
Therapeutic Options for Systemic Sclerosis: Current and Future Perspectives in Tackling Immune-Mediated Fibrosis

 ,
,  ,
,
Abstract
:1. Introduction
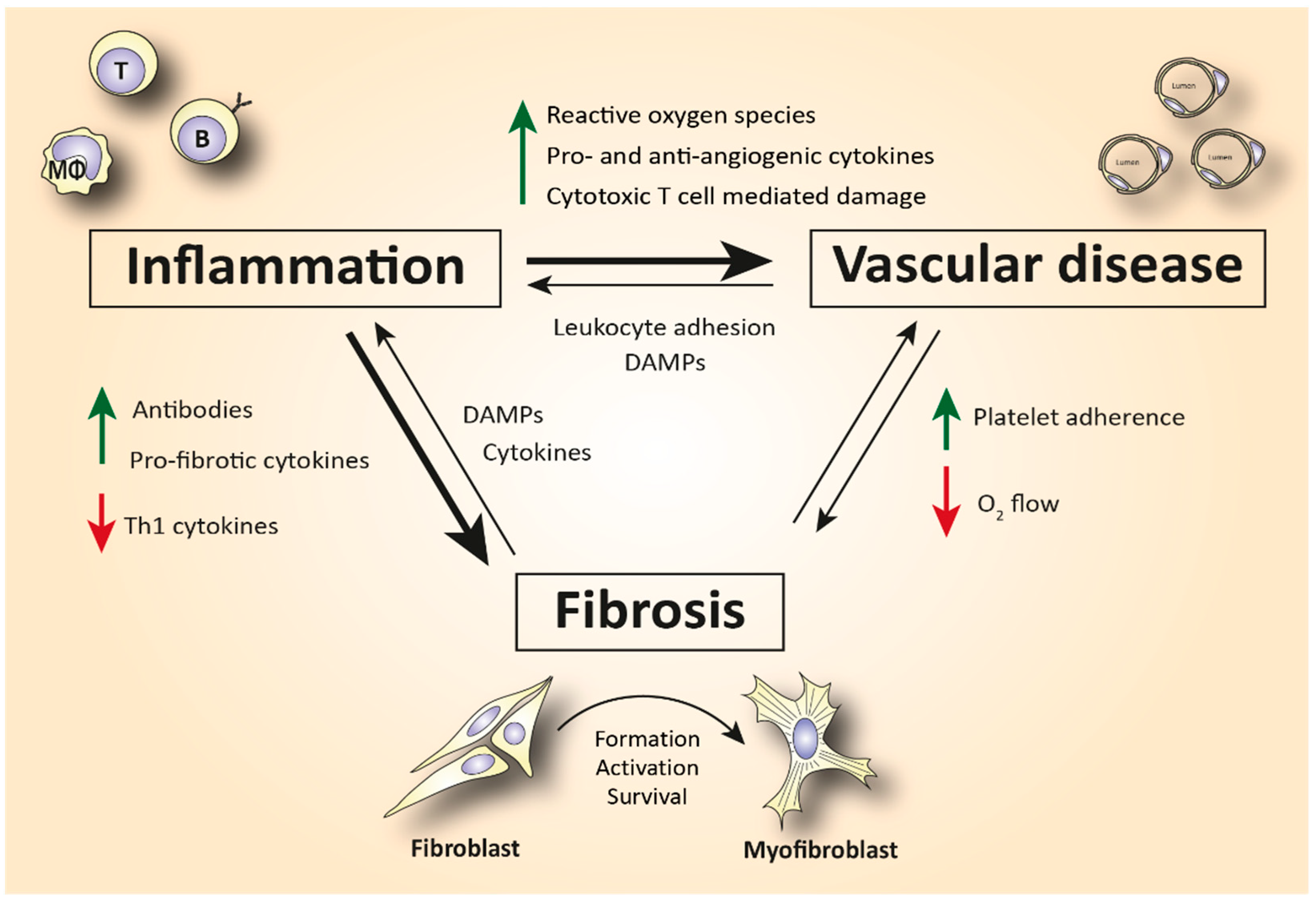
2. The Role of Immune Cells in SSc-Related Inflammation and Fibrosis
2.1. The Role of Innate Immunity in SSc
2.1.1. Macrophages
2.1.2. Eosinophils and Mast Cells
2.1.3. New Players: Innate Lymphoid Cells and Plasmacytoid Dendritic Cells
2.2. The Role of Adaptive Immunity in SSc
2.2.1. T Lymphocytes
2.2.2. B Lymphocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Function |
|---|---|
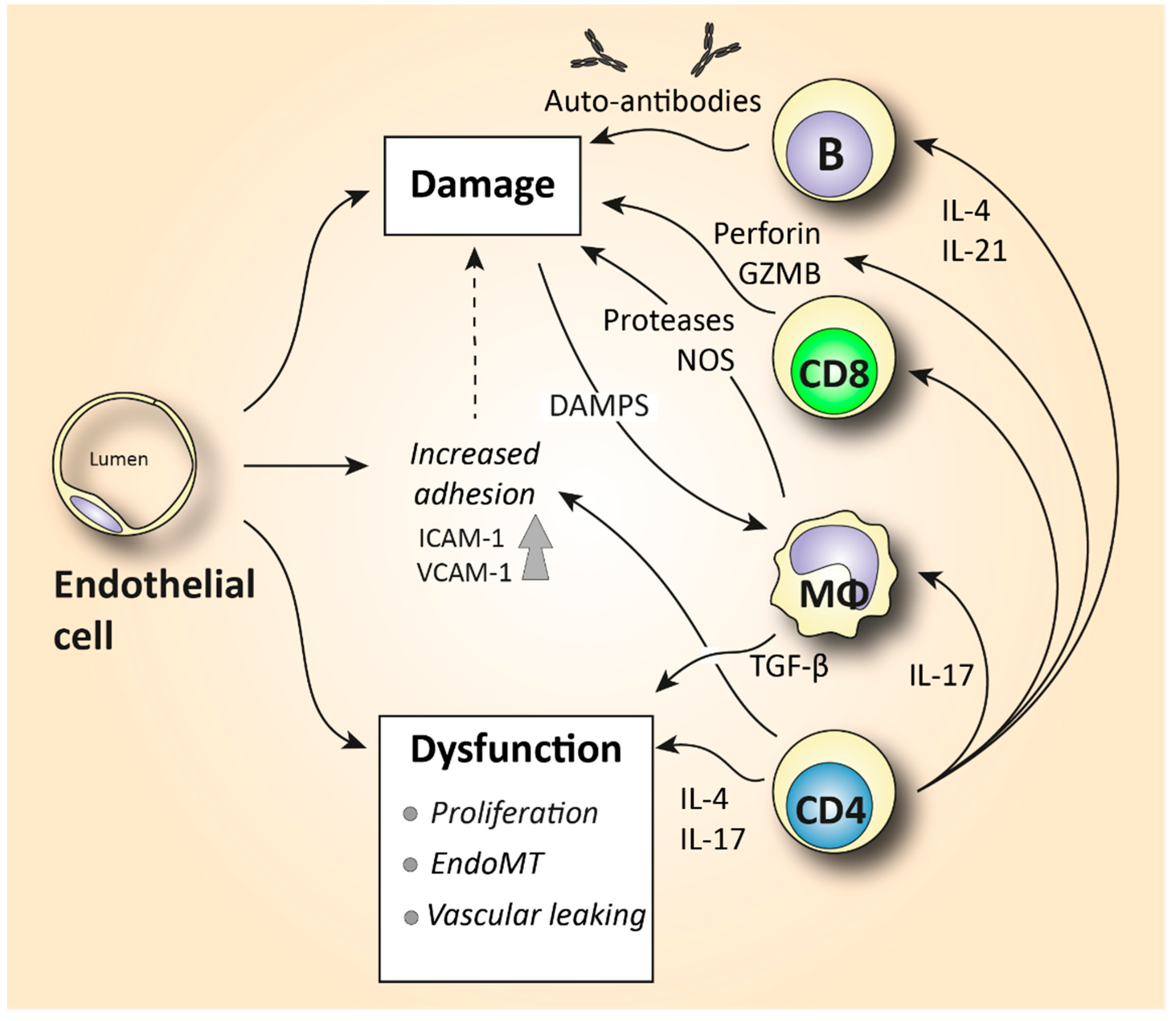
| Endothelial cell | Platelet adhesion activates fibrotic pathways. Increased microvascular permeability causes leukocyte adhesion to the endothelium, leading to increased inflammation [7]. |
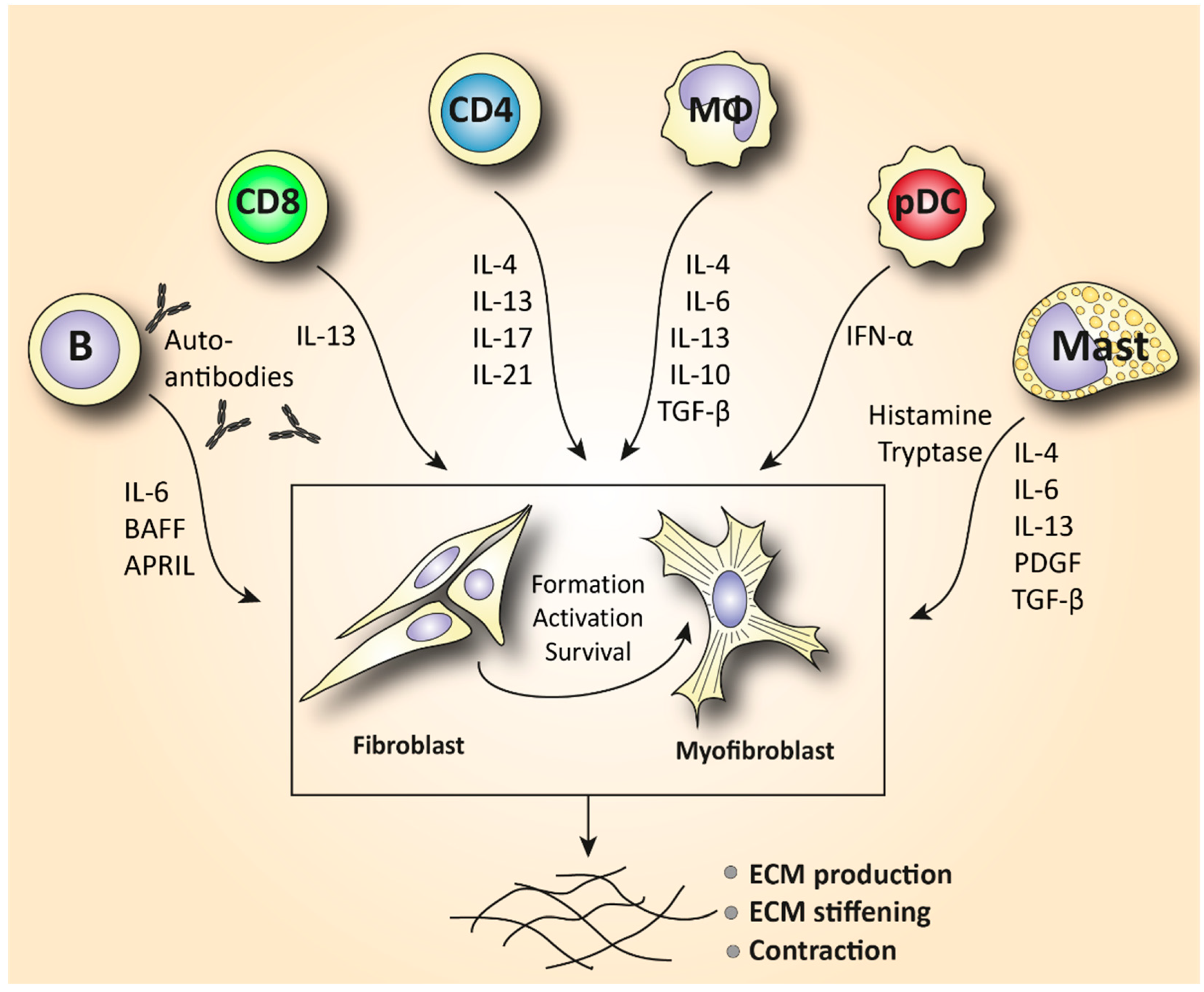
| Monocyte/Macrophage | A prominent M2 macrophage signature increases levels of pro-fibrotic cytokines such as IL-4, IL-6, and IL-13 and correlates with elevated tissue fibrosis [17,18,19,20]. |
| Eosinophil | Elevated eosinophil counts in the peripheral blood are associated with severe lung disease and presence of skin ulcers [31,32,33]. |
| Mast cell | Release of cytokines and growth factors such as IL-4, IL-6, IL-13, TNF-a, PDGF, and TGF-β activates myofibroblasts to produce collagen [37]. Tryptase and histamine release triggers fibroblast proliferation [38,39]. |
| Innate lymphoid cell | Increased production of the ILC2 cytokines IL-25, IL-33, and TSLP in serum and skin mediates fibrosis in a TGF-β-dependent manner [43,44,45,46,47,48,49]. |
| Plasmacytoid dendritic cell | Elevated numbers of CXCL4+- and IFN-a-producing pDCs in skin and lungs are involved in increased fibrotic manifestations mediated by TLR8 activation [28,50,52]. |
| T lymphocyte | Skin is predominantly infiltrated by CD4+ and CD8+ cytotoxic T cells that produce pro-fibrotic cytokines and cause apoptosis to epithelial cells [6,72]. Increased IL-21-producing Tph cells promote plasmablast differentiation and increase activation of myofibroblasts [75,77]. In the peripheral blood, SSc patients are characterized by increased Th2 and Th17 numbers compared to healthy donors [4,60,63]. |
| B lymphocyte | Elevated BAFF and APRIL are correlated with skin thickening [81,83]. Increased IL-6-producing Beffs increase inflammation, while decreased IL-10-producing Bregs exhibit a reduced capacity for immunosuppression [82]. In SSc peripheral blood, an increase in naïve and a decrease in activated memory B cells is observed compared to healthy controls [4,85,87,91]. |
3. SSc First-Line Anti-Inflammatory Treatment
3.1. Synthetic Corticosteroids
3.2. Methotrexate
3.3. Cyclophosphamide
3.4. Mycophenolate Mofetil
3.5. Autologous Hematopoietic Stem Cell Transplantation
| Drug | Target | Type of Trial(s) | Duration (Months) | Patients | Results |
|---|---|---|---|---|---|
| Methotrexate (MTX) | Exact anti-inflammatory role is unknown | Multicenter, double-blind | |||
| 1. RCT [108] | 1. 6 | 1. 29 early dSSc | 1. Mean TSS 21.61 at baseline, 19.96 (p = 0.135) 6 months after | ||
| 2. RCT [107] | 2. 12 | 2. 71 early dSSc | 2. Mean TSS 18.3 at baseline, 14.5 (p = 0.027) 12 months post-treatment | ||
| Cyclophosphamide (CYC) | Inhibition and suppression of T helper and regulatory T cells | Double-blind, RCT (SLC) [112] | 12 | 158 SSc-ILD | 2. 53% (p < 0.03) improvement in predicted FVC and 3.02 (p = 0.08) unit improvement in mRSS in CYC’s favor |
| Mycophenolate mofetil (MMF) | T and B cell depletion | 1. Double-blind, RCT (SLC II) [118] | 1. 24 | 1. 69 SSc-ILD | 1. Percentage of predicted FVC improved from 67 to 75, and mRSS decreased from 14.5 at baseline to 10 24 months post-treatment |
| 2. Double-blind RCT [119] | 2. 6 | 2. 41 mild SSc-ILD | 2. No statistically significant improvement in mRSS and FVC scores | ||
| Autologous hematopoietic stem cell transplantation (ASCT) | Depletion of T and B cells, followed by stem cell transplantation | 1. Open-label, multicenter RCT (SCOT) (NCT00860548) | 1. 54 | 1. 75 severe SSc | 1. ASCT more effective in diminishing skin fibrosis compared to CYC (−19.9 vs. −8.8, p < 0.001) and shows greater event-free survival |
| 2. Open-label, multicenter RCT (ASTIS) [131] | 2. 24 | 2. 156 early dcSSc | 2. Overall survival, mRSS, and FVC significantly improved with ASCT compared to CYC (67% of 1404 pairwise comparisons in favor of ASCT vs. 33% in CYC, p = 0.01) |
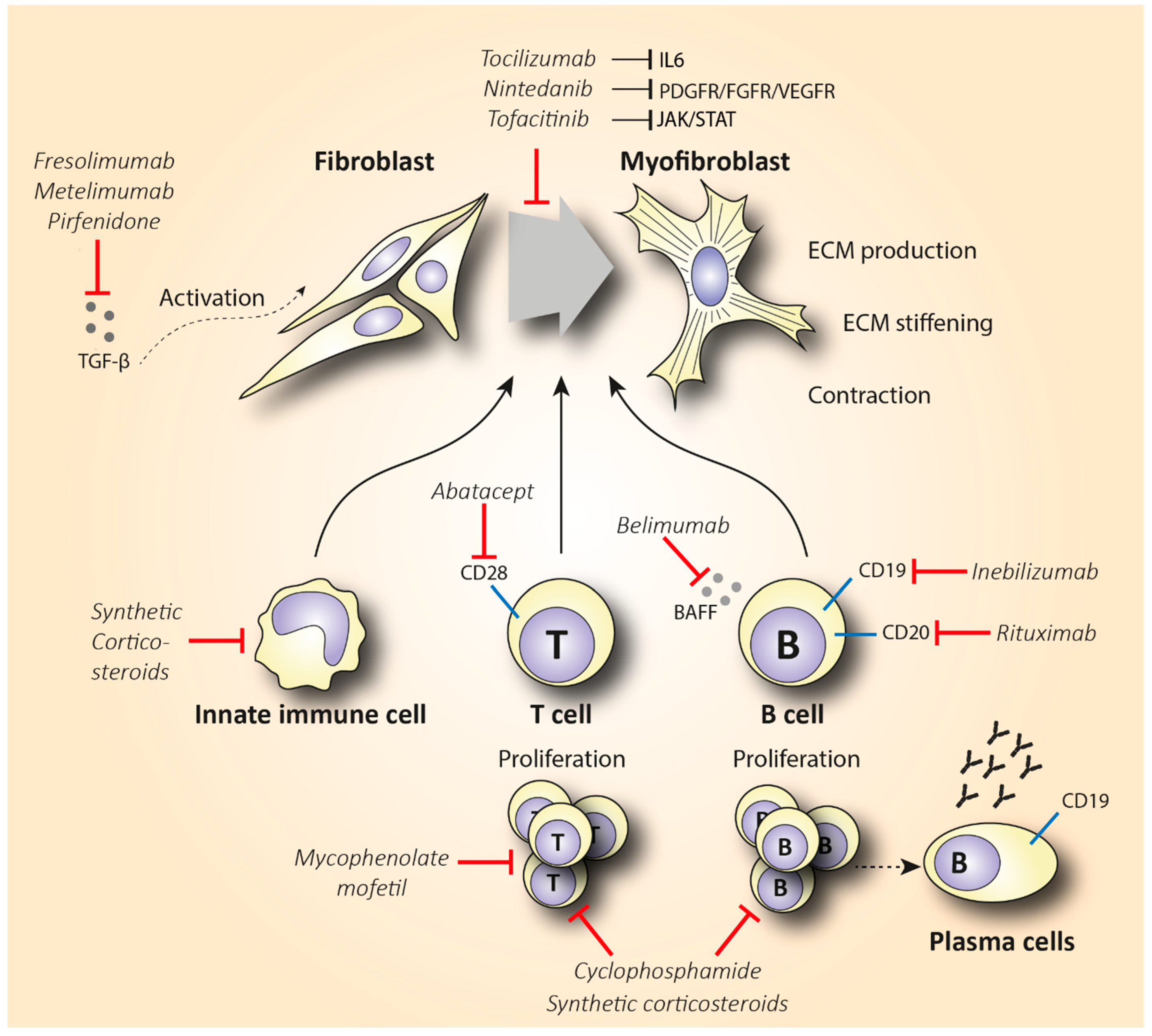
4. Evaluation of Cell-Specific Anti-Inflammatory Treatment
4.1. B Cell-Specific Treatment
4.1.1. Rituximab
4.1.2. Inebilizumab and Belimumab
4.2. T Cell-Specific Treatment
| Drug | Target | Type of Trial(s) | Duration (Months) | Patients | Results |
|---|---|---|---|---|---|
| Rituximab (RTX) | Anti-CD20 B cell depletion | 1. Double-blind, RCT [137] | 1. 24 | 1. 16 early SSc | 1. B cell depletion in blood did not improve mRSS and FVC scores |
| 2. Open-label trial [139] | 2. 54 | 2. 9 dcSSc | 2. Median decrease in mRSS of 43.3% (p = 0.001) accompanied by reduction in IL-6 levels | ||
| 3. Open-label RCT [140] | 3. 24 | 3. 14 SSc | 3. FVC improved by 10.25% in RTX and reduced by 5.04% in the placebo group (p = 0.0018). RTX arm: mRSS improvement of 13.5 vs. 8.37 in placebo, 12 months post-treatment (p < 0.001) | ||
| 4. Multicenter, open-label trial [141] | 4. 48 | 4. 51 SSc-ILD | 4. No significant change in mRSS, but lung function improved significantly | ||
| 5. Open-label RCT [142] | 5. 6 | 5. 60 early dSSc | 5. Significant improvement in RTX vs. CYC in median percentage of FVC (67.52 vs. 58.06, p = 0.003), but not in mRSS scores | ||
| 6. Multicenter, double-blind RCT [143] | 6. 6 | 6. 57 SSc-PAH | 6. RTX arm: the improvement in median 6MWD was 25.5 m compared to 0.4 m in placebo (p = 0.03) after 48 weeks | ||
| Inebilizumab (MEDI-551) | Anti-CD19 B cell depletion | Multicenter, double-blind RCT [149] | 3 | 28 SSc | Depletion of B and plasma cells was correlated with improved mRSS and reduced expression of fibrotic genes in skin biopsies |
| Belimumab | Inhibition of B cell survival by blocking BAFF | Double-blind RCT [152] | 13 | 20 dcSSc | mRSS score improved from 27 to 18 (p = 0.039), while in placebo group from 28 to 21 (p = 0.023) |
| Abatacept | CD28 blocking T cell depletion | 1. RCT [156] | 1. 10 | 1. 6 dcSSc | 1. 5/7 patients and 1/3 controls showed >30% improved mRSS |
| 2.Multicenter, double-blind RCT [157] | 2. 12 | 2. 88 dcSSc | 2. No significant improvement in mRSS, but secondary outcomes related to quality of life improved significantly |
5. Targeting Cytokine Production
| Drug | Target | Type of Trial(s) | Duration (Months) | Patients | Results |
|---|---|---|---|---|---|
| Tocilizumab (TCZ) | Inhibits IL-6 signaling | 1. Open-label RCT (faSScinate) [162] | 1. 24 | 1. 87 early SSc | 1. Insignificant change in mRSS, but IL-6 reduction was correlated with decreased TGF-β expression |
| 2. EUSTAR observational study [158] | 2. 5 | 2. 189 SSc-polyarthritis and myopathy | 2. No statistically significant change in mRSS, but remarkable improvement in joint function | ||
| 3. Multicenter, double-blind RCT (focuSSed) [169] | 3. 12 | 3. 210 dcSSc | 3. Change of −1.73 in mRSS between treated and placebo groups was not statistically significant (p = 0.10), but the 4.2% improvement in predicted FVC was (p = 0.0002) | ||
| Rilonacept | Blocks IL-1b signaling | Double-blind RCT [172] | 5 weeks | 19 dcSSc | No changes in mRSS between treatment and placebo |
| Basiliximab | Anti-CD25-mediated inhibition of IL-2 inhibits T cell activation and proliferation | Small-scale, open-label study [175] | 17 | 10 dcSSc | Median mRSS reduced from 26/51 to 11/51 at week 68 (p = 0.015) Median predicted FVC between baseline and 44 weeks after treatment increased from 82.1% to 88.4% |
| Fresolimumab | Blocks TGF-β signaling | Small-scale, open-label study [176] | 6 | 15 dcSSc | Reduction of 8 units in mRSS score (p < 0.001) Reduced expression of TGF-β-regulated genes in skin biopsies (p < 0.049) |
| Metelimumab | Blocks TGF-β signaling | Double-blind RCT [177] | 6 | 45 early SSc | No statistically significant change in mRSS scores |
| Pirfenidone | Reduces fibroblast proliferation and TGF-β-induced collagen production in primary skin fibroblasts | Open-label, phase II study (LOTUSS) [180] | 4 | 63 SSc-ILD | No difference in the predicted FVC between baseline and post-treatment |
6. Emerging Therapies with Tyrosine Kinase Inhibitors
6.1. Indolinone-Derived Small Molecule Tyrosine Kinase Inhibitors
6.2. Tofacitinib
| Drug | Target | Type of Trial(s) | Duration (Months) | Patients | Results |
|---|---|---|---|---|---|
| Imatinib mesylate | Inhibits PDGFR and TGF-β signaling | Multicenter, open-label RCT [193] | 6 | 27 dcSSc | Mean decrease in mRSS was 21% compared to baseline (p < 0.001) |
| Nilotinib | Same as imatinib, but 20–30-fold more potent | Open-label, single-arm trial [195] | 8 | 10 early dcSSc | Promising reduction of 23% in mRSS 12 months post-treatment (p = 0.01) |
| Nintedanib | Inhibits PDGFR, FGFR, and VEGFR signaling | Double-blind RCT (SENSCIS) [199] | 13 | 576 SSc-ILD | Adjusted annual rate of decline in FVC −52.4 mL compared to −93.3 mL in the placebo group (p = 0.04) |
| Tofacitinib | Inhibits JAK/STAT signaling | Double-blind RCT (NCT03274076) | 6 | 15 dcSSc | Preliminary data show a trend towards improved fibrosis |
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herrick, A.L.; Pan, X.; Peytrignet, S.; Lunt, M.; Hesselstrand, R.; Mouthon, L.; Silman, A.; Brown, E.; Czirják, L.; Distler, J.H.W.; et al. Treatment outcome in early diffuse cutaneous systemic sclerosis: The European Scleroderma Observational Study (ESOS). Ann. Rheum. Dis. 2017, 76, 1207–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellar, R.E.; Pope, J.E. Evidence-based management of systemic sclerosis: Navigating recommendations and guidelines. Semin. Arthritis Rheum. 2017, 46, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Pope, J.E. Emerging drugs and therapeutics for systemic sclerosis. Expert Opin. Emerg. Drugs 2016, 21, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.A.; Lundy, S.K.; Whitfield, M.L.; Berrocal, V.; Campbell, P.; Rasmussen, S.; Ohara, R.; Stinson, A.; Gurrea-Rubio, M.; Wiewiora, E.; et al. Lymphocyte subset abnormalities in early diffuse cutaneous systemic sclerosis. Arthritis Res. Ther. 2021, 23, 10. [Google Scholar] [CrossRef]
- Liu, M.; Wu, W.; Sun, X.; Yang, J.; Xu, J.; Fu, W.; Li, M. New insights into CD4+ T cell abnormalities in systemic sclerosis. Cytokine Growth Factor Rev. 2016, 28, 31–36. [Google Scholar] [CrossRef]
- Maehara, T.; Kaneko, N.; Perugino, C.A.; Mattoo, H.; Kers, J.; Allard-Chamard, H.; Mahajan, V.S.; Liu, H.; Murphy, S.J.; Ghebremichael, M.; et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J. Clin. Investig. 2020, 130, 2451–2464. [Google Scholar] [CrossRef] [Green Version]
- Henderson, J.; Bhattacharyya, S.; Varga, J.; O’Reilly, S. Targeting TLRs and the inflammasome in systemic sclerosis. Pharmacol. Ther. 2018, 192, 163–169. [Google Scholar] [CrossRef]
- Fuschiotti, P. T cells and cytokines in systemic sclerosis. Curr. Opin. Rheumatol. 2018, 30, 594–599. [Google Scholar] [CrossRef]
- Romano, E.; Rosa, I.; Fioretto, B.S.; Matucci-Cerinic, M.; Manetti, M. New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy. Life 2021, 11, 610. [Google Scholar] [CrossRef]
- Bruni, C.; Frech, T.; Manetti, M.; Rossi, F.W.; Furst, D.E.; De Paulis, A.; Rivellese, F.; Guiducci, S.; Matucci-Cerinic, M.; Bellando-Randone, S. Vascular Leaking, a Pivotal and Early Pathogenetic Event in Systemic Sclerosis: Should the Door Be Closed? Front. Immunol. 2018, 9, 2045. [Google Scholar] [CrossRef]
- Host, L.; Nikpour, M.; Calderone, A.; Cannell, P.; Roddy, J. Autologous stem cell transplantation in systemic sclerosis: A systematic review. Clin. Exp. Rheumatol. 2017, 35, S198–S207. [Google Scholar]
- Laurent, P.; Sisirak, V.; Lazaro, E.; Richez, C.; Duffau, P.; Blanco, P.; Truchetet, M.-E.; Contin-Bordes, C. Innate Immunity in Systemic Sclerosis Fibrosis: Recent Advances. Front. Immunol. 2018, 9, 1702. [Google Scholar] [CrossRef] [Green Version]
- van Caam, A.; Vonk, M.; van den Hoogen, F.; van Lent, P.; van der Kraan, P. Unraveling SSc Pathophysiology; The Myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesce, J.T.; Ramalingam, T.R.; Mentink-Kane, M.M.; Wilson, M.S.; El Kasmi, K.C.; Smith, A.M.; Thompson, R.W.; Cheever, A.W.; Murray, P.J.; Wynn, T.A. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009, 5, e1000371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Christmann, R.B.; Sampaio-Barros, P.; Stifano, G.; Borges, C.L.; de Carvalho, C.R.; Kairalla, R.; Parra, E.R.; Spira, A.; Simms, R.; Capellozzi, V.L.; et al. Association of Interferon- and transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. 2014, 66, 714–725. [Google Scholar] [CrossRef]
- Assassi, S.; Swindell, W.R.; Wu, M.; Tan, F.D.; Khanna, D.; Furst, D.E.; Tashkin, D.P.; Jahan-Tigh, R.R.; Mayes, M.D.; Gudjonsson, J.E.; et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol. 2015, 67, 3016–3026. [Google Scholar] [CrossRef]
- Rudnik, M.; Hukara, A.; Kocherova, I.; Jordan, S.; Schniering, J.; Milleret, V.; Ehrbar, M.; Klingel, K.; Feghali-Bostwick, C.; Distler, O.; et al. Elevated Fibronectin Levels in Profibrotic CD14(+) Monocytes and CD14(+) Macrophages in Systemic Sclerosis. Front. Immunol. 2021, 12, 642891. [Google Scholar] [CrossRef]
- Soldano, S.; Trombetta, A.C.; Contini, P.; Tomatis, V.; Ruaro, B.; Brizzolara, R.; Montagna, P.; Sulli, A.; Paolino, S.; Pizzorni, C.; et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1842–1845. [Google Scholar] [CrossRef]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.T.; Van Rooijen, N.; Haslett, C.; Howie, S.E.; Simpson, A.J.; et al. Ly6Chimonocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Soldano, S.; Smith, V. Pathophysiology of systemic sclerosis: Current understanding and new insights. Expert Rev. Clin. Immunol. 2019, 15, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Higashi-Kuwata, N.; Jinnin, M.; Makino, T.; Fukushima, S.; Inoue, Y.; Muchemwa, F.C.; Yonemura, Y.; Komohara, Y.; Takeya, M.; Mitsuya, H.; et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res. Ther. 2010, 12, R128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manetti, M. Deciphering the alternatively activated (M2) phenotype of macrophages in scleroderma. Exp. Dermatol. 2015, 24, 576–578. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Wei, J.; Tourtellotte, W.G.; Hinchcliff, M.; Gottardi, C.G.; Varga, J. Fibrosis in systemic sclerosis: Common and unique pathobiology. Fibrogenesis Tissue Repair 2012, 5 (Suppl. S1), S18. [Google Scholar] [CrossRef] [Green Version]
- Borie, R.; Quesnel, C.; Phin, S.; Debray, M.-P.; Marchal-Somme, J.; Tiev, K.; Bonay, M.; Fabre, A.; Soler, P.; Dehoux, M.; et al. Detection of alveolar fibrocytes in idiopathic pulmonary fibrosis and systemic sclerosis. PLoS ONE 2013, 8, e53736. [Google Scholar] [CrossRef]
- Ruaro, B.; Soldano, S.; Smith, V.; Paolino, S.; Contini, P.; Montagna, P.; Pizzorni, C.; Casabella, A.; Tardito, S.; Sulli, A.; et al. Correlation between circulating fibrocytes and dermal thickness in limited cutaneous systemic sclerosis patients: A pilot study. Rheumatol. Int. 2019, 39, 1369–1376. [Google Scholar] [CrossRef]
- Xue, D.; Tabib, T.; Morse, C.; Yang, Y.; Domsic, R.; Khanna, D.; Lafyatis, R. Expansion of FCGR3A(+) macrophages, FCN1(+) mo-DC, and plasmacytoid dendritic cells associated with severe skin disease in systemic sclerosis. Arthritis Rheumatol. 2021, 74, 329–341. [Google Scholar] [CrossRef]
- Pope, S.M.; Brandt, E.B.; Mishra, A.; Hogan, S.P.; Zimmermann, N.; Matthaei, K.I.; Foster, P.S.; Rothenberg, M.E. IL-13 induces eosinophil recruitment into the lung by an IL-5– and eotaxin-dependent mechanism. J. Allergy Clin. Immunol. 2001, 108, 594–601. [Google Scholar] [CrossRef]
- Chen, L.; Grabowski, K.A.; Xin, J.-P.; Coleman, J.; Huang, Z.; Espiritu, B.; Alkan, S.; Xie, H.B.; Zhu, Y.; White, F.A.; et al. IL-4 induces differentiation and expansion of Th2 cytokine-producing eosinophils. J. Immunol. 2004, 172, 2059–2066. [Google Scholar] [CrossRef]
- Falanga, V.; Medsger, T.A.J. Frequency, levels, and significance of blood eosinophilia in systemic sclerosis, localized scleroderma, and eosinophilic fasciitis. J. Am. Acad. Dermatol. 1987, 17, 648–656. [Google Scholar] [CrossRef]
- Ando, K.; Nakashita, T.; Kaneko, N.; Takahashi, K.; Motojima, S. Associations between peripheral blood eosinophil counts in patients with systemic sclerosis and disease severity. Springerplus 2016, 5, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Ikeda, T.; Inaba, Y.; Kunimoto, K.; Mikita, N.; Kaminaka, C.; Kanazawa, N.; Yamamoto, Y.; Tabata, K.; Fujii, T.; et al. Peripheral blood eosinophilia is associated with the presence of skin ulcers in patients with systemic sclerosis. J. Dermatol. 2019, 46, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Asano, Y.; Yamashita, T.; Takahashi, T.; Nakamura, K.; Miura, S.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Sumida, H.; et al. Systemic sclerosis complicated with localized scleroderma-like lesions induced by Köbner phenomenon. J. Dermatol. Sci. 2018, 89, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebbar, M.; Gillot, J.M.; Hachulla, E.; Lassalle, P.; Hatron, P.Y.; Devulder, B.; Janin, A. Early expression of E-selectin, tumor necrosis factor alpha, and mast cell infiltration in the salivary glands of patients with systemic sclerosis. Arthritis Rheum. 1996, 39, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, G.; Roberts, W.N.; Sciortino, D.; Sangari, D.; Cirmi, S.; Ravenell, R.L.; Navarra, M.; Bagnato, G.; Gangemi, S. Mastocytosis and systemic sclerosis: A clinical association. Clin. Mol. Allergy 2016, 14, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yukawa, S.; Yamaoka, K.; Sawamukai, N.; Shimajiri, S.; Kubo, S.; Miyagawa, I.; Sonomoto, K.; Saito, K.; Tanaka, Y. Dermal mast cell density in fingers reflects severity of skin sclerosis in systemic sclerosis. Mod. Rheumatol. 2013, 23, 1151–1157. [Google Scholar] [CrossRef]
- Dees, C.; Akhmetshina, A.; Zerr, P.; Reich, N.; Palumbo, K.; Horn, A.; Jüngel, A.; Beyer, C.; Krönke, G.; Zwerina, J.; et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. J. Exp. Med. 2011, 208, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Prieto-García, A.; Zheng, D.; Adachi, R.; Xing, W.; Lane, W.S.; Chung, K.; Anderson, P.; Hansbro, P.M.; Castells, M.; Stevens, R.L. Mast cell restricted mouse and human tryptase·heparin complexes hinder thrombin-induced coagulation of plasma and the generation of fibrin by proteolytically destroying fibrinogen. J. Biol. Chem. 2012, 287, 7834–7844. [Google Scholar] [CrossRef] [Green Version]
- Jordana, M.; Befus, A.D.; Newhouse, M.T.; Bienenstock, J.; Gauldie, J. Effect of histamine on proliferation of normal human adult lung fibroblasts. Thorax 1988, 43, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Karpec, D.; Rudys, R.; Leonaviciene, L.; Mackiewicz, Z.; Bradunaite, R.; Kirdaite, G.; Venalis, A. The safety and efficacy of light emitting diodes-based ultraviolet A1 phototherapy in bleomycin-induced scleroderma in mice. Adv. Med. Sci. 2018, 63, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Kabata, H.; Moro, K.; Koyasu, S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol. Rev. 2018, 286, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- McHedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.J.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlfahrt, T.; Usherenko, S.; Englbrecht, M.; Dees, C.; Weber, S.; Beyer, C.; Gelse, K.; Distler, O.; Schett, G.; Distler, J.H.W.; et al. Type 2 innate lymphoid cell counts are increased in patients with systemic sclerosis and correlate with the extent of fibrosis. Ann. Rheum. Dis. 2016, 75, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Truchetet, M.-E.; Demoures, B.; Eduardo Guimaraes, J.; Bertrand, A.; Laurent, P.; Jolivel, V.; Douchet, I.; Jacquemin, C.; Khoryati, L.; Duffau, P.; et al. Platelets Induce Thymic Stromal Lymphopoietin Production by Endothelial Cells: Contribution to Fibrosis in Human Systemic Sclerosis. Arthritis Rheumatol. 2016, 68, 2784–2794. [Google Scholar] [CrossRef]
- Manetti, M.; Guiducci, S.; Ceccarelli, C.; Romano, E.; Bellando-Randone, S.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Increased circulating levels of interleukin 33 in systemic sclerosis correlate with early disease stage and microvascular involvement. Ann. Rheum. Dis. 2011, 70, 1876–1878. [Google Scholar] [CrossRef]
- La Barbera, L.; Lo Pizzo, M.; DI Liberto, D.; Schinocca, C.; Ruscitti, P.; Giacomelli, R.; Dieli, F.; Ciccia, F.; Guggino, G. POS0326 role of the IL-25 / IL-17RB Axis in TH9 Polarization in Patients with Progressive Systemic Sclerosis. Ann. Rheum. Dis. 2021, 80, 390–391. [Google Scholar] [CrossRef]
- Laurent, P.; Allard, B.; Manicki, P.; Jolivel, V.; Levionnois, E.; Jeljeli, M.; Henrot, P.; Izotte, J.; Leleu, D.; Groppi, A.; et al. TGFβ promotes low IL10-producing ILC2 with profibrotic ability involved in skin fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2021, 80, 1594–1603. [Google Scholar] [CrossRef]
- Ah Kioon, M.D.; Tripodo, C.; Fernandez, D.; Kirou, K.A.; Spiera, R.F.; Crow, M.K.; Gordon, J.K.; Barrat, F.J. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med. 2018, 10, 8458. [Google Scholar] [CrossRef] [Green Version]
- van Bon, L.; Affandi, A.J.; Broen, J.; Christmann, R.B.; Marijnissen, R.J.; Stawski, L.; Farina, G.A.; Stifano, G.; Mathes, A.L.; Cossu, M.; et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med. 2014, 370, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafaja, S.; Valera, I.; Divekar, A.A.; Saggar, R.; Abtin, F.; Furst, D.E.; Khanna, D.; Singh, R.R. pDCs in lung and skin fibrosis in a bleomycin-induced model and patients with systemic sclerosis. JCI Insight 2018, 3, 98380. [Google Scholar] [CrossRef] [PubMed]
- Valenzi, E.; Tabib, T.; Papazoglou, A.; Sembrat, J.; Trejo Bittar, H.E.; Rojas, M.; Lafyatis, R. Disparate Interferon Signaling and Shared Aberrant Basaloid Cells in Single-Cell Profiling of Idiopathic Pulmonary Fibrosis and Systemic Sclerosis-Associated Interstitial Lung Disease. Front. Immunol. 2021, 12, 595811. [Google Scholar] [CrossRef] [PubMed]
- Skaug, B.; Assassi, S. Type I interferon dysregulation in Systemic Sclerosis. Cytokine 2020, 132, 154635. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Assassi, S. The Role of Type 1 Interferon in Systemic Sclerosis. Front. Immunol. 2013, 4, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Kroef, M.; van den Hoogen, L.L.; Mertens, J.S.; Blokland, S.L.M.; Haskett, S.; Devaprasad, A.; Carvalheiro, T.; Chouri, E.; Vazirpanah, N.; Cossu, M.; et al. Cytometry by time of flight identifies distinct signatures in patients with systemic sclerosis, systemic lupus erythematosus and Sjögrens syndrome. Eur. J. Immunol. 2020, 50, 119–129. [Google Scholar] [CrossRef]
- Alkema, W.; Koenen, H.; Kersten, B.E.; Kaffa, C.; Dinnissen, J.W.B.; Damoiseaux, J.G.M.C.; Joosten, I.; Driessen-Diks, S.; van der Molen, R.G.; Vonk, M.C.; et al. Autoantibody profiles in systemic sclerosis; a comparison of diagnostic tests. Autoimmunity 2021, 54, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Mehta, H.; Goulet, P.-O.; Nguyen, V.; Pérez, G.; Koenig, M.; Senécal, J.-L.; Sarfati, M. Topoisomerase I peptide-loaded dendritic cells induce autoantibody response as well as skin and lung fibrosis. Autoimmunity 2016, 49, 503–513. [Google Scholar] [CrossRef]
- Asano, Y. Systemic sclerosis. J. Dermatol. 2018, 45, 128–138. [Google Scholar] [CrossRef]
- O’Reilly, S.; Hugle, T.; van Laar, J.M. T cells in systemic sclerosis: A reappraisal. Rheumatology 2012, 51, 1540–1549. [Google Scholar] [CrossRef] [Green Version]
- Boin, F.; De Fanis, U.; Bartlett, S.J.; Wigley, F.M.; Rosen, A. T Cell polarization indentifies distinct clinical phenotypes in scleroderma lung disease. Arththritis Rheum. 2008, 58, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Lonati, P.A.; Brembilla, N.C.; Montanari, E.; Fontao, L.; Gabrielli, A.; Vettori, S.; Valentini, G.; Laffitte, E.; Kaya, G.; Meroni, P.L.; et al. High IL-17E and Low IL-17C dermal expression identifies a fibrosis-specific motif common to morphea and systemic sclerosis. PLoS ONE 2014, 9, e0105008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budzyńska-Włodarczyk, J.; Michalska-Jakubus, M.M.; Kowal, M.; Krasowska, D. Evaluation of serum concentrations of the selected cytokines in patients with localized scleroderma. Postep. Dermatol. I Alergol. 2016, 33, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Ayers, N.B.; Sun, C.-M.; Chen, S.-Y. Transforming growth factor-β signaling in systemic sclerosis. J. Biomed. Res. 2018, 32, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, R.S.G.; Pereira, M.C.; Dantas, A.T.; de Almeida, A.R.; Marques, C.D.L.; Rego, M.J.B.M.; Pitta, I.R.; Duarte, A.L.B.P.; Pitta, M.G.R. IL-17 and related cytokines involved in systemic sclerosis: Perspectives. Autoimmunity 2018, 51, 1–9. [Google Scholar] [CrossRef]
- Rafael-Vidal, C.; Pérez, N.; Altabás, I.; Garcia, S.; Pego-Reigosa, J.M. Blocking IL-17: A Promising Strategy in the Treatment of Systemic Rheumatic Diseases. Int. J. Mol. Sci. 2020, 21, 7100. [Google Scholar] [CrossRef]
- Kalfin, R.; Righi, A.; Del Rosso, A.; Bagchi, D.; Generini, S.; Guiducci, S.; Cerinic, M.M.; Das, D.K. Activin, a grape seed-derived proanthocyanidin extract, reduces plasma levels of oxidative stress and adhesion molecules (ICAM-1, VCAM-1 and E-selectin) in systemic sclerosis. Free Radic. Res. 2002, 36, 819–825. [Google Scholar] [CrossRef]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Auffray, C.; Avouac, J.; Allanore, Y. Regulatory T Cells in Systemic Sclerosis. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef]
- Milano, A.; Pendergrass, S.A.; Sargent, J.L.; George, L.K.; McCalmont, T.H.; Connolly, M.K.; Whitfield, M.L. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS ONE 2008, 3, e2696. [Google Scholar] [CrossRef]
- Rice, L.M.; Stifano, G.; Ziemek, J.; Lafyatis, R. Local skin gene expression reflects both local and systemic skin disease in patients with systemic sclerosis. Rheumatology 2016, 55, 377–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuschiotti, P. Current perspectives on the role of CD8+ T cells in systemic sclerosis. Immunol. Lett. 2018, 195, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Casciola-Rosen, L.; Andrade, F.; Ulanet, D.; Wong, W.B.; Rosen, A. Cleavage by granzyme B is strongly predictive of autoantigen status: Implications for initiation of autoimmunity. J. Exp. Med. 1999, 190, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Schmitt, N.; Bentebibel, S.-E.; Ranganathan, R.; Bourdery, L.; Zurawski, G.; Foucat, E.; Dullaers, M.; Oh, S.; Sabzghabaei, N.; et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011, 34, 108–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricard, L.; Jachiet, V.; Malard, F.; Ye, Y.; Stocker, N.; Rivière, S.; Senet, P.; Monfort, J.-B.; Fain, O.; Mohty, M.; et al. Circulating follicular helper T cells are increased in systemic sclerosis and promote plasmablast differentiation through the IL-21 pathway which can be inhibited by ruxolitinib. Ann. Rheum. Dis. 2019, 78, 539–550. [Google Scholar] [CrossRef]
- Taylor, D.K.; Mittereder, N.; Kuta, E.; Delaney, T.; Burwell, T.; Dacosta, K.; Zhao, W.; Cheng, L.I.; Brown, C.; Boutrin, A.; et al. T follicular helper-like cells contribute to skin fibrosis. Sci. Transl. Med. 2018, 10, 5307. [Google Scholar] [CrossRef] [Green Version]
- Gaydosik, A.M.; Tabib, T.; Domsic, R.; Khanna, D.; Lafyatis, R.; Fuschiotti, P. Single-cell transcriptome analysis identifies skin-specific T-cell responses in systemic sclerosis. Ann. Rheum. Dis. 2021, 80, 1453–1460. [Google Scholar] [CrossRef]
- Mahler, M.; Hudson, M.; Bentow, C.; Roup, F.; Beretta, L.; Pilar Simeón, C.; Guillén-Del-Castillo, A.; Casas, S.; Fritzler, M.J. Autoantibodies to stratify systemic sclerosis patients into clinically actionable subsets. Autoimmun. Rev. 2020, 19, 102583. [Google Scholar] [CrossRef]
- Melissaropoulos, K.; Daoussis, D. B cells in systemic sclerosis: From pathophysiology to treatment. Clin. Rheumatol. 2021, 40, 2621–2631. [Google Scholar] [CrossRef]
- Skaug, B.; Khanna, D.; Swindell, W.R.; Hinchcliff, M.E.; Frech, T.M.; Steen, V.D.; Hant, F.N.; Gordon, J.K.; Shah, A.A.; Zhu, L.; et al. Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann. Rheum. Dis. 2020, 79, 379–386. [Google Scholar] [CrossRef]
- Bosello, S.; Angelucci, C.; Lama, G.; Alivernini, S.; Proietti, G.; Tolusso, B.; Sica, G.; Gremese, E.; Ferraccioli, G. Characterization of inflammatory cell infiltrate of scleroderma skin: B cells and skin score progression. Arthritis Res. Ther. 2018, 20, 75. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Kobayashi, T.; Mizumaki, K.; Kano, M.; Sawada, T.; Tennichi, M.; Okamura, A.; Hamaguchi, Y.; Iwakura, Y.; Hasegawa, M.; et al. BAFF inhibition attenuates fibrosis in scleroderma by modulating the regulatory and effector B cell balance. Sci. Adv. 2018, 4, eaas9944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, T.; Hasegawa, M.; Yanaba, K.; Kodera, M.; Takehara, K.; Sato, S. Elevated serum BAFF levels in patients with systemic sclerosis: Enhanced BAFF signaling in systemic sclerosis B lymphocytes. Arthritis Rheum. 2006, 54, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Fujimoto, M.; Hasegawa, M.; Matsushita, Y.; Komura, K.; Ogawa, F.; Watanabe, R.; Takehara, K.; Sato, S. BAFF antagonist attenuates the development of skin fibrosis in tight-skin mice. J. Investig. Dermatol. 2007, 127, 2772–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, L.; Ferrier, A.; Aravena, O.; Fonseca, E.; Berendsen, J.; Biere, A.; Bueno, D.; Ramos, V.; Aguillón, J.C.; Catalán, D. Systemic Sclerosis Patients Present Alterations in the Expression of Molecules Involved in B-Cell Regulation. Front. Immunol. 2015, 6, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumoitier, N.; Chaigne, B.; Régent, A.; Lofek, S.; Mhibik, M.; Dorfmüller, P.; Terrier, B.; London, J.; Bérezné, A.; Tamas, N.; et al. Scleroderma Peripheral B Lymphocytes Secrete Interleukin-6 and Transforming Growth Factor β and Activate Fibroblasts. Arthritis Rheumatol. 2017, 69, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forestier, A.; Guerrier, T.; Jouvray, M.; Giovannelli, J.; Lefèvre, G.; Sobanski, V.; Hauspie, C.; Hachulla, E.; Hatron, P.-Y.; Zéphir, H.; et al. Altered B lymphocyte homeostasis and functions in systemic sclerosis. Autoimmun. Rev. 2018, 17, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Fujimoto, M.; Hasegawa, M.; Takehara, K. Altered blood B lymphocyte homeostasis in systemic sclerosis: Expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum. 2004, 50, 1918–1927. [Google Scholar] [CrossRef]
- Sato, S.; Hasegawa, M.; Fujimoto, M.; Tedder, T.F.; Takehara, K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J. Immunol. 2000, 165, 6635–6643. [Google Scholar] [CrossRef] [Green Version]
- Wilfong, E.M.; Vowell, K.N.; Bunn, K.E.; Rizzi, E.; Annapureddy, N.; Dudenhofer, R.B.; Barnado, A.; Bonami, R.H.; Johnson, J.E.; Crofford, L.J.; et al. CD19 + CD21lo/neg cells are increased in systemic sclerosis-associated interstitial lung disease. Clin. Exp. Med. 2021. [Google Scholar] [CrossRef]
- Asano, N.; Fujimoto, M.; Yazawa, N.; Shirasawa, S.; Hasegawa, M.; Okochi, H.; Tamaki, K.; Tedder, T.F.; Sato, S. B Lymphocyte signaling established by the CD19/CD22 loop regulates autoimmunity in the tight-skin mouse. Am. J. Pathol. 2004, 165, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Ono, N.; Steeber, D.A.; Pisetsky, D.S.; Tedder, T.F. CD19 regulates B lymphocyte signaling thresholds critical for the development of B-1 lineage cells and autoimmunity. J. Immunol. 1996, 157, 4371–4378. [Google Scholar] [PubMed]
- van Zelm, M.C.; Reisli, I.; van der Burg, M.; Castaño, D.; van Noesel, C.J.M.; van Tol, M.J.D.; Woellner, C.; Grimbacher, B.; Patiño, P.J.; van Dongen, J.J.M.; et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N. Engl. J. Med. 2006, 354, 1901–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrier, B.; Tamby, M.C.; Camoin, L.; Guilpain, P.; Bérezné, A.; Tamas, N.; Broussard, C.; Hotellier, F.; Humbert, M.; Simonneau, G.; et al. Antifibroblast antibodies from systemic sclerosis patients bind to {alpha}-enolase and are associated with interstitial lung disease. Ann. Rheum. Dis. 2010, 69, 428–433. [Google Scholar] [CrossRef]
- Baroni, S.S.; Santillo, M.; Bevilacqua, F.; Luchetti, M.; Spadoni, T.; Mancini, M.; Fraticelli, P.; Sambo, P.; Funaro, A.; Kazlauskas, A.; et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N. Engl. J. Med. 2006, 354, 2667–2676. [Google Scholar] [CrossRef] [Green Version]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Denton, C.P.; Hughes, M.; Gak, N.; Vila, J.; Buch, M.H.; Chakravarty, K.; Fligelstone, K.; Gompels, L.L.; Griffiths, B.; Herrick, A.L.; et al. BSR and BHPR guideline for the treatment of systemic sclerosis. Rheumatology 2016, 55, 1906–1910. [Google Scholar] [CrossRef] [Green Version]
- Iudici, M. What should clinicians know about the use of glucocorticoids in systemic sclerosis? Mod. Rheumatol. 2017, 27, 919–923. [Google Scholar] [CrossRef]
- Shah, A.A.; Wigley, F.M. My approach to the treatment of scleroderma. Mayo Clin. Proc. 2013, 88, 377–393. [Google Scholar] [CrossRef] [Green Version]
- Uy, G.L.; Duncavage, E.J.; Chang, G.S.; Jacoby, M.A.; Miller, C.A.; Shao, J.; Heath, S.; Elliott, K.; Fulton, R.S.; Fronick, C.C.; et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. HHS Public Access 2017, 31, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, I.; Kratz, A.K.; Alexanderson, H.; Patarroyo, M. Decreased expression of interleukin-1α, interleukin-1β, and cell adhesion molecules in muscle tissue following corticosteroid treatment in patients with polymyositis and dermatomyositis. Arthritis Rheum. 2000, 43, 336–348. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A. Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998, 41, 1613–1619. [Google Scholar] [CrossRef]
- Cozzi, F.; Marson, P.; Cardarelli, S.; Favaro, M.; Tison, T.; Tonello, M.; Pigatto, E.; De Silvestro, G.; Punzi, L.; Doria, A. Prognosis of scleroderma renal crisis: A long-term observational study. Nephrol. Dial. Transplant. 2012, 27, 4398–4403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanatta, E.; Polito, P.; Favaro, M.; Larosa, M.; Marson, P.; Cozzi, F.; Doria, A. Therapy of scleroderma renal crisis: State of the art. Autoimmun. Rev. 2018, 17, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, V.; Pavlovic-Popovic, Z.; Vukmirovic, S.; Cvejic, J.; Mooranian, A.; Al-Salami, H.; Mikov, M.; Golocorbin-Kon, S. Molecular mechanism of action and pharmacokinetic properties of methotrexate. Mol. Biol. Rep. 2020, 47, 4699–4708. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Aune, T.M. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef]
- Pope, J.E.; Bellamy, N.; Seibold, J.R.; Baron, M.; Ellman, M.; Carette, S.; Smith, C.D.; Chalmers, I.M.; Hong, P.; O’Hanlon, D.; et al. A randomized, controlled trial of methotrexate versus placebo in early diffuse scleroderma. Arthritis Rheum. 2001, 44, 1351–1358. [Google Scholar] [CrossRef]
- van den Hoogen, F.H.; Boerbooms, A.M.; Swaak, A.J.; Rasker, J.J.; van Lier, H.J.; van de Putte, L.B. Comparison of methotrexate with placebo in the treatment of systemic sclerosis: A 24 week randomized double-blind trial, followed by a 24 week observational trial. Br. J. Rheumatol. 1996, 35, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Wachsmuth, L.P.; Patterson, M.T.; Eckhaus, M.A.; Venzon, D.J.; Gress, R.E.; Kanakry, C.G. Posttransplantation cyclophosphamide prevents graft-versus-host disease by inducing alloreactive T cell dysfunction and suppression. J. Clin. Investig. 2019, 129, 2357–2373. [Google Scholar] [CrossRef]
- Bruni, C.; Shirai, Y.; Kuwana, M.; Matucci-Cerinic, M. Cyclophosphamide: Similarities and differences in the treatment of SSc and SLE. Lupus 2019, 28, 571–574. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Elashoff, R.; Clements, P.J.; Goldin, J.; Roth, M.D.; Furst, D.E.; Arriola, E.; Silver, R.; Strange, C.; Bolster, M.; et al. Cyclophosphamide versus Placebo in Scleroderma Lung Disease. N. Engl. J. Med. 2006, 354, 2655–2666. [Google Scholar] [CrossRef] [Green Version]
- Goldin, J.; Elashoff, R.; Kim, H.J.; Yan, X.; Lynch, D.; Strollo, D.; Roth, M.D.; Clements, P.; Furst, D.E.; Khanna, D.; et al. Treatment of scleroderma-interstitial lung disease with cyclophosphamide is associated with less progressive fibrosis on serial thoracic high-resolution CT scan than placebo: Findings from the scleroderma lung study. Chest 2009, 136, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Broen, J.C.A.; van Laar, J.M. Mycophenolate mofetil, azathioprine and tacrolimus: Mechanisms in rheumatology. Nat. Rev. Rheumatol. 2020, 16, 167–178. [Google Scholar] [CrossRef]
- Gernert, M.; Tony, H.-P.; Schwaneck, E.C.; Gadeholt, O.; Fröhlich, M.; Portegys, J.; Strunz, P.-P.; Schmalzing, M. Lymphocyte subsets in the peripheral blood are disturbed in systemic sclerosis patients and can be changed by immunosuppressive medication. Rheumatol. Int. 2021, 1–9. [Google Scholar] [CrossRef]
- Hinchcliff, M.; Toledo, D.M.; Taroni, J.N.; Wood, T.A.; Franks, J.M.; Ball, M.S.; Hoffmann, A.; Amin, S.M.; Tan, A.U.; Tom, K.; et al. Mycophenolate Mofetil Treatment of Systemic Sclerosis Reduces Myeloid Cell Numbers and Attenuates the Inflammatory Gene Signature in Skin. J. Investig. Dermatol. 2018, 138, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Morath, C.; Reuter, H.; Simon, V.; Krautkramer, E.; Muranyi, W.; Schwenger, V.; Goulimari, P.; Grosse, R.; Hahn, M.; Lichter, P.; et al. Effects of mycophenolic acid on human fibroblast proliferation, migration and adhesion in vitro and in vivo. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2008, 8, 1786–1797. [Google Scholar] [CrossRef]
- Volkmann, E.R.; Tashkin, D.P.; Li, N.; Roth, M.D.; Khanna, D.; Hoffmann-Vold, A.-M.; Kim, G.; Goldin, J.; Clements, P.J.; Furst, D.E.; et al. Mycophenolate Mofetil Versus Placebo for Systemic Sclerosis-Related Interstitial Lung Disease: An Analysis of Scleroderma Lung Studies I and II. Arthritis Rheumatol. 2017, 69, 1451–1460. [Google Scholar] [CrossRef] [Green Version]
- Naidu, G.S.R.S.N.K.; Sharma, S.K.; Adarsh, M.B.; Dhir, V.; Sinha, A.; Dhooria, S.; Jain, S. Effect of mycophenolate mofetil (MMF) on systemic sclerosis-related interstitial lung disease with mildly impaired lung function: A double-blind, placebo-controlled, randomized trial. Rheumatol. Int. 2020, 40, 207–216. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Roth, M.D.; Clements, P.J.; Furst, D.E.; Khanna, D.; Kleerup, E.C.; Goldin, J.; Arriola, E.; Volkmann, E.R.; Kafaja, S.; et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): A randomised controlled, double-blind, parallel group trial. Lancet. Respir. Med. 2016, 4, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Gerbino, A.J.; Goss, C.H.; Molitor, J.A. Effect of mycophenolate mofetil on pulmonary function in scleroderma-associated interstitial lung disease. Chest 2008, 133, 455–460. [Google Scholar] [CrossRef]
- Morganroth, P.A.; Kreider, M.E.; Werth, V.P. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res. 2010, 62, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Stratton, R.J.; Wilson, H.; Black, C.M. Pilot study of anti-thymocyte globulin plus mycophenolate mofetil in recent-onset diffuse scleroderma. Rheumatology 2001, 40, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Simeón-Aznar, C.P.; Fonollosa-Plá, V.; Tolosa-Vilella, C.; Selva-O’Callaghan, A.; Solans-Laqué, R.; Vilardell-Tarrés, M. Effect of mycophenolate sodium in scleroderma-related interstitial lung disease. Clin. Rheumatol. 2011, 30, 1393–1398. [Google Scholar] [CrossRef]
- Yilmaz, N.; Can, M.; Kocakaya, D.; Karakurt, S.; Yavuz, S. Two-year experience with mycophenolate mofetil in patients with scleroderma lung disease: A case series. Int. J. Rheum. Dis. 2014, 17, 923–928. [Google Scholar] [CrossRef]
- Binks, M.; Passweg, J.R.; Furst, D.; McSweeney, P.; Sullivan, K.; Besenthal, C.; Finke, J.; Peter, H.H.; van Laar, J.; Breedveld, F.C.; et al. Phase I/II trial of autologous stem cell transplantation in systemic sclerosis: Procedure related mortality and impact on skin disease. Ann. Rheum. Dis. 2001, 60, 577–584. [Google Scholar] [CrossRef]
- Walker, U.A.; Saketkoo, L.A.; Distler, O. Haematopoietic stem cell transplantation in systemic sclerosis. RMD Open 2018, 4, e000533. [Google Scholar] [CrossRef] [Green Version]
- Zeher, M.; Papp, G.; Nakken, B.; Szodoray, P. Hematopoietic stem cell transplantation in autoimmune disorders: From immune-regulatory processes to clinical implications. Autoimmun. Rev. 2017, 16, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Farge, D.; Passweg, J.; van Laar, J.M.; Marjanovic, Z.; Besenthal, C.; Finke, J.; Peter, H.H.; Breedveld, F.C.; Fibbe, W.E.; Black, C.; et al. Autologous stem cell transplantation in the treatment of systemic sclerosis: Report from the EBMT/EULAR Registry. Ann. Rheum. Dis. 2004, 63, 974–981. [Google Scholar] [CrossRef]
- Burt, R.K.; Shah, S.J.; Dill, K.; Grant, T.; Gheorghiade, M.; Schroeder, J.; Craig, R.; Hirano, I.; Marshall, K.; Ruderman, E.; et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): An open-label, randomised phase 2 trial. Lancet 2011, 378, 498–506. [Google Scholar] [CrossRef]
- van Laar, J.M.; Farge, D.; Sont, J.K.; Naraghi, K.; Marjanovic, Z.; Larghero, J.; Schuerwegh, A.J.; Marijt, E.W.A.; Vonk, M.C.; Schattenberg, A.V.; et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: A randomized clinical trial. JAMA 2014, 311, 2490–2498. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Goldmuntz, E.A.; Keyes-Elstein, L.; McSweeney, P.A.; Pinckney, A.; Welch, B.; Mayes, M.D.; Nash, R.A.; Crofford, L.J.; Eggleston, B.; et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma. N. Engl. J. Med. 2018, 378, 35–47. [Google Scholar] [CrossRef]
- Katsiari, C.G.; Simopoulou, T.; Alexiou, I.; Sakkas, L.I. Immunotherapy of systemic sclerosis. Hum. Vaccin. Immunother. 2018, 14, 2559–2567. [Google Scholar] [CrossRef]
- Ramwadhdoebe, T.H.; van Baarsen, L.G.M.; Boumans, M.J.H.; Bruijnen, S.T.G.; Safy, M.; Berger, F.H.; Semmelink, J.F.; van der Laken, C.J.; Gerlag, D.M.; Thurlings, R.M.; et al. Effect of rituximab treatment on T and B cell subsets in lymph node biopsies of patients with rheumatoid arthritis. Rheumatology 2019, 58, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Thurlings, R.M.; Vos, K.; Wijbrandts, C.A.; Zwinderman, A.H.; Gerlag, D.M.; Tak, P.P. Synovial tissue response to rituximab: Mechanism of action and identification of biomarkers of response. Ann. Rheum. Dis. 2008, 67, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Bergantini, L.; d’Alessandro, M.; Cameli, P.; Vietri, L.; Vagaggini, C.; Perrone, A.; Sestini, P.; Frediani, B.; Bargagli, E. Effects of rituximab therapy on B cell differentiation and depletion. Clin. Rheumatol. 2020, 39, 1415–1421. [Google Scholar] [CrossRef]
- Boonstra, M.; Meijs, J.; Dorjée, A.L.; Marsan, N.A.; Schouffoer, A.; Ninaber, M.K.; Quint, K.D.; Bonte-Mineur, F.; Huizinga, T.W.J.; Scherer, H.U.; et al. Rituximab in early systemic sclerosis. RMD Open 2017, 3, e000384. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.; Van Praet, J.T.; Vandooren, B.; Van Der Cruyssen, B.; Naeyaert, J.M.; Decuman, S.; Elewaut, D.; De Keyser, F. Rituximab in diffuse cutaneous systemic sclerosis: An open-label clinical and histopathological study. Ann. Rheum. Dis. 2010, 69, 193–197. [Google Scholar] [CrossRef]
- Bosello, S.; De Santis, M.; Lama, G.; Spanò, C.; Angelucci, C.; Tolusso, B.; Sica, G.; Ferraccioli, G. B cell depletion in diffuse progressive systemic sclerosis: Safety, skin score modification and IL-6 modulation in an up to thirty-six months follow-up open-label trial. Arthritis Res. Ther. 2010, 12, R54. [Google Scholar] [CrossRef] [Green Version]
- Daoussis, D.; Liossis, S.-N.C.; Tsamandas, A.C.; Kalogeropoulou, C.; Kazantzi, A.; Sirinian, C.; Karampetsou, M.; Yiannopoulos, G.; Andonopoulos, A.P. Experience with rituximab in scleroderma: Results from a 1-year, proof-of-principle study. Rheumatology 2010, 49, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Daoussis, D.; Melissaropoulos, K.; Sakellaropoulos, G.; Antonopoulos, I.; Markatseli, T.E.; Simopoulou, T.; Georgiou, P.; Andonopoulos, A.P.; Drosos, A.A.; Sakkas, L.; et al. A multicenter, open-label, comparative study of B-cell depletion therapy with Rituximab for systemic sclerosis-associated interstitial lung disease. Semin. Arthritis Rheum. 2017, 46, 625–631. [Google Scholar] [CrossRef]
- Sircar, G.; Goswami, R.P.; Sircar, D.; Ghosh, A.; Ghosh, P. Intravenous cyclophosphamide vs rituximab for the treatment of early diffuse scleroderma lung disease: Open label, randomized, controlled trial. Rheumatology 2018, 57, 2106–2113. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Badesch, D.; Chung, L.; Domsic, R.T.; Medsger, T.; Pinckney, A.; Keyes-Elstein, L.; D’Aveta, C.; Spychala, M.; White, R.J.; et al. Safety and Efficacy of B-Cell Depletion with Rituximab for the Treatment of Systemic Sclerosis-associated Pulmonary Arterial Hypertension: A Multicenter, Double-Blind, Randomized, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2021, 204, 209–221. [Google Scholar] [CrossRef]
- Lafyatis, R.; Kissin, E.; York, M.; Farina, G.; Viger, K.; Fritzler, M.J.; Merkel, P.A.; Simms, R.W. B cell depletion with rituximab in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2009, 60, 578–583. [Google Scholar] [CrossRef] [Green Version]
- Jordan, S.; Distler, J.H.W.; Maurer, B.; Huscher, D.; Van Laar, J.M.; Allanore, Y.; Distler, O.; Kvien, T.K.; Airo, P.; Sancho, J.J.A.; et al. Effects and safety of rituximab in systemic sclerosis: An analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann. Rheum. Dis. 2015, 74, 1188–1194. [Google Scholar] [CrossRef]
- Moazedi-Fuerst, F.C.; Kielhauser, S.M.; Brickmann, K.; Hermann, J.; Lutfi, A.; Meilinger, M.; Brezinschek, H.P.; Graninger, W.B. Rituximab for systemic sclerosis: Arrest of pulmonary disease progression in five cases. Results of a lower dosage and shorter interval regimen. Scand. J. Rheumatol. 2014, 43, 257–258. [Google Scholar] [CrossRef] [Green Version]
- Yılmaz, D.D.; Borekci, S.; Musellim, B. Comparison of the effectiveness of cyclophosphamide and rituximab treatment in patients with systemic sclerosis-related interstitial lung diseases: A retrospective, observational cohort study. Clin. Rheumatol. 2021, 40, 4071–4079. [Google Scholar] [CrossRef]
- Giuggioli, D.; Lumetti, F.; Colaci, M.; Fallahi, P.; Antonelli, A.; Ferri, C. Rituximab in the treatment of patients with systemic sclerosis. Our experience and review of the literature. Autoimmun. Rev. 2015, 14, 1072–1078. [Google Scholar] [CrossRef]
- Schiopu, E.; Chatterjee, S.; Hsu, V.; Flor, A.; Cimbora, D.; Patra, K.; Yao, W.; Li, J.; Streicher, K.; McKeever, K.; et al. Safety and tolerability of an anti-CD19 monoclonal antibody, MEDI-551, in subjects with systemic sclerosis: A phase I, randomized, placebo-controlled, escalating single-dose study. Arthritis Res. Ther. 2016, 18, 131. [Google Scholar] [CrossRef] [Green Version]
- Streicher, K.; Sridhar, S.; Kuziora, M.; Morehouse, C.A.; Higgs, B.W.; Sebastian, Y.; Groves, C.J.; Pilataxi, F.; Brohawn, P.Z.; Herbst, R.; et al. Baseline Plasma Cell Gene Signature Predicts Improvement in Systemic Sclerosis Skin Scores Following Treatment With Inebilizumab (MEDI-551) and Correlates with Disease Activity in Systemic Lupus Erythematosus and Chronic Obstructive Pulmonary Disease. Arthritis Rheumatol. 2018, 70, 2087–2095. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro Castro, S.; Isenberg, D.A. Belimumab in systemic lupus erythematosus (SLE): Evidence-to-date and clinical usefulness. Ther. Adv. Musculoskelet. Dis. 2017, 9, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.K.; Martyanov, V.; Franks, J.M.; Bernstein, E.J.; Szymonifka, J.; Magro, C.; Wildman, H.F.; Wood, T.A.; Whitfield, M.L.; Spiera, R.F. Belimumab for the Treatment of Early Diffuse Systemic Sclerosis: Results of a Randomized, Double-Blind, Placebo-Controlled, Pilot Trial. Arthritis Rheumatol. 2018, 70, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell. Physiol. 2019, 234, 10018–10031. [Google Scholar] [CrossRef]
- Nie, J.; Li, Y.Y.; Zheng, S.G.; Tsun, A.; Li, B. FOXP3+ Treg Cells and Gender Bias in Autoimmune Diseases. Front. Immunol. 2015, 6, 493. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Nakao, A.; Nakano, H.; Takahashi, F.; Takahashi, K.; Shimozato, O.; Takeda, K.; Yagita, H.; Okumura, K. Impairment of Bleomycin-Induced Lung Fibrosis in CD28-Deficient Mice. J. Immunol. 2001, 167, 1977–1981. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, E.F.; Martyanov, V.; Fiorentino, D.; Wood, T.A.; Haddon, D.J.; Jarrell, J.A.; Utz, P.J.; Genovese, M.C.; Whitfield, M.L.; Chung, L. Gene expression changes reflect clinical response in a placebo-controlled randomized trial of abatacept in patients with diffuse cutaneous systemic sclerosis. Arthritis Res. Ther. 2015, 17, 159. [Google Scholar] [CrossRef] [Green Version]
- Khanna, D.; Spino, C.; Johnson, S.; Chung, L.; Whitfield, M.L.; Denton, C.P.; Berrocal, V.; Franks, J.; Mehta, B.; Molitor, J.; et al. Abatacept in Early Diffuse Cutaneous Systemic Sclerosis: Results of a Phase II Investigator-Initiated, Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial. Arthritis Rheumatol. 2020, 72, 125–136. [Google Scholar] [CrossRef]
- Elhai, M.; Meunier, M.; Matucci-Cerinic, M.; Maurer, B.; Riemekasten, G.; Leturcq, T.; Pellerito, R.; Von Mühlen, C.A.; Vacca, A.; Airo, P.; et al. Outcomes of patients with systemic sclerosis-associated polyarthritis and myopathy treated with tocilizumab or abatacept: A EUSTAR observational study. Ann. Rheum. Dis. 2013, 72, 1217–1220. [Google Scholar] [CrossRef]
- Castellví, I.; Elhai, M.; Bruni, C.; Airò, P.; Jordan, S.; Beretta, L.; Codullo, V.; Montecucco, C.M.; Bokarewa, M.; Iannonne, F.; et al. Safety and effectiveness of abatacept in systemic sclerosis: The EUSTAR experience. Semin. Arthritis Rheum. 2020, 50, 1489–1493. [Google Scholar] [CrossRef]
- Turnier, J.L.; Brunner, H.I. Tocilizumab for treating juvenile idiopathic arthritis. Expert Opin. Biol. Ther. 2016, 16, 559–566. [Google Scholar] [CrossRef]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef] [Green Version]
- Khanna, D.; Denton, C.P.; Lin, C.J.F.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: Results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann. Rheum. Dis. 2018, 77, 212–220. [Google Scholar] [CrossRef]
- Denton, C.P.; Ong, V.H.; Xu, S.; Chen-Harris, H.; Modrusan, Z.; Lafyatis, R.; Khanna, D.; Jahreis, A.; Siegel, J.; Sornasse, T. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: Insights from the faSScinate clinical trial in systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1362–1371. [Google Scholar] [CrossRef]
- Kono, M.; Yasuda, S.; Kono, M.; Atsumi, T. Tocilizumab reduced production of systemic sclerosis-related autoantibodies and anti-cyclic citrullinated protein antibodies in two patients with overlapping systemic sclerosis and rheumatoid arthritis. Scand. J. Rheumatol. 2018, 47, 248–250. [Google Scholar] [CrossRef]
- Shima, Y.; Hosen, N.; Hirano, T.; Arimitsu, J.; Nishida, S.; Hagihara, K.; Narazaki, M.; Ogata, A.; Tanaka, T.; Kishimoto, T.; et al. Expansion of range of joint motion following treatment of systemic sclerosis with tocilizumab. Mod. Rheumatol. 2013, 25, 134–137. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M.; Murakawa, Y.; Matsumura, T.; Matsumoto, O.; Taira, M.; Moriyama, M.; Sumita, Y.; Yamaguchi, S. A case of overlap syndrome successfully treated with tocilizumab: A hopeful treatment strategy for refractory dermatomyositis? Rheumatology 2014, 53, 1907–1908. [Google Scholar] [CrossRef] [Green Version]
- Fernandes das Neves, M.; Oliveira, S.; Amaral, M.C.; Delgado Alves, J. Treatment of systemic sclerosis with tocilizumab. Rheumatology 2015, 54, 371–372. [Google Scholar] [CrossRef] [Green Version]
- Saito, E.; Sato, S.; Nogi, S.; Sasaki, N.; Chinen, N.; Honda, K.; Wakabayashi, T.; Yamada, C.; Nakamura, N.; Suzuki, Y. A case of rheumatoid arthritis and limited systemic sclerosis overlap successfully treated with tocilizumab for arthritis and concomitant generalized lymphadenopathy and primary biliary cirrhosis. Case Rep. Rheumatol. 2014, 2014, 386328. [Google Scholar] [CrossRef] [Green Version]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef]
- Roofeh, D.; Lin, C.J.F.; Goldin, J.; Kim, G.H.; Furst, D.E.; Denton, C.P.; Huang, S.; Khanna, D. Tocilizumab Prevents Progression of Early Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2021, 73, 1301–1310. [Google Scholar] [CrossRef]
- Hoffman, H.M. Rilonacept for the treatment of cryopyrin-associated periodic syndromes (CAPS). Expert Opin. Biol. Ther. 2009, 9, 519–531. [Google Scholar] [CrossRef]
- Mantero, J.C.; Kishore, N.; Ziemek, J.; Stifano, G.; Zammitti, C.; Khanna, D.; Gordon, J.K.; Spiera, R.; Zhang, Y.; Simms, R.W.; et al. Randomised, double-blind, placebo-controlled trial of IL1-trap, rilonacept, in systemic sclerosis. A phase I/II biomarker trial. Clin. Exp. Rheumatol. 2018, 36 (Suppl. S1), 146–149. [Google Scholar]
- Tan, J.; Yang, S.; Wu, W. Basiliximab (Simulect) reduces acute rejection among sensitized kidney allograft recipients. Transplant. Proc. 2005, 37, 903–905. [Google Scholar] [CrossRef]
- Scherer, H.U.; Burmester, G.-R.; Riemekasten, G. Targeting activated T cells: Successful use of anti-CD25 monoclonal antibody basiliximab in a patient with systemic sclerosis. Ann. Rheum. Dis. 2006, 65, 1245–1247. [Google Scholar] [CrossRef]
- Becker, M.O.; Brückner, C.; Scherer, H.U.; Wassermann, N.; Humrich, J.Y.; Hanitsch, L.G.; Schneider, U.; Kawald, A.; Hanke, K.; Burmester, G.R.; et al. The monoclonal anti-CD25 antibody basiliximab for the treatment of progressive systemic sclerosis: An open-label study. Ann. Rheum. Dis. 2011, 70, 1340–1341. [Google Scholar] [CrossRef]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef]
- Denton, C.P.; Merkel, P.A.; Furst, D.E.; Khanna, D.; Emery, P.; Hsu, V.M.; Silliman, N.; Streisand, J.; Powell, J.; Akesson, A.; et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: A multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007, 56, 323–333. [Google Scholar] [CrossRef]
- Gerber, E.E.; Gallo, E.M.; Fontana, S.C.; Davis, E.C.; Wigley, F.M.; Huso, D.L.; Dietz, H.C. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature 2013, 503, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, S.T.; Veijola, A.; Karvonen, H.; Lappi-Blanco, E.; Sormunen, R.; Korpela, S.; Zagai, U.; Sköld, M.C.; Kaarteenaho, R. Pirfenidone and nintedanib modulate properties of fibroblasts and myofibroblasts in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 14. [Google Scholar] [CrossRef] [Green Version]
- Khanna, D.; Albera, C.; Fischer, A.; Khalidi, N.; Raghu, G.; Chung, L.; Chen, D.; Schiopu, E.; Tagliaferri, M.; Seibold, J.R.; et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: The LOTUSS trial. J. Rheumatol. 2016, 43, 1672–1679. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Feng, R.E.; Li, S.; Xu, K.; Bi, Y.L.; Xu, Z.J. A case report: The efficacy of pirfenidone in a Chinese patient with progressive systemic sclerosis-associated interstitial lung disease: A CARE-compliant article. Medicine 2016, 95, e4113. [Google Scholar] [CrossRef]
- Miura, Y.; Saito, T.; Fujita, K.; Tsunoda, Y.; Tanaka, T.; Takoi, H.; Yatagai, Y.; Rin, S.; Sekine, A.; Hayashihara, K.; et al. Clinical experience with pirfenidone in five patients with scleroderma-related interstitial lung disease. Sarcoidosis Vasc. Diffus. Lung Dis. 2014, 31, 235–238. [Google Scholar]
- Zarrin, A.A.; Bao, K.; Lupardus, P.; Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 2021, 20, 39–63. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Tamura, T.; Koch, A. Tyrosine Kinases as Targets for Anti-Inflammatory Therapy. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2007, 6, 47–60. [Google Scholar] [CrossRef]
- Iwamoto, N.; Distler, J.H.W.; Distler, O. Tyrosine kinase inhibitors in the treatment of systemic sclerosis: From animal models to clinical trials. Curr. Rheumatol. Rep. 2011, 13, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Gajski, G.; Gerić, M.; Domijan, A.-M.; Golubović, I.; Garaj-Vrhovac, V. Evaluation of oxidative stress responses in human circulating blood cells after imatinib mesylate treatment—Implications to its mechanism of action. Saudi Pharm. J. 2019, 27, 1216–1221. [Google Scholar] [CrossRef]
- Haddon, D.J.; Wand, H.E.; Jarrell, J.A.; Spiera, R.F.; Utz, P.J.; Gordon, J.K.; Chung, L.S. Proteomic Analysis of Sera from Individuals with Diffuse Cutaneous Systemic Sclerosis Reveals a Multianalyte Signature Associated with Clinical Improvement during Imatinib Mesylate Treatment. J. Rheumatol. 2017, 44, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Spiera, R.F.; Gordon, J.K.; Mersten, J.N.; Magro, C.M.; Mehta, M.; Wildman, H.F.; Kloiber, S.; Kirou, K.A.; Lyman, S.; Crow, M.K. Imatinib mesylate (Gleevec) in the treatment of diffuse cutaneous systemic sclerosis: Results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann. Rheum. Dis. 2011, 70, 1003–1009. [Google Scholar] [CrossRef] [Green Version]
- Divekar, A.A.; Khanna, D.; Abtin, F.; Maranian, P.; Saggar, R.; Saggar, R.; Furst, D.E.; Singh, R.R. Treatment with imatinib results in reduced IL-4-producing T cells, but increased CD4(+) T cells in the broncho-alveolar lavage of patients with systemic sclerosis. Clin. Immunol. 2011, 141, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.; Fiorentino, D.F.; Benbarak, M.J.; Adler, A.S.; Mariano, M.M.; Paniagua, R.T.; Milano, A.; Connolly, M.K.; Ratiner, B.D.; Wiskocil, R.L.; et al. Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheum. 2009, 60, 584–591. [Google Scholar] [CrossRef]
- Fraticelli, P.; Gabrielli, B.; Pomponio, G.; Valentini, G.; Bosello, S.; Riboldi, P.; Gerosa, M.; Faggioli, P.; Giacomelli, R.; Del Papa, N.; et al. Low-dose oral imatinib in the treatment of systemic sclerosis interstitial lung disease unresponsive to cyclophosphamide: A phase II pilot study. Arthritis Res. Ther. 2014, 16, R144. [Google Scholar] [CrossRef] [Green Version]
- Khanna, D.; Saggar, R.; Mayes, M.D.; Abtin, F.; Clements, P.J.; Maranian, P.; Assassi, S.; Saggar, R.; Singh, R.R.; Furst, D.E. A one-year, phase I/IIa, open-label pilot trial of imatinib mesylate in the treatment of systemic sclerosis-associated active interstitial lung disease. Arthritis Rheum. 2011, 63, 3540–3546. [Google Scholar] [CrossRef]
- Bradeen, H.A.; Eide, C.A.; O’Hare, T.; Johnson, K.J.; Willis, S.G.; Lee, F.Y.; Druker, B.J.; Deininger, M.W. Comparison of imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: High efficacy of drug combinations. Blood 2006, 108, 2332–2338. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.K.; Martyanov, V.; Magro, C.; Wildman, H.F.; Wood, T.A.; Huang, W.-T.; Crow, M.K.; Whitfield, M.L.; Spiera, R.F. Nilotinib (TasignaTM) in the treatment of early diffuse systemic sclerosis: An open-label, pilot clinical trial. Arthritis Res. Ther. 2015, 17, 213. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Distler, J.H.W.; Redente, E.F.; Riches, D.W.H.; Stowasser, S.; Schlenker-Herceg, R.; Maher, T.M.; Kolb, M. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2019, 54, 1900161. [Google Scholar] [CrossRef]
- Sato, S.; Shinohara, S.; Hayashi, S.; Morizumi, S.; Abe, S.; Okazaki, H.; Chen, Y.; Goto, H.; Aono, Y.; Ogawa, H.; et al. Anti-fibrotic efficacy of nintedanib in pulmonary fibrosis via the inhibition of fibrocyte activity. Respir. Res. 2017, 18, 172. [Google Scholar] [CrossRef]
- Cutolo, M.; Gotelli, E.; Montagna, P.; Tardito, S.; Paolino, S.; Pizzorni, C.; Sulli, A.; Smith, V.; Soldano, S. Nintedanib downregulates the transition of cultured systemic sclerosis fibrocytes into myofibroblasts and their pro-fibrotic activity. Arthritis Res. Ther. 2021, 23, 205. [Google Scholar] [CrossRef]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef]
- Bruni, T.; Varone, F. The adoption of nintedanib in systemic sclerosis: The SENSCIS study. Breathe 2020, 16, 200005. [Google Scholar] [CrossRef]
- Wang, W.; Bhattacharyya, S.; Marangoni, R.G.; Carns, M.; Dennis-Aren, K.; Yeldandi, A.; Wei, J.; Varga, J. The JAK/STAT pathway is activated in systemic sclerosis and is effectively targeted by tofacitinib. J. Scleroderma Relat. Disord. 2019, 5, 40–50. [Google Scholar] [CrossRef]
- Kucharz, E.J.; Stajszczyk, M.; Kotulska-Kucharz, A.; Batko, B.; Brzosko, M.; Jeka, S.; Leszczyński, P.; Majdan, M.; Olesińska, M.; Samborski, W.; et al. Tofacitinib in the treatment of patients with rheumatoid arthritis: Position statement of experts of the Polish Society for Rheumatology. Reumatologia 2018, 56, 203–211. [Google Scholar] [CrossRef]
- You, H.; Xu, D.; Hou, Y.; Zhou, J.; Wang, Q.; Li, M.; Zeng, X. Tofacitinib as a possible treatment for skin thickening in diffuse cutaneous systemic sclerosis. Rheumatology 2021, 60, 2472–2477. [Google Scholar] [CrossRef]
- Karalilova, R.V.; Batalov, Z.A.; Sapundzhieva, T.L.; Matucci-Cerinic, M.; Batalov, A.Z. Tofacitinib in the treatment of skin and musculoskeletal involvement in patients with systemic sclerosis, evaluated by ultrasound. Rheumatol. Int. 2021, 41, 1743–1753. [Google Scholar] [CrossRef]
- Vonk, M.C.; Smith, V.; Sfikakis, P.P.; Cutolo, M.; del Galdo, F.; Seibold, J.R. Pharmacological treatments for SSc-ILD: Systematic review and critical appraisal of the evidence. Autoimmun. Rev. 2021, 20, 102978. [Google Scholar] [CrossRef]
- Lepri, G.; Hughes, M.; Bruni, C.; Cerinic, M.M.; Randone, S.B. Recent advances steer the future of systemic sclerosis toward precision medicine. Clin. Rheumatol. 2020, 39, 1–4. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadimitriou, T.-I.; van Caam, A.; van der Kraan, P.M.; Thurlings, R.M. Therapeutic Options for Systemic Sclerosis: Current and Future Perspectives in Tackling Immune-Mediated Fibrosis. Biomedicines 2022, 10, 316. https://doi.org/10.3390/biomedicines10020316
Papadimitriou T-I, van Caam A, van der Kraan PM, Thurlings RM. Therapeutic Options for Systemic Sclerosis: Current and Future Perspectives in Tackling Immune-Mediated Fibrosis. Biomedicines. 2022; 10(2):316. https://doi.org/10.3390/biomedicines10020316
Chicago/Turabian StylePapadimitriou, Theodoros-Ioannis, Arjan van Caam, Peter M. van der Kraan, and Rogier M. Thurlings. 2022. "Therapeutic Options for Systemic Sclerosis: Current and Future Perspectives in Tackling Immune-Mediated Fibrosis" Biomedicines 10, no. 2: 316. https://doi.org/10.3390/biomedicines10020316