
Teenage-Onset Colorectal Cancers in a Digenic Cancer Predisposition Syndrome Provide Clues for the Interaction between Mismatch Repair and Polymerase δ Proofreading Deficiency in Tumorigenesis

 , , and
, , and 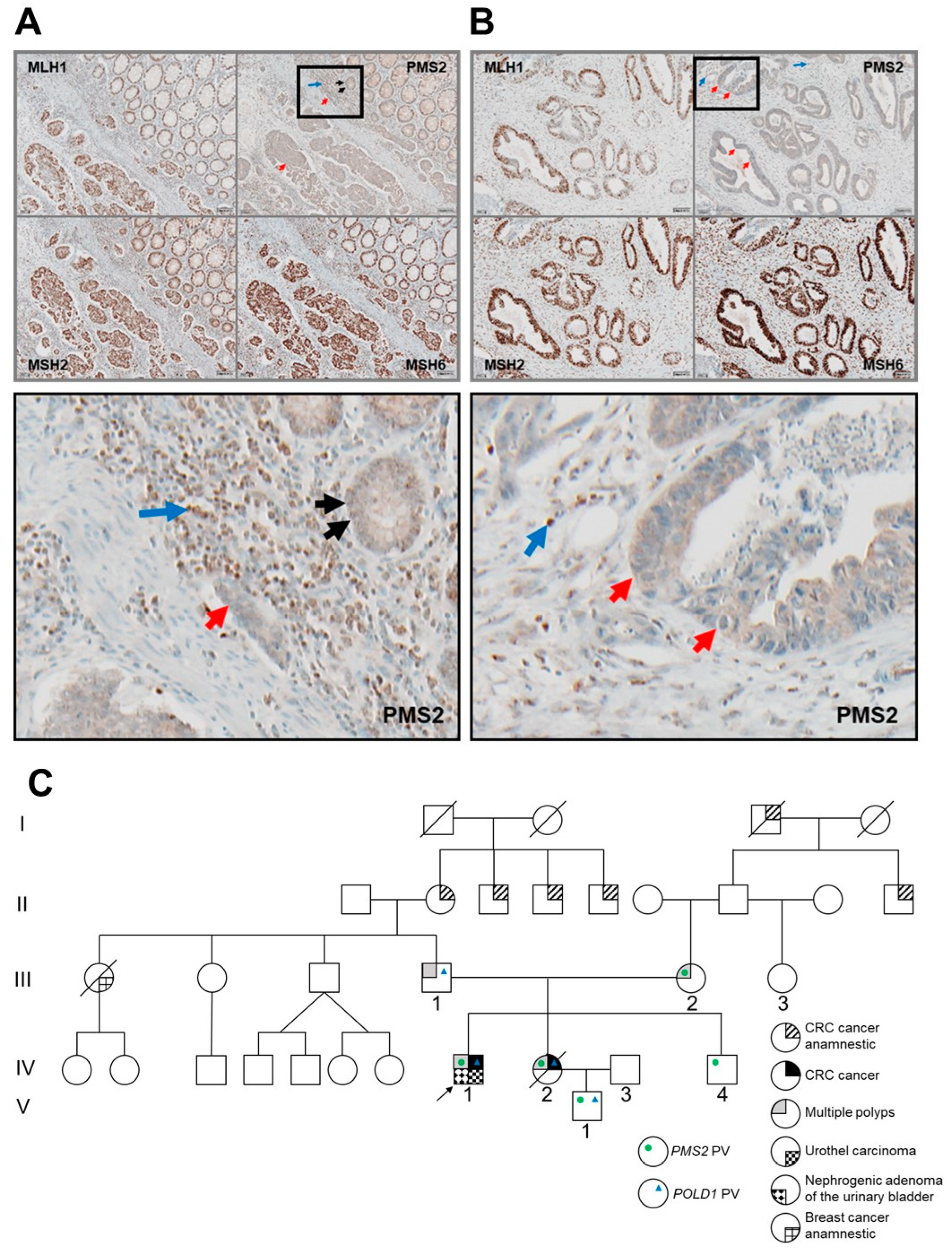{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials & Methods
2.1. Ethics
2.2. Immunohistochemistry
2.3. Tumor Microsatellite Instability (MSI) Analysis
2.4. DNA Extraction for Sequencing
2.5. RNA Extraction
2.6. Constitutional MSI Analysis
2.7. Multiplex-Ligation-Dependent Probe Amplification Analysis
2.8. Transcript Analysis
2.9. Deletion-Spanning PCR and Sequencing
2.10. Determination of Constitutional Variants by Panel Next-Generation Sequencing (NGS) of Blood DNA
2.11. Determination of Somatic Variants by Whole-Exome NGS of Tumor and Blood DNA
2.12. Calculation of Tumor Mutational Burden
2.13. Mutational Signature Analysis
3. Results
3.1. Clinical History of Two AYA-CRC Siblings
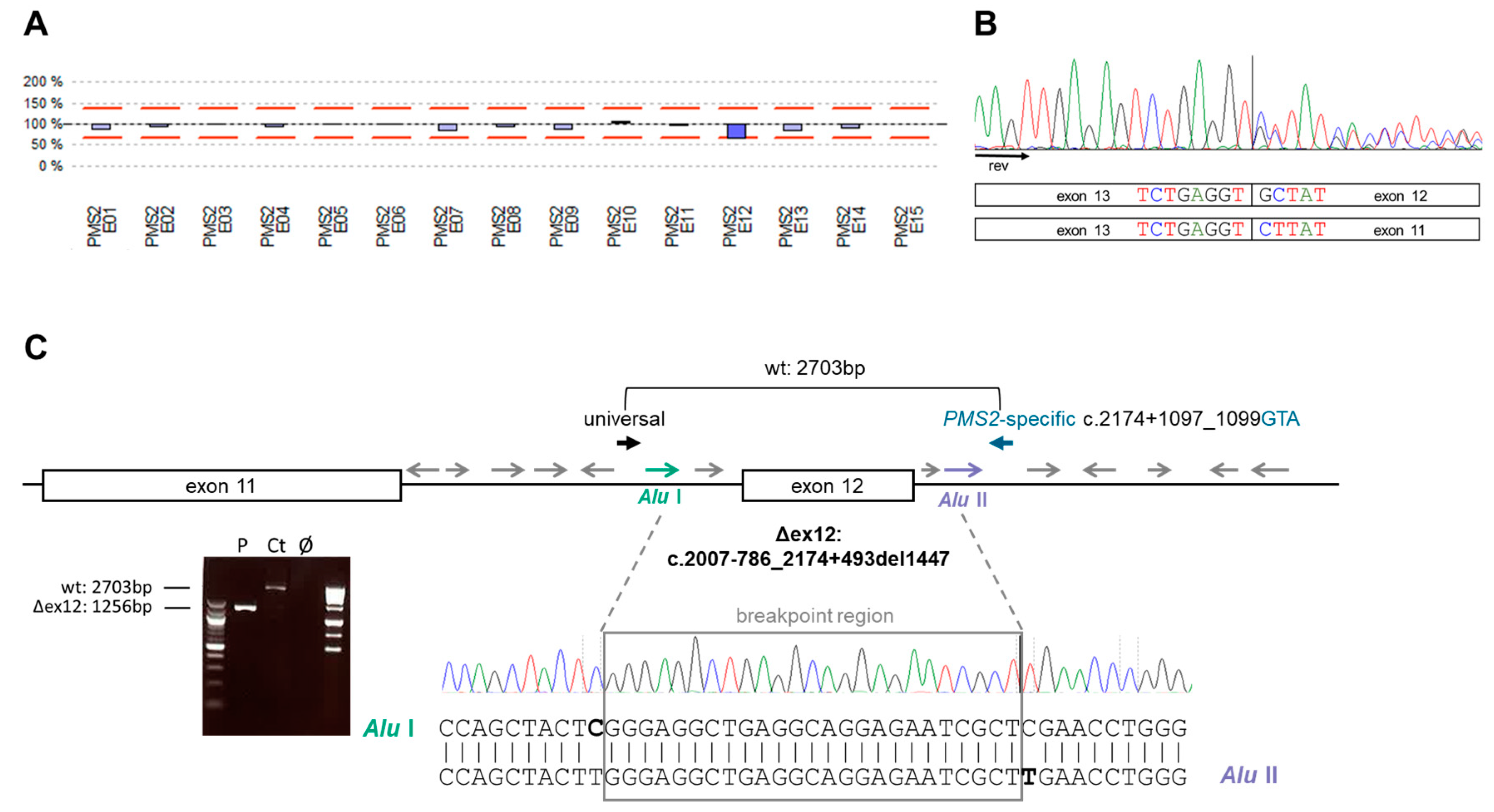
3.2. Identification of Germline PMS2 and POLD1 Variants Causing a Digenic Inheritance of AYA-CRC
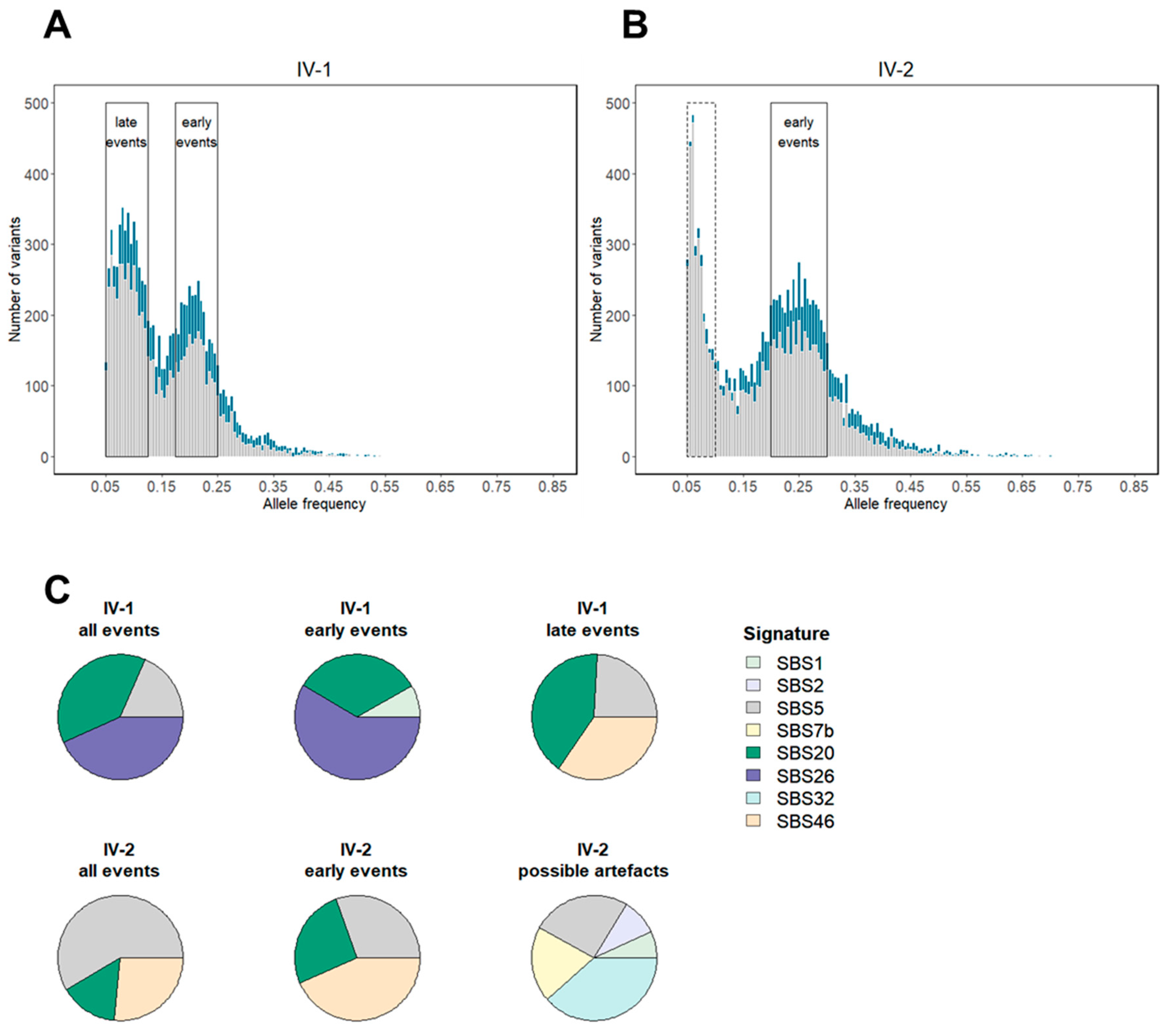
3.3. Tumor Mutation Characterization Identifies MMRd as an Early Event in Combined MMR and PP Deficiency-Driven Colorectal Tumorigenesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- American Cancer Society. Colorectal Cancer Facts & Figures 2017–2019. Available online: https://www.cancer.org/research/cancer-facts-statistics/colorectal-cancer-facts-figures.html (accessed on 26 April 2022).
- Sultan, I.; Rodriguez-Galindo, C.; El-Taani, H.; Pastore, G.; Casanova, M.; Gallino, G.; Ferrari, A. Distinct features of colorectal cancer in children and adolescents: A population-based study of 159 cases. Cancer 2010, 116, 758–765. [Google Scholar] [CrossRef]
- De Voer, R.M.; Diets, I.J.; van der Post, R.S.; Weren, R.D.A.; Kamping, E.J.; de Bitter, T.J.J.; Elze, L.; Verhoeven, R.H.A.; Vink-Börger, E.; Eijkelenboom, A.; et al. Clinical, Pathology, Genetic, and Molecular Features of Colorectal Tumors in Adolescents and Adults 25 Years or Younger. Clin. Gastroenterol. Hepatol. 2021, 19, 1642–1651.e81648. [Google Scholar] [CrossRef]
- Burt, R. Inheritance of Colorectal Cancer. Drug Discov. Today Dis. Mech. 2007, 4, 293–300. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Mork, M.E.; You, Y.N.; Ying, J.; Bannon, S.A.; Lynch, P.M.; Rodriguez-Bigas, M.A.; Vilar, E. High Prevalence of Hereditary Cancer Syndromes in Adolescents and Young Adults with Colorectal Cancer. J. Clin. Oncol. 2015, 33, 3544–3549. [Google Scholar] [CrossRef]
- Khan, S.A.; Morris, M.; Idrees, K.; Gimbel, M.I.; Rosenberg, S.; Zeng, Z.; Li, F.; Gan, G.; Shia, J.; LaQuaglia, M.P.; et al. Colorectal cancer in the very young: A comparative study of tumor markers, pathology and survival in early onset and adult onset patients. J. Pediatr. Surg. 2016, 51, 1812–1817. [Google Scholar] [CrossRef]
- Fernandez-Rozadilla, C.; Alvarez-Barona, M.; Schamschula, E.; Bodo, S.; Lopez-Novo, A.; Dacal, A.; Calviño-Costas, C.; Lancho, A.; Amigo, J.; Bello, X.; et al. Early Colorectal Cancers Provide New Evidence for a Lynch Syndrome-to-CMMRD Phenotypic Continuum. Cancers 2019, 11, 1081. [Google Scholar] [CrossRef]
- Talseth-Palmer, B.A. The genetic basis of colonic adenomatous polyposis syndromes. Hered. Cancer Clin. Pract. 2017, 15, 5. [Google Scholar] [CrossRef]
- Jongmans, M.C.J.; Zhang, J.; Hoogerbrugge, N.; Ligtenberg, M.J.L.; De Voer, R.M. Genetic Cancer Susceptibility in Adolescents and Adults 25 Years or Younger with Colorectal Cancer. Gastroenterology 2021, 162, 969–974. [Google Scholar] [CrossRef]
- Salo-Mullen, E.E.; Maio, A.; Mukherjee, S.; Bandlamudi, C.; Shia, J.; Kemel, Y.; Cadoo, K.A.; Liu, Y.; Carlo, M.; Ranganathan, M.; et al. Prevalence and Characterization of Biallelic and Monoallelic NTHL1 and MSH3 Variant Carriers from a Pan-Cancer Patient Population. JCO Precis. Oncol. 2021, 5, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J. Med. Genet. 2014, 51, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Rosenbaum, T.; Messiaen, L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin. Genet. 2017, 91, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Martin, L.; Domingo, E.; Chegwidden, L.; McGuire, J.; Cuthill, V.; Heitzer, E.; Kerr, R.; Kerr, D.; Kearsey, S.; et al. The clinical features of polymerase proof-reading associated polyposis (PPAP) and recommendations for patient management. Fam. Cancer 2021, 21, 197–209. [Google Scholar] [CrossRef]
- Sehested, A.; Meade, J.; Scheie, D.; Østrup, O.; Bertelsen, B.; Misiakou, M.A.; Sarosiek, T.; Kessler, E.; Melchior, L.C.; Munch-Petersen, H.F.; et al. Constitutional POLE variants causing a phenotype reminiscent of constitutional mismatch repair deficiency. Hum. Mutat. 2022, 43, 85–96. [Google Scholar] [CrossRef]
- Wimmer, K.; Beilken, A.; Nustede, R.; Ripperger, T.; Lamottke, B.; Ure, B.; Steinmann, D.; Reineke-Plaass, T.; Lehmann, U.; Zschocke, J.; et al. A novel germline POLE mutation causes an early onset cancer prone syndrome mimicking constitutional mismatch repair deficiency. Fam. Cancer 2017, 16, 67–71. [Google Scholar] [CrossRef]
- Mur, P.; García-Mulero, S.; del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef]
- Park, V.S.; Pursell, Z.F. POLE proofreading defects: Contributions to mutagenesis and cancer. DNA Repair 2019, 76, 50–59. [Google Scholar] [CrossRef]
- Heydt, C.; Fassunke, J.; Künstlinger, H.; Ihle, M.A.; König, K.; Heukamp, L.C.; Schildhaus, H.U.; Odenthal, M.; Büttner, R.; Merkelbach-Bruse, S. Comparison of pre-analytical FFPE sample preparation methods and their impact on massively parallel sequencing in routine diagnostics. PLoS ONE 2014, 9, e104566. [Google Scholar] [CrossRef]
- Etzler, J.; Peyrl, A.; Zatkova, A.; Schildhaus, H.U.; Ficek, A.; Merkelbach-Bruse, S.; Kratz, C.P.; Attarbaschi, A.; Hainfellner, J.A.; Yao, S.; et al. RNA-based mutation analysis identifies an unusual MSH6 splicing defect and circumvents PMS2 pseudogene interference. Hum. Mutat. 2008, 29, 299–305. [Google Scholar] [CrossRef]
- Gallon, R.; Mühlegger, B.; Wenzel, S.S.; Sheth, H.; Hayes, C.; Aretz, S.; Dahan, K.; Foulkes, W.; Kratz, C.P.; Ripperger, T.; et al. A sensitive and scalable microsatellite instability assay to diagnose constitutional mismatch repair deficiency by sequencing of peripheral blood leukocytes. Hum. Mutat. 2019, 40, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wernstedt, A.; Valtorta, E.; Armelao, F.; Togni, R.; Girlando, S.; Baudis, M.; Heinimann, K.; Messiaen, L.; Staehli, N.; Zschocke, J.; et al. Improved multiplex ligation-dependent probe amplification analysis identifies a deleterious PMS2 allele generated by recombination with crossover between PMS2 and PMS2CL. Genes Chromosomes Cancer 2012, 51, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- The InSiGHT Variant Interpretation Committee. Mismatch Repair Gene Variant Classification Criteria 2018. Available online: https://www.insight-group.org/content/uploads/2018/08/2018-06_InSiGHT_VIC_v2.4.pdf (accessed on 8 July 2022).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Köster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2017, 34, 867–868. [Google Scholar] [CrossRef]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019. [Google Scholar] [CrossRef]
- Andrews, S. S-andrews/FastQC [Java]. 2020. Available online: https://github.com/s-andrews/FastQC (accessed on 29 July 2021).
- Kandoth, C. Mskcc/vcf2maf [Perl]. 2020. Available online: https://github.com/mskcc/vcf2maf (accessed on 29 July 2021).
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers ofpathogenic mismatch repair variants: Findings from the Prospective Lynch SyndromeDatabase. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Haradhvala, N.J.; Kim, J.; Maruvka, Y.E.; Polak, P.; Rosebrock, D.; Livitz, D.; Hess, J.M.; Leshchiner, I.; Kamburov, A.; Mouw, K.W.; et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 2018, 9, 1746. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Bajwa-Ten Broeke, S.W.; Ballhausen, A.; Ahadova, A.; Suerink, M.; Bohaumilitzky, L.; Seidler, F.; Morreau, H.; van Wezel, T.; Krzykalla, J.; Benner, A.; et al. The coding microsatellite mutation profile of PMS2-deficient colorectal cancer. Exp. Mol. Pathol. 2021, 122, 104668. [Google Scholar] [CrossRef]
- Zou, X.; Koh, G.C.C.; Nanda, A.S.; Degasperi, A.; Urgo, K.; Roumeliotis, T.I.; Agu, C.A.; Badja, C.; Momen, S.; Young, J.; et al. A systematic CRISPR screen defines mutational mechanisms underpinning signatures caused by replication errors and endogenous DNA damage. Nat. Cancer 2021, 2, 643–657. [Google Scholar] [CrossRef]
- Koh, G.; Degasperi, A.; Zou, X.; Momen, S.; Nik-Zainal, S. Mutational signatures: Emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 2021, 21, 619–637. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Bläker, H.; et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef]
- Stanich, P.P.; Pearlman, R.; Hinton, A.; Gutierrez, S.; LaDuca, H.; Hampel, H.; Jasperson, K. Prevalence of Germline Mutations in Polyposis and Colorectal Cancer-Associated Genes in Patients with Multiple Colorectal Polyps. Clin. Gastroenterol. Hepatol. 2019, 17, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Jelsig, A.M.; Byrjalsen, A.; Busk Madsen, M.; Kuhlmann, T.P.; van Overeem Hansen, T.; Wadt, K.A.W.; Karstensen, J.G. Novel Genetic Causes of Gastrointestinal Polyposis Syndromes. Appl. Clin. Genet. 2021, 14, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Song, I.H.; Lee, A.; Kang, J.; Lee, Y.S.; Lee, I.K.; Song, Y.S.; Lee, S.H. Enhancing the landscape of colorectal cancer using targeted deep sequencing. Sci. Rep. 2021, 11, 8154. [Google Scholar] [CrossRef] [PubMed]
- Bläker, H.; Haupt, S.; Morak, M.; Holinski-Feder, E.; Arnold, A.; Horst, D.; Sieber-Frank, J.; Seidler, F.; von Winterfeld, M.; Alwers, E.; et al. Age-dependent performance of BRAF mutation testing in Lynch syndrome diagnostics. Int. J. Cancer 2020, 147, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Schmidt, R.J.; Ankala, A.; McDonald Gibson, K.; Bowser, M.; Sharma, H.; Duffy, E.; Hegde, M.; Santani, A.; Lebo, M.; et al. Navigating highly homologous genes in a molecular diagnostic setting: A resource for clinical next-generation sequencing. Genet. Med. 2016, 18, 1282–1289. [Google Scholar] [CrossRef]
- van der Klift, H.M.; Jansen, A.M.; van der Steenstraten, N.; Bik, E.C.; Tops, C.M.; Devilee, P.; Wijnen, J.T. Splicing analysis for exonic and intronic mismatch repair gene variants associated with Lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol. Genet. Genom. Med. 2015, 3, 327–345. [Google Scholar] [CrossRef]
- Berrino, E.; Filippi, R.; Visintin, C.; Peirone, S.; Fenocchio, E.; Farinea, G.; Veglio, F.; Aglietta, M.; Sapino, A.; Cereda, M.; et al. Collision of germline POLE and PMS2 variants in a young patient treated with immune checkpoint inhibitors. NPJ Precis. Oncol. 2022, 6, 15. [Google Scholar] [CrossRef]
- Michaeli, O.; Ladany, H.; Erez, A.; Ben Shachar, S.; Izraeli, S.; Lidzbarsky, G.; Basel-Salmon, L.; Biskup, S.; Maruvka, Y.E.; Toledano, H.; et al. Di-genic inheritance of germline POLE and PMS2 pathogenic variants causes a unique condition associated with pediatric cancer predisposition. Clin. Genet. 2022, 101, 442–447. [Google Scholar] [CrossRef]
- Lonati, C.; Necchi, A.; Gómez Rivas, J.; Afferi, L.; Laukhtina, E.; Martini, A.; Ventimiglia, E.; Colombo, R.; Gandaglia, G.; Salonia, A.; et al. Upper Tract Urothelial Carcinoma in the Lynch Syndrome Tumour Spectrum: A Comprehensive Overview from the European Association of Urology-Young Academic Urologists and the Global Society of Rare Genitourinary Tumors. Eur. Urol. Oncol. 2022, 5, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, I.T.; Myrhøj, T. Surveillance for urinary tract cancer in Lynch syndrome. Fam. Cancer 2013, 12, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Maruvka, Y.E.; Sudhaman, S.; Kelly, J.; Haradhvala, N.J.; Bianchi, V.; Edwards, M.; Forster, V.J.; Nunes, N.M.; Galati, M.A.; et al. DNA Polymerase and Mismatch Repair Exert Distinct Microsatellite Instability Signatures in Normal and Malignant Human Cells. Cancer Discov. 2021, 11, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Andrianova, M.A.; Bazykin, G.A.; Nikolaev, S.I.; Seplyarskiy, V.B. Human mismatch repair system balances mutation rates between strands by removing more mismatches from the lagging strand. Genome Res. 2017, 27, 1336–1343. [Google Scholar] [CrossRef]
- Hodel, K.P.; de Borja, R.; Henninger, E.E.; Campbell, B.B.; Ungerleider, N.; Light, N.; Wu, T.; LeCompte, K.G.; Goksenin, A.Y.; Bunnell, B.A.; et al. Explosive mutation accumulation triggered by heterozygous human Pol ε proofreading-deficiency is driven by suppression of mismatch repair. Elife 2018, 7, 32692. [Google Scholar] [CrossRef]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- Kloor, M.; Huth, C.; Voigt, A.Y.; Benner, A.; Schirmacher, P.; von Knebel Doeberitz, M.; Bläker, H. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: A pathological study. Lancet Oncol. 2012, 13, 598–606. [Google Scholar] [CrossRef]
- Sekine, S.; Mori, T.; Ogawa, R.; Tanaka, M.; Yoshida, H.; Taniguchi, H.; Nakajima, T.; Sugano, K.; Yoshida, T.; Kato, M.; et al. Mismatch repair deficiency commonly precedes adenoma formation in Lynch Syndrome-Associated colorectal tumorigenesis. Mod. Pathol. 2017, 30, 1144–1151. [Google Scholar] [CrossRef] [Green Version]
- Helderman, N.C.; Bajwa-Ten Broeke, S.W.; Morreau, H.; Suerink, M.; Terlouw, D.; van der Werf, T.L.A.S.; van Wezel, T.; Nielsen, M. The diverse molecular profiles of lynch syndrome-associated colorectal cancers are (highly) dependent on underlying germline mismatch repair mutations. Crit. Rev. Oncol. Hematol. 2021, 163, 103338. [Google Scholar] [CrossRef] [PubMed]
- Ten Broeke, S.W.; van Bavel, T.C.; Jansen, A.M.L.; Gómez-García, E.; Hes, F.J.; van Hest, L.P.; Letteboer, T.G.W.; Olderode-Berends, M.J.W.; Ruano, D.; Spruijt, L.; et al. Molecular Background of Colorectal Tumors from Patients with Lynch Syndrome Associated with Germline Variants in PMS2. Gastroenterology 2018, 155, 844–851. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schamschula, E.; Kinzel, M.; Wernstedt, A.; Oberhuber, K.; Gottschling, H.; Schnaiter, S.; Friedrichs, N.; Merkelbach-Bruse, S.; Zschocke, J.; Gallon, R.; et al. Teenage-Onset Colorectal Cancers in a Digenic Cancer Predisposition Syndrome Provide Clues for the Interaction between Mismatch Repair and Polymerase δ Proofreading Deficiency in Tumorigenesis. Biomolecules 2022, 12, 1350. https://doi.org/10.3390/biom12101350
Schamschula E, Kinzel M, Wernstedt A, Oberhuber K, Gottschling H, Schnaiter S, Friedrichs N, Merkelbach-Bruse S, Zschocke J, Gallon R, et al. Teenage-Onset Colorectal Cancers in a Digenic Cancer Predisposition Syndrome Provide Clues for the Interaction between Mismatch Repair and Polymerase δ Proofreading Deficiency in Tumorigenesis. Biomolecules. 2022; 12(10):1350. https://doi.org/10.3390/biom12101350
Chicago/Turabian StyleSchamschula, Esther, Miriam Kinzel, Annekatrin Wernstedt, Klaus Oberhuber, Hendrik Gottschling, Simon Schnaiter, Nicolaus Friedrichs, Sabine Merkelbach-Bruse, Johannes Zschocke, Richard Gallon, and et al. 2022. "Teenage-Onset Colorectal Cancers in a Digenic Cancer Predisposition Syndrome Provide Clues for the Interaction between Mismatch Repair and Polymerase δ Proofreading Deficiency in Tumorigenesis" Biomolecules 12, no. 10: 1350. https://doi.org/10.3390/biom12101350