liu, W.; Hu, J.-X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.-C. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef]
- Edvardson, S.; Cinnamon, Y.; Ta-Shma, A.; Shaag, A.; Yim, Y.-I.; Zenvirt, S.; Jalas, C.; Lesage, S.; Brice, A.; Taraboulos, A.; et al. A Deleterious Mutation in DNAJC6 Encoding the Neuronal-Specific Clathrin-Uncoating Co-Chaperone Auxilin, Is Associated with Juvenile Parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amodio, G.; Moltedo, O.; Fasano, D.; Zerillo, L.; Oliveti, M.; Di Pietro, P.; Faraonio, R.; Barone, P.; Pellecchia, M.T.; De Rosa, A.; et al. PERK-Mediated Unfolded Protein Response Activation and Oxidative Stress in PARK20 Fibroblasts. Front. Neurosci. 2019, 13, 673. [Google Scholar] [CrossRef] [Green Version]
- Vilariño-Güell, C.; Rajput, A.; Milnerwood, A.J.; Shah, B.; Szu-Tu, C.; Trinh, J.; Yu, I.; Encarnacion, M.; Munsie, L.N.; Tapia, L.; et al. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 2014, 23, 1794–1801. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Hasegawa, T.; Suzuki, M.; Sugeno, N.; Kobayashi, J.; Ueyama, M.; Fukuda, M.; Ido-Fujibayashi, A.; Sekiguchi, K.; Ezura, M.; et al. Parkinson’s disease-linked DNAJC13 mutation aggravates alpha-synuclein-induced neurotoxicity through perturbation of endosomal trafficking. Hum. Mol. Genet. 2018, 27, 823–836. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, G. Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Hüttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [Green Version]
- Tsao, R. Chemistry and Biochemistry of Dietary Polyphenols. Nutrients 2010, 2, 1231–1246. [Google Scholar] [CrossRef]
- Brglez Mojzer, E.; Knez Hrnčič, M.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction Methods, Antioxidative Action, Bioavailability and Anticarcinogenic Effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef]
- Marín, L.; Miguélez, E.M.; Villar, C.J.; Lombó, F. Bioavailability of Dietary Polyphenols and Gut Microbiota Metabolism: Antimicrobial Properties. BioMed Res. Int. 2015, 2015, 905215. [Google Scholar] [CrossRef] [Green Version]
- Li, A.-N.; Li, S.; Zhang, Y.-J.; Xu, X.-R.; Chen, Y.-M.; Li, H.-B. Resources and Biological Activities of Natural Polyphenols. Nutrients 2014, 6, 6020–6047. [Google Scholar] [CrossRef]
- Vauzour, D.; Rodriguez-Mateos, A.; Corona, G.; Oruna-Concha, M.J.; Spencer, J.P.E. Polyphenols and Human Health: Prevention of Disease and Mechanisms of Action. Nutrients 2010, 2, 1106–1131. [Google Scholar] [CrossRef] [Green Version]
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 2018, 5, 87. [Google Scholar] [CrossRef] [Green Version]
- Williamson, G. The role of polyphenols in modern nutrition. Nutr. Bull. 2017, 42, 226–235. [Google Scholar] [CrossRef]
- Franceschi, S.; Parpinel, M.; La Vecchia, C.; Favero, A.; Talamini, R.; Negri, E. Role of Different Types of Vegetables and Fruit in the Prevention of Cancer of the Colon, Rectum, and Breast. Epidemiology 1998, 9, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Van Erk, M.J.; Roepman, P.; van der Lende, T.R.; Stierum, R.H.; Aarts, G.; van Bladeren, P.J.; van Ommen, B. Integrated assessment by multiple gene expression analysis of quercetin bioactivity on anticancer-related mechanisms in colon cancer cells in vitro. Eur. J. Nutr. 2005, 44, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Plaumann, B.; Fritsche, M.; Rimpler, H.; Brandner, G.; Hess, R.D. Flavonoids activate wild-type p53. Oncogene 1996, 13, 1605–1614. [Google Scholar] [PubMed]
- Owen, R.; Giacosa, A.; Hull, W.; Haubner, R.; Spiegelhalder, B.; Bartsch, H. The antioxidant/anticancer potential of phenolic compounds isolated from olive oil. Eur. J. Cancer 2000, 36, 1235–1247. [Google Scholar] [CrossRef]
- Fini, L.; Hotchkiss, E.; Fogliano, V.; Graziani, G.; Romano, M.; De Vol, E.B.; Qin, H.; Selgrad, M.; Boland, C.R.; Ricciardiello, L. Chemopreventive properties of pinoresinol-rich olive oil involve a selective activation of the ATM–p53 cascade in colon cancer cell lines. Carcinogenesis 2008, 29, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Quiñones, M.; Miguel, M.; Aleixandre, A. Beneficial effects of polyphenols on cardiovascular disease. Pharmacol. Res. 2013, 68, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Peters, U.; Poole, C.; Arab, L. Does Tea Affect Cardiovascular Disease? A Meta-Analysis. Am. J. Epidemiol. 2001, 154, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Di Castelnuovo, A.; Rotondo, S.; Iacoviello, L.; Donati, M.B.; De Gaetano, G. Meta-Analysis of Wine and Beer Consumption in Relation to Vascular Risk. Circulation 2002, 105, 2836–2844. [Google Scholar] [CrossRef] [Green Version]
- Taubert, D.; Roesen, R.; Lehmann, C.; Jung, N.; Schömig, E. Effects of low habitual cocoa intake on blood pressure and bioactive nitric oxide: A randomized controlled trial. JAMA 2007, 298, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, J.; Keevil, J.G.; Wiebe, D.A.; Aeschlimann, S.; Folts, J.D. Purple Grape Juice Improves Endothelial Function and Reduces the Susceptibility of LDL Cholesterol to Oxidation in Patients with Coronary Artery Disease. Circulation 1999, 100, 1050–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, C.; Dejam, A.; Kleinbongard, P.; Schewe, T.; Sies, H.; Kelm, M. Vascular Effects of Cocoa Rich in Flavan-3-ols. JAMA 2003, 290, 1030–1031. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Borenstein, A.R.; Wu, Y.; Jackson, J.C.; Larson, E.B. Fruit and Vegetable Juices and Alzheimer’s Disease: The Kame Project. Am. J. Med. 2006, 119, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checkoway, H.; Powers, K.; Smith-Weller, T.; Franklin, G.M.; Longstreth, W.T.; Swanson, P.D. Parkinson’s disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am. J. Epidemiol. 2002, 155, 732–738. [Google Scholar] [CrossRef]
- Youdim, K.; Qaiser, M.; Begley, D.J.; Rice-Evans, C.; Abbott, N. Flavonoid permeability across an in situ model of the blood–brain barrier. Free Radic Biol. Med. 2004, 36, 592–604. [Google Scholar] [CrossRef]
- Atawodi, S.E.; Atawodi, J.C.; Idakwo, G.A.; Pfundstein, B.; Haubner, R.; Wurtele, G.; Bartsch, H.; Owen, R.W. Evaluation of the Polyphenol Content and Antioxidant Properties of Methanol Extracts of the Leaves, Stem, and Root Barks of Moringa oleiferaLam. J. Med. Food 2010, 13, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Gorzynik-Debicka, M.; Przychodzen, P.; Cappello, F.; Kuban-Jankowska, A.; Marino Gammazza, A.; Knap, N.; Wozniak, M.; Gorska-Ponikowska, M. Potential Health Benefits of Olive Oil and Plant Polyphenols. Int. J. Mol. Sci. 2018, 19, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perron, N.R.; Brumaghim, J.L. A Review of the Antioxidant Mechanisms of Polyphenol Compounds Related to Iron Binding. Cell Biochem. Biophys. 2009, 53, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Pietta, P.-G. Flavonoids as Antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Cherrak, S.A.; Mokhtari-Soulimane, N.; Berroukeche, F.; Bensenane, B.; Cherbonnel, A.; Merzouk, H.; Elhabiri, M. In Vitro Antioxidant versus Metal Ion Chelating Properties of Flavonoids: A Structure-Activity Investigation. PLoS ONE 2016, 11, e0165575. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.A.; Zucca, P.; Varoni, E. Plant-Derived Bioactives and Oxidative Stress-Related Disorders: A Key Trend towards Healthy Aging and Longevity Promotion. Appl. Sci. 2020, 10, 947. [Google Scholar] [CrossRef] [Green Version]
- Hasima, N.; Ozpolat, B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014, 5, e1509. [Google Scholar] [CrossRef] [Green Version]
- Kiruthiga, C.D.; Nabavi, S.M.; Bishayee, M. Autophagy: A Potential Therapeutic Target of Polyphenols in Hepatocellular Carcinoma. Cancers 2020, 12, 562. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.F.; Sureda, A.; Dehpour, A.R.; Shirooie, S.; Silva, A.S.; Devi, K.P.; Ahmed, T.; Ishaq, N.; Hashim, R.; Sobarzo-Sánchez, E.; et al. Regulation of autophagy by polyphenols: Paving the road for treatment of neurodegeneration. Biotechnol. Adv. 2018, 36, 1768–1778. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Gambini, J.; Inglés, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Borras, C. Properties of Resveratrol: In Vitro and In Vivo Studies about Metabolism, Bioavailability, and Biological Effects in Animal Models and Humans. Oxid. Med. Cell Longev. 2015, 2015, 837042. [Google Scholar] [CrossRef] [Green Version]
- Jeandet, P.; Douillet-Breuil, A.-C.; Bessis, R.; Debord, S.; Sbaghi, M.; Adrian, M. Phytoalexins from the Vitaceae: Biosynthesis, Phytoalexin Gene Expression in Transgenic Plants, Antifungal Activity, and Metabolism. J. Agric. Food Chem. 2002, 50, 2731–2741. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Choi, H.J.; Zhu, B.T. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic Biol. Med. 2010, 49, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Belguendouz, L.; Fremont, L.; Linard, A. Resveratrol inhibits metal ion-dependent and independent peroxidation of porcine low-density lipoproteins. Biochem. Pharmacol. 1997, 53, 1347–1355. [Google Scholar] [CrossRef]
- Leonard, S.S.; Xia, C.; Jiang, B.-H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem. Biophys. Res. Commun. 2003, 309, 1017–1026. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High Absorption but Very Low Bioavailability of Oral Resveratrol in Humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, S.; Ramalho, M.J.; Pereira, M.D.C.; Loureiro, J.A. Resveratrol Brain Delivery for Neurological Disorders Prevention and Treatment. Front. Pharmacol. 2018, 9, 1261. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-C.; Tsai, H.-I.; Yu, H.-P. Organ-Protective Effects of Red Wine Extract, Resveratrol, in Oxidative Stress-Mediated Reperfusion Injury. Oxid. Med. Cell. Longev. 2015, 2015, 568634. [Google Scholar] [CrossRef]
- Li, Y.; Cao, Z.; Zhu, H. Upregulation of endogenous antioxidants and phase 2 enzymes by the red wine polyphenol, resveratrol in cultured aortic smooth muscle cells leads to cytoprotection against oxidative and electrophilic stress. Pharmacol. Res. 2006, 53, 6–15. [Google Scholar] [CrossRef]
- Park, D.-W.; Baek, K.; Kim, J.-R.; Lee, J.-J.; Ryu, S.-H.; Chin, B.-R.; Baek, S.-H. Resveratrol inhibits foam cell formation via NADPH oxidase 1-mediated reactive oxygen species and monocyte chemotactic protein-1. Exp. Mol. Med. 2009, 41, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Rege, S.; Kumar, S.; Wilson, D.N.; Tamura, L.; Geetha, T.; Mathews, S.T.; Huggins, K.W.; Broderick, T.L.; Babu, J.R. Resveratrol Protects the Brain of Obese Mice from Oxidative Damage. Oxid. Med. Cell. Longev. 2013, 2013, 419092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadi, G.; Konat, D. Resveratrol regulates oxidative biomarkers and antioxidant enzymes in the brain of streptozotocin-induced diabetic rats. Pharm. Biol. 2015, 54, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanier, G.; Xu, H.; Xia, N.; Tobias, S.; Deng, S.; Wojnowski, L.; Forstermann, U.; Li, H. Resveratrol reduces endothelial oxidative stress by modulating the gene expression of superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1) and NADPH oxidase subunit (Nox4). J. Physiol. Pharmacol. 2009, 60, 111–116. [Google Scholar]
- Zini, R.; Morin, C.; Bertelli, A.; Tillement, J.-P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar]
- Lin, T.-K.; Chen, S.-D.; Chuang, Y.-C.; Lin, H.-Y.; Wang, P.-W.; Huang, S.-T.; Tiao, M.-M.; Chen, J.-B.; Liou, C.-W. Resveratrol Partially Prevents Rotenone-Induced Neurotoxicity in Dopaminergic SH-SY5Y Cells through Induction of Heme Oxygenase-1 Dependent Autophagy. Int. J. Mol. Sci. 2014, 15, 1625–1646. [Google Scholar] [CrossRef]
- Sakata, Y.; Zhuang, H.; Kwansa, H.; Koehler, R.C.; Doré, S. Resveratrol protects against experimental stroke: Putative neuroprotective role of heme oxygenase 1. Exp. Neurol. 2010, 224, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Bastianetto, S.; Quirion, R. Heme oxygenase 1: Another possible target to explain the neuroprotective action of resveratrol, a multifaceted nutrient-based molecule. Exp. Neurol. 2010, 225, 237–239. [Google Scholar] [CrossRef]
- Wang, N.; He, J.; Pan, C.; Wang, J.; Ma, M.; Shi, X.; Xu, Z. Resveratrol Activates Autophagy via the AKT/mTOR Signaling Pathway to Improve Cognitive Dysfunction in Rats with Chronic Cerebral Hypoperfusion. Front. Neurosci. 2019, 13, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, X.; Chen, N. Resveratrol as a Natural Autophagy Regulator for Prevention and Treatment of Alzheimer’s Disease. Nutrients 2017, 9, 927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.-G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Polzella, M.; Frati, A.; Fornai, F. Phytochemicals Bridging Autophagy Induction and Alpha-Synuclein Degradation in Parkinsonism. Int. J. Mol. Sci. 2019, 20, 3274. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, srep45465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-L.; Yi, L.; Jin, X.; Liang, X.-Y.; Zhou, Y.; Zhang, T.; Xie, Q.; Zhou, X.; Chang, H.; Fu, Y.-J.; et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 2013, 9, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Mi, M. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS ONE 2014, 9, e102535. [Google Scholar]
- Holczer, M.; Hajdú, B.; Lőrincz, T.; Szarka, A.; Bánhegyi, G.; Kapuy, O. A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress. Int. J. Mol. Sci. 2019, 20, 5543. [Google Scholar] [CrossRef] [Green Version]
- Pineda-Ramírez, N.; Burgos, I.M.A.; Ortiz-Plata, A.; Ruiz-Tachiquín, M.-E.; Espinoza-Rojo, M.; Aguilera, P. Resveratrol Activates Neuronal Autophagy Through AMPK in the Ischemic Brain. Mol. Neurobiol. 2019, 57, 1055–1069. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, B.; Milbrandt, J. Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Tao, Y.; Zhang, J.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Zhao, J.; Wu, Q.; et al. Resveratrol Regulates Mitochondrial Biogenesis and Fission/Fusion to Attenuate Rotenone-Induced Neurotoxicity. Oxid. Med. Cell. Longev. 2016, 2016, 6705621. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Sonntag, W.; de Cabo, R.; Baur, J.; Csiszar, A. Mitochondrial Protection by Resveratrol. Exerc. Sport Sci. Rev. 2011, 39, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, A.; Labinskyy, N.; Pinto, J.T.; Ballabh, P.; Zhang, H.; Losonczy, G.; Pearson, K.J.; de Cabo, R.; Pacher, P.; Zhang, C.; et al. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am. J. Physiol. Circ. Physiol. 2009, 297, H13–H20. [Google Scholar] [CrossRef] [Green Version]
- Chuang, Y.-C.; Chen, S.-D.; Hsu, C.-Y.; Chen, S.-F.; Chen, N.-C.; Jou, S.-B. Resveratrol Promotes Mitochondrial Biogenesis and Protects against Seizure-Induced Neuronal Cell Damage in the Hippocampus Following Status Epilepticus by Activation of the PGC-1α Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 998. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.L.; Lin, K.J.; Wang, P.W.; Chuang, J.H.; Lin, H.Y.; Chen, S.D.; Lin, T.K. Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy. Free Radic Res. 2018, 52, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-Activated AMPK/SIRT1/Autophagy in Cellular Models of Parkinson’s Disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Arbo, B.D.; André-Miral, C.; Nasre-Nasser, R.G.; Schimith, L.E.; Santos, M.G.; Costa-Silva, D.; Muccillo-Baisch, A.L.; Hort, M.A. Resveratrol Derivatives as Potential Treatments for Alzheimer’s and Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Bastianetto, S.; Ménard, C.; Quirion, R. Neuroprotective action of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Albani, D.; Polito, L.; Batelli, S.; De Mauro, S.; Fracasso, C.; Martelli, G.; Forloni, G. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1–42) peptide. J. Neurochem. 2009, 110, 1445–1456. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, T.; Dong, S.-Y.; Guo, Y.-J.; Jankovic, J.; Xu, H.; Wu, Y.-C. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J. Neurochem. 2015, 134, 668–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.-J.; Dong, S.-Y.; Cui, X.-X.; Feng, Y.; Liu, T.; Yin, M.; Kuo, S.-H.; Tan, E.-K.; Zhao, W.-J.; Wu, Y.-C. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of α-synuclein via SIRT1-deacetylated LC3. Mol. Nutr. Food Res. 2016, 60, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Dunlap, T.L.; Dietz, B.M. Formation and biological targets of botanical o-quinones. Food Chem. Toxicol. 2018, 120, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Fujiki, Y.; Matsui, N.; Ojika, M.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of resveratrol produces a highly reactive ortho -quinone: Implications for melanocyte toxicity. Pigment. Cell Melanoma Res. 2019, 32, 766–776. [Google Scholar] [CrossRef]
- Crowell, J.A.; Korytko, P.J.; Morrissey, R.L.; Booth, T.D.; Levine, B.S. Resveratrol-Associated Renal Toxicity. Toxicol. Sci. 2004, 82, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Guha, P.; Chattopadhyay, S.; Bandyopadhyay, S.K. Biphasic activity of resveratrol on indomethacin-induced gastric ulcers. Biochem. Biophys. Res. Commun. 2009, 381, 90–95. [Google Scholar] [CrossRef]
- Posadino, A.M.; Giordo, R.; Cossu, A.; Nasrallah, G.K.; Shaito, A.; Abou-Saleh, H.; Eid, A.H.; Pintus, G. Flavin Oxidase-Induced ROS Generation Modulates PKC Biphasic Effect of Resveratrol on Endothelial Cell Survival. Biomolecules 2019, 9, 209. [Google Scholar] [CrossRef] [Green Version]
- Posadino, A.M.; Cossu, A.; Giordo, R.; Zinellu, A.; Sotgia, S.; Vardeu, A.; Hoa, P.T.; Van Nguyen, L.H.; Carru, C.; Pintus, G. Resveratrol alters human endothelial cells redox state and causes mitochondrial-dependent cell death. Food Chem. Toxicol. 2015, 78, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Gadacha, W.; Ben-Attia, M.; Bonnefont-Rousselot, D.; Aouani, E.; Ghanem-Boughanmi, N.; Touitou, Y. Resveratrol opposite effects on rat tissue lipoperoxidation: Pro-oxidant during day-time and antioxidant at night. Redox Rep. 2009, 14, 154–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordo, R.; Cossu, A.; Pasciu, V.; Hoa, P.T.; Posadino, A.M.; Pintus, G. Different Redox Response Elicited by Naturally Occurring Antioxidants in Human Endothelial Cells. Open Biochem. J. 2013, 7, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Rocha, K.; Souza, G.; Ebaid, G.; Seiva, F.; Cataneo, A.; Novelli, E. Resveratrol toxicity: Effects on risk factors for atherosclerosis and hepatic oxidative stress in standard and high-fat diets. Food Chem. Toxicol. 2009, 47, 1362–1367. [Google Scholar] [CrossRef]
- Bolton, J.L.; Dunlap, T. Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Shin, J.-W.; Lee, H.-S.; Na, J.-I.; Huh, C.-H.; Park, K.-C.; Choi, H.-R. Resveratrol Inhibits Particulate Matter-Induced Inflammatory Responses in Human Keratinocytes. Int. J. Mol. Sci. 2020, 21, 3446. [Google Scholar] [CrossRef]
- Fontecave, M.; Lepoivre, M.; Elleingand, E.; Gerez, C.; Guittet, O. Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Lett. 1998, 421, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, G.A.; Savio, M.; Forti, L.; Shevelev, I.; Ramadan, K.; Stivala, L.A.; Vannini, V.; Hübscher, U.; Spadari, S.; Maga, G. Inhibition of mammalian DNA polymerases by resveratrol: Mechanism and structural determinants. Biochem. J. 2005, 389, 259–268. [Google Scholar] [CrossRef] [PubMed]
- De la Lastra, C.A.; Villegas, I. Resveratrol as an antioxidant and pro-oxidant agent: Mechanisms and clinical implications. Biochem. Soc. Trans. 2007, 35, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, X.; Hu, X.; Chen, Z.; Liu, H.; Takeda, S.; Qing, Y. Multiple repair pathways mediate cellular tolerance to resveratrol-induced DNA damage. Toxicol. Vitr. 2017, 42, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Park, J.H.; Woo, J.S. Resveratrol induces cell death through ROS-dependent downregulation of Notch1/PTEN/Akt signaling in ovarian cancer cells. Mol. Med. Rep. 2019, 19, 3353–3360. [Google Scholar] [CrossRef]
- Heo, J.; Kim, S.; Hwang, K.; Kang, J.; Choi, K. Resveratrol induced reactive oxygen species and endoplasmic reticulum stress-mediated apoptosis, and cell cycle arrest in the A375SM malignant melanoma cell line. Int. J. Mol. Med. 2018, 42, 1427–1435. [Google Scholar] [CrossRef]
- Zhu, C.W.; Grossman, H.; Neugroschl, J.; Parker, S.; Burden, A.; Luo, X.; Sano, M. A randomized, double-blind, placebo-controlled trial of resveratrol with glucose and malate (RGM) to slow the progression of Alzheimer’s disease: A pilot study. Alzheimers Dement. 2018, 4, 609–616. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; Van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S.; et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Scott, E.; Brown, V.A.; Gescher, A.J.; Steward, W.P.; Brown, K. Clinical trials of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Walle, T. Bioavailability of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 9–15. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Symbol | Locus | Gene Name | Inheritance | Disease | Pathological Effects on the Mitochondria | Ref. |
|---|---|---|---|---|---|---|
| PARK1 PARK4 | 4q21-22 | SNCA | AD | EOPD | Mutant SNCA aggregates more easily, binds to mitochondrial membranes, inhibits Complex I activity damages mitochondrial structures, and causes mitochondrial toxicity. | [138,139,140] |
| PARK2 | 6q25.2-q27 | Parkin | AR | EOPD | Point mutations in parkin can inhibit its ability to interact with E2 and other protein substrates, ubiquitinate substrates, and translocate to depolarized mitochondria and induce mitophagy. | [141,142] |
| PARK3 | 2p13 | Unknown | AD | Classical PD | Unconfirmed, but may be a risk factor | [143] |
| PARK5 | 4p13 | UCHL1 | AD | Classical PD | Mutations in UCHL1 can lead to impaired ubiquitin proteasome system (UPS), accumulation of damaged proteins, and formation of Lewy bodies. | [144,145,146,147] |
| PARK6 | 1p35-p36 | PINK1 | AR | EOPD | Mutations at PINK1 impair mitophagy and mitochondrial quality control by disrupting activation and recruitment of parkin to the mitochondria and the normal phosphorylation of proteins that facilitate mitophagy. | [142,148] |
| PARK7 | 1p36 | DJ-1 | AR | EOPD | Mutations in DJ-1 cause mitochondria damage from oxidative stress, loss of ability to prevent α-synuclein fibrillation, and increased likelihood of mitochondria depolarization and fragmentation. | [149] |
| PARK8 | 12q12 | LRRK2 | AD | Classical PD | Mutations in LRRK2 result in increased mitochondrial fragmentation, increased basal activity, increased susceptibility to oxidative damage, and the disruption of mitophagy. | [150,151] |
| PARK9 | 1p36 | ATP13A2 | AR | Kufor-Rakeb syndrome; atypical dementia with spasticity, dementia, and supranuclear glaze palsy | Mutations in ATP13A2 have been associated with reduced ATP production, increased mitochondrial fragmentation, increased ROS production, increased glycolysis (which aggravates mitochondrial dysfunction), and defective mitophagy. | [132,144,152,153,154,155,156] |
| PARK10 | 1p32 | Unknown | Risk factor | Classical PD | Confirmed susceptible locus, but unknown pathology | [143] |
| PARK11 | 2q36-27 | Unknown, not GIGYF2 | AD | Late-onset PD | May be a risk factor, but not independently confirmed | [143] |
| PARK12 | Xq21-q25 | Unknown | Risk factor | Classical PD | Confirmed susceptible locus; may be possible risk factor; pathology unknown | [143] |
| PARK13 | 2p12 | HTRA2 | AD or risk factor | Classical PD | HTRA2 mutations could possibly lead to insufficient protein degradation, atypical mitochondrial morphology and function, and increased mitochondrial susceptibility to oxidative stress. | [144,157,158,159] |
| PARK14 | 22q13.1 | PLA2G6 | AR | Early-onset dystonia–parkinsonism | PLA2G6 participates in the regulation of Ca2+ within the cell. Impaired PLA2G6-dependent store-operated Ca2+ signaling causes autophagy dysfunction, while increased influx of Ca2+ into the mitochondria is associated with oxidative stress. | [144,160] |
| PARK15 | 22q12-q13 | FBXO7 | AR | Early-onset parkinsonian-pyramidal syndrome | Mutations in the FBXO7 gene can cause protein aggregation in the mitochondria and inhibition of mitophagy and ROS generation. | [143,161,162] |
| PARK16 | 1q32 | Unknown | Risk factor | Classical PD | Confirmed susceptibility locus | [143] |
| PARK17 | 16q11.2 | VPS35 | AD | Classical PD | Mutations in VPS35 lead to increased mitochondrial fission/fragmentation. | [161,163] |
| PARK18 | 3q27.1 | EIF4G1 | AD | Classical PD | The exact mechanism of this mutation has yet to be understood. | [143,161] |
| PARK19 | 1p31.3 | DNAJC6 | AR | Juvenile onset, atypical PD | DNAJC6 encodes HPS40 Auxilin, but the mechanism of the mutation is not yet understood. | [164,165] |
| PARK20 | 21q22.11 | SYNJ1 | AR | Juvenile onset, atypical PD | SYNJ1 results in an increase in oxidative stress and change in mitochondrial morphology | [164,166] |
| PARK21 | 3q22.1 | DNAJC13 | AD | Late-onset PD | Mutations in DNAJC13 disrupts normal endosomal trafficking and results in α-synuclein aggregation in the lysosomes. | [164,167,168] |
| PARK22 | 7p11.2 | CHCHD2 | AD | Late-onset PD | Deficiency in CHCHD2 leads to reduced cytochrome c oxidase (COX) activity, decreased mitochondrial membrane potential, increased ROS production, and increased mitochondrial fragmentation. | [169,170] |
| PARK23 | 15q22 | VPS13C | AR | EOPD, rapid progression | Mutations in the VPS13C gene have been associated with reduced mitochondrial membrane potential, increased mitochondrial fragmentation, and upregulated PINK1/parkin-dependent mitophagy. | [161,171] |
| Type of Study | Sample | Purpose | Dose | Duration | Completion Date | Main Results | Ref. |
|---|---|---|---|---|---|---|---|
| DBRCT, crossover, placebo-controlled phase I | 20 healthy part. | To study resveratrol pharmacokinetics when taken together with levodopa | BIA 6-512 (trans-RSV) 25 mg, 50 mg, 100 mg dose | 11 weeks | 23 July 2004 | Not Posted | NCT: NCT03091543 |
| DBRCT, placebo-controlled phase I | 80 healthy part. | To study the tolerability and pharmacokinetics of resveratrol and its effects on levodopa | Oral BIA 6-512 (trans-RSV) 25 mg, 50 mg, 100 mg dose | 17 weeks | 28 February 2005 | Not Posted | NCT: NCT03091868 |
| Single-center, open-label, RCT, two-way crossover | 24 healthy part. | To study the effect of food on resveratrol pharmacokinetics | Oral BIA 6-512 400 mg dose following a breakfast (Test) or at least 8 h of fasting (Reference) | 7 weeks | 7 July 2005 | Not Posted | NCT: NCT03095092 |
| DBRCT, crossover, placebo-controlled phase I | 40 healthy part. | To study the safety and tolerability of different doses of BIA 6-512 six times a day and to characterize the pharmacokinetics of BIA 6-512 | Oral BIA 6-512 (25, 50, 100, or 150 mg dose) six times a day/4 h intervals | 11 weeks | 29 July 2005 | Not Posted | NCT: NCT03093389 |
| DBRCT, placebo-controlled phase I | 25 part. | To compare the pharmacokinetic profile of BIA 6-512 in healthy young and old subjects | Oral BIA 6-512 200 mg every 8 h | 5 weeks | 2 March 2006 | Not Posted | NCT: NCT03095105 |
| Single-center, open-label, RCT, two-way crossover | 39 healthy part. | To investigate the effects of BIA 6-512 at steady state on the pharmacokinetics of levodopa when administered with levodopa/benserazide with or without entacapone | Oral BIA 6-512 (25, 50, 75, and 100 mg) plus a single dose of immediate release levodopa/benserazide 200/50 mg with or without a single dose of entacapone 200 mg | 7 weeks | 11 July 2006 | Not Posted | NCT: NCT03094156 |
| DBRCT, crossover, placebo-controlled phase I | 38 healthy part. | To investigate the effects of BIA 6-512 at steady state on the pharmacokinetics of levodopa when administered with levodopa/benserazide with or without nebicapone | Oral BIA 6-512 (25, 50, 75, and 100 mg) plus a single dose of immediate release levodopa/benserazide 200/50 mg with or without a single dose of nebicapone 150 mg | 13 weeks | 20 October 2006 | Not Posted | NCT: NCT03097211 |
| Type of Study | Sample | Purpose | Dose | Duration | Main Results | Completion Date | Ref. |
|---|---|---|---|---|---|---|---|
| DBRCT, placebo-controlled parallel | 102 early affected Huntington disease (HD) patients | To study the therapeutic potential of RSV on the caudate volume of HD patients | RSV 40 mg twice a day | 1 year | Not Posted | October 2019 | NCT: NCT02336633 |
| DBRCT, placebo-controlled Phase II | 120 patients with mild to moderate dementia most likely due to AD | To study the impact on biomarkers of RSV treatment in patients with mild to moderate AD | Oral RSV 500 mg OD with dose escalation of up to 1000 mg BID | 52 weeks | RSV is safe and well tolerated with nausea, weight loss, and diarrhea as side effects. No benefit on biomarkers CSF Aβ40 and Aβ42, etc. [239,263] Increased brain volume loss | March 2014 | NCT: NCT01504854 |
| DBRCT, placebo-controlled 2-period crossover, Phase II | 40 Friederich ataxia (FRDA) patients | To study the efficacy of RSV as a treatment for FRDA | 1 g micronized RSV or placebo twice daily for two 24 week periods | 52 weeks | Recruiting | Ongoing | NCT: NCT03933163 |
| Non-randomized, parallel assignment, open label clinical Phase I and II | 27 FRDA patients (n = 15 will receive RSV) | To study the effects of RSV on frataxin levels in FRDA patients and to measure RSV’s effects on markers of oxidative stress, clinical measures of ataxia, and cardiac parameters | RSV 40 mg twice a day | 12 weeks | Not Posted | August 2012 | NCT: NCT01339884 |
| Single center, multi-site, DBRCT, placebo-controlled Phase-3 Trial | 27 mild to moderate AD patients | To investigate the efficacy of RSV in delaying the progression of AD | RSV, glucose, and malate supp. delivered in grape juice | 12 months | RSV is safe and well-tolerated at low dose. No significant changes in AD Assessment Scale-cognitive subscale, Mini-Mental State Exam, etc. [239,262] | December 2010 | NCT: NCT00678431 |
| Prospective, longitudinal, mixed, analytical, experimental, double-blind, placebo-controlled study | 100 amyotrophic lateral sclerosis (ALS) patients | To assess the clinical improvement of ALS patients treated with curcumin and RSV liposomed polyphenols with dutasteride | RSV 75 mg, curcumin 200 mg, and dutasteride 0.5 mg | 6 months | Not Yet Recruiting | Not Yet Recruiting | NCT: NCT04654689 |
| RCT, parallel assignment, quadruple-blind, Phase I | 48 part. | To study the safety and CSF penetration of oral BDPP (grape seed polyphonic extract, RSV) in humans to assess possible benefits of BDPP to MCI | Low, moderate, and high dose of BDPP | 4 months | Recruiting | Ongoing | NCT: NCT02502253 |
| RCT, crossover assignment, open label | 12 patients with hereditary spastic paraplegia (SPG5) | To study the efficacy of Xenbilox, Tahor, and RSV in decreasing oxysterols synthesis, reducing cholesterol proudction, regulating bile production, and/or providing neuroprotection | Xenbilox, Tahor, or resveratrol (80 mg for 2 months) | 2 months | Not Posted | 27 September 2017 | NCT: NCT02314208 |
| RCT, crossover assignment, open label, Phase I | 12 patients with mild to moderate AD | To study the efficacy and safety of administering etanercept with nutritional supp. versus administering nutritional supp. alone | Nutritional supp. (curcum., luteol., theaflav., lip., acid, fish oil, quercet., resveratr.) with or without etanercept | 16 weeks | Not Posted | October 2015 | NCT: NCT01716637 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kung, H.-C.; Lin, K.-J.; Kung, C.-T.; Lin, T.-K. Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’s Disease. Biomedicines 2021, 9, 918. https://doi.org/10.3390/biomedicines9080918
Kung H-C, Lin K-J, Kung C-T, Lin T-K. Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’s Disease. Biomedicines. 2021; 9(8):918. https://doi.org/10.3390/biomedicines9080918
Chicago/Turabian StyleKung, Heng-Chung, Kai-Jung Lin, Chia-Te Kung, and Tsu-Kung Lin. 2021. "Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’s Disease" Biomedicines 9, no. 8: 918. https://doi.org/10.3390/biomedicines9080918