
Molecular and Metabolic Phenotyping of Hepatocellular Carcinoma for Biomarker Discovery: A Meta-Analysis

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
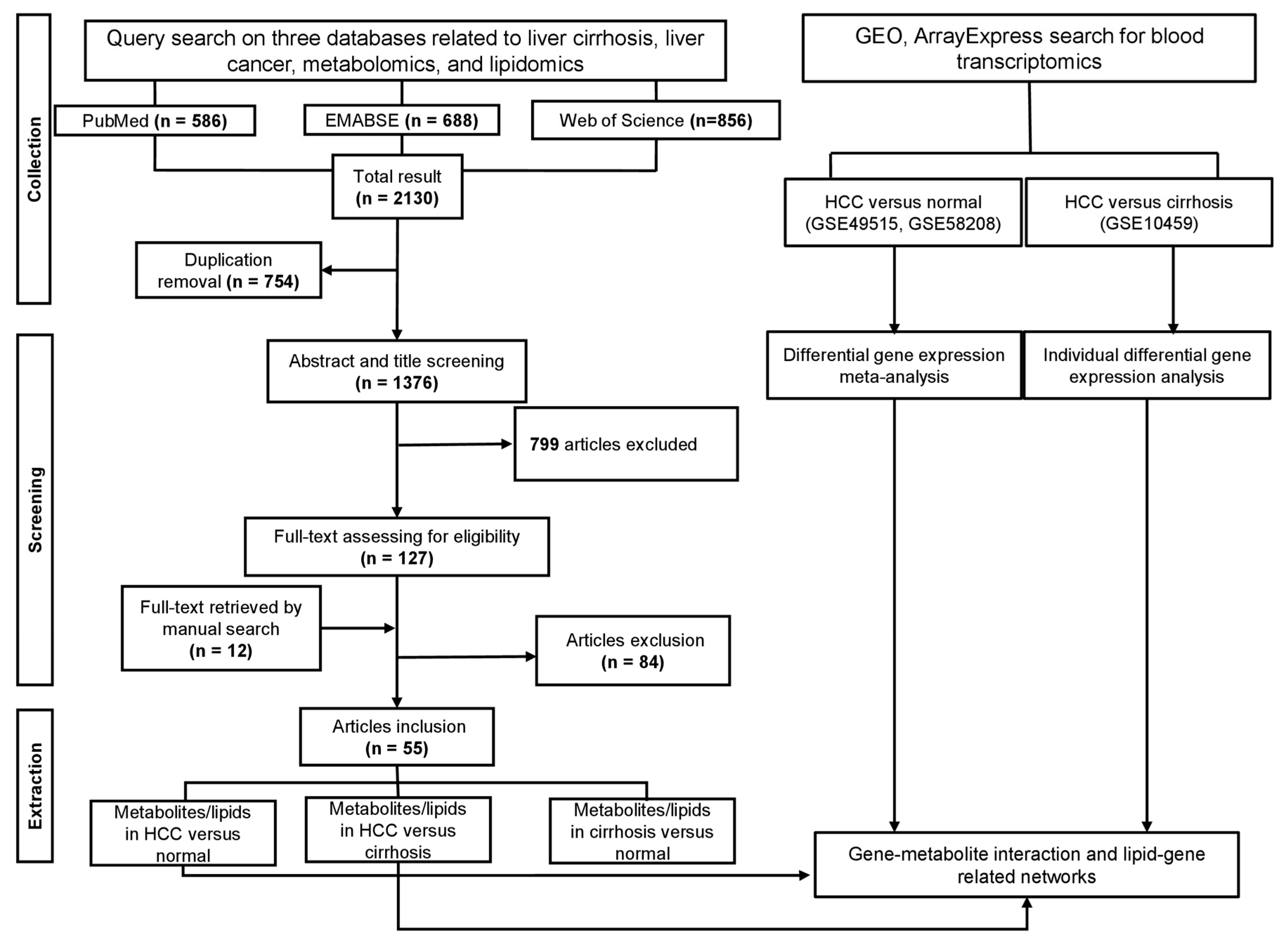
2.1. Literature Search
2.2. Data Selection
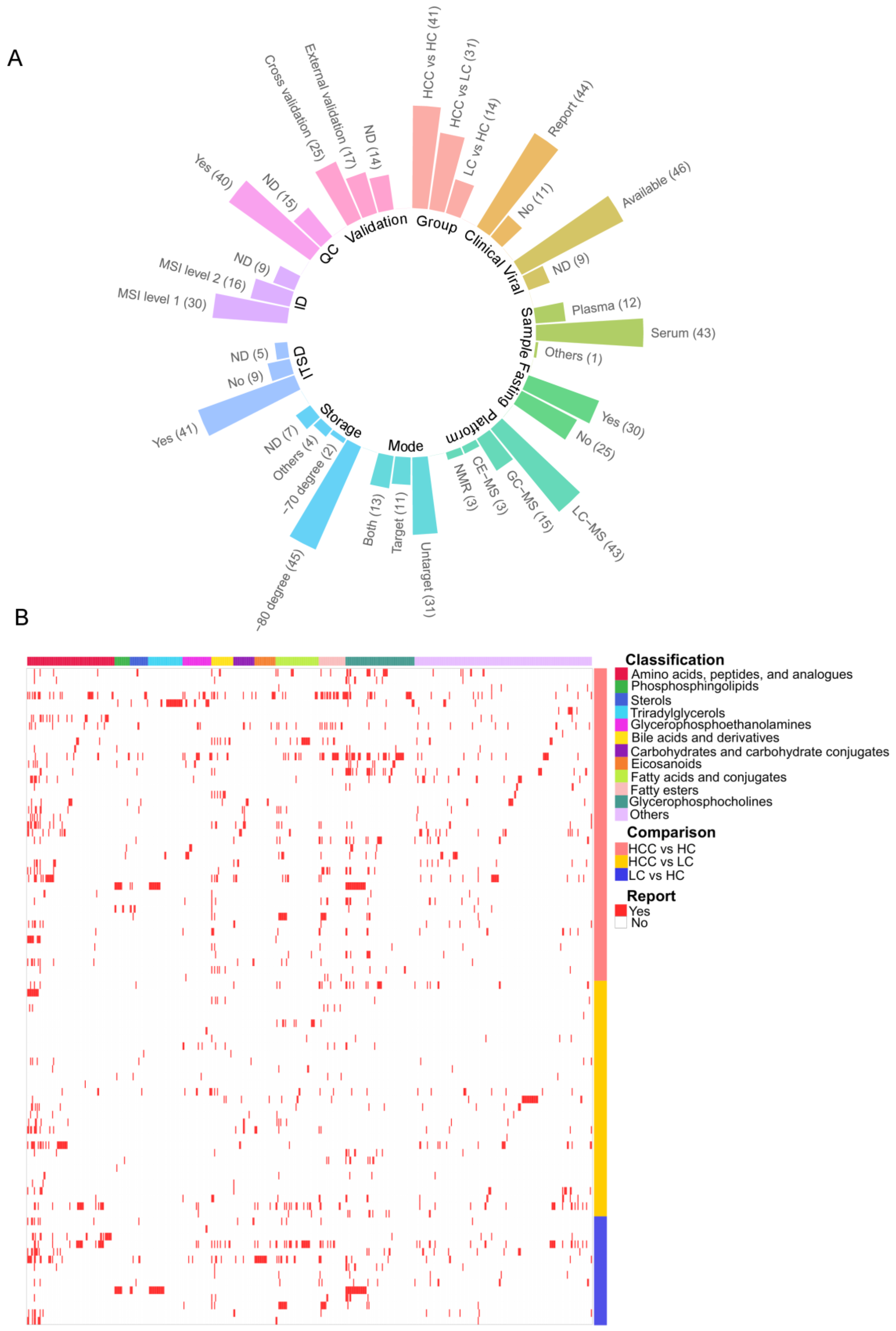
2.3. Data Extraction
2.4. Metabolite and Lipid Identification
2.5. Transcriptomics Meta-Analysis
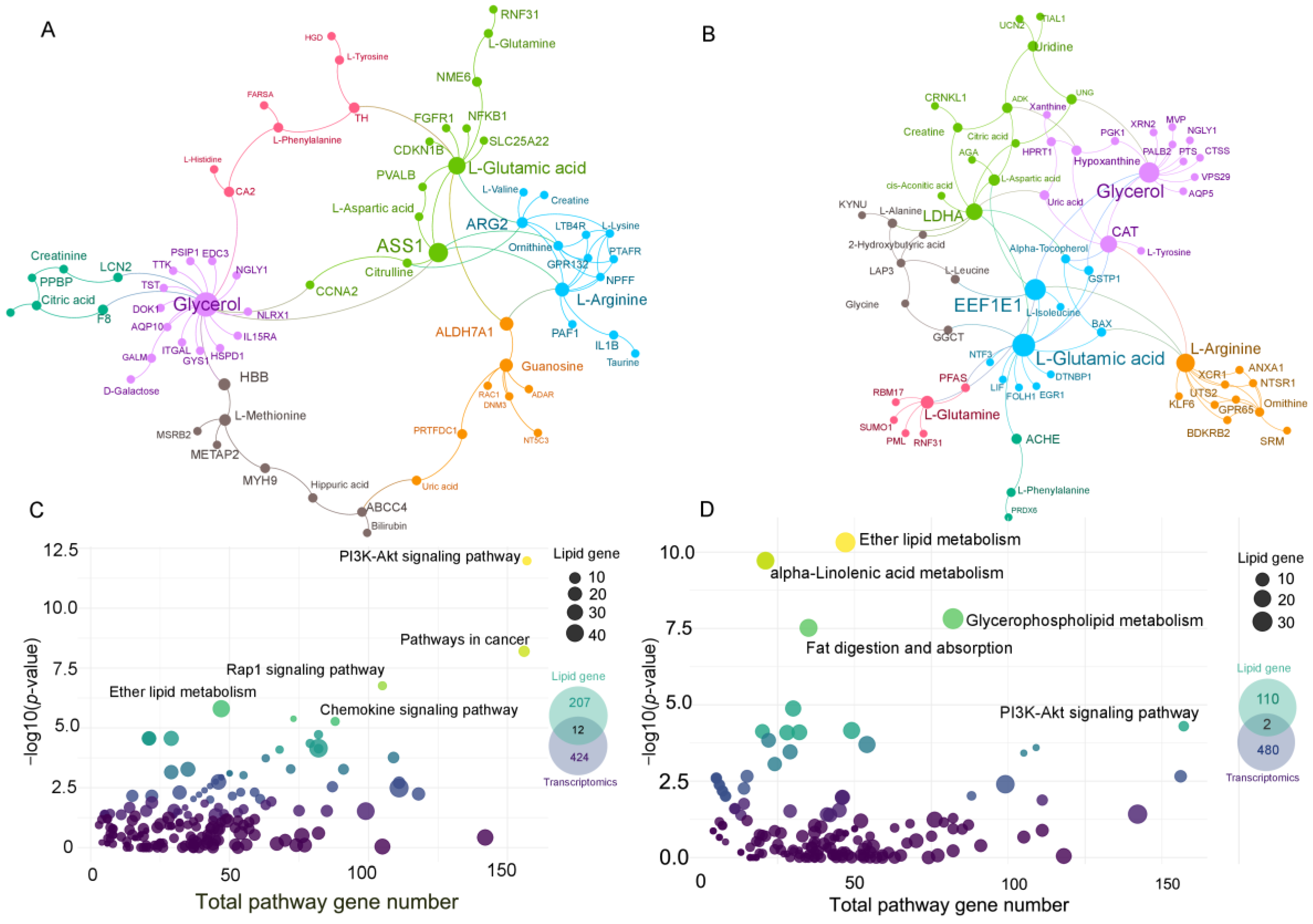
2.6. Pathway Enrichment Analysis
2.7. Gene–Metabolite Interaction Network and Lipid-Related Gene Network
2.8. Bioinformatics, Survival Analysis, Immunohistochemistry (IHC), and Machine Learning Model
2.9. Exploratory Data Analysis and Visualization
2.10. Statistical Analysis
3. Results
3.1. Compendium Biomarker Report for HCC
3.2. Vote-Counting Meta-Analysis for Robust Reported Compounds
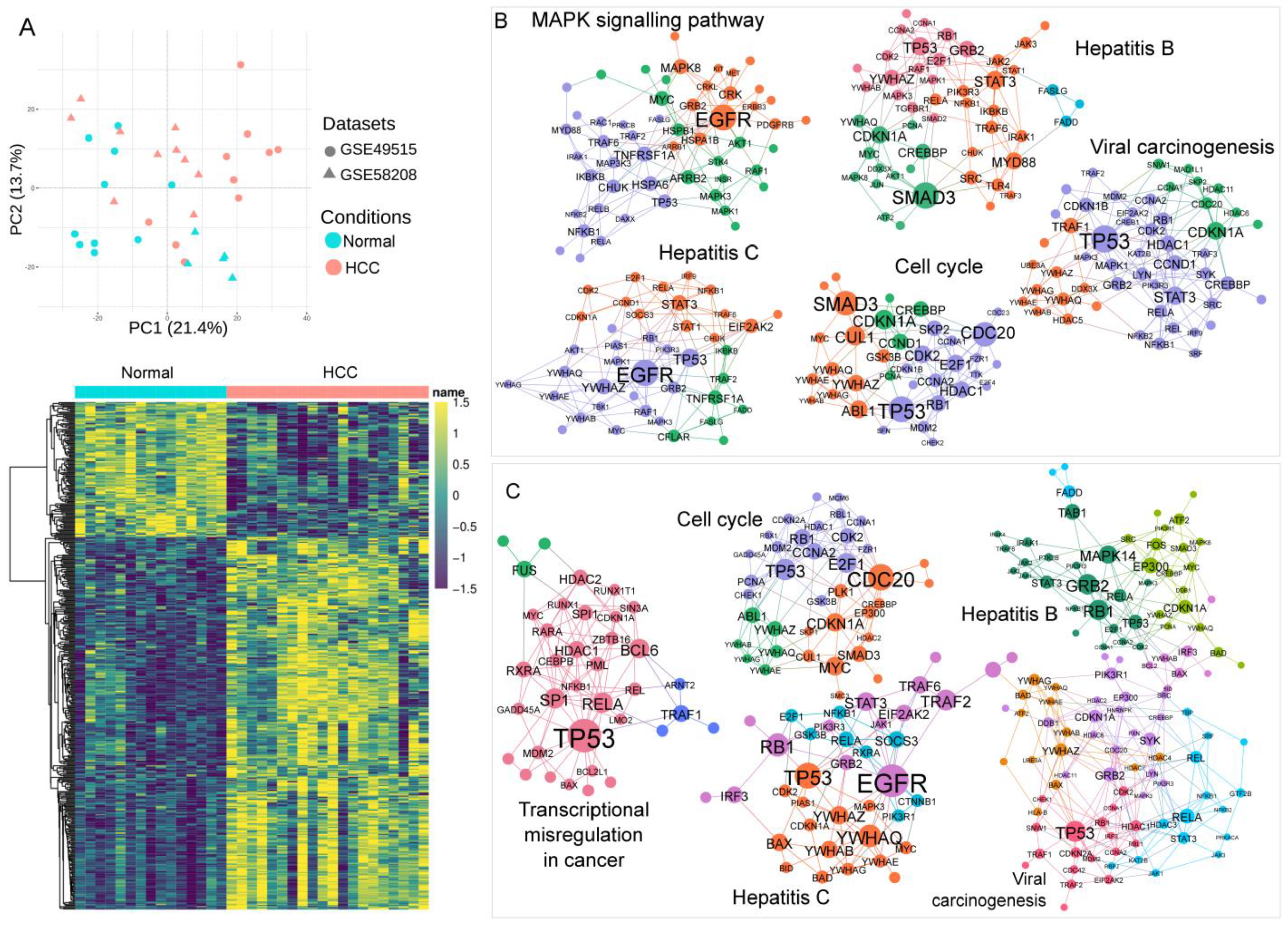
3.3. Association of Blood Transcriptomics with HCC Pathogenesis
3.4. Gene–Metabolite Network Analysis and Lipid-Related Gene Network
3.5. Bioinformatic Analysis and Prediction Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Wheeler, D.A.; Roberts, L.R. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Jiang, L.; Chen, Y. Recent advances in systemic therapy for hepatocellular carcinoma. Biomark. Res. 2022, 10, 3. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Ng, D.J.Y.; Pasikanti, K.K.; Chan, E.C.Y. Trend analysis of metabonomics and systematic review of metabonomics-derived cancer marker metabolites. Metabolomics 2011, 7, 155–178. [Google Scholar] [CrossRef]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef]
- Ulmer, C.Z.; Maus, A.; Hines, J.; Singh, R. Challenges in Translating Clinical Metabolomics Data Sets from the Bench to the Bedside. Clin. Chem. 2021, 67, 1581–1583. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ (Clin. Res. Ed.) 2021, 372, n71. [Google Scholar] [CrossRef]
- Zhixu, N.; Maria, F. LipidLynxX: Lipid annotations converter for large scale lipidomics and epilipidomics datasets. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shi, M.; Chen, M.S.; Sekar, K.; Tan, C.K.; Ooi, L.L.; Hui, K.M. A blood-based three-gene signature for the non-invasive detection of early human hepatocellular carcinoma. Eur. J. Cancer 2014, 50, 928–936. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Honda, M.; Fujinaga, H.; Tatsumi, I.; Mizukoshi, E.; Nakamoto, Y.; Kaneko, S. Common transcriptional signature of tumor-infiltrating mononuclear inflammatory cells and peripheral blood mononuclear cells in hepatocellular carcinoma patients. Cancer Res. 2008, 68, 10267–10279. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-J.; Shen, P.-C.; Liu, H.-C.; Cho, Y.-C.; Hsu, M.-K.; Lin, I.C.; Chen, F.-H.; Yang, J.-C.; Ma, W.-L.; Cheng, W.-C. LipidSig: A web-based tool for lipidomic data analysis. Nucleic Acids Res. 2021, 49, W336–W345. [Google Scholar] [CrossRef] [PubMed]
- CONSORTIUM, T.G.; Ardlie Kristin, G.; Deluca David, S.; Segrè Ayellet, V.; Sullivan Timothy, J.; Young Taylor, R.; Gelfand Ellen, T.; Trowbridge Casandra, A.; Maller Julian, B.; Tukiainen, T.; et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Lee, J.; Choi, C. Oncopression: Gene expression compendium for cancer with matched normal tissues. Bioinformatics 2017, 33, 2068–2070. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Menyhárt, O.; Nagy, Á.; Győrffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 2018, 5, 181006. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.-X.; Wang, Z.-L. GraphBio: A shiny web app to easily perform popular visualization analysis for omics data. bioRxiv 2022, 13, 957317. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. Proc. Int. AAAI Conf. Web Soc. Media 2009, 3, 361–362. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Llambrich, M.; Correig, E.; Gumà, J.; Brezmes, J.; Cumeras, R. Amanida: An R package for meta-analysis of metabolomics non-integral data. Bioinformatics 2021, 38, 583–585. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Huang, C.; Li, N.; Zou, L.; Chia, S.E.; Chen, S.; Yu, K.; Ling, Q.; Cheng, Q.; et al. Comparison of hepatic and serum lipid signatures in hepatocellular carcinoma patients leads to the discovery of diagnostic and prognostic biomarkers. Oncotarget 2018, 9, 5032–5043. [Google Scholar] [CrossRef]
- Soga, T.; Sugimoto, M.; Honma, M.; Mori, M.; Igarashi, K.; Kashikura, K.; Ikeda, S.; Hirayama, A.; Yamamoto, T.; Yoshida, H.; et al. Serum metabolomics reveals γ-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J. Hepatol. 2011, 55, 896–905. [Google Scholar] [CrossRef]
- Zhou, P.C.; Sun, L.Q.; Shao, L.; Yi, L.Z.; Li, N.; Fan, X.G. Establishment of a pattern recognition metabolomics model for the diagnosis of hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 4607–4623. [Google Scholar] [CrossRef]
- Fitian, A.I.; Nelson, D.R.; Liu, C.; Xu, Y.; Ararat, M.; Cabrera, R. Integrated metabolomic profiling of hepatocellular carcinoma in hepatitis C cirrhosis through GC/MS and UPLC/MS-MS. Liver Int. Off. J. Int. Assoc. Study Liver 2014, 34, 1428–1444. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.G.; Zhao, W.; Zhang, J.; Wu, X.; Hu, J.; Yin, G.C.; Xu, Y.J. Metabolomics and eicosanoid analysis identified serum biomarkers for distinguishing hepatocellular carcinoma from hepatitis B virus-related cirrhosis. Oncotarget 2017, 8, 63890–63900. [Google Scholar] [CrossRef]
- Wang, B.; Chen, D.; Chen, Y.; Hu, Z.; Cao, M.; Xie, Q.; Chen, Y.; Xu, J.; Zheng, S.; Li, L. Metabonomic profiles discriminate hepatocellular carcinoma from liver cirrhosis by ultraperformance liquid chromatography-mass spectrometry. J. Proteome Res. 2012, 11, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Wan, D.; Zhao, C.; Chen, J.; Zhao, X.; Wang, W.; Lu, X.; Yang, S.; Gu, J.; Xu, G. A metabonomic study of hepatitis B-induced liver cirrhosis and hepatocellular carcinoma by using RP-LC and HILIC coupled with mass spectrometry. Mol. Biosyst. 2009, 5, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hong, Z.; Tan, G.; Dong, X.; Yang, G.; Zhao, L.; Chen, X.; Zhu, Z.; Lou, Z.; Qian, B.; et al. NMR and LC/MS-based global metabolomics to identify serum biomarkers differentiating hepatocellular carcinoma from liver cirrhosis. Int. J. Cancer 2014, 135, 658–668. [Google Scholar] [CrossRef]
- Chen, S.; Yin, P.; Zhao, X.; Xing, W.; Hu, C.; Zhou, L.; Xu, G. Serum lipid profiling of patients with chronic hepatitis B, cirrhosis, and hepatocellular carcinoma by ultra fast LC/IT-TOF MS. Electrophoresis 2013, 34, 2848–2856. [Google Scholar]
- Zhou, L.; Ding, L.; Yin, P.; Lu, X.; Wang, X.; Niu, J.; Gao, P.; Xu, G. Serum metabolic profiling study of hepatocellular carcinoma infected with hepatitis B or hepatitis C virus by using liquid chromatography-mass spectrometry. J. Proteome Res. 2012, 11, 5433–5442. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Q.; Yin, P.; Xing, W.; Wu, Z.; Chen, S.; Lu, X.; Zhang, Y.; Lin, X.; Xu, G. Serum metabolomics reveals the deregulation of fatty acids metabolism in hepatocellular carcinoma and chronic liver diseases. Anal. Bioanal. Chem. 2012, 403, 203–213. [Google Scholar] [CrossRef]
- Gao, R.; Cheng, J.; Fan, C.; Shi, X.; Cao, Y.; Sun, B.; Ding, H.; Hu, C.; Dong, F.; Yan, X. Serum Metabolomics to Identify the Liver Disease-Specific Biomarkers for the Progression of Hepatitis to Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 18175. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Cho, E.J.; Yu, K.S.; Jang, I.J.; Yoon, J.H.; Park, T.; Cho, J.Y. Comprehensive Metabolomic Search for Biomarkers to Differentiate Early Stage Hepatocellular Carcinoma from Cirrhosis. Cancers 2019, 11, 1497. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.D.; Maurhofer, O.; Beyoglu, D.; Lanz, C.; Krausz, K.W.; Pabst, T.; Gonzalez, F.J.; Dufour, J.F.; Idle, J.R. Aberrant lipid metabolism in hepatocellular carcinoma revealed by plasma metabolomics and lipid profiling. Cancer Res. 2011, 71, 6590–6600. [Google Scholar] [CrossRef]
- Lin, X.; Yang, F.; Zhou, L.; Yin, P.; Kong, H.; Xing, W.; Lu, X.; Jia, L.; Wang, Q.; Xu, G. A support vector machine-recursive feature elimination feature selection method based on artificial contrast variables and mutual information. J. Chromatogr. 2012, 910, 149–155. [Google Scholar] [CrossRef]
- Luo, P.; Yin, P.; Hua, R.; Tan, Y.; Li, Z.; Qiu, G.; Yin, Z.; Xie, X.; Wang, X.; Chen, W.; et al. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma. Hepatology 2018, 67, 662–675. [Google Scholar] [CrossRef]
- Cai, F.F.; Song, Y.N.; Lu, Y.Y.; Zhang, Y.; Hu, Y.Y.; Su, S.B. Analysis of plasma metabolic profile, characteristics and enzymes in the progression from chronic hepatitis B to hepatocellular carcinoma. Aging 2020, 12, 14949–14965. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Yin, P.; Tan, Y.; Dong, L.; Hu, C.; Huang, Q.; Lu, X.; Wang, H.; Xu, G. Metabolomics study of hepatocellular carcinoma: Discovery and validation of serum potential biomarkers by using capillary electrophoresis-mass spectrometry. J. Proteome Res. 2014, 13, 3420–3431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, G.Y.; Wu, Y.J.; Liu, S.Y. The serum metabolic profiles of different Barcelona stages hepatocellular carcinoma associated with hepatitis B virus. Oncol. Lett. 2018, 15, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Qin, W.X.; Li, Z.L.; Xu, A.J.; Xing, H.; Wu, H.; Zhang, H.; Wang, M.D.; Li, C.; Liang, L.; et al. Tissue and serum metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Clin. Chim. Acta 2019, 488, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, Y.; Ye, G.; Li, X.; Yin, P.; Ruan, Q.; Xu, G. Classification and differential metabolite discovery of liver diseases based on plasma metabolic profiling and support vector machines. J. Sep. Sci. 2011, 34, 3029–3036. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, J.; Wang, J.; Chen, L.; Huang, H.; Xiong, Y.; Xie, B. Quantification of serum purine metabolites for distinguishing patients with hepatitis B from hepatocellular carcinoma. Bioanalysis 2019, 11, 1003–1013. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Ma, S.; Zhang, C.; Liu, H.; Sun, D. Clinical significance of small molecule metabolites in the blood of patients with different types of liver injury. Sci. Rep. 2021, 11, 11642. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, C.; Gao, L.; Xu, Y.-J.; Chia, S.E.; Chen, S.; Li, N.; Yu, K.; Ling, Q.; Cheng, Q.; et al. Identification of serum biomarkers associated with hepatitis B virus-related hepatocellular carcinoma and liver cirrhosis using mass-spectrometry-based metabolomics. Metabolomics 2015, 11, 1526–1538. [Google Scholar] [CrossRef]
- Lu, X.; Nie, H.; Li, Y.; Zhan, C.; Liu, X.; Shi, X.; Shi, M.; Zhang, Y.; Li, Y. Comprehensive characterization and evaluation of hepatocellular carcinoma by LC–MS based serum metabolomics. Metabolomics 2015, 11, 1381–1393. [Google Scholar] [CrossRef]
- Chen, F.; Xue, J.; Zhou, L.; Wu, S.; Chen, Z. Identification of serum biomarkers of hepatocarcinoma through liquid chromatography/mass spectrometry-based metabonomic method. Anal. Bioanal. Chem. 2011, 401, 1899–1904. [Google Scholar] [CrossRef]
- Luo, P.; Dai, W.; Yin, P.; Zeng, Z.; Kong, H.; Zhou, L.; Wang, X.; Chen, S.; Lu, X.; Xu, G. Multiple Reaction Monitoring-Ion Pair Finder: A Systematic Approach To Transform Nontargeted Mode to Pseudotargeted Mode for Metabolomics Study Based on Liquid Chromatography–Mass Spectrometry. Anal. Chem. 2015, 87, 5050–5055. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Ali, A.; Roome, T.; Razzak, A.; Iqbal, A.; Jabbar Siddiqui, A.; Muhammad Zahid Azam, S.; Shaikh, H.; El-Seedi, H.R.; Musharraf, S.G. Metabolomics approach to understand the hepatitis C virus induced hepatocellular carcinoma using LC-ESI-MS/MS. Arab. J. Chem. 2021, 14, 102907. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Y.; Qin, L.; Li, K.; Zhao, Y.; Sun, H.; Zhang, T.; Zhang, Y. Metabolomic Profiles for HBV Related Hepatocellular Carcinoma Including Alpha-Fetoproteins Positive and Negative Subtypes. Front. Oncol. 2019, 9, 1069. [Google Scholar] [CrossRef]
- Stepien, M.; Keski-Rahkonen, P.; Kiss, A.; Robinot, N.; Duarte-Salles, T.; Murphy, N.; Perlemuter, G.; Viallon, V.; Tjønneland, A.; Rostgaard-Hansen, A.L.; et al. Metabolic perturbations prior to hepatocellular carcinoma diagnosis: Findings from a prospective observational cohort study. Int. J. Cancer 2021, 148, 609–625. [Google Scholar] [CrossRef]
- Alsaleh, M.; Leftley, Z.; Barbera, T.A.; Koomson, L.K.; Zabron, A.; Crossey, M.M.E.; Reeves, H.L.; Cramp, M.; Ryder, S.; Greer, S.; et al. Characterisation of the Serum Metabolic Signature of Cholangiocarcinoma in a United Kingdom Cohort. J. Clin. Exp. Hepatol. 2020, 10, 17–29. [Google Scholar] [CrossRef]
- Thomas, C.E.; Luu, H.N.; Wang, R.; Xie, G.; Adams-Haduch, J.; Jin, A.; Koh, W.P.; Jia, W.; Behari, J.; Yuan, J.M. Association between Pre-Diagnostic Serum Bile Acids and Hepatocellular Carcinoma: The Singapore Chinese Health Study. Cancers 2021, 13, 2648. [Google Scholar] [CrossRef]
- Li, Y.-F.; Qiu, S.; Gao, L.-J.; Zhang, A.-H. Metabolomic estimation of the diagnosis of hepatocellular carcinoma based on ultrahigh performance liquid chromatography coupled with time-of-flight mass spectrometry. RSC Adv. 2018, 8, 9375–9382. [Google Scholar] [CrossRef] [PubMed]
- Fages, A.; Duarte-Salles, T.; Stepien, M.; Ferrari, P.; Fedirko, V.; Pontoizeau, C.; Trichopoulou, A.; Aleksandrova, K.; Tjønneland, A.; Olsen, A.; et al. Metabolomic profiles of hepatocellular carcinoma in a European prospective cohort. BMC Med. 2015, 13, 242. [Google Scholar] [CrossRef] [PubMed]
- Jee, S.H.; Kim, M.; Kim, M.; Yoo, H.J.; Kim, H.; Jung, K.J.; Hong, S.; Lee, J.H. Metabolomics Profiles of Hepatocellular Carcinoma in a Korean Prospective Cohort: The Korean Cancer Prevention Study-II. Cancer Prev. Res. 2018, 11, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Cotte, A.K.; Cottet, V.; Aires, V.; Mouillot, T.; Rizk, M.; Vinault, S.; Binquet, C.; de Barros, J.P.; Hillon, P.; Delmas, D. Phospholipid profiles and hepatocellular carcinoma risk and prognosis in cirrhotic patients. Oncotarget 2019, 10, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bai, Y.; Han, X.; Shi, Y.; Liu, H. Plasma Lipidomic Analysis to Identify Novel Biomarkers for Hepatocellular Carcinoma. J. Anal. Test. 2017, 1, 223–232. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, J.; Li, J.; Feng, J.; Chen, Z.; Wang, X. Plasma metabolomic analysis of human hepatocellular carcinoma: Diagnostic and therapeutic study. Oncotarget 2016, 7, 47332–47342. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Kong, H.; Lu, X.; Li, Y.; Yin, P.; Zeng, Z.; Xu, G. Pseudotargeted metabolomics method and its application in serum biomarker discovery for hepatocellular carcinoma based on ultra high-performance liquid chromatography/triple quadrupole mass spectrometry. Anal. Chem. 2013, 85, 8326–8333. [Google Scholar] [CrossRef]
- Chen, T.; Xie, G.; Wang, X.; Fan, J.; Qiu, Y.; Zheng, X.; Qi, X.; Cao, Y.; Su, M.; Wang, X.; et al. Serum and urine metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Mol. Cell. Proteom. 2011, 10, M110.004945. [Google Scholar] [CrossRef]
- Banales, J.M.; Iñarrairaegui, M.; Arbelaiz, A.; Milkiewicz, P.; Muntané, J.; Muñoz-Bellvis, L.; La Casta, A.; Gonzalez, L.M.; Arretxe, E.; Alonso, C.; et al. Serum Metabolites as Diagnostic Biomarkers for Cholangiocarcinoma, Hepatocellular Carcinoma, and Primary Sclerosing Cholangitis. Hepatology 2019, 70, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Ressom, H.W.; Xiao, J.F.; Tuli, L.; Varghese, R.S.; Zhou, B.; Tsai, T.H.; Ranjbar, M.R.; Zhao, Y.; Wang, J.; Di Poto, C.; et al. Utilization of metabolomics to identify serum biomarkers for hepatocellular carcinoma in patients with liver cirrhosis. Anal. Chim. Acta 2012, 743, 90–100. [Google Scholar] [CrossRef]
- Bowers, J.; Hughes, E.; Skill, N.; Maluccio, M.; Raftery, D. Detection of hepatocellular carcinoma in hepatitis C patients: Biomarker discovery by LC-MS. J. Chromatogr. 2014, 966, 154–162. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, N.; Cao, Y.; Zhu, Z.; Gao, P. Differential diagnosis between hepatocellular carcinoma and cirrhosis by serum amino acids and acylcarnitines. Int. J. Clin. Exp. Pathol. 2018, 11, 1763–1769. [Google Scholar]
- Xiao, J.; Zhao, Y.; Varghese, R.S.; Zhou, B.; Di Poto, C.; Zhang, L.; Tadesse, M.G.; Ziada, D.H.; Shetty, K.; Ressom, H.W. Evaluation of metabolite biomarkers for hepatocellular carcinoma through stratified analysis by gender, race, and alcoholic cirrhosis. Cancer Epidemiol. Biomark. Prev. 2014, 23, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Nezami Ranjbar, M.R.; Luo, Y.; Di Poto, C.; Varghese, R.S.; Ferrarini, A.; Zhang, C.; Sarhan, N.I.; Soliman, H.; Tadesse, M.G.; Ziada, D.H.; et al. GC-MS Based Plasma Metabolomics for Identification of Candidate Biomarkers for Hepatocellular Carcinoma in Egyptian Cohort. PLoS ONE 2015, 10, e0127299. [Google Scholar] [CrossRef] [PubMed]
- Nomair, A.M.; Madkour, M.A.; Shamseya, M.M.; Elsheredy, H.G.; Shokr, A. Profiling of plasma metabolomics in patients with hepatitis C-related liver cirrhosis and hepatocellular carcinoma. Clin. Exp. Hepatol. 2019, 5, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Di Poto, C.; He, S.; Varghese, R.S.; Zhao, Y.; Ferrarini, A.; Su, S.; Karabala, A.; Redi, M.; Mamo, H.; Rangnekar, A.S.; et al. Identification of race-associated metabolite biomarkers for hepatocellular carcinoma in patients with liver cirrhosis and hepatitis C virus infection. PLoS ONE 2018, 13, e0192748. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.M.; Gjuka, D.; Jiao, J.; Song, X.; Wang, Y.; Wang, J.; Wei, P.; El-Serag, H.B.; Marrero, J.A.; Beretta, L. A Novel Biomarker Panel for the Early Detection and Risk Assessment of Hepatocellular Carcinoma in Patients with Cirrhosis. Cancer Prev. Res. 2021, 14, 667–674. [Google Scholar] [CrossRef]
- Pan, H.Y.; Wu, Q.Q.; Yin, Q.Q.; Dai, Y.N.; Huang, Y.C.; Zheng, W.; Hui, T.C.; Chen, M.J.; Wang, M.S.; Zhang, J.J.; et al. LC/MS-Based Global Metabolomic Identification of Serum Biomarkers Differentiating Hepatocellular Carcinoma from Chronic Hepatitis B and Liver Cirrhosis. ACS Omega 2021, 6, 1160–1170. [Google Scholar] [CrossRef]
- Di Poto, C.; Ferrarini, A.; Zhao, Y.; Varghese, R.S.; Tu, C.; Zuo, Y.; Wang, M.; Nezami Ranjbar, M.R.; Luo, Y.; Zhang, C.; et al. Metabolomic Characterization of Hepatocellular Carcinoma in Patients with Liver Cirrhosis for Biomarker Discovery. Cancer Epidemiol. Biomark. Prev. 2017, 26, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Huang, X.; Zhou, L.; Tan, Y.; Hu, C.; Wang, X.; Niu, J.; Wang, H.; Lin, X.; Yin, P. Metabolomics Identifies Biomarker Pattern for Early Diagnosis of Hepatocellular Carcinoma: From Diethylnitrosamine Treated Rats to Patients. Sci. Rep. 2015, 5, 16101. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zheng, X.; Yang, M.; Zhao, A.; Li, M.; Chen, T.; Panee, J.; Jia, W.; Ji, G. Serum lipid alterations identified in chronic hepatitis B, hepatitis B virus-associated cirrhosis and carcinoma patients. Sci. Rep. 2017, 7, 42710. [Google Scholar] [CrossRef] [PubMed]
- Passos-Castilho, A.M.; Carvalho, V.M.; Cardozo, K.H.M.; Kikuchi, L.; Chagas, A.L.; Gomes-Gouvêa, M.S.; Malta, F.; de Seixas-Santos Nastri, A.C.; Pinho, J.R.R.; Carrilho, F.J.; et al. Serum lipidomic profiling as a useful tool for screening potential biomarkers of hepatitis B-related hepatocellular carcinoma by ultraperformance liquid chromatography–mass spectrometry. BMC Cancer 2015, 15, 985. [Google Scholar] [CrossRef] [PubMed]
- Baniasadi, H.; Gowda, G.A.; Gu, H.; Zeng, A.; Zhuang, S.; Skill, N.; Maluccio, M.; Raftery, D. Targeted metabolic profiling of hepatocellular carcinoma and hepatitis C using LC-MS/MS. Electrophoresis 2013, 34, 2910–2917. [Google Scholar] [CrossRef] [PubMed]
- Long, N.P.; Yoon, S.J.; Anh, N.H.; Nghi, T.D.; Lim, D.K.; Hong, Y.J.; Hong, S.-S.; Kwon, S.W. A systematic review on metabolomics-based diagnostic biomarker discovery and validation in pancreatic cancer. Metabolomics 2018, 14, 109. [Google Scholar] [CrossRef]
- Sivanand, S.; Vander Heiden, M.G. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 2020, 37, 147–156. [Google Scholar] [CrossRef]
- Pataskar, A.; Champagne, J.; Nagel, R.; Kenski, J.; Laos, M.; Michaux, J.; Pak, H.S.; Bleijerveld, O.B.; Mordente, K.; Navarro, J.M.; et al. Tryptophan depletion results in tryptophan-to-phenylalanine substitutants. Nature 2022, 603, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.-K.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881. [Google Scholar] [CrossRef]
- Stepien, M.; Lopez-Nogueroles, M.; Lahoz, A.; Kühn, T.; Perlemuter, G.; Voican, C.; Ciocan, D.; Boutron-Ruault, M.-C.; Jansen, E.; Viallon, V.; et al. Prediagnostic alterations in circulating bile acid profiles in the development of hepatocellular carcinoma. Int. J. Cancer 2022, 150, 1255–1268. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Scialpi, N.; Peres, C.; Cariello, M.; Ko, B.; Luo, J.; Porru, E.; Roda, A.; Sabbà, C.; Moschetta, A. Suppression of Hepatic Bile Acid Synthesis by a non-tumorigenic FGF19 analogue Protects Mice from Fibrosis and Hepatocarcinogenesis. Sci. Rep. 2018, 8, 17210. [Google Scholar] [CrossRef]
- Wang, M.-D.; Wang, N.-Y.; Zhang, H.-L.; Sun, L.-Y.; Xu, Q.-R.; Liang, L.; Li, C.; Huang, D.-S.; Zhu, H.; Yang, T. Fatty acid transport protein-5 (FATP5) deficiency enhances hepatocellular carcinoma progression and metastasis by reprogramming cellular energy metabolism and regulating the AMPK-mTOR signaling pathway. Oncogenesis 2021, 10, 74. [Google Scholar] [CrossRef]
- Luo, X.; Zheng, E.; Wei, L.; Zeng, H.; Qin, H.; Zhang, X.; Liao, M.; Chen, L.; Zhao, L.; Ruan, X.Z.; et al. The fatty acid receptor CD36 promotes HCC progression through activating Src/PI3K/AKT axis-dependent aerobic glycolysis. Cell Death Dis. 2021, 12, 328. [Google Scholar] [CrossRef] [PubMed]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Quan, Y.; Zeng, J.; Lyu, X.; Wang, H.; Lei, J.H.; Feng, Y.; Xu, J.; Chen, Q.; Sun, H.; et al. Cullin3 deficiency shapes tumor microenvironment and promotes cholangiocarcinoma in liver-specific Smad4/Pten mutant mice. Int. J. Biol. Sci. 2021, 17, 4176–4191. [Google Scholar] [CrossRef] [PubMed]
- Che, Z.; Liu, F.; Zhang, W.; McGrath, M.; Hou, D.; Chen, P.; Song, C.; Yang, D. Targeting CAND1 promotes caspase-8/RIP1-dependent apoptosis in liver cancer cells. Am. J. Transl. Res. 2018, 10, 1357–1372. [Google Scholar]
- Xu, K.; Zhang, Z.; Qian, J.; Wang, S.; Yin, S.; Xie, H.; Zhou, L.; Zheng, S. LncRNA FOXD2-AS1 plays an oncogenic role in hepatocellular carcinoma through epigenetically silencing CDKN1B(p27) via EZH2. Exp. Cell Res. 2019, 380, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-J.; Chiu, F.-Y.; Wang, S.-C.; Li, C.-Y. The oncogenic role of ARG2 in hepatocellular carcinoma. J. Clin. Oncol. 2020, 38, e16713. [Google Scholar] [CrossRef]
- Han, R.; Feng, P.; Pang, J.; Zou, D.; Li, X.; Geng, C.; Li, L.; Min, J.; Shi, J. A Novel HCC Prognosis Predictor EEF1E1 Is Related to Immune Infiltration and May Be Involved in EEF1E1/ATM/p53 Signaling. Front. Oncol. 2021, 11, 700972. [Google Scholar] [CrossRef] [PubMed]
- Faloppi, L.; Scartozzi, M.; Bianconi, M.; Svegliati Baroni, G.; Toniutto, P.; Giampieri, R.; Del Prete, M.; De Minicis, S.; Bitetto, D.; Loretelli, C.; et al. The role of LDH serum levels in predicting global outcome in HCC patients treated with sorafenib: Implications for clinical management. BMC Cancer 2014, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.; Mak, T.W. Targeting PI3K Signaling in Cancer: A Cautionary Tale of Two AKTs. Cancer Cell 2016, 29, 429–431. [Google Scholar] [CrossRef]
- Khattar, E.; Maung, K.Z.Y.; Chew, C.L.; Ghosh, A.; Mok, M.M.H.; Lee, P.; Zhang, J.; Chor, W.H.J.; Cildir, G.; Wang, C.Q.; et al. Rap1 regulates hematopoietic stem cell survival and affects oncogenesis and response to chemotherapy. Nat. Commun. 2019, 10, 5349. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Luo, J.; Gu, T.; Yu, X.; Song, Z.; Jun, Y.; Gu, H.; Han, K.; Huang, X.; Yu, W.; et al. LPCAT1 reprogramming cholesterol metabolism promotes the progression of esophageal squamous cell carcinoma. Cell Death Dis. 2021, 12, 845. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Ichu, T.-A.; Zanca, C.; Yang, H.; Zhang, W.; Gu, Y.; Chowdhry, S.; Reed, A.; Ikegami, S.; Turner, K.M.; et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers Dependent on Membrane Lipid Remodeling. Cell Metabolism 2019, 30, 525–538.e8. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Peng, Z.; Sun, B.; Chen, L.; Zhang, Q.; Guo, M.; Su, C. LpCat1 Promotes Malignant Transformation of Hepatocellular Carcinoma Cells by Directly Suppressing STAT1. Front. Oncol. 2021, 11, 678714. [Google Scholar] [CrossRef]
- Cannon, A.C.; Uribe-Alvarez, C.; Chernoff, J. RAC1 as a Therapeutic Target in Malignant Melanoma. Trends Cancer 2020, 6, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Floerchinger, A.; Murphy, K.J.; Latham, S.L.; Warren, S.C.; McCulloch, A.T.; Lee, Y.-K.; Stoehr, J.; Mélénec, P.; Guaman, C.S.; Metcalf, X.L.; et al. Optimizing metastatic-cascade-dependent Rac1 targeting in breast cancer: Guidance using optical window intravital FRET imaging. Cell Rep. 2021, 36, 109689. [Google Scholar] [CrossRef]
- Lopez, A.; Reyna, D.E.; Gitego, N.; Kopp, F.; Zhou, H.; Miranda-Roman, M.A.; Nordstrøm, L.U.; Narayanagari, S.-R.; Chi, P.; Vilar, E.; et al. Co-targeting of BAX and BCL-XL proteins broadly overcomes resistance to apoptosis in cancer. Nat. Commun. 2022, 13, 1199. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polar Metabolite/Lipid | Votes a | Number of Articles | Vote Counting a | FDR | Type |
|---|---|---|---|---|---|
| HCC versus HC | |||||
| L-phenylalanine | 6 | 14 | 0.43 | 0.54 | Metabolite |
| L-tyrosine | 7 | 13 | 0.54 | 0.83 | Metabolite |
| L-leucine | 0 | 8 | 0.00 | 1.00 | Metabolite |
| L-serine | 0 | 8 | 0.00 | 1.00 | Metabolite |
| L-tryptophan | −5 | 7 | −0.71 | 0.56 | Metabolite |
| L-glutamic acid | 3 | 7 | 0.43 | 1.00 | Metabolite |
| L-proline | 3 | 7 | 0.43 | 0.82 | Metabolite |
| Ornithine | 2 | 6 | 0.33 | 1.00 | Metabolite |
| Taurine | −2 | 6 | −0.33 | 0.88 | Metabolite |
| Creatine | −1 | 5 | −0.20 | NA | Metabolite |
| Creatinine | −3 | 5 | −0.60 | NA | Metabolite |
| L-alanine | 1 | 5 | 0.20 | NA | Metabolite |
| L-methionine | 3 | 5 | 0.60 | NA | Metabolite |
| Uric acid | 1 | 5 | 0.20 | NA | Metabolite |
| D-glucose | 0 | 4 | 0.00 | NA | Metabolite |
| Glycerol | −2 | 4 | −0.50 | NA | Metabolite |
| Hypoxanthine | 0 | 4 | 0.00 | NA | Metabolite |
| L-aspartic acid | −2 | 4 | −0.50 | NA | Metabolite |
| L-isoleucine | −2 | 4 | −0.50 | NA | Metabolite |
| L-valine | −2 | 4 | −0.50 | NA | Metabolite |
| Myo-inositol | −2 | 4 | −0.50 | NA | Metabolite |
| Oxoproline | 0 | 4 | 0.00 | NA | Metabolite |
| Phenylalanyl phenylalanine | −4 | 4 | −1.00 | NA | Metabolite |
| Uridine | 0 | 4 | 0.00 | NA | Metabolite |
| LPC (16:0) | −12 | 12 | −1.00 | 0.005 b | Lipid |
| Glycocholic acid | 13 | 15 | 0.87 | 0.007 b | Lipid |
| LPC (18:0) | −11 | 11 | −1.00 | 0.004 b | Lipid |
| Glycochenodeoxycholic acid | 9 | 9 | 1.00 | 0.013 b | Lipid |
| LPC (18:1) | −9 | 9 | −1.00 | 0.010 b | Lipid |
| LPC (20:4) | −9 | 9 | −1.00 | 0.009 b | Lipid |
| LPC (18:2) | −10 | 12 | −0.83 | 0.012 b | Lipid |
| LPC (14:0) | −8 | 8 | −1.00 | 0.013 b | Lipid |
| LPC (20:3) | −7 | 7 | −1.00 | 0.023 b | Lipid |
| LPC (20:5) | −7 | 7 | −1.00 | 0.020 b | Lipid |
| LPC (22:6) | −7 | 7 | −1.00 | 0.018 b | Lipid |
| Taurocholic acid | 6 | 6 | 1.00 | 0.034 b | Lipid |
| CAR (10:0) | −6 | 6 | −1.00 | 0.031 b | Lipid |
| CAR (18:1) | 5 | 5 | 1.00 | NA | Lipid |
| PC (32:1) | 5 | 5 | 1.00 | NA | Lipid |
| LPC (17:0) | −5 | 5 | −1.00 | NA | Lipid |
| PC (38:6) | −5 | 5 | −1.00 | NA | Lipid |
| FA (18:1) | 4 | 6 | 0.67 | NA | Lipid |
| FA (18:2) | 4 | 6 | 0.67 | NA | Lipid |
| CAR (2:0) | 3 | 5 | 0.60 | NA | Lipid |
| CAR (8:0) | −3 | 5 | −0.60 | NA | Lipid |
| FA (20:4) | 0 | 8 | 0.00 | NA | Lipid |
| CAR (16:1) | 4 | 4 | 1.00 | NA | Lipid |
| CAR (18:2) | 4 | 4 | 1.00 | NA | Lipid |
| FA (16:1) | 4 | 4 | 1.00 | NA | Lipid |
| PC (32:0) | 4 | 4 | 1.00 | NA | Lipid |
| Taurochenodesoxycholic acid | 4 | 4 | 1.00 | NA | Lipid |
| FA (22:6) | 2 | 4 | 0.50 | NA | Lipid |
| CAR (16:0) | 0 | 4 | 0.00 | NA | Lipid |
| Oleamide | 0 | 4 | 0.00 | NA | Lipid |
| PE (38:6) | 0 | 4 | 0.00 | NA | Lipid |
| FA (20:5) | −2 | 4 | −0.50 | NA | Lipid |
| LPC (15:0) | −4 | 4 | −1.00 | NA | Lipid |
| LPC (18:3) | −4 | 4 | −1.00 | NA | Lipid |
| HCC versus LC | |||||
| L-glutamic acid | 7 | 7 | 1.00 | 0.0312 b | Metabolite |
| L-phenylalanine | −1 | 7 | −0.14 | 1 | Metabolite |
| L-serine | 3 | 5 | 0.60 | NA | Metabolite |
| L-valine | 5 | 5 | 1.00 | NA | Metabolite |
| L-isoleucine | 2 | 4 | 0.50 | NA | Metabolite |
| L-methionine | 0 | 4 | 0.00 | NA | Metabolite |
| L-proline | 0 | 4 | 0.00 | NA | Metabolite |
| L-tyrosine | −2 | 4 | −0.50 | NA | Metabolite |
| 1-methyladenosine | 3 | 3 | 1.00 | NA | Metabolite |
| 2-hydroxybutyric acid | 3 | 3 | 1.00 | NA | Metabolite |
| Citric acid | −1 | 3 | −0.33 | NA | Metabolite |
| Creatine | 1 | 3 | 0.33 | NA | Metabolite |
| Glycerol | −1 | 3 | −0.33 | NA | Metabolite |
| Glycine | 1 | 3 | 0.33 | NA | Metabolite |
| Hypoxanthine | 3 | 3 | 1.00 | NA | Metabolite |
| L-alanine | 3 | 3 | 1.00 | NA | Metabolite |
| L-aspartic acid | 3 | 3 | 1.00 | NA | Metabolite |
| Ornithine | 3 | 3 | 1.00 | NA | Metabolite |
| Uric acid | −1 | 3 | −0.33 | NA | Metabolite |
| Xanthine | −1 | 3 | −0.33 | NA | Metabolite |
| FA (18:2) | 4 | 6 | 0.67 | NA | Lipid |
| LPC (18:0) | 1 | 5 | 0.20 | NA | Lipid |
| CAR (2:0) | 2 | 4 | 0.50 | NA | Lipid |
| FA (18:1) | 4 | 4 | 1.00 | NA | Lipid |
| Glycocholic acid | −4 | 4 | −1.00 | NA | Lipid |
| LPC (16:0) | 2 | 4 | 0.50 | NA | Lipid |
| LPC (18:2) | −4 | 4 | −1.00 | NA | Lipid |
| CAR (0:0) | 3 | 3 | 1.00 | NA | Lipid |
| CAR (18:1) | −1 | 3 | −0.33 | NA | Lipid |
| FA (18:3) | 1 | 3 | 0.33 | NA | Lipid |
| FA (20:4) | 1 | 3 | 0.33 | NA | Lipid |
| LPE (16:0) | 1 | 3 | 0.33 | NA | Lipid |
| LC versus HC | |||||
| L-phenylalanine | 5 | 9 | 0.56 | 0.36 | Metabolite |
| L-serine | 0 | 6 | 0.00 | 1 | Metabolite |
| L-tyrosine | 4 | 6 | 0.67 | NA | Metabolite |
| L-glutamic acid | −3 | 5 | −0.60 | NA | Metabolite |
| L-methionine | 4 | 4 | 1.00 | NA | Metabolite |
| Bilirubin | 3 | 3 | 1.00 | NA | Metabolite |
| Glycine | −1 | 3 | −0.33 | NA | Metabolite |
| L-aspartic acid | −1 | 3 | −0.33 | NA | Metabolite |
| L-proline | −1 | 3 | −0.33 | NA | Metabolite |
| Ornithine | 3 | 3 | 1.00 | NA | Metabolite |
| FA (18:2) | 1 | 5 | 0.2 | NA | Lipid |
| FA (20:4) | −1 | 5 | −0.2 | NA | Lipid |
| FA (18:0) | 2 | 4 | 0.5 | NA | Lipid |
| Glycochenodeoxycholic acid | 4 | 4 | 1 | NA | Lipid |
| Glycocholic acid | 4 | 4 | 1 | NA | Lipid |
| CAR (2:0) | 3 | 3 | 1 | NA | Lipid |
| FA (16:1) | 3 | 3 | 1 | NA | Lipid |
| FA (18:1) | 3 | 3 | 1 | NA | Lipid |
| LPC (16:0) | −3 | 3 | −1 | NA | Lipid |
| LPC (18:0) | −3 | 3 | −1 | NA | Lipid |
| LPC (18:2) | −3 | 3 | −1 | NA | Lipid |
| LPC (22:6) | −3 | 3 | −1 | NA | Lipid |
| Pathway Name | Significantly Enriched Pathways a | ||
|---|---|---|---|
| HCC vs. Control | HCC vs. LC | LC vs. Control | |
| Alanine, aspartate, and glutamate metabolism | o | o | o |
| Aminoacyl-tRNA biosynthesis | o | o | o |
| Arginine and proline metabolism | o | o | o |
| Arginine biosynthesis | o | o | o |
| D-glutamine and D-glutamate metabolism | o | o | x |
| Glyoxylate and dicarboxylate metabolism | o | o | o |
| Nitrogen metabolism | o | o | x |
| Phenylalanine metabolism | o | o | o |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | o | o | o |
| Primary bile acid biosynthesis | o | o | o |
| Valine, leucine, and isoleucine biosynthesis | o | o | x |
| Glutathione metabolism | x | o | o |
| Ascorbate and aldarate metabolism | o | x | x |
| Butanoate metabolism | o | x | o |
| Citrate cycle (TCA cycle) | o | x | o |
| Glycine, serine, and threonine metabolism | o | x | x |
| Histidine metabolism | o | x | o |
| Porphyrin and chlorophyll metabolism | o | x | o |
| Pyruvate metabolism | o | x | x |
| Taurine and hypotaurine metabolism | o | x | x |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anh, N.H.; Long, N.P.; Min, Y.J.; Ki, Y.; Kim, S.J.; Jung, C.W.; Park, S.; Kwon, S.W.; Lee, S.J. Molecular and Metabolic Phenotyping of Hepatocellular Carcinoma for Biomarker Discovery: A Meta-Analysis. Metabolites 2023, 13, 1112. https://doi.org/10.3390/metabo13111112
Anh NH, Long NP, Min YJ, Ki Y, Kim SJ, Jung CW, Park S, Kwon SW, Lee SJ. Molecular and Metabolic Phenotyping of Hepatocellular Carcinoma for Biomarker Discovery: A Meta-Analysis. Metabolites. 2023; 13(11):1112. https://doi.org/10.3390/metabo13111112
Chicago/Turabian StyleAnh, Nguyen Hoang, Nguyen Phuoc Long, Young Jin Min, Yujin Ki, Sun Jo Kim, Cheol Woon Jung, Seongoh Park, Sung Won Kwon, and Seul Ji Lee. 2023. "Molecular and Metabolic Phenotyping of Hepatocellular Carcinoma for Biomarker Discovery: A Meta-Analysis" Metabolites 13, no. 11: 1112. https://doi.org/10.3390/metabo13111112