
The Biological Significance of Targeting Acetylation-Mediated Gene Regulation for Designing New Mechanistic Tools and Potential Therapeutics



Abstract
:1. Introduction
2. The Biochemistry and Epigenetics of Lysine Acetylation on Chromatin
2.1. Cellular Localization of KATs
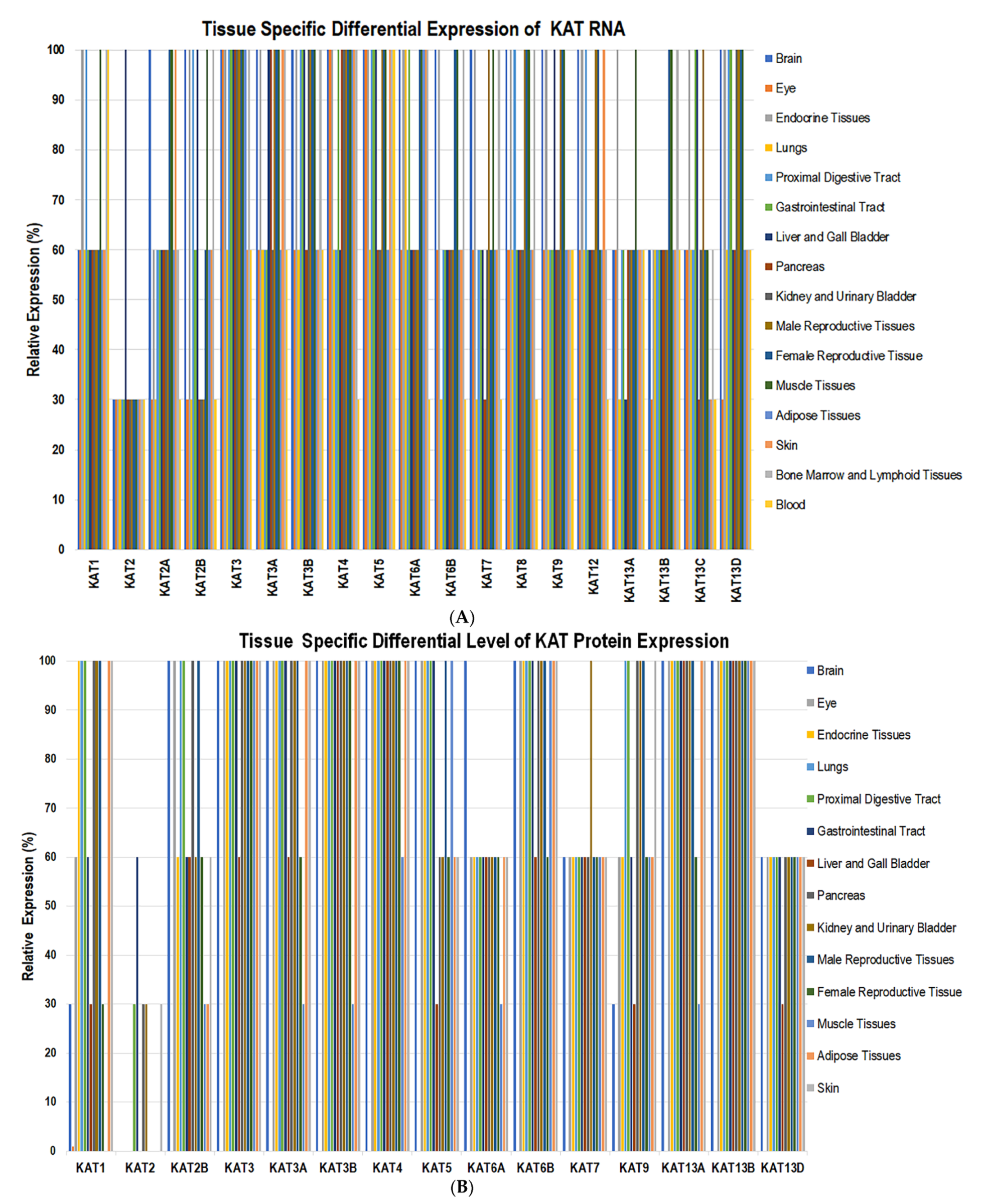
2.2. Genome-Wide Expression of KATs
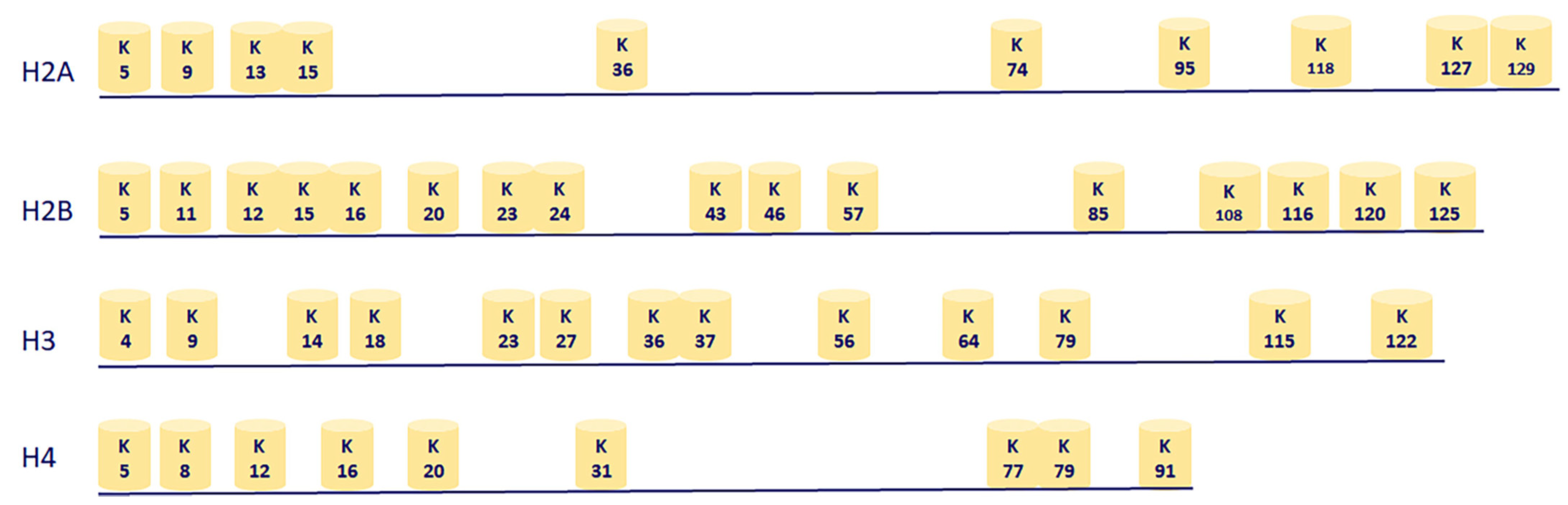
2.3. Genome-Wide Acetylation Marks on Chromatin
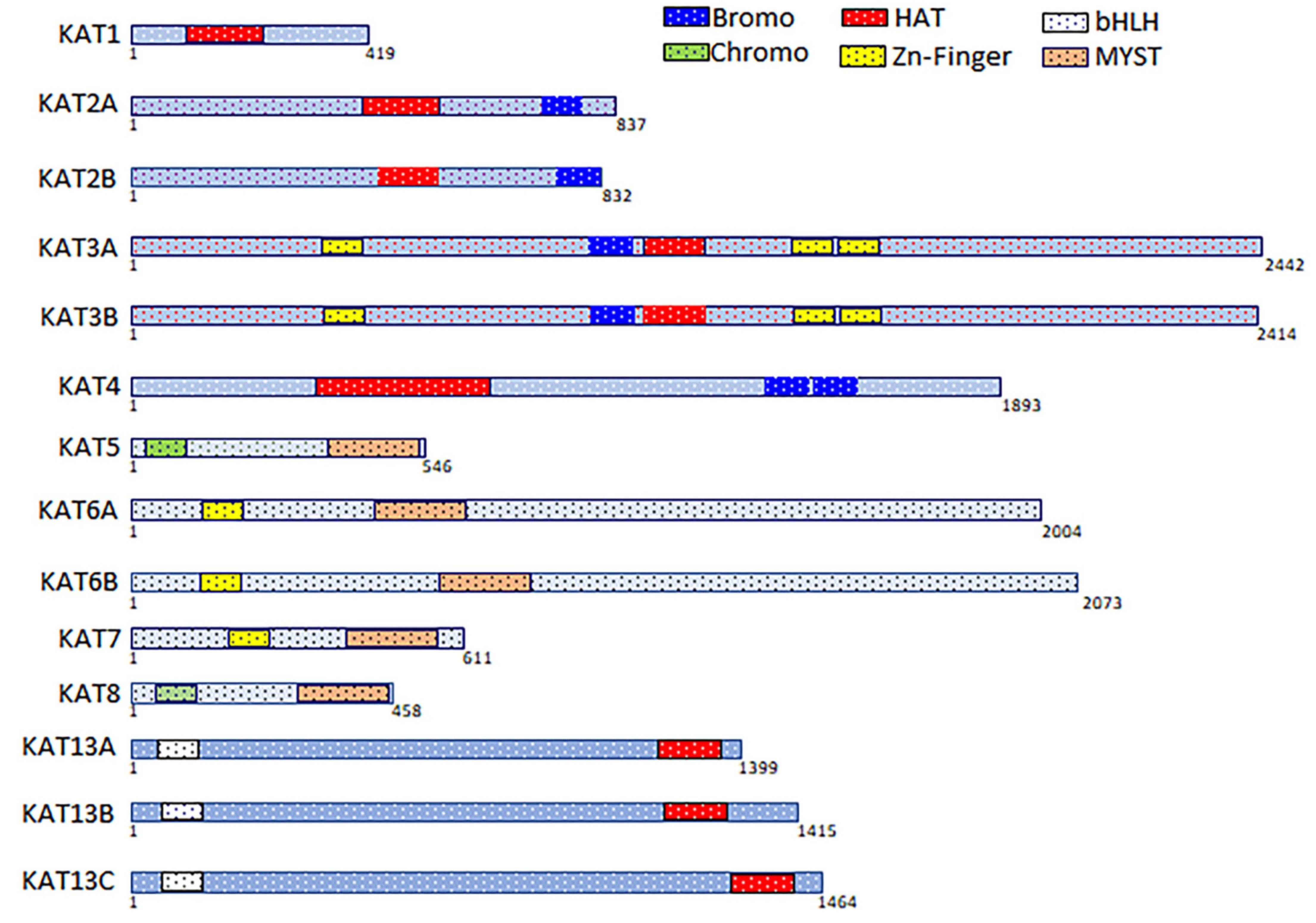
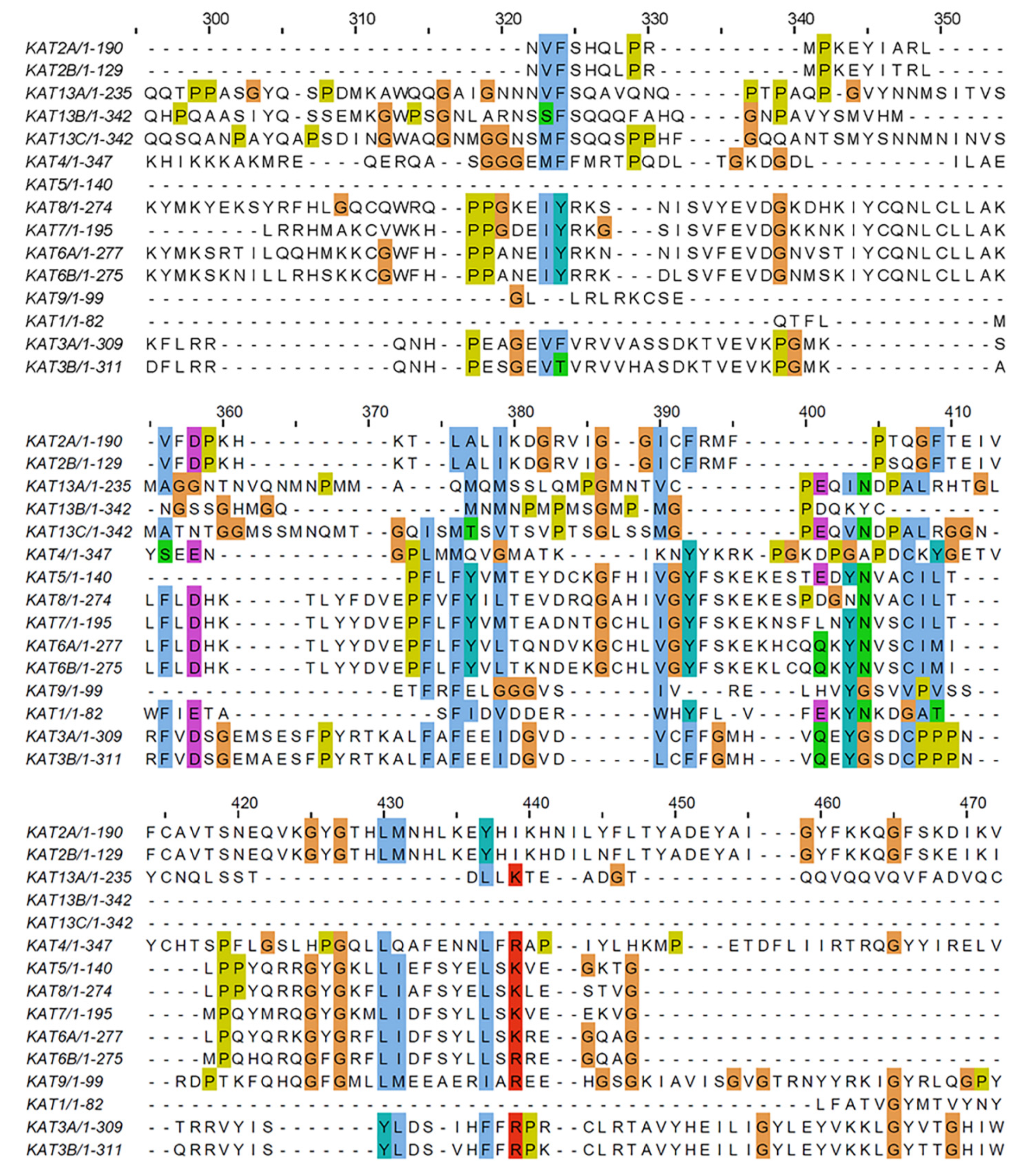
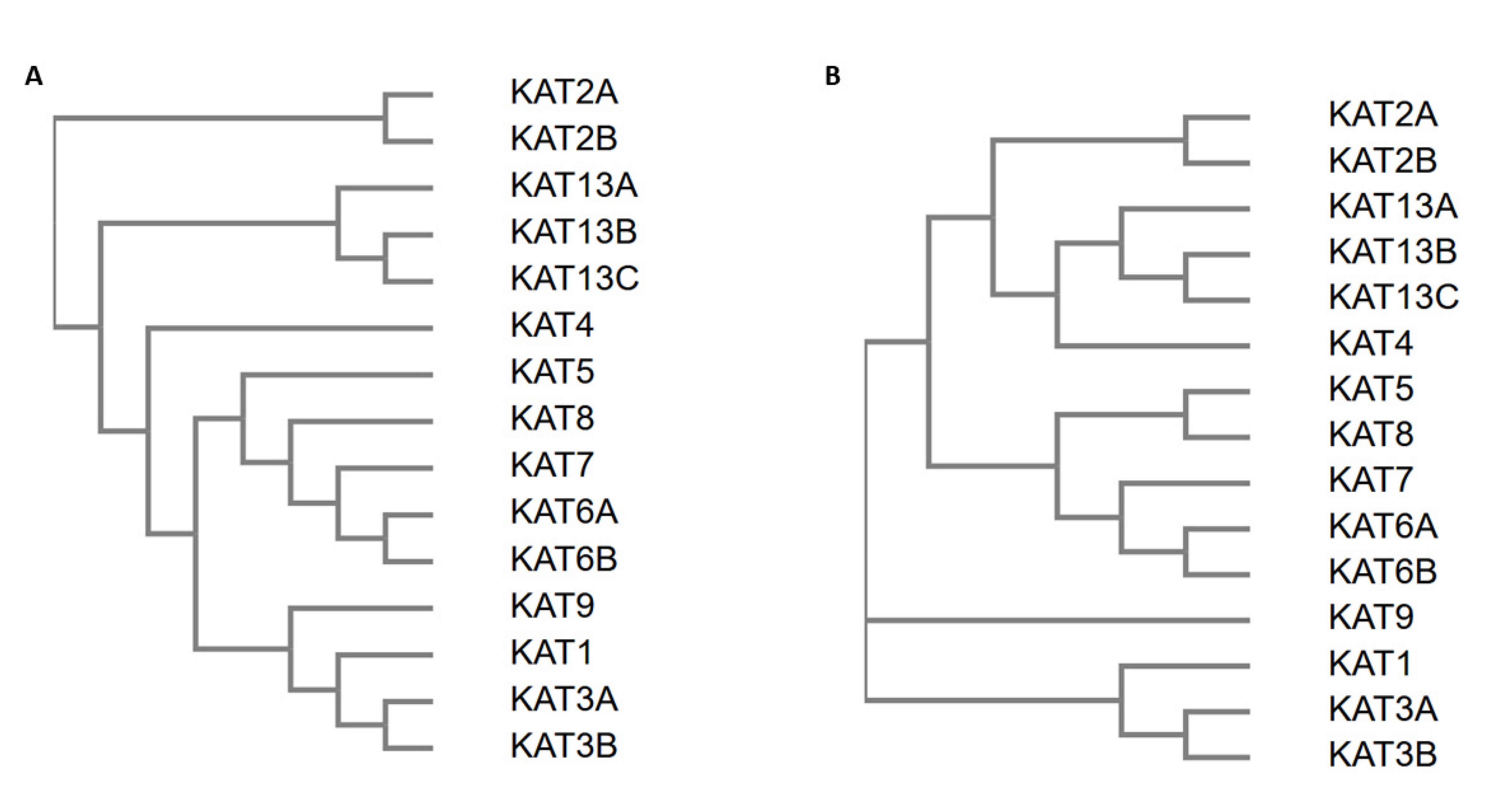
2.4. Sequence Analysis of Acetyltransferase Domain of KATs
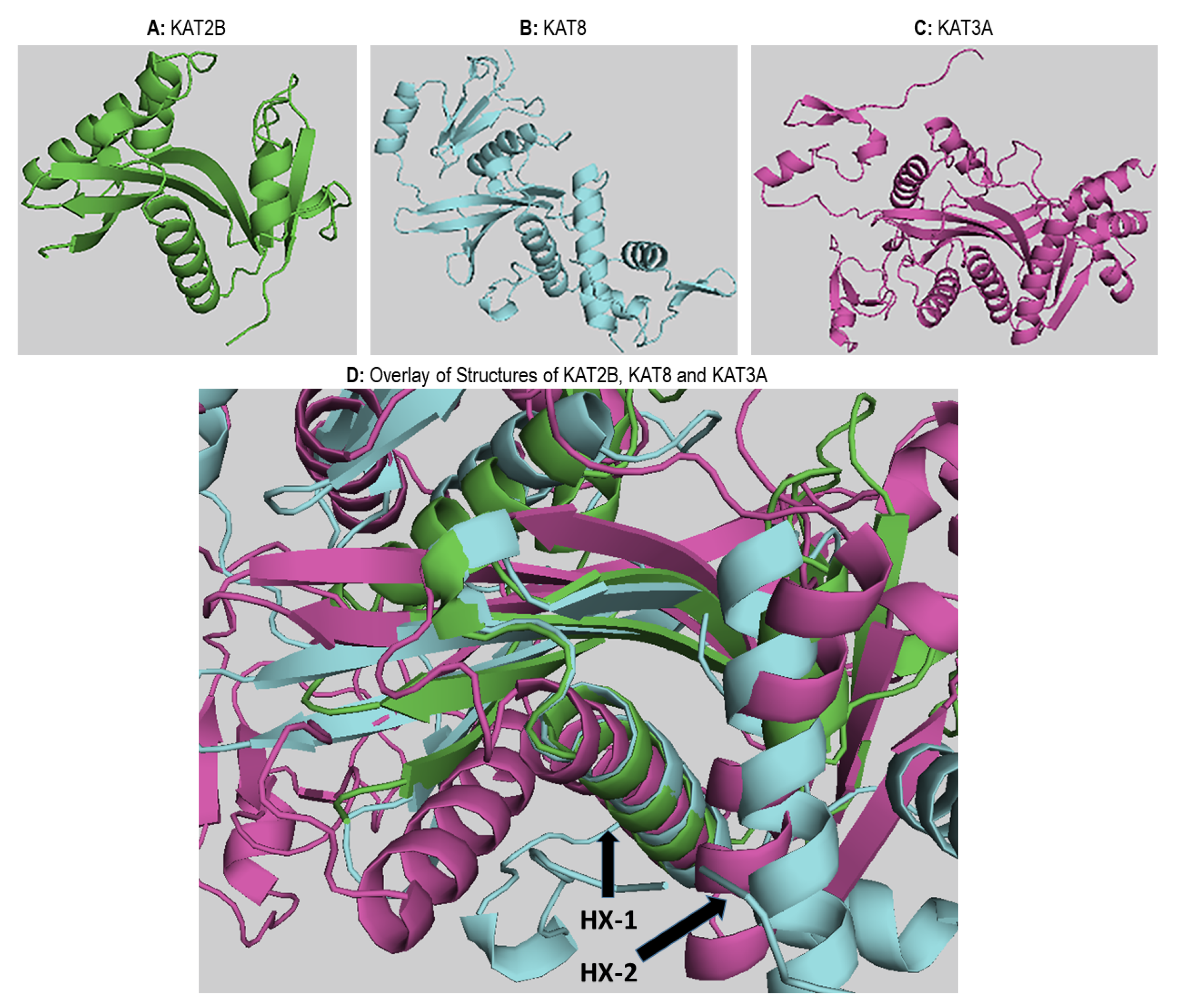
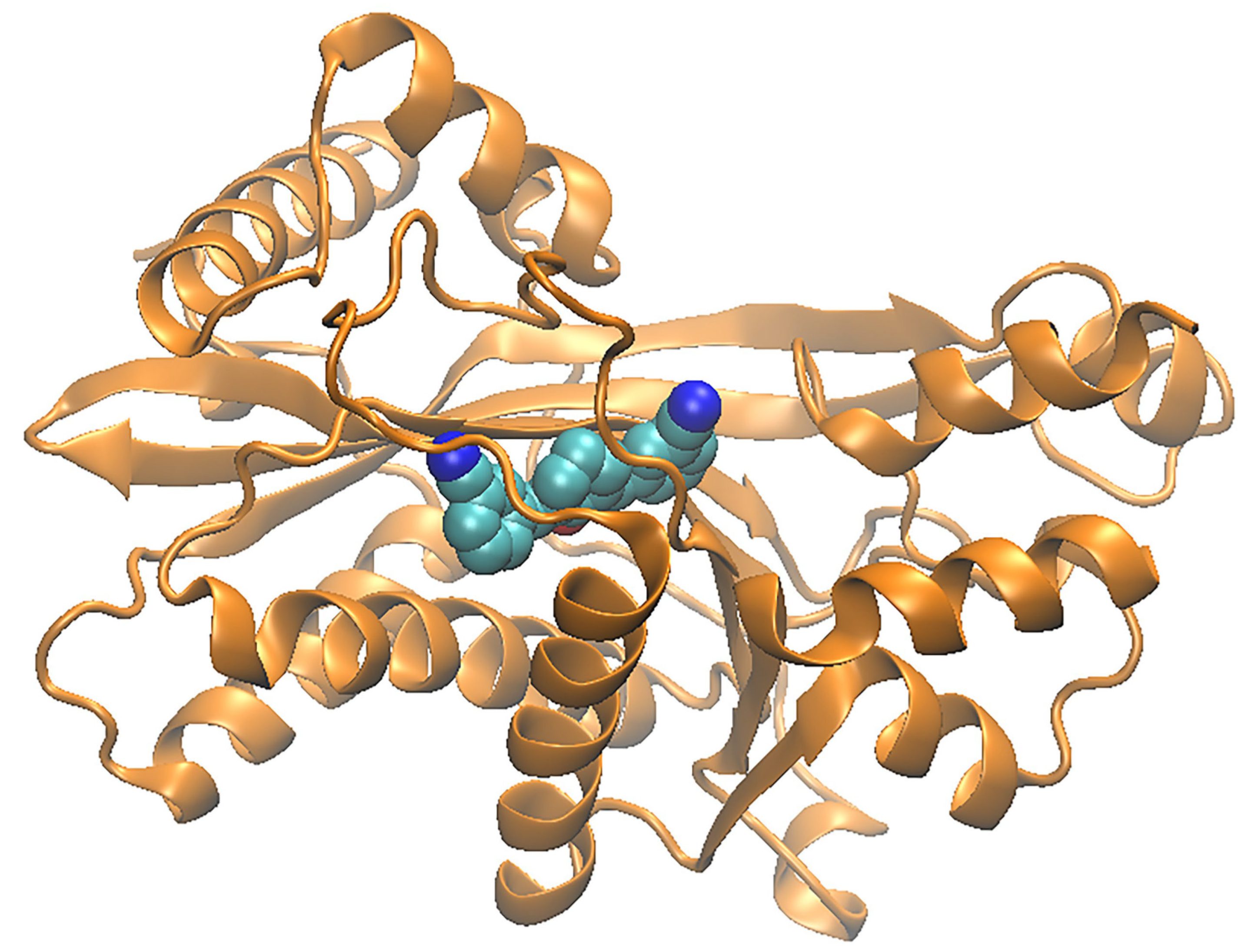
2.5. Structural Analysis of Acetyltransferase Domain of KATs
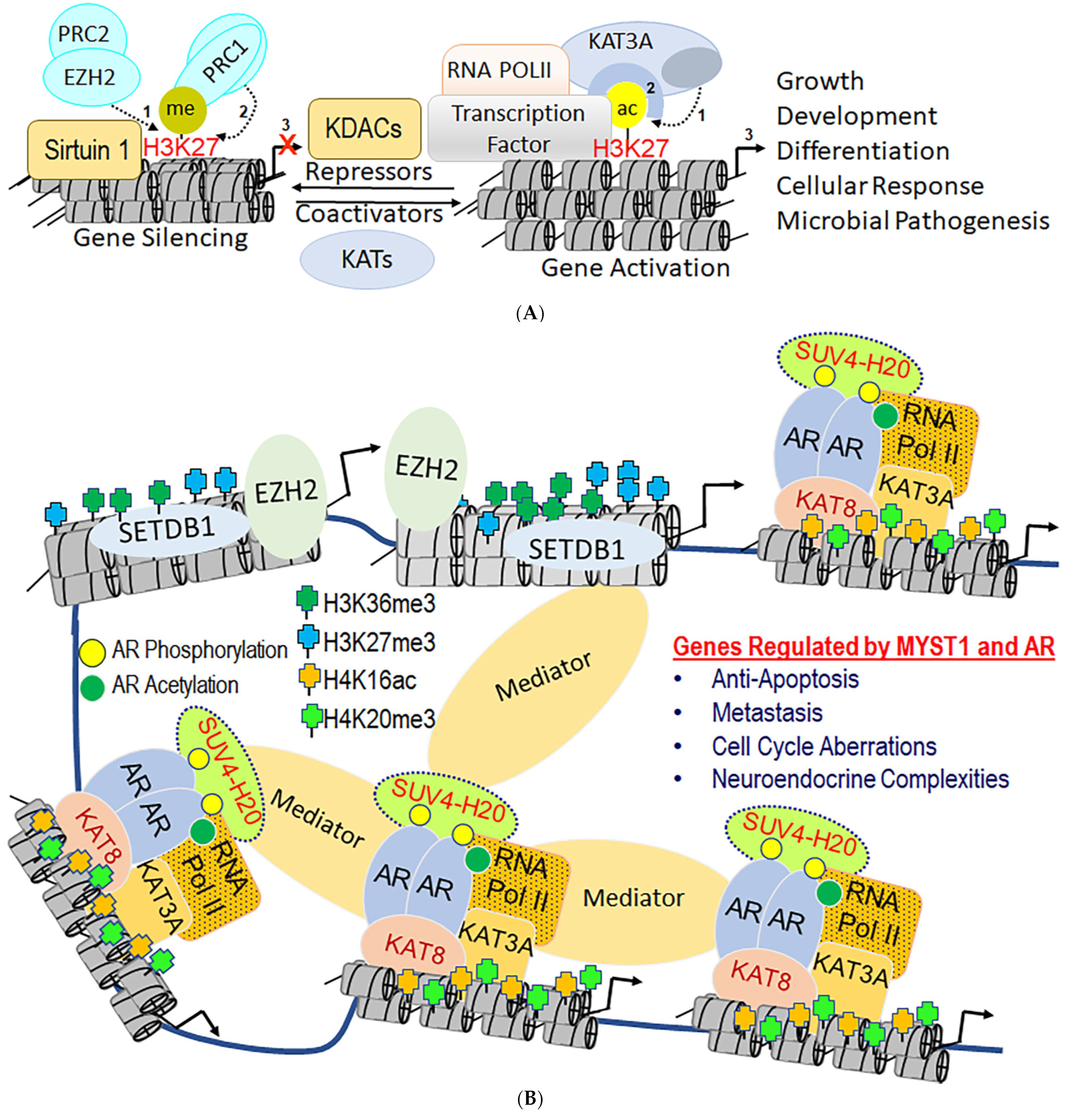
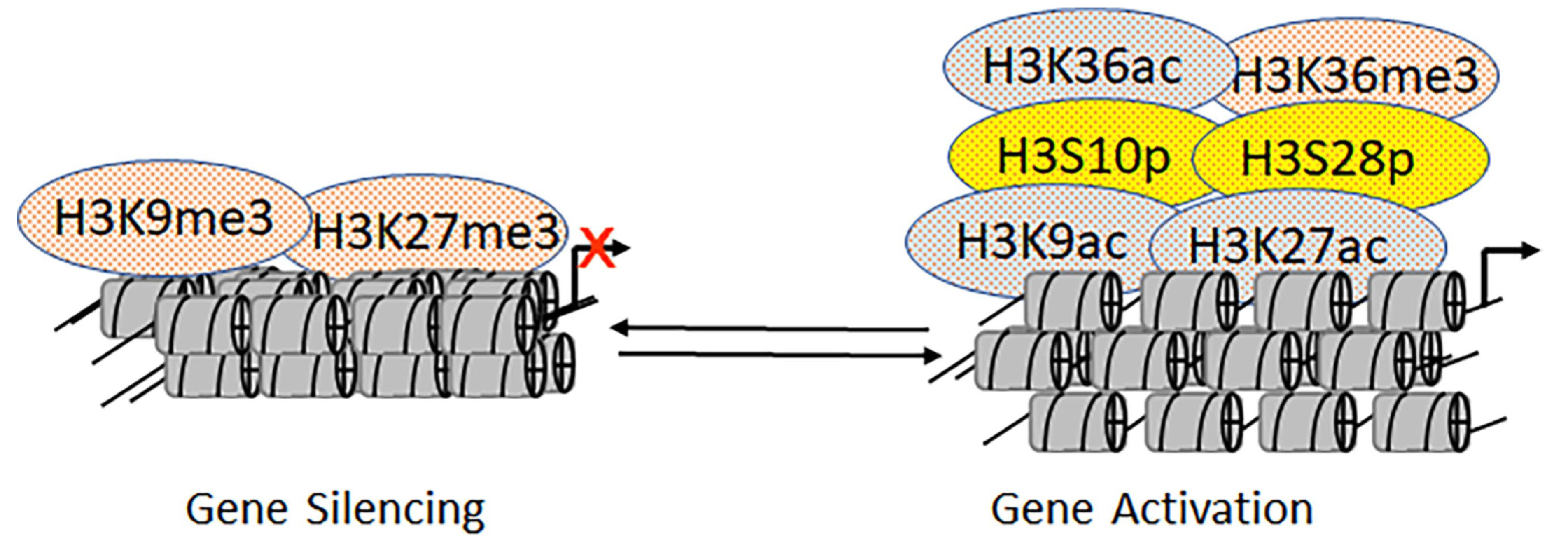
2.6. Mutually Exclusive Crosstalk between Epigenetic Marks
3. Acetylation of Nonhistone Proteins
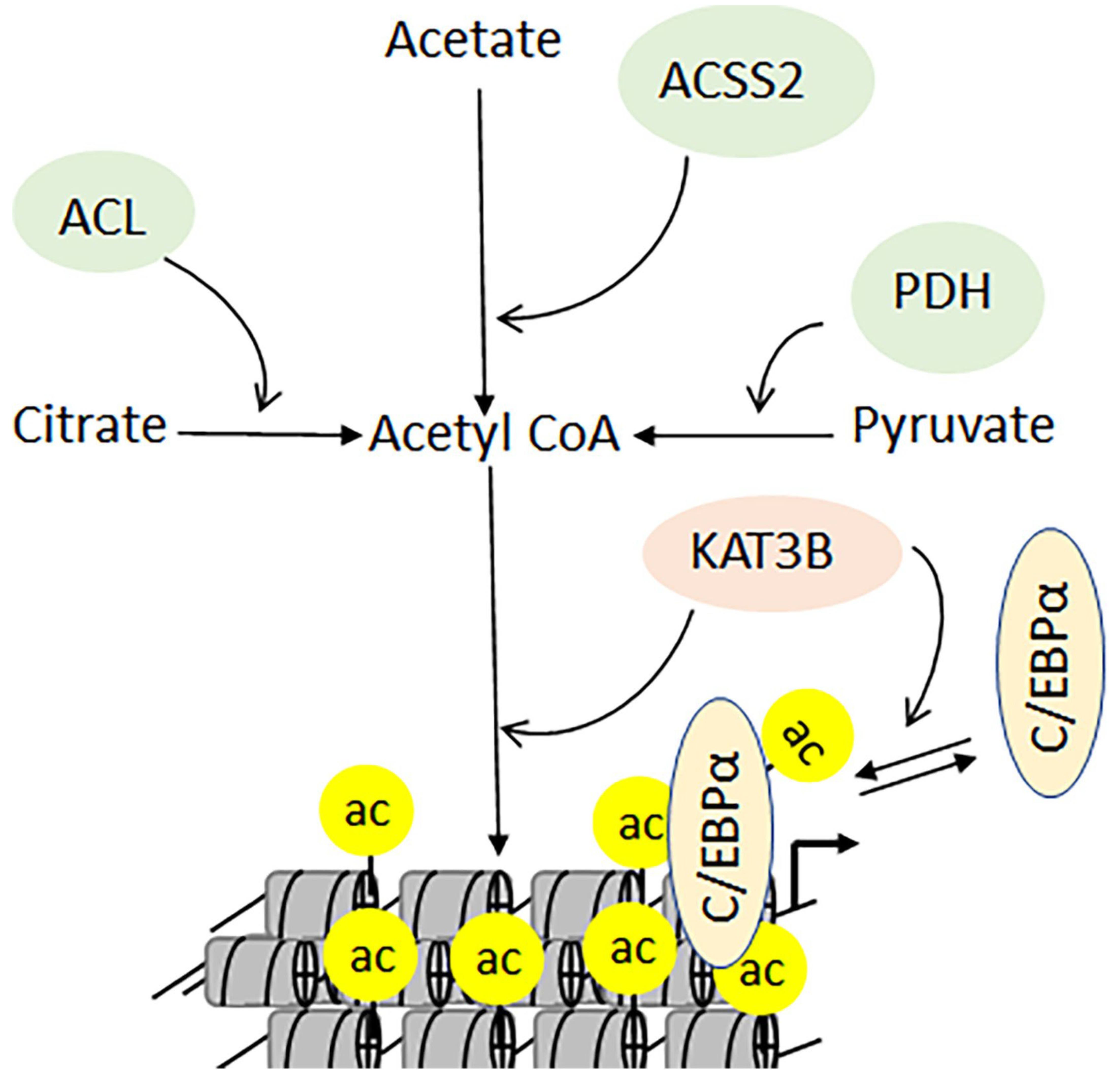
3.1. Metabolism, Developmental Disorders, and Cancers
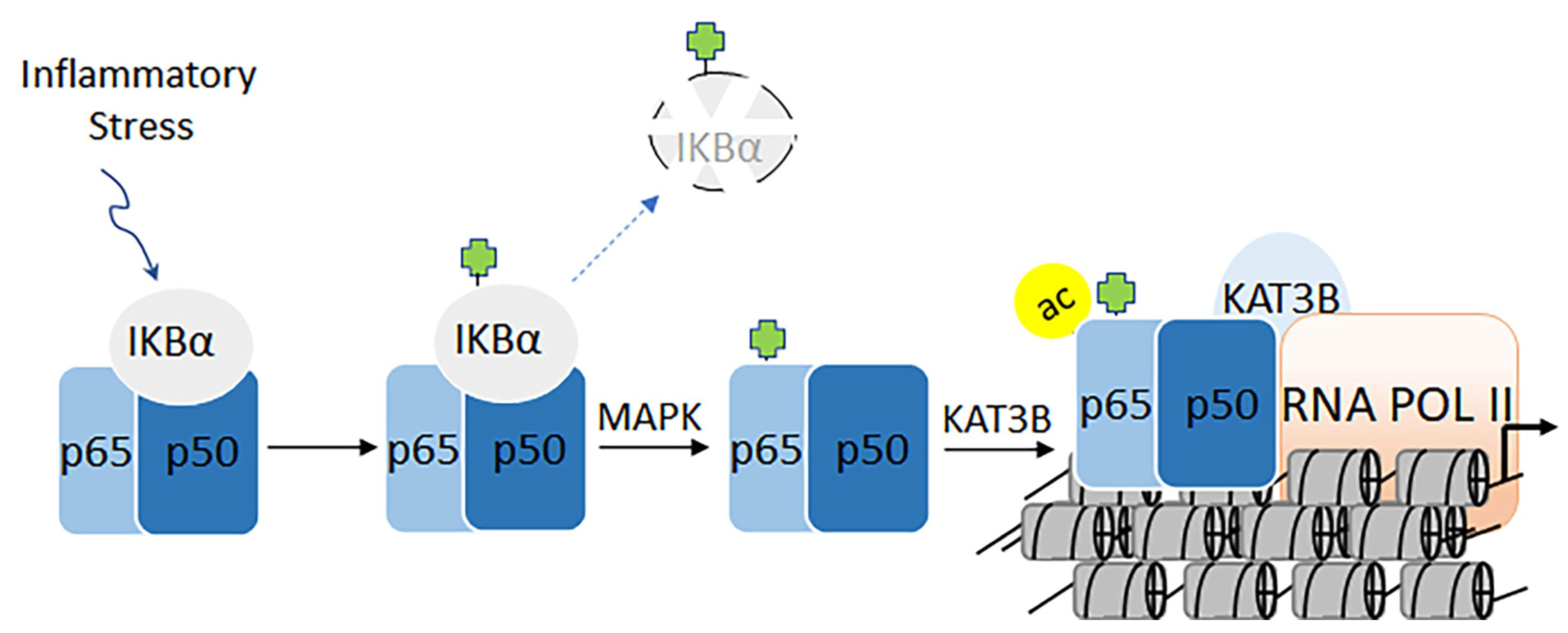
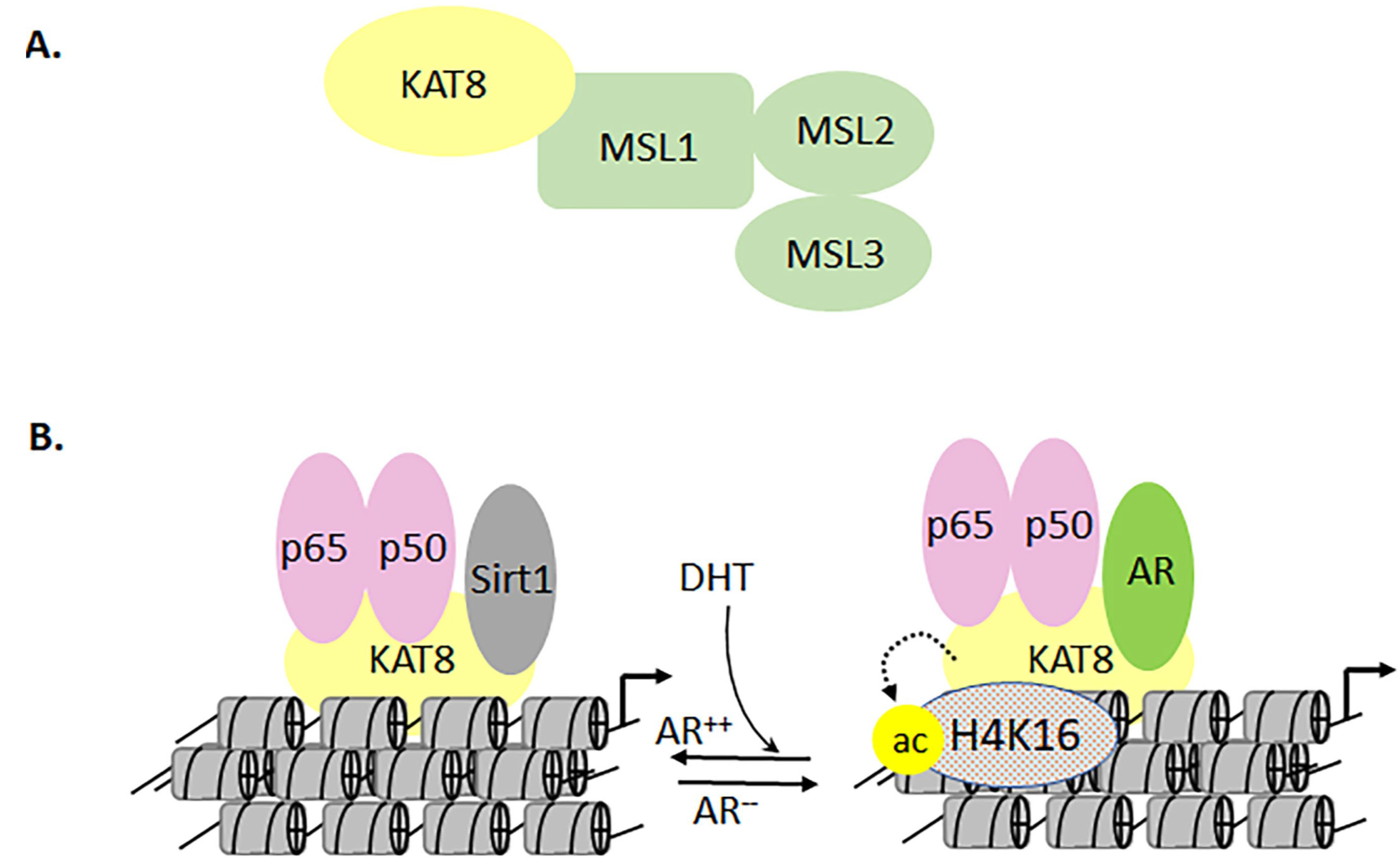
3.2. Target Acetylation Pathway in Nuclear Factor-Kappa B-directed Chronic Diseases
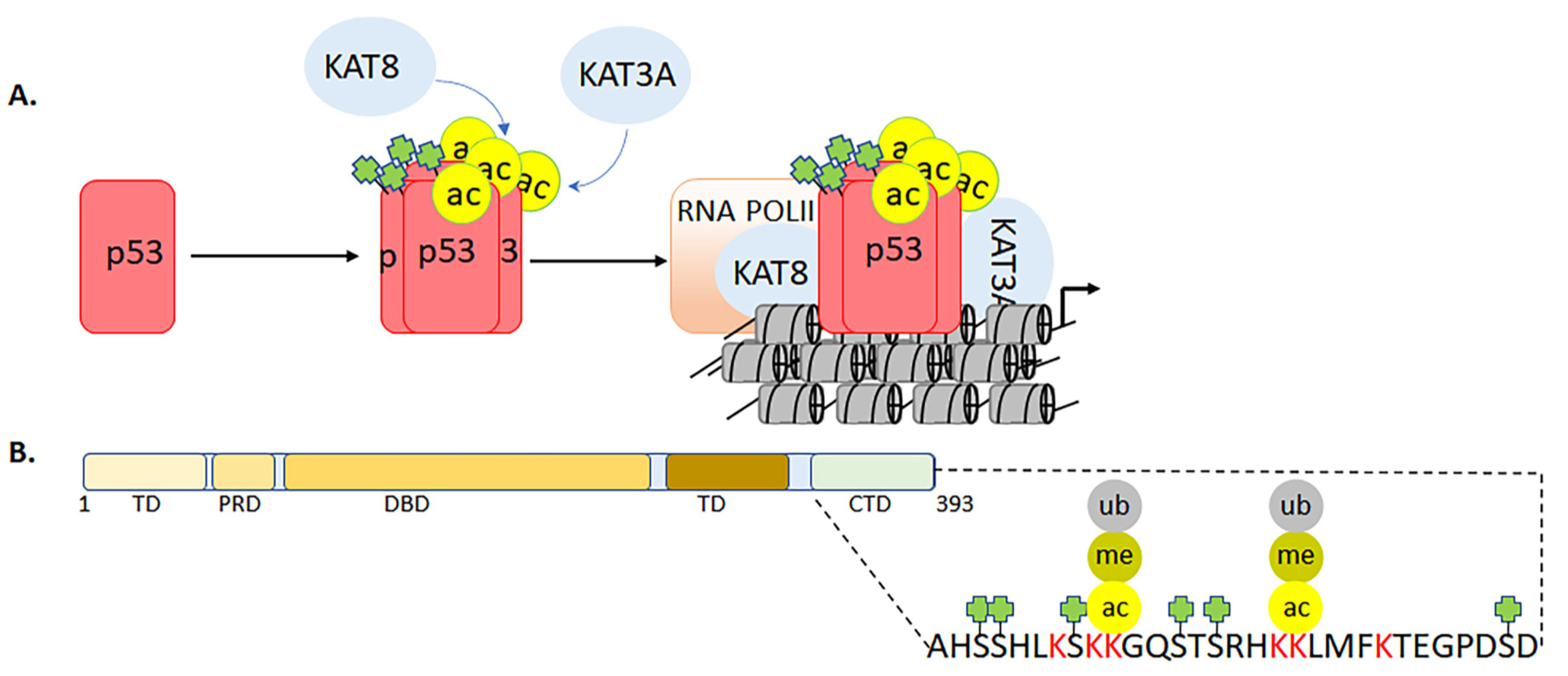
3.3. Multifaceted Roles of p53 C-Terminal Domain during Genotoxic Stress
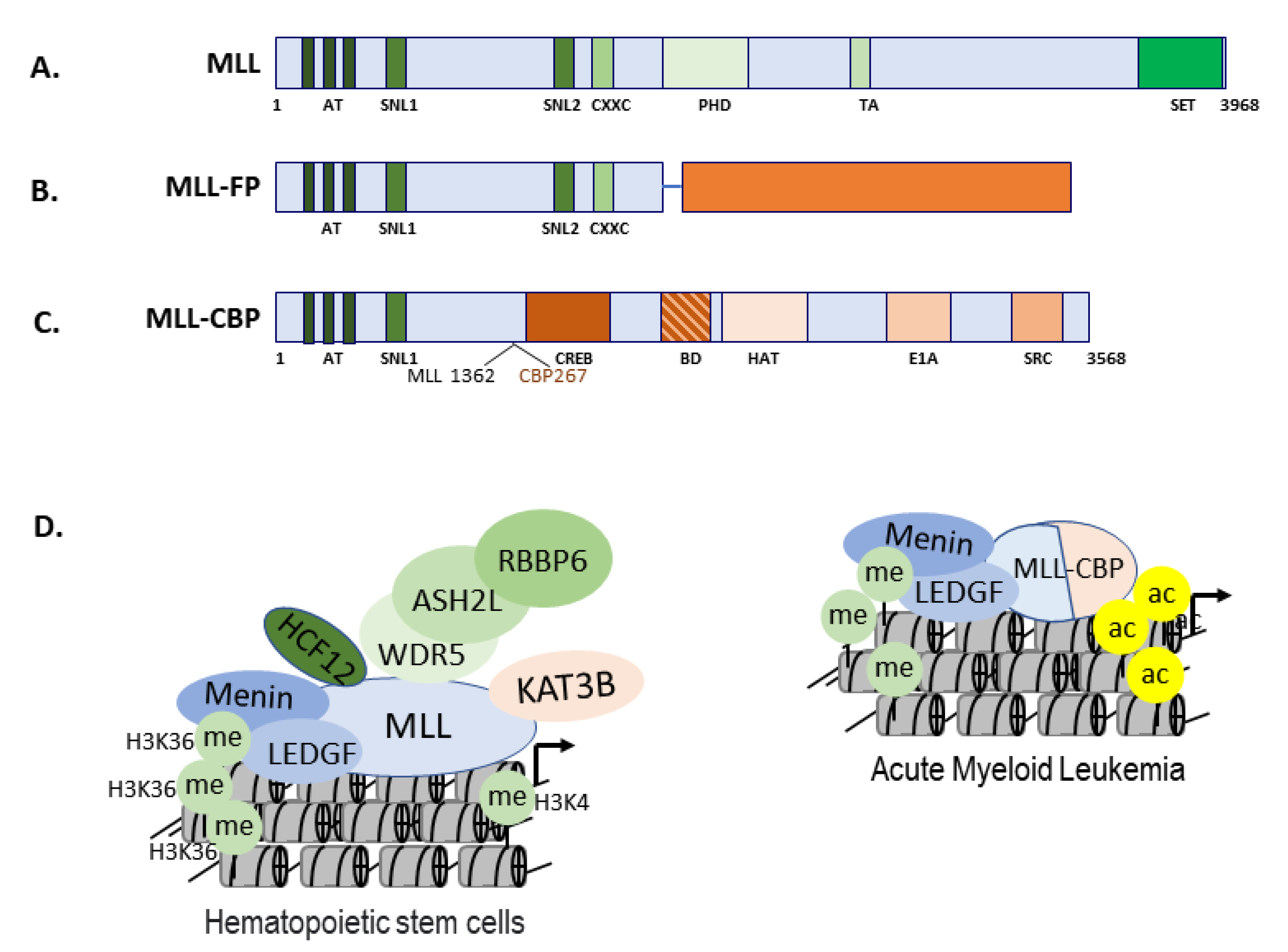
3.4. The Role of MYST Family in Gene Regulation and Cellular Response
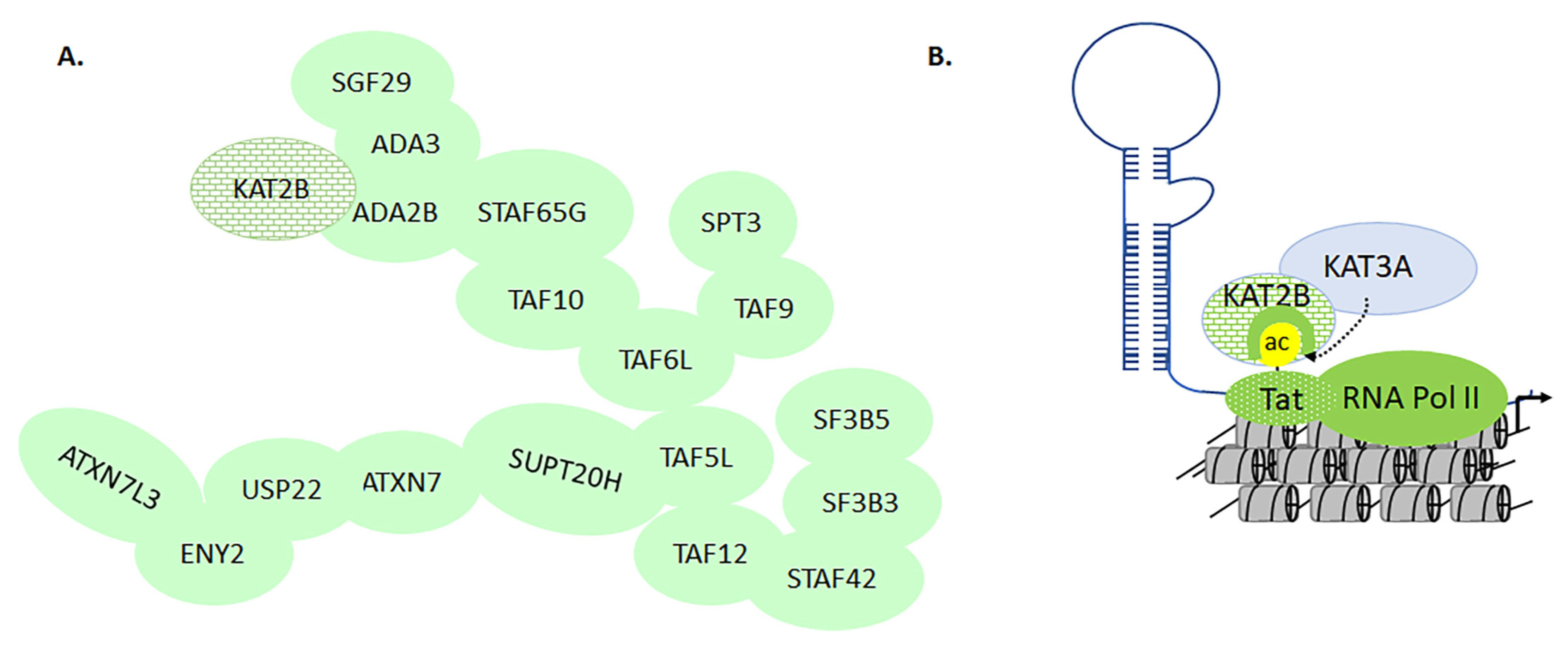
3.5. KAT2A and -2B in Transcriptional Regulation of HIV Activation
3.6. The Dynamics of KATs and KDACs in Models of Traumatic Brain Injury
4. Chemical Biology of Acetylation-Mediated Mechanisms Directed by KAT3A/-3B
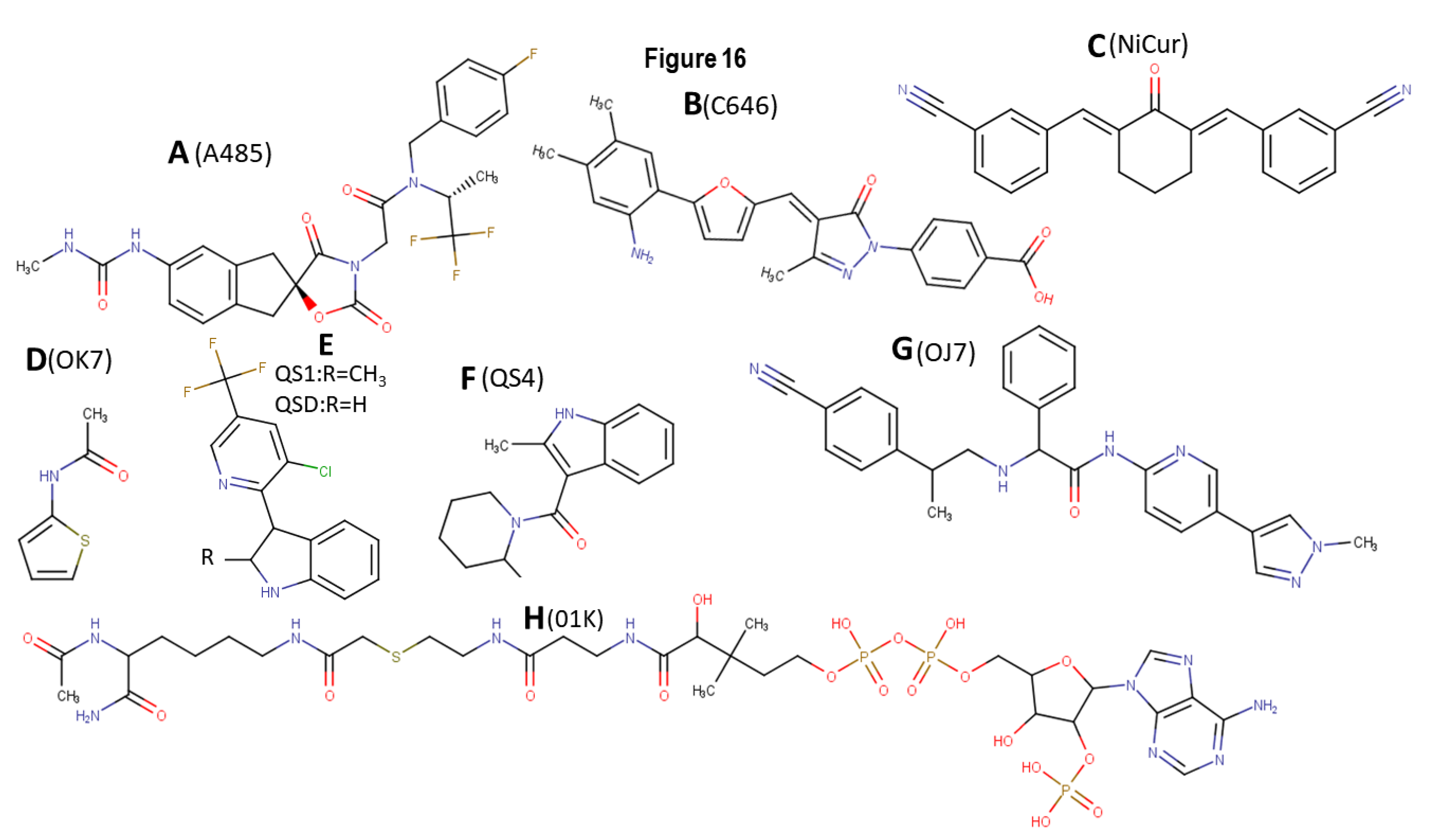
4.1. A485
4.2. C646
4.3. NiCur
4.4. Structural Analysis of Ligands Binding to KAT3A/-3B Acetyltransferase Domain
5. Material and Methods
Amino Acid Homology Alignment of KATs
6. Discussion and Future Perspectives
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartl, F.U. Cellular Homeostasis and Aging. Annu. Rev. Biochem. 2016, 85, 1–4. [Google Scholar] [CrossRef]
- Engel, M.L.; Gunnar, M.R. The development of stress reactivity and regulation during human development. Int. Rev. Neurobiol. 2020, 150, 41–76. [Google Scholar] [CrossRef]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B. Mechanisms underlying epigenetically mediated gene silencing in cancer. Semin. Cancer Biol. 2002, 12, 331–337. [Google Scholar] [CrossRef]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular Signals of Epigenetic States. Science 2010, 330, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Shi, Y. Epigenetic regulation: Methylation of histone and non-histone proteins. Sci. China Ser. C Life Sci. 2009, 52, 311–322. [Google Scholar] [CrossRef]
- Lotterman, C.; Karikari, C.; Omura, N.; Feldmann, G.; Habbe, N.; Goggins, M.G.; Mendell, J.T.; Maitra, A. Epigenetic silencing of MicroRNA miR-107 regulates cyclin-dependent kinase 6 expression in pancreatic cancer. Pancreatology 2009, 9, 293–301. [Google Scholar]
- Jiang, Y.-H.; Bressler, J.; Beaudet, A.L. Epigenetics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2004, 5, 479–510. [Google Scholar] [CrossRef] [Green Version]
- Rodenhiser, D.; Mann, M. Epigenetics and human disease: Translating basic biology into clinical applications. Can. Med. Assoc. J. 2006, 174, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigen. 2016, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belotserkovskaya, R.; Saunders, A.; Lis, J.T.; Reinberg, D. Transcription through chromatin: Understanding a complex FACT. Biochim. Biophys. Acta 2004, 1677, 87–99. [Google Scholar] [CrossRef]
- Sarma, K.; Reinberg, D. Histone variants meet their match. Nat. Rev. Mol. Cell Biol. 2005, 6, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Becker, P.B.; Workman, J.L. Nucleosome Remodeling and Epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017905. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Li, Q.; Mohan, A.L.; Baylin, S.B.; Issa, J.P. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998, 58, 5489–5494. [Google Scholar]
- Bannister, A.J.; Kouzarides, T. Reversing histone methylation. Nat. Cell Biol. 2005, 436, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2, S4–S11. [Google Scholar] [CrossRef]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New Nomenclature for Chromatin-Modifying Enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Wang, Y.; Allis, C.D. Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 2003, 15, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, I.; Roatsch, M.; Schmitt, M.L.; Carlino, L.; Pippel, M.; Sippl, W.; Jung, M. The role of histone demethylases in cancer therapy. Mol. Oncol. 2012, 6, 683–703. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.Y.; Sun, Z.W.; Li, X.; Reuben, M.; Tatchell, K.; Bishop, D.K.; Grushcow, J.M.; Brame, C.J.; Caldwell, J.A. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in bud-ding yeast and nematodes. Cell 2000, 102, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.A.; Torok, M.S.; Sun, Z.-W.; Schieltz, D.; Allis, C.; Yates, J.R.; Grant, P.A. Deubiquitination of Histone H2B by a Yeast Acetyltransferase Complex Regulates Transcription. J. Biol. Chem. 2004, 279, 1867–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Zhang, L.; Qu, Y.; Zhou, Y.; Zhu, J.; Li, Y.; Zhu, T.; Zhao, F.; Tang, J.; Mu, D. Histone acetylation of oligodendrocytes protects against white matter injury induced by inflammation and hypoxia-ischemia through activation of BDNF-TrkB signaling pathway in neonatal rats. Brain Res. 2018, 1688, 33–46. [Google Scholar] [CrossRef]
- Zaini, M.A.; Müller, C.; de Jong, T.V.; Ackermann, T.; Hartleben, G.; Kortman, G.; Gührs, K.-H.; Fusetti, F.; Krämer, O.H.; Guryev, V.; et al. A p300 and SIRT1 Regulated Acetylation Switch of C/EBPalpha Controls Mitochondrial Function. Cell Rep. 2018, 22, 497–511. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2003, 10, 33–39. [Google Scholar] [CrossRef]
- Chiao, P.; Zhang, X.; Zhang, X.; Lazar, M.; Seto, E.; Young, H.A.; Ye, J. Coactivators and corepressors of NF-kappaB in IkappaB alpha gene promoter. J. Biol. Chem. 2005, 280, 21091–21098. [Google Scholar]
- Liu, Z.; Auboeuf, D.; Wong, J.; Chen, J.D.; Tsai, S.Y.; Tsai, M.-J.; O’Malley, B.W. Coactivator/corepressor ratios modulate PR-mediated transcription by the selective receptor modulator RU486. Proc. Natl. Acad. Sci. USA 2002, 99, 7940–7944. [Google Scholar] [CrossRef] [Green Version]
- Andersen, C.L.; Asmar, F.; Klausen, T.; Hasselbalch, H.; Gronbaek, K. Somatic mutations of the CREBBP and EP300 genes affect response to histone deacetylase inhibition in malignant DLBCL clones. Leuk. Res. Rep. 2012, 2, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, A.; Chaurasia, P.; Xiao, G.Q.; Philizaire, M.; Lv, X.; Yao, S.; Burnstein, K.L.; Liu, D.-P.; Levine, A.C.; Mujtaba, S. Coactivator MYST1 regulates nuclear factor-kappaB and androgen receptor functions during proliferation of prostate cancer cells. Mol. Endocrinol. 2014, 28, 872–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debes, J.D.; Sebo, T.J.; Heemers, H.V.; Kipp, B.R.; De Anna, L.H.; Lohse, C.M.; Tindall, D.J. p300 modulates nuclear morphology in prostate cancer. Cancer Res. 2005, 65, 708–712. [Google Scholar] [PubMed]
- Debes, J.D.; Sebo, T.J.; Lohse, C.M.; Murphy, L.M.; De Anna, L.H.; Tindall, D.J. p300 in prostate cancer proliferation and progression. Cancer Res. 2003, 63, 7638–7640. [Google Scholar] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.H. Genome-Editing Technologies: Concept, Pros, and Cons of Various Genome-Editing Techniques and Bioethical Concerns for Clinical Application. Mol. Ther. Nucleic Acids 2019, 16, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation And Methylation Of Histones And Their Possible Role In The Regulation Of Rna Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, S.Y.; Allis, C. Histone Acetylation and Chromatin Assembly: A Single Escort, Multiple Dances? Cell 1996, 87, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, R. Structural and chemical basis of histone acetylation. Novartis Found Symp. 2004, 259, 78–98. [Google Scholar]
- Marmorstein, R.; Zhou, M.-M. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [Green Version]
- Crawford, G.E.; Davis, S.; Scacheri, P.C.; Renaud, G.; Halawi, M.J.; Erdos, M.R.; Green, R.; Meltzer, P.S.; Wolfsberg, T.G.; Collins, F. DNase-chip: A high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat. Methods 2006, 3, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Sandmann, T.; Jakobsen, J.S.; Furlong, E.E.M. ChIP-on-chip protocol for genome-wide analysis of transcription factor binding in Drosophila melanogaster embryos. Nat. Protoc. 2006, 1, 2839–2855. [Google Scholar] [CrossRef]
- Edmondson, D.; Zhang, W.; Watson, A.; Xu, W.; Bone, J.; Yu, Y.; Stillman, D.; Roth, S. In Vivo Functions of Histone Acetylation/Deacetylation in Tup1p Repression and Gcn5p Activation. Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 459–468. [Google Scholar] [CrossRef]
- Munoz-Galvan, S.; Jimeno, S.; Rothstein, R.; Aguilera, A. Histone H3K56 acetylation, Rad52, and non-DNA repair factors con-trol double-strand break repair choice with the sister chromatid. PLoS Genet. 2013, 9, e1003237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, G.; Stefanovsky, V.Y.; Faubladier, M.; Hirschler-Laszkiewicz, I.; Savard, J.; Rothblum, L.I.; Côté, J.; Moss, T. Competitive Recruitment of CBP and Rb-HDAC Regulates UBF Acetylation and Ribosomal Transcription. Mol. Cell 2000, 6, 1059–1066. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Akhtar, A. The many lives of KATs—Detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet. 2019, 20, 7–23. [Google Scholar] [CrossRef]
- Patel, J.H.; Du, Y.; Ard, P.G.; Phillips, C.; Carella, B.; Chen, C.-J.; Rakowski, C.; Chatterjee, C.; Lieberman, P.M.; Lane, W.S.; et al. The c-MYC Oncoprotein Is a Substrate of the Acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Dekker, F.J.; Haisma, H.J. Histone acetyl transferases as emerging drug targets. Drug Discov. Today 2009, 14, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Dekker, F.J.; van den Bosch, T.; Martin, N.I. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov. Today 2014, 19, 654–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujtaba, S.; He, Y.; Zeng, L.; Farooq, A.; Carlson, J.E.; Ott, M.; Verdin, E.; Zhou, M.-M. Structural Basis of Lysine-Acetylated HIV-1 Tat Recognition by PCAF Bromodomain. Mol. Cell 2002, 9, 575–586. [Google Scholar] [CrossRef]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.-M. Structural Mechanism of the Bromodomain of the Coactivator CBP in p53 Transcriptional Activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Mujtaba, S.F.; Zeng, L.; Zhou, M.-M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007, 26, 5521–5527. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Yap, K.L.; Ivanov, A.V.; Wang, X.; Mujtaba, S.; Plotnikova, O.; Iii, F.J.R.; Zhou, M.-M. Structural insights into human KAP1 PHD finger–bromodomain and its role in gene silencing. Nat. Struct. Mol. Biol. 2008, 15, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Parthun, M.R. Histone acetyltransferase 1: More than just an enzyme? Biochim. Biophys. Acta 2012, 1819, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Shi, L.; Guo, Q.; Sun, W.; Yu, Y.; Yang, L.; Zhang, X.; Zhang, M. HAT1 induces lung cancer cell apoptosis via up regulating Fas. Oncotarget 2017, 8, 89970–89977. [Google Scholar] [CrossRef]
- Fan, P.; Zhao, J.; Meng, Z.; Wu, H.; Wang, B.; Wu, H.; Jin, X. Overexpressed histone acetyltransferase 1 regulates cancer immunity by increasing programmed death-ligand 1 expression in pancreatic cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Yuan, Y.; Yuan, H.; Wang, J.; Yun, H.; Geng, Y.; Zhao, M.; Li, L.; Weng, Y.; Liu, Z.; et al. Histone acetyltransferase 1 is a succinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. 2020, 22, e50967. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Atanassov, B.S.; Evrard, Y.A.; Multani, A.S.; Zhang, Z.; Tora, L.; Devys, D.; Chang, S.; Dent, S.Y. Gcn5 and SAGA Regulate Shelterin Protein Turnover and Telomere Maintenance. Mol. Cell 2009, 35, 352–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farria, A.T.; Mustachio, L.M.; Akdemir, Z.H.C.; Dent, S.Y. GCN5 HAT inhibition reduces human Burkitt lymphoma cell survival through reduction of MYC target gene expression and impeding BCR signaling pathways. Oncotarget 2019, 10, 5847–5858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farria, A.T.; Plummer, J.B.; Salinger, A.P.; Shen, J.; Lin, K.; Lu, Y.; McBride, K.M.; Koutelou, E.; Dent, S.Y. Transcriptional Activation of MYC-Induced Genes by GCN5 Promotes B-cell Lymphomagenesis. Cancer Res. 2020, 80, 5543–5553. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Lemen, J.M.; Chan, V.; Wey, A.; Lin, W.; Dent, S.R.; Knoepfler, P.S. N-Myc and GCN5 Regulate Significantly Overlapping Transcriptional Programs in Neural Stem Cells. PLoS ONE 2012, 7, e39456. [Google Scholar] [CrossRef]
- Mustachio, L.M.; Roszik, J.; Farria, A.T.; Guerra, K.; Dent, S.Y. Repression of GCN5 expression or activity attenuates c-MYC expression in non-small cell lung cancer. Am. J. Cancer Res. 2019, 9, 1830–1845. [Google Scholar]
- Liu, T.; Wang, X.; Hu, W.; Fang, Z.; Jin, Y.; Fang, X.; Miao, Q.R. Epigenetically Down-Regulated Acetyltransferase PCAF Increases the Resistance of Colorectal Cancer to 5-Fluorouracil. Neoplasia 2019, 21, 557–570. [Google Scholar] [CrossRef]
- Wang, L.; Liu, K.; Jeng, W.; Chiang, C.; Chai, C.; Chiou, S.; Huang, M.; Yokoyama, K.K.; Wang, S.; Huang, S.; et al. PCAF -mediated acetylation of ISX recruits BRD 4 to promote epithelial-mesenchymal transition. EMBO Rep. 2020, 21, e48795. [Google Scholar] [CrossRef]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med. 2016, 7, a026534. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.E.; Huhn, A.; Gardberg, A.S.; Poy, F.; Brucelle, F.; Vivat, V.; Cantone, N.; Patel, G.; Patel, C.; Cummings, R.; et al. Early Drug-Discovery Efforts towards the Identification of EP300/CBP Histone Acetyltransferase (HAT) In-hibitors. ChemMedChem 2020, 15, 955–960. [Google Scholar]
- Iyer, N.G.; Ozdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.G.; Xian, J.; Chin, S.F.; Bannister, A.J.; Daigo, Y.; Aparicio, S.; Kouzarides, T.; Caldas, C. p300 is required for orderly G1/S transition in human cancer cells. Oncogene 2007, 26, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Rühlmann, F.; Windhof-Jaidhauser, I.M.; Menze, C.; Beißbarth, T.; Bohnenberger, H.; Ghadimi, M.; Dango, S. The prognostic capacities of CBP and p300 in locally advanced rectal cancer. World J. Surg. Oncol. 2019, 17, 1–8. [Google Scholar] [CrossRef]
- Scolnick, D.M.; Chehab, N.H.; Stavridi, E.S.; Lien, M.C.; Caruso, L.; Moran, E.; Berger, S.L.; Halazonetis, T.D. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 1997, 57, 3693–3696. [Google Scholar]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Li, J.; Huang, C.; Xiong, T.; Zhuang, C.; Zhuang, C.; Li, Y.; Ye, J.; Gui, Y. A CRISPR Interference of CBP and p300 Selectively Induced Synthetic Lethality in Bladder Cancer Cells In Vitro. Int. J. Biol. Sci. 2019, 15, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Tavassoli, P.; Wafa, L.A.; Cheng, H.; Zoubeidi, A.; Fazli, L.; Gleave, M.; Snoek, R.; Rennie, P.S. TAF1 differentially enhances androgen receptor transcriptional activity via its N-terminal kinase and ubiq-uitin-activating and -conjugating domains. Mol. Endocrinol. 2010, 24, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Man, N.; Karl, D.; Martinez, C.; Liu, F.; Sun, J.; Martinez, C.J.; Martin, G.M.; Beckedorff, F.; Lai, F.; et al. TAF1 plays a critical role in AML1-ETO driven leukemogenesis. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Ribeiro, J.R.; Lovasco, L.A.; Vanderhyden, B.C.; Freiman, R.N. Targeting TBP-Associated Factors in Ovarian Cancer. Front. Oncol. 2014, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Cregan, S.; McDonagh, L.; Gao, Y.; Barr, M.P.; O’Byrne, K.J.; Finn, S.P.; Cuffe, S.; Gray, S.G. KAT5 (Tip60) is a potential therapeutic target in malignant pleural mesothelioma. Int. J. Oncol. 2016, 48, 1290–1296. [Google Scholar] [CrossRef] [Green Version]
- Halkidou, K.; Gnanapragasam, V.J.; Mehta, P.B.; Logan, I.R.; E Brady, M.; Cook, S.; Leung, H.Y.; E Neal, D.; Robson, C.N. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene 2003, 22, 2466–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idrissou, M.; Rifaï, K.; Daures, M.; Penault-Llorca, F.; Bignon, Y.-J.; Bernard-Gallon, D. Exciting History of Tip60 and Its Companions in Carcinogenesis Across the Heterochromatin Landscapes. OMICS A J. Integr. Biol. 2018, 22, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Judes, G.; Dubois, L.; Rifaï, K.; Idrissou, M.; Mishellany, F.; Pajon, A.; Besse, S.; Daures, M.; Degoul, F.; Bignon, Y.-J.; et al. TIP60: An actor in acetylation of H3K4 and tumor development in breast cancer. Epigenomics 2018, 10, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Judes, G.; Rifaï, K.; Ngollo, M.; Daures, M.; Bignon, Y.-J.; Penault-Llorca, F.; Bernard-Gallon, D. A bivalent role of TIP60 histone acetyl transferase in human cancer. Epigenomics 2015, 7, 1351–1363. [Google Scholar] [CrossRef]
- Sakuraba, K.; Yasuda, T.; Sakata, M.; Kitamura, Y.-H.; Shirahata, A.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; et al. Down-regulation of Tip60 gene as a potential marker for the malignancy of colorectal cancer. Anticancer Res. 2009, 29, 3953–3955. [Google Scholar] [PubMed]
- Sakuraba, K.; Yokomizo, K.; Shirahata, A.; Goto, T.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Hibi, K. TIP60 as a potential marker for the malignancy of gastric cancer. Anticancer Res. 2011, 31, 77–79. [Google Scholar]
- Shiota, M.; Yokomizo, A.; Masubuchi, D.; Tada, Y.; Inokuchi, J.; Eto, M.; Uchiumi, T.; Fujimoto, N.; Naito, S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2009, 70, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, K.; Ayton, P.M.; Carapeti, M.; Kutok, J.L.; Snyder, C.S.; Williams, I.R.; Cross, N.C.; Glass, C.K.; Cleary, M.L.; Gilliland, D. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell 2003, 3, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Huntly, B.J.; Shigematsu, H.; Deguchi, K.; Lee, B.H.; Mizuno, S.; Duclos, N.; Rowan, R.; Amaral, S.; Curley, D.; Williams, I.R.; et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004, 6, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Kitabayashi, I.; Aikawa, Y.; Nguyen, L.A.; Yokoyama, A.; Ohki, M. Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J. 2001, 20, 7184–7196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitabayashi, I.; Aikawa, Y.; Yokoyama, A.; Hosoda, F.; Nagai, M.; Kakazu, N.; Abe, T.; Ohki, M. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 2001, 15, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Largeot, A.; Perez-Campo, F.M.; Marinopoulou, E.; Lie-A-Ling, M.; Kouskoff, V.; Lacaud, G. Expression of the MOZ-TIF2 oncoprotein in mice represses senescence. Exp. Hematol. 2016, 44, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, K.; Safaralizadeh, R.; Hosseinpour-Feizi, M.; Dastmalchi, N.; Moaddab, Y. Investigation of the changes in the ex-pression levels of MOZ gene in colorectal cancer tissues. J. Gastrointest Oncol. 2019, 10, 68–73. [Google Scholar] [CrossRef]
- Ullah, M.; Pelletier, N.; Xiao, L.; Zhao, S.P.; Wang, K.; Degerny, C.; Tahmasebi, S.; Cayrou, C.; Doyon, Y.; Goh, S.-L.; et al. Molecular Architecture of Quartet MOZ/MORF Histone Acetyltransferase Complexes. Mol. Cell. Biol. 2008, 28, 6828–6843. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-J.; Ullah, M.F. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene 2007, 26, 5408–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.L.; Sheu JJ, C.; Lin, H.P.; Jeng, Y.M.; Chang CY, Y.; Chen, C.M.; Cheng, J.; Mao, T.L. The overexpression of MYST4 in human solid tumors is associated with increased aggressiveness and de-creased overall survival. Int. J. Clin. Exp. Pathol. 2019, 12, 431–442. [Google Scholar]
- Guo, L.-L.; Yu, S.-Y.; Li, M. Functional analysis of HBO1 in tumor development and inhibitor screening. Int. J. Mol. Med. 2016, 38, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, M.; Takahashi, Y.; Mizzen, C.A.; Cook, R.G.; Fujita, M.; Allis, C.D.; Frierson, H.F.; Fukusato, T.; Smith, M.M. Histone acetyltransferase Hbo1: Catalytic activity, cellular abundance, and links to primary cancers. Gene 2009, 436, 108–114. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, L.; Anokye, J.; Yeung, M.M.; Lam, E.Y.N.; Chan, Y.-C.; Weng, C.-F.; Yeh, P.; Knezevic, K.; Butler, M.S.; Hoegl, A.; et al. HBO1 is required for the maintenance of leukaemia stem cells. Nat. Cell Biol. 2020, 577, 266–270. [Google Scholar] [CrossRef]
- Song, B.; Liu, X.S.; Rice, S.J.; Kuang, S.; Elzey, B.D.; Konieczny, S.F.; Ratliff, T.L.; Hazbun, T.; Chiorean, E.G. Plk1 Phosphorylation of Orc2 and Hbo1 Contributes to Gemcitabine Resistance in Pancreatic Cancer. Mol. Cancer Ther. 2013, 12, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Ye, X.; Tang, N.; Shen, S.; Li, Z.; Niu, X.; Lu, S.; Xu, L. The histone acetylranseferase hMOF acetylates Nrf2 and regulates anti-drug responses in human non-small cell lung cancer. Br. J. Pharmacol. 2014, 171, 3196–3211. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltrans-ferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.; Rea, S.; Taipale, M.; Mendrzyk, F.; Straub, B.; Ittrich, C.; Thuerigen, O.; Sinn, H.P.; Akhtar, A.; Lichter, P. The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medullo-blastoma and constitutes a biomarker for clinical outcome in medulloblastoma. Int. J. Cancer 2008, 122, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Song, J.S.; Chun, S.M.; Lee, J.Y.; Kim, D.K.; Kim, Y.H.; Jang, S.J. The Histone Acetyltransferase hMOF is Overexoressed in Non-small Cell Lung Carcinoma. Korean J. Pathol. 2011, 45, 386–396. [Google Scholar] [CrossRef]
- Wu, L.; Zee, B.M.; Wang, Y.; Garcia, B.A.; Dou, Y. The RING finger protein MSL2 in the MOF complex is an E3 ubiquitin ligase for H2B K34 and is involved in crosstalk with H3 K4 and K79 methylation. Mol. Cell 2011, 43, 132–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaunay, S.; Rapino, F.; Tharun, L.; Zhou, Z.; Heukamp, L.; Termathe, M.; Shostak, K.; Klevernic, I.; Florin, A.; Desmecht, H.; et al. Elp3 links tRNA modification to IRES-dependent translation of LEF1 to sustain metastasis in breast cancer. J. Exp. Med. 2016, 213, 2503–2523. [Google Scholar] [CrossRef] [Green Version]
- Li, M.-T.; Liang, J.-Y.; Sun, Y.-P.; Jin, J.; Xiong, Y.; Guan, K.-L.; Yuan, H.-X. ELP3 Acetyltransferase is phosphorylated and regulated by the oncogenic anaplastic lymphoma kinase (ALK). Biochem. J. 2019, 476, 2239–2254. [Google Scholar] [CrossRef]
- Rosu, A.; El Hachem, N.; Rapino, F.; Rouault-Pierre, K.; Jorssen, J.; Somja, J.; Remery, E.; Thiry, M.; Nguyen, L.; Jacquemyn, M.; et al. Loss of tRNA-modifying enzyme Elp3 activates a p53-dependent antitumor checkpoint in hematopoiesis. J. Exp. Med. 2021, 218, e20200662. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jin, C.; Zhang, X.; Sun, M.; Zhang, Y.; Zhang, G. Clinical implications of the coexpression of SRC1 and NANOG in HER-2-overexpressing breast cancers. OncoTargets Ther. 2016, 9, 5483–5488. [Google Scholar] [CrossRef] [Green Version]
- Berns, E.M.; Van Staveren, I.L.; Klijn, J.G.; Foekens, J.A. Predictive value of SRC-1 for tamoxifen response of recurrent breast cancer. Breast Cancer Res. Treat. 1998, 48, 87–92. [Google Scholar] [CrossRef]
- Browne, A.L.; Charmsaz, S.; Varešlija, D.; Fagan, A.; Cosgrove, N.; Cocchiglia, S.; Purcell, S.; Ward, E.; Bane, F.; Hudson, L.; et al. Network analysis of SRC-1 reveals a novel transcription factor hub which regulates endocrine resistant breast cancer. Oncogene 2018, 37, 2008–2021. [Google Scholar] [CrossRef]
- Deblacam, C.; Byrne, C.; Hughes, E.; McIlroy, M.; Bane, F.; Hill AD, K.; Young, L.S. HOXC11-SRC-1 regulation of S100beta in cutaneous melanoma: New targets for the kinase inhibitor da-satinib. Br. J. Cancer 2011, 105, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Fleming, F.J.; Myers, E.R.; Kelly, G.M.; Crotty, T.B.; McDermott, E.W.; O’Higgins, N.J.; Hill, A.D.K.; Young, L.S. Expression of SRC-1, AIB1, and PEA3 in HER2 mediated endocrine resistant breast cancer; a predictive role for SRC-1. J. Clin. Pathol. 2004, 57, 1069–1074. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wong, J.; Tsai, S.Y.; Tsai, M.-J.; O’Malley, B.W. Steroid receptor coactivator-1 (SRC-1) enhances ligand-dependent and receptor-dependent cell-free transcription of chromatin. Proc. Natl. Acad. Sci. USA 1999, 96, 9485–9490. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wong, J.; Tsai, S.Y.; Tsai, M.-J.; O’Malley, B.W. Sequential recruitment of steroid receptor coactivator-1 (SRC-1) and p300 enhances progesterone receptor-dependent initiation and reinitiation of transcription from chromatin. Proc. Natl. Acad. Sci. USA 2001, 98, 12426–12431. [Google Scholar] [CrossRef] [Green Version]
- McBryan, J.; Theissen, S.M.; Byrne, C.; Hughes, E.; Cocchiglia, S.; Sande, S.; O’Hara, J.; Tibbitts, P.; Hill, A.D.; Young, L.S. Metastatic Progression with Resistance to Aromatase Inhibitors Is Driven by the Steroid Receptor Coactivator SRC-1. Cancer Res. 2012, 72, 548–559. [Google Scholar] [CrossRef] [Green Version]
- McCartan, D.; Bolger, J.C.; Fagan, A.; Byrne, C.; Hao, Y.; Qin, L.; McIlroy, M.; Xu, J.; Hill, A.D.; Gaora, P.Ó.; et al. Global Characterization of the SRC-1 Transcriptome Identifies ADAM22 as an ER-Independent Mediator of Endocrine-Resistant Breast Cancer. Cancer Res. 2012, 72, 220–229. [Google Scholar] [CrossRef] [Green Version]
- McIlroy, M.; McCartan, D.; Early, S.; Ogaora, P.; Pennington, S.; Hill, A.; Young, L. Interaction of developmental transcription factor HOXC11 with steroid receptor coactivator SRC-1 mediates resistance to endocrine therapy in breast cancer [corrected]. Cancer Res. 2010, 70, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Myers, E.; Fleming, F.J.; Crotty, T.B.; Kelly, G.; McDermott, E.W.; O’Higgins, N.J.; Hill, A.D.K.; Young, L.S. Inverse relationship between ER-beta and SRC-1 predicts outcome in endocrine-resistant breast cancer. Br. J. Cancer 2004, 91, 1687–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, E.; Hill, A.D.; Kelly, G.; McDermott, E.W.; O’Higgins, N.J.; Buggy, Y.; Young, L.S. Associations and Interactions between Ets-1 and Ets-2 and Coregulatory Proteins, SRC-1, AIB1, and NCoR in Breast Cancer. Clin. Cancer Res. 2005, 11, 2111–2122. [Google Scholar] [CrossRef] [Green Version]
- Gojis, O.; Rudraraju, B.; Alifrangis, C.; Krell, J.; Libalova, P.; Palmieri, C. The role of steroid receptor coactivator-3 (SRC-3) in human malignant disease. Eur. J. Surg. Oncol. 2010, 36, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gojis, O.; Rudraraju, B.; Gudi, M.; Hogben, K.; Sousha, S.; Coombes, C.R.; Cleator, S.; Palmieri, C. The role of SRC-3 in human breast cancer. Nat. Rev. Clin. Oncol. 2009, 7, 83–89. [Google Scholar] [CrossRef]
- Lonard, D.M.; O’Malley, B.W. SRC-3 Transcription-Coupled Activation, Degradation, and the Ubiquitin Clock: Is There Enough Coactivator to Go Around in Cells? Sci. Signal. 2008, 1, pe16. [Google Scholar] [CrossRef]
- Song, X.; Chen, H.; Zhang, C.; Yu, Y.; Chen, Z.; Liang, H.; Van Buren, G.; McElhany, A.L.; Fisher, W.E.; Lonard, D.M.; et al. SRC-3 inhibition blocks tumor growth of pancreatic ductal adenocarcinoma. Cancer Lett. 2019, 442, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhang, C.; Zhao, M.; Chen, H.; Liu, X.; Chen, J.; Lonard, D.M.; Qin, L.; Xu, J.; Wang, X.; et al. Steroid Receptor Coactivator-3 (SRC-3/AIB1) as a Novel Therapeutic Target in Triple Negative Breast Cancer and Its Inhibition with a Phospho-Bufalin Prodrug. PLoS ONE 2015, 10, e0140011. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liao, L.; Ning, G.; Yoshida-Komiya, H.; Deng, C.; O’Malley, B.W. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puber-ty, female reproductive function, and mammary gland development. Proc. Natl. Acad. Sci. USA 2000, 97, 6379–6384. [Google Scholar] [CrossRef] [Green Version]
- Morales-Santana, S.; Morell, S.; Leon, J.; Carazo-Gallego, A.; Jimenez-Lopez, J.C.; Morell, M. An Overview of the Polymorphisms of Circadian Genes Associated With Endocrine Cancer. Front. Endocrinol. 2019, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.J.; Man, N.; Tan, Y.; Nimer, S.D.; Wang, L. The Role of Histone Acetyltransferases in Normal and Malignant Hemato-poiesis. Front. Oncol. 2015, 5, 108. [Google Scholar] [CrossRef] [Green Version]
- Vu, L.P.; Luciani, L.; Nimer, S.D. Histone-modifying enzymes: Their role in the pathogenesis of acute leukemia and their ther-apeutic potential. Int. J. Hematol. 2013, 97, 198–209. [Google Scholar] [CrossRef] [Green Version]
- UniProt. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Björling, E.; Uhlén, M. Antibodypedia, a Portal for Sharing Antibody and Antigen Validation Data. Mol. Cell. Proteom. 2008, 7, 2028–2037. [Google Scholar] [CrossRef] [Green Version]
- Pornputtapong, N.; Nookaew, I.; Nielsen, J. Human metabolic atlas: An online resource for human metabolism. Database 2015, 2015, bav068. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, P.; Argoud-Puy, G.; Cusin, I.; Duek, P.; Evalet, O.; Gateau, A.; Gleizes, A.; Pereira, M.; Zahn-Zabal, M.; Zwahlen, C.; et al. neXtProt: Organizing Protein Knowledge in the Context of Human Proteome Projects. J. Proteome Res. 2012, 12, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Zahn-Zabal, M.; Lane, L. What will neXtProt help us achieve in 2020 and beyond? Expert Rev. Proteom. 2020, 17, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.L.; Onate, S.A.; Tsai, M.J.; O’Malley, B.W. CREB binding protein acts synergistically with steroid receptor coactiva-tor-1 to enhance steroid receptor-dependent transcription. Proc. Natl. Acad. Sci. USA 1996, 93, 8884–8888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menet, J.S.; Pescatore, S.; Rosbash, M. CLOCK:BMAL1 is a pioneer-like transcription factor. Genes Dev. 2014, 28, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, F.; Pedrazzoli, M.; Coelho, F.M.D.S.; Pradella-Hallinan, M.; Da Conceição, M.C.L.; Peregrino, A.J.P.; De Oliveira, E.C.; Tufik, S. Clock gene polymorphisms and narcolepsy in positive and negative HLA-DQB1*0602 patients. Mol. Brain Res. 2005, 140, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nat. Cell Biol. 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Viotti, M.; Wilson, C.; McCleland, M.; Koeppen, H.; Haley, B.; Jhunjhunwala, S.; Klijn, C.; Modrusan, Z.; Arnott, D.; Classon, M.; et al. SUV420H2 is an epigenetic regulator of epithelial/mesenchymal states in pancreatic cancer. J. Cell Biol. 2017, 217, 763–777. [Google Scholar] [CrossRef]
- Kanz, C. The EMBL Nucleotide Sequence Database. Nucleic Acids Res. 2004, 33, D29–D33. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Tivey, A.R.N.; Lopez, R. Using EMBL-EBI Services via Web Interface and Program-matically via Web Services. Curr. Protoc. Bioinform. 2019, 66, e74. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M. The Protein Data Bank: A historical perspective. Acta Crystallogr. 2007, 64, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Kleywegt, G.J.; Nakamura, H.; Markley, J.L. The Protein Data Bank at 40: Reflecting on the Past to Prepare for the Future. Structure 2012, 20, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zang, C.; A Rosenfeld, J.; E Schones, D.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Conway, E.; Healy, E.; Bracken, A.P. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr. Opin. Cell Biol. 2015, 37, 42–48. [Google Scholar] [CrossRef]
- Deevy, O.; Bracken, A.P. PRC2 functions in development and congenital disorders. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latham, J.A.; Dent, S.Y.R. Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 2007, 14, 1017–1024. [Google Scholar] [CrossRef]
- Bell, O.; Wirbelauer, C.; Hild, M.; Scharf, A.N.; Schwaiger, M.; MacAlpine, D.M.; Zilbermann, F.; van Leeuwen, F.; Bell, S.P.; Imhof, A.; et al. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J. 2007, 26, 4974–4984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-J.; Ou, Y.; Jiang, L.; Gu, W. Ferroptosis: A missing puzzle piece in the p53 blueprint? Mol. Cell. Oncol. 2016, 3, e1046581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gorospe, M.; Barnes, J.; Liu, Y. Tumor Promoter Arsenite Stimulates Histone H3 Phosphoacetylation of Proto-oncogenes c-fos and c-jun Chromatin in Human Diploid Fibroblasts. J. Biol. Chem. 2003, 278, 13183–13191. [Google Scholar] [CrossRef] [Green Version]
- Maksimoska, J.; Segura-Peña, D.; Cole, P.A.; Marmorstein, R. Structure of the p300 Histone Acetyltransferase Bound to Acetyl-Coenzyme A and Its Analogues. Biochemistry 2014, 53, 3415–3422. [Google Scholar] [CrossRef]
- Poux, A.N.; Marmorstein, R. Molecular Basis for Gcn5/PCAF Histone Acetyltransferase Selectivity for Histone and Nonhistone Substrates. Biochemistry 2003, 42, 14366–14374. [Google Scholar] [CrossRef]
- Rojas, J.R.; Trievel, R.C.; Zhou, J.; Mo, Y.; Li, X.; Berger, S.L.; Allis, C.D.; Marmorstein, R. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nat. Cell Biol. 1999, 401, 93–98. [Google Scholar] [CrossRef]
- Daujat, S.; Bauer, U.-M.; Shah, V.; Turner, B.; Berger, S.; Kouzarides, T. Crosstalk between CARM1 Methylation and CBP Acetylation on Histone H3. Curr. Biol. 2002, 12, 2090–2097. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.L.; Turner, F.B.; Krishnamoorthy, T.; Wolner, B.; Ahn, S.-H.; Foley, M.; Dorsey, J.A.; Peterson, C.L.; Berger, S.L.; Allis, C.D. Phosphorylation of Histone H4 Serine 1 during DNA Damage Requires Casein Kinase II in S. cerevisiae. Curr. Biol. 2005, 15, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.; Pathak, R.R.; Mujtaba, S. The biology of lysine acetylation integrates transcriptional programming and metabolism. Nutr. Metab. 2011, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Küttner, V.; Bhukel, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. Nucleocytosolic Depletion of the Energy Metabolite Acetyl-Coenzyme A Stimulates Autophagy and Prolongs Lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Lai, B.; Lee, J.E.; Jang, Y.; Wang, L.; Peng, W.; Ge, K. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogene-sis. Nucleic Acids Res. 2017, 45, 6388–6403. [Google Scholar] [CrossRef] [PubMed]
- Demetriadou, C.; Kirmizis, A. Histone Acetyltransferases in Cancer: Guardians or Hazards? Crit. Rev. Oncog. 2017, 22, 195–218. [Google Scholar] [CrossRef]
- Han, M.; Jia, L.; Lv, W.; Wang, L.; Cui, W. Epigenetic Enzyme Mutations: Role in Tumorigenesis and Molecular Inhibitors. Front. Oncol. 2019, 9, 194. [Google Scholar] [CrossRef]
- Lavau, C.; Du, C.; Thirman, M.; Zeleznik-Le, N. Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J. 2000, 19, 4655–4664. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Luo, R.T.; Mi, S.; Sun, M.; Chen, P.; Bao, J.; Neilly, M.B.; Jayathilaka, N.; Johnson, D.S.; Wang, L.; et al. Consistent Deregulation of Gene Expression between Human and Murine MLL Rearrangement Leukemias. Cancer Res. 2009, 69, 1109–1116. [Google Scholar] [CrossRef] [Green Version]
- Der Poel, S.Z.-V.; McCabe, N.R.; Gill, H.J.; Espinosa, R.; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; Lebeau, M.M.; Rowley, J.D. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739. [Google Scholar] [CrossRef] [Green Version]
- Jakovcevski, M.; Ruan, H.; Shen, E.Y.; Dincer, A.; Javidfar, B.; Ma, Q.; Peter, C.J.; Cheung, I.; Mitchell, A.C.; Jiang, Y.; et al. Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and work-ing memory. J. Neurosci. 2015, 35, 5097–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santillan, D.A.; Theisler, C.M.; Ryan, A.S.; Popovic, R.; Stuart, T.; Zhou, M.M.; Alkan, S.; Zeleznik-Le, N.J. Bromodomain and histone acetyltransferase domain specificities control mixed lineage leukemia pheno-type. Cancer Res. 2006, 66, 10032–10039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobulo, O.M.; Borrow, J.; Tomek, R.; Reshmi, S.; Harden, A.; Schlegelberger, B.; Housman, D.; Doggett, N.A.; Rowley, J.D.; Zeleznik-Le, N.J. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc. Natl. Acad. Sci. USA 1997, 94, 8732–8737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesel-Motiuk, N.; Assaraf, Y.G. The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology. Drug Resist. Update 2020, 53, 100729. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Karin, M. NF-kappaB and cancer: Mechanisms and targets. Mol. Carcinog. 2006, 45, 355–361. [Google Scholar] [CrossRef]
- Karin, M.; Lawrence, T.; Nizet, V. Innate Immunity Gone Awry: Linking Microbial Infections to Chronic Inflammation and Cancer. Cell 2006, 124, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Durand, J.K.; Baldwin, A.S. Targeting IKK and NF-kappaB for Therapy. Adv. Protein Chem. Struct. Biol. 2017, 107, 77–115. [Google Scholar] [PubMed]
- Brasier, A.R.; Tian, B.; Jamaluddin, M.; Kalita, M.K.; Garofalo, R.P.; Lu, M. RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflamma-tory cytokines in respiratory syncytial virus infection. J. Virol. 2011, 85, 11752–11769. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer. 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Lu, T.; Stark, G.R. NF-kappaB: Regulation by Methylation. Cancer Res. 2015, 75, 3692–3695. [Google Scholar] [CrossRef] [Green Version]
- Stark, G.R.; Wang, Y.; Lu, T. Lysine methylation of promoter-bound transcription factors and relevance to cancer. Cell Res. 2010, 21, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Smale, S.T. Hierarchies of NF-kappaB target-gene regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Brd4 and JMJD6-Associated Anti-Pause Enhancers in Regulation of Transcriptional Pause Release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef] [Green Version]
- Hah, N.; Benner, C.; Chong, L.-W.; Yu, R.T.; Downes, M.; Evans, R.M. Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proc. Natl. Acad. Sci. USA 2015, 112, E297–E302. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Blatt, K.; Shi, J.; Gleixner, K.V.; Cerny-Reiterer, S.; Müllauer, L.; Vakoc, C.R.; Sperr, W.R.; Horny, H.-P.; Bradner, J.E.; et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia (AML). Oncotarget 2012, 3, 1588–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Vakoc, C.R. Brd4 is on the move during inflammation. Trends Cell Biol. 2014, 24, 615–616. [Google Scholar] [CrossRef] [Green Version]
- Laptenko, O.; Tong, D.R.; Manfredi, J.; Prives, C. The Tail That Wags the Dog: How the Disordered C-Terminal Domain Con-trols the Transcriptional Activities of the p53 Tumor-Suppressor Protein. Trends Biochem. Sci. 2016, 41, 1022–1034. [Google Scholar] [CrossRef] [Green Version]
- Senturk, E.; Manfredi, J.J. Determine the Effect of p53 on Chemosensitivity. Methods Mol. Biol. 2013, 962, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Senturk, E.; Manfredi, J.J. p53 and cell cycle effects after DNA damage. Breast Cancer 2012, 962, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Mujtaba, S.; Zeng, L.; Zhoug, M.-M. Modulating Molecular Functions of p53 with Small Molecules. Cell Cycle 2006, 5, 2575–2578. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Shi, X.-L.; Roeder, R.G. Synergistic activation of transcription by CBP and p53. Nat. Cell Biol. 1997, 387, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Banck, M.; Mujtaba, S.; Zhou, M.-M.; Sugrue, M.M.; Walsh, M.J. p53-Induced Growth Arrest Is Regulated by the Mitochondrial SirT3 Deacetylase. PLoS ONE 2010, 5, e10486. [Google Scholar] [CrossRef]
- Prives, C.; Manley, J.L. Why Is p53 Acetylated? Cell 2001, 107, 815–818. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Turner-Ivey, B.; Guest, S.T.; Irish, J.C.; Kappler, C.S.; Garrett-Mayer, E.; Wilson, R.C.; Ethier, S.P. KAT6A, a Chromatin Modifier from the 8p11-p12 Amplicon is a Candidate Oncogene in Luminal Breast Cancer. Neoplasia 2014, 16, 644–655. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, M.S.; Oses, C.; Echegaray, C.V.; Solari, C.; Waisman, A.; Álvarez, Y.; Petrone, M.V.; Francia, M.; Schultz, M.; Sevlever, G.; et al. Kat6b Modulates Oct4 and Nanog Binding to Chromatin in Embryonic Stem Cells and Is Required for Efficient Neural Differentiation. J. Mol. Biol. 2019, 431, 1148–1159. [Google Scholar] [CrossRef]
- Sapountzi, V.; Logan, I.R.; Robson, C.N. Cellular functions of TIP60. Int. J. Biochem. Cell Biol. 2006, 38, 1496–1509. [Google Scholar] [CrossRef]
- Gupta, A.; Guerin-Peyrou, T.G.; Sharma, G.G.; Park, C.; Agarwal, M.; Ganju, R.K.; Pandita, S.; Choi, K.; Sukumar, S.; Pandita, R.K.; et al. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogene-sis and oncogenesis. Mol. Cell Biol. 2008, 28, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Corsa, C.A.S.; Pan, P.W.; Wu, L.; Ferguson, D.; Yu, X.; Min, J.; Dou, Y. MOF and H4 K16 Acetylation Play Important Roles in DNA Damage Repair by Modulating Recruitment of DNA Damage Repair Protein Mdc1. Mol. Cell Biol. 2010, 30, 5335–5347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.G.; So, S.; Gupta, A.; Kumar, R.; Cayrou, C.; Avvakumov, N.; Bhadra, U.; Pandita, R.K.; Porteus, M.H.; Chen, D.J.; et al. MOF and Histone H4 Acetylation at Lysine 16 Are Critical for DNA Damage Response and Double-Strand Break Repair. Mol. Cell Biol. 2010, 30, 3582–3595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.; Dehm, S.M. Androgen Receptor Rearrangement and Splicing Variants in Resistance to Endocrine Therapies in Prostate Cancer. Endocrinology 2017, 158, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Jentzmik, F.; Azoitei, A.; Zengerling, F.; Damjanoski, I.; Cronauer, M.V. Androgen receptor aberrations in the era of abiraterone and enzalutamide. World J. Urol. 2015, 34, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.; Kundu, T.K.; Fu, J.; Roeder, R.G. A Human SPT3-TAFII31-GCN5-L Acetylase Complex Distinct from Transcription Factor IID. J. Biol. Chem. 1998, 273, 23781–23785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terreni, M.; Valentini, P.; Liverani, V.; Gutierrez, M.I.; Di Primio, C.; Di Fenza, A.; Tozzini, V.; Allouch, A.; Albanese, A.; Giacca, M.; et al. GCN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology 2010, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Mujtaba, S.; Zhou, M.-M. Anti-viral opportunities during transcriptional activation of latent HIV in the host chromatin. Methods 2011, 53, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Li, J.; Müller, M.; Yan, S.; Mujtaba, S.; Pan, C.; Wang, Z.; Zhou, M.-M. Selective Small Molecules Blocking HIV-1 Tat and Coactivator PCAF Association. J. Am. Chem. Soc. 2005, 127, 2376–2377. [Google Scholar] [CrossRef]
- Bryant, R.A. Posttraumatic stress disorder and traumatic brain injury: Can they co-exist? Clin. Psychol. Rev. 2001, 21, 931–948. [Google Scholar] [CrossRef]
- Bryant, R.A. Disentangling Mild Traumatic Brain Injury and Stress Reactions. N. Engl. J. Med. 2008, 358, 525–527. [Google Scholar] [CrossRef]
- Dash, P.K.; Orsi, S.A.; Zhang, M.; Grill, R.J.; Pati, S.; Zhao, J.; Moore, A.N. Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive func-tion in rats. PLoS ONE 2010, 5, e11383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; West, E.J.; Van, K.C.; Gurkoff, G.G.; Zhou, J.; Zhang, X.-M.; Kozikowski, A.P.; Lyeth, B.G. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 2008, 1226, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.-M.; Chadha, M.S.; Kline, A.E.; Clark, R.S.; Kochanek, P.M.; Dixon, C.E.; Jenkins, L.W. Immunohistochemical analysis of histone H3 acetylation and methylation—Evidence for altered epigenetic signaling following traumatic brain injury in immature rats. Brain Res. 2006, 1070, 31–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
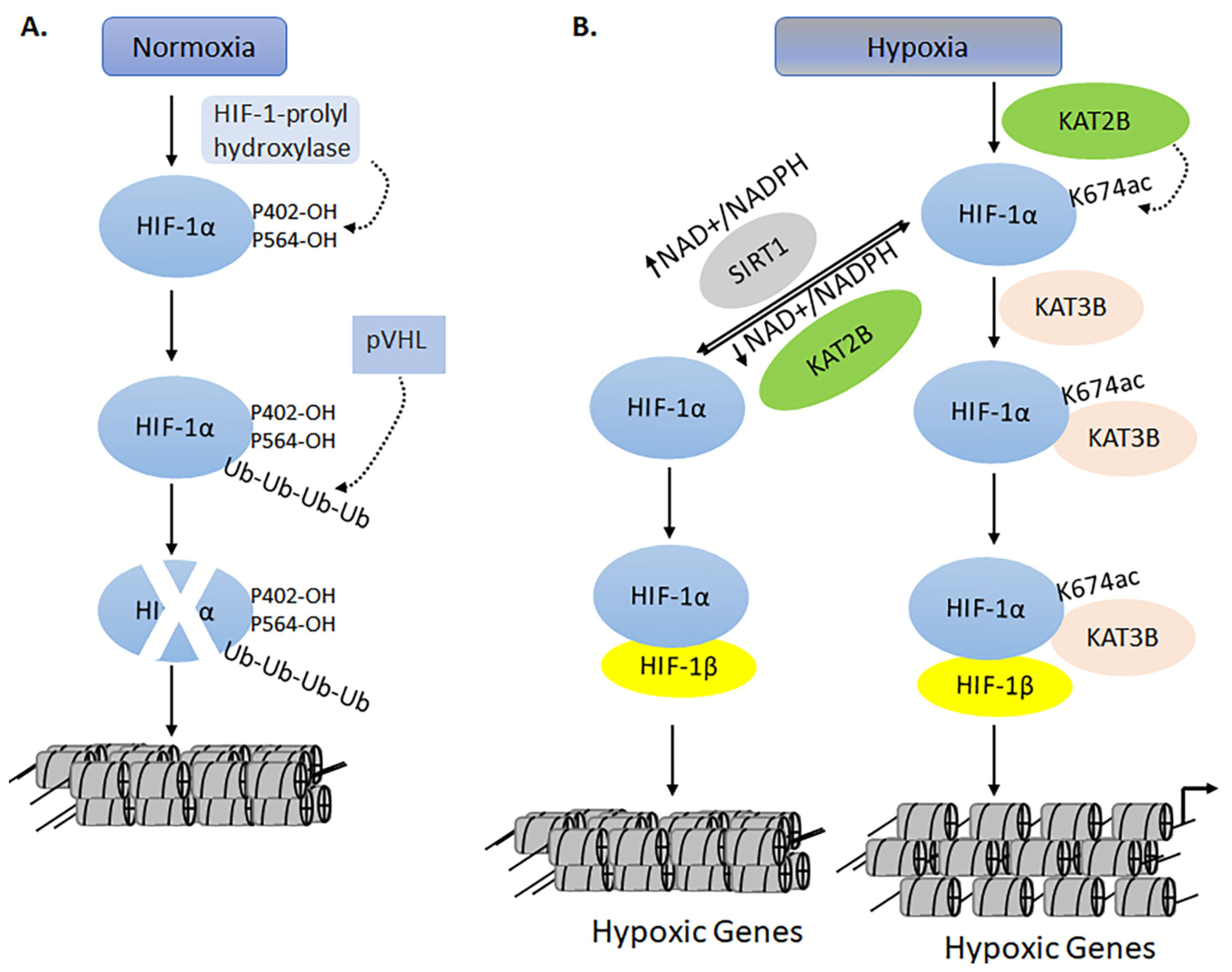
- Leiser, S.F.; Kaeberlein, M. A Role for SIRT1 in the Hypoxic Response. Mol. Cell 2010, 38, 779–780. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Lopez, M.S.; Kliegman, J.I.; Shokat, K.M. The Logic and Design of Analog-Sensitive Kinases and Their Small Molecule Inhibitors. Methods Enzymol. 2014, 548, 189–213. [Google Scholar] [CrossRef]
- Weiss, W.A.; Taylor, S.S.; Shokat, K.M. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat. Chem. Biol. 2007, 3, 739–744. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gurevich, V.V. Therapeutic Potential of Small Molecules and Engineered Proteins. Snake Venoms 2014, 219, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gerona-Navarro, G.; Yoel-Rodríguez; Mujtaba, S.; Frasca, A.; Patel, J.; Zeng, L.; Plotnikov, A.N.; Osman, R.; Zhou, M.-M. Rational Design of Cyclic Peptide Modulators of the Transcriptional Coactivator CBP: A New Class of p53 Inhibitors. J. Am. Chem. Soc. 2011, 133, 2040–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincek, A.S.; Patel, J.; Jaganathan, A.; Green, A.; Pierre-Louis, V.; Arora, V.; Rehmann, J.; Mezei, M.; Zhou, M.-M.; Ohlmeyer, M.; et al. Inhibitor of CBP Histone Acetyltransferase Downregulates p53 Activation and Facilitates Methylation at Lysine 27 on Histone H3. Molecules 2018, 23, 1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, F.; Banerjee, R.; Conrad, P.A.; Scacheri, P.C.; Harte, P.J. Histone Demethylase UTX and Chromatin Remodeler BRM Bind Directly to CBP and Modulate Acetylation of Histone H3 Lysine 27. Mol. Cell. Biol. 2012, 32, 2323–2334. [Google Scholar] [CrossRef] [Green Version]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, D.; A Okan, N.; Bales, E.; Nascimento, L.; A Cole, P.; E Medrano, E. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 2002, 62, 6231–6239. [Google Scholar] [PubMed]
- Katsumoto, T.; Yoshida, N.; Kitabayashi, I. Roles of the histone acetyltransferase monocytic leukemia zinc finger protein in normal and malignant hematopoiesis. Cancer Sci. 2008, 99, 1523–1527. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; DiGiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nat. Cell Biol. 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Michaelides, M.R.; Kluge, A.; Patane, M.; Van Drie, J.H.; Wang, C.; Hansen, T.M.; Risi, R.M.; Mantei, R.; Hertel, C.; Karukurichi, K.; et al. Discovery of Spiro Oxazolidinediones as Selective, Orally Bioavailable Inhibitors of p300/CBP Histone Acetyltransferases. ACS Med. Chem. Lett. 2018, 9, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small mole-cule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Li, G.; Jing, M.; Xu, L.; Li, Z.; Li, M.; Yang, C.; Liu, Y.; Qian, G.; Hu, X.; et al. C646 modulates inflammatory response and antibacterial activity of macrophage. Int. Immunopharmacol. 2019, 74, 105736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Gu, M.-L.; Meng, F.-S.; Jiao, W.-R.; Zhou, X.-X.; Yao, H.-P.; Ji, F. Histone acetyltransferase p300/CBP inhibitor C646 blocks the survival and invasion pathways of gastric cancer cell lines. Int. J. Oncol. 2017, 51, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, L.; Zhao, K.; Thompson, P.R.; Hwang, Y.; Marmorstein, R.; Cole, P.A. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nat. Cell Biol. 2008, 451, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Gardberg, A.S.; Huhn, A.J.; Cummings, R.; Bommi-Reddy, A.; Poy, F.; Setser, J.; Vivat, V.; Brucelle, F.; Wilson, J. Make the right measurement: Discovery of an allosteric inhibition site for p300-HAT. Struct. Dyn. 2019, 6, 054702. [Google Scholar] [CrossRef] [PubMed]
- Mezei, M.; Zhou, M.-M. Dockres: A computer program that analyzes the output of virtual screening of small molecules. Source Code Biol. Med. 2010, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Generic Name | Chromosomal Location | Number of Amino Acids | Cellular Location |
|---|---|---|---|---|
| KAT1 | HAT1 | 2 | 419 | Nucleus |
| KAT2A | GCN5 | 17 | 837 | Nucleus |
| KAT2B | PCAF | 3 | 832 | Nucleus |
| KAT3A | CBP | 16 | 2404 | Nucleus |
| KAT3B | p300 | 22 | 2414 | Nucleus |
| KAT4 | TAF1 | X | 1893 | Nucleus |
| KAT5 | TIP60 | 11 | 546 | Nucleus |
| KAT6A | MYST3 | 8 | 2004 | Nucleus and Cytosol |
| KAT6B | MYST4 | 10 | 2073 | Nucleus |
| KAT7 | MYST2 | 17 | 611 | Nucleus |
| KAT8 | MYST1 | 16 | 458 | Nucleus |
| KAT9 | ELP3 | 8 | 547 | Nucleus |
| KAT12 | TFIIIC90 | 9 | 822 | Nucleus |
| KAT13A | NCOA1 | 2 | 1399 | Nucleus, Plasma Membrane, and Cytosol |
| KAT13B | NCOA3 | 20 | 1415 | Nucleus and Cytosol |
| KAT13C | NCOA2 | 8 | 1464 | Nucleus |
| KAT13D | CLOCK | 4 | 846 | Nucleus |
| Name | Tissue | Cancer Type | References |
|---|---|---|---|
| KAT1 | Appendix, bone marrow, lymph node, tonsil, nasopharynx, esophagus, stomach, duodenum, small intestine, colon, rectum, urinary bladder, testis, epididymis, vagina, cervix, uterine, endometrium, placenta, skin | Liver, ovarian, cervical, skin, testis | [59,60,61,62] |
| KAT2A | Skin, spleen, cerebral cortex, parathyroid | Renal, colorectal, melanoma, testis, thyroid | [48,62,63,64,65,66,67] |
| KAT2B | Cerebellum, thyroid, salivary, stomach, urinary bladder, placenta | Glioma, thyroid, melanoma | [48,62,68,69] |
| KAT3A | Cerebellum, thyroid, nasopharynx, gallbladder, oral mucosa, esophagus, small intestines, colon, rectum, urinary bladder, testis, fallopian tubes, vagina, cervix, uterine, endometrium, placenta, skin | Renal, thyroid, lung, head, neck, testis, breast | [62,70,71,72,73,74,75] |
| KAT3B | Cerebral cortex, parathyroid, adrenal, bone marrow, esophagus, colon, rectum, placenta, skin | Renal, thyroid, carcinoid, stomach, renal, head, neck | [62,70,71,72,73,74,75,76,77] |
| KAT4 | Cerebellum, thyroid, salivary, stomach, urinary bladder, placenta, hippocampus, caudate, adrenal, appendix, tonsil, skeletal muscle, lung, nasopharynx, bronchus, gallbladder, esophagus, duodenum, small intestines, colon, rectum, kidney, testis, epididymis, fallopian tube, vagina, cervix, uterine, endometrium, ovary, placenta, soft tissues, skin, hippocampus, heart muscle, skeletal muscle, lung, bronchus, seminal vesicle, breast | Lung, glioma, thyroid, lymphoma, pancreatic, carcinoids | [62,78,79,80] |
| KAT5 | Cerebral cortex, parathyroid, adrenal, caudate, cerebellum, thyroid, nasopharynx, colon, rectum, placenta, stomach, duodenum, ovary, cervix, uterine, oral mucosa, gallbladder | Renal, melanoma, testis, lymphoma | [62,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96] |
| KAT6A | Cerebral cortex, caudate, cerebellum | Glioma, thyroid, carcinoid | [62,89,90,91,92,93,94,95,96] |
| KAT6B | Cerebellum, thyroid, salivary, stomach, urinary bladder, placenta, hippocampus, caudate, adrenal, appendix, tonsil, skeletal muscle, lung, nasopharynx, bronchus, gallbladder, esophagus, duodenum, small intestines, colon, rectum, kidney, testis, epididymis, fallopian tube, vagina, cervix, uterine, endometrium, ovary, placenta, soft tissues, skin | Renal, glioma, thyroid, lung, head, skin, neck | [62,96,97] |
| KAT7 | Testis | Glioma, thyroid, melanoma | [62,98,99,100,101] |
| KAT8 | - | Lung, breast, endometrial | [62,102,103,104,105,106] |
| KAT9 | Appendix, duodenum, small intestine, colon, rectum, kidney, urinary bladder, prostate, endometrium, placenta | Renal, colorectal, thyroid, prostate, liver | [62,107,108,109] |
| KAT13A | Cerebral cortex, hippocampus, cerebellum, thyroid, parathyroid, adrenal, lymph node, nasopharynx, bronchus, gallbladder, pancreas, oral mucosa, esophagus, stomach, duodenum, small intestines, colon, rectum, kidney, urinary bladder, testis, fallopian tube, breast, vagina, placenta | Thyroid, carcinoid, head, neck | [62,110,111,112,113,114,115,116,117,118,119,120,121] |
| KAT13B | Cerebral cortex, hippocampus, cerebellum, thyroid, parathyroid, adrenal, lymph node, nasopharynx, bronchus, gallbladder, pancreas, oral mucosa, esophagus, stomach, duodenum, small intestines, colon, rectum, kidney, urinary bladder, testis, fallopian tube, breast, vagina, placenta, soft tissue, skin | Thyroid, carcinoid, head, neck | [122,123,124,125,126,127] |
| KAT13D | Cerebral cortex, hippocampus, cerebellum, thyroid, parathyroid, adrenal, lymph node, nasopharynx, bronchus, gallbladder, pancreas, oral mucosa, esophagus, stomach, duodenum, small intestines, colon, rectum, kidney, urinary bladder, testis, fallopian tube, breast, vagina, placenta, soft tissue, skin | Thyroid, breast, cervical, head, neck | [62,128] |
| PDB id: | 3biy | 4bhw | 5kj2 | 6pf1 | 6pgu | 6v8k | 6v8n | 6v90 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligand: | 01K | 01K | 6TF | OJ7 | OK7 | QS4 | QS1 | QSD | NiCur | ||
| 1374 | PHE | 2.99 | |||||||||
| 1379 | GLN | 3.68 | |||||||||
| 1394 | TYR | 3.52 | 3.23 | ||||||||
| 1395 | ILE | 2.95 | |||||||||
| 1396 | SER | 3.34 | 3.17 | 2.78 | |||||||
| 1397 | TYR | 3.22 | 2.49 | 3.23 | |||||||
| 1398 | LEU | 2.91 | 2.9 | 3.21 | 3.24 | ||||||
| 1399 | ASP | 3.35 | 3.32 | 3.25 | 3.1 | ||||||
| 1400 | SER | 2.62 | 2.45 | 2.94 | 2.69 | 2.89 | |||||
| 1407 | LYS | HX2 | 3.65 | ||||||||
| 1410 | ARG | HX2 | 2.89 | 2.93 | |||||||
| 1411 | THR | HX2 | 2.68 | ||||||||
| 1414 | TYR | HX2 | 3.47 | 3.37 | 3.7 | 3.86 | |||||
| 1434 | HIS | 3.33 | 3.18 | ||||||||
| 1435 | ILE | 3.08 | |||||||||
| 1436 | TRP | 2.96 | 2.78 | 2.87 | 3.06 | ||||||
| 1438 | CYS | 3.68 | 3.7 | 2.7 | |||||||
| 1440 | PRO | 3.24 | 3.51 | 3.07 | 3.87 | ||||||
| 1443 | GLY | 3.41 | 3.53 | ||||||||
| 1444 | ASP | ||||||||||
| 1446 | TYR | 3.68 | 3.44 | 2.92 | |||||||
| 1451 | HIS | 3.24 | 3.39 | 3.31 | |||||||
| 1452 | PRO | ||||||||||
| 1453 | PRO | ||||||||||
| 1455 | GLN | 3.05 | 3.21 | 3.23 | 3.07 | ||||||
| 1456 | LYS | 3.48 | |||||||||
| 1457 | ILE | 2.91 | 2.69 | ||||||||
| 1458 | PRO | 3.17 | 3.37 | 3.09 | 3.89 | 3.34 | |||||
| 1462 | ARG | HX1 | 3.22 | 3.26 | 2.99 | ||||||
| 1463 | LEU | HX1 | 3.93 | 4.07 | 3.85 | 3.27 | |||||
| 1466 | TRP | HX1 | 2.78 | 2.9 | 3.19 | 3.33 | 3.8 | ||||
| 1467 | PHE | HX1 | 3.11 | 3.44 | 2.39 | ||||||
| 1486 | ILE | 3.25 | |||||||||
| 1489 | GLN | 3.21 | |||||||||
| 1490 | ALA | 3.06 | |||||||||
| 1495 | LEU | 3.4 | |||||||||
| 1501 | LEU | 3.32 | |||||||||
| 1502 | PRO | 3.25 | 3.11 | ||||||||
| 1505 | GLU | 3.53 | |||||||||
| 1507 | ASP | 2.87 | 2.8 | ||||||||
| 1509 | TRP | 3.53 | |||||||||
| 1591 | HIS | 3.29 | |||||||||
| 1595 | PHE | 3.56 | |||||||||
| 1596 | PHE | 3.2 | |||||||||
| 1597 | VAL | 3.79 | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Garro, C.; Igbineweka, L.; Ali, Z.; Mezei, M.; Mujtaba, S. The Biological Significance of Targeting Acetylation-Mediated Gene Regulation for Designing New Mechanistic Tools and Potential Therapeutics. Biomolecules 2021, 11, 455. https://doi.org/10.3390/biom11030455
O’Garro C, Igbineweka L, Ali Z, Mezei M, Mujtaba S. The Biological Significance of Targeting Acetylation-Mediated Gene Regulation for Designing New Mechanistic Tools and Potential Therapeutics. Biomolecules. 2021; 11(3):455. https://doi.org/10.3390/biom11030455
Chicago/Turabian StyleO’Garro, Chenise, Loveth Igbineweka, Zonaira Ali, Mihaly Mezei, and Shiraz Mujtaba. 2021. "The Biological Significance of Targeting Acetylation-Mediated Gene Regulation for Designing New Mechanistic Tools and Potential Therapeutics" Biomolecules 11, no. 3: 455. https://doi.org/10.3390/biom11030455