
The Interplay of Hypoxia Signaling on Mitochondrial Dysfunction and Inflammation in Cardiovascular Diseases and Cancer: From Molecular Mechanisms to Therapeutic Approaches


 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
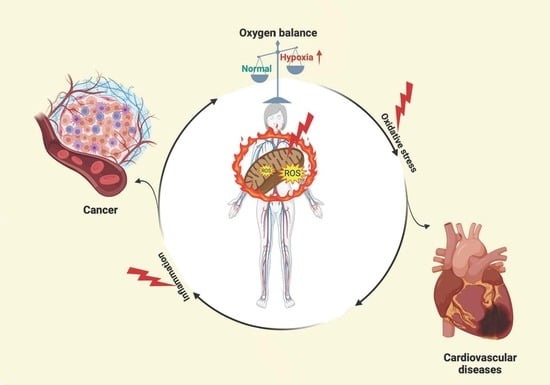
1. Introduction
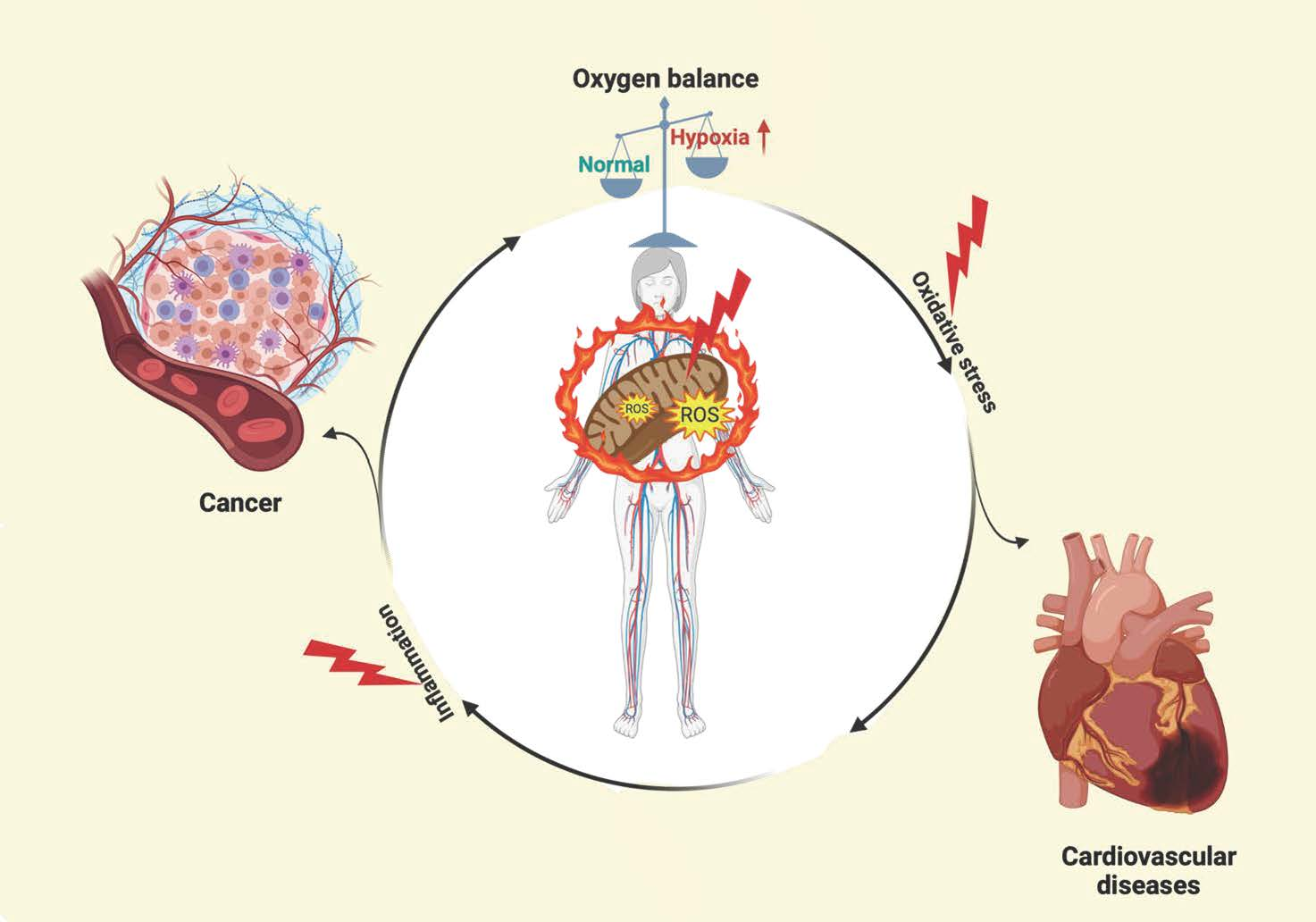
2. Molecular Characteristic and Regulation of HIF-1
3. Role of Hypoxia Signaling and Mitochondria in Cardiovascular Diseases (CVDs) and Cancer
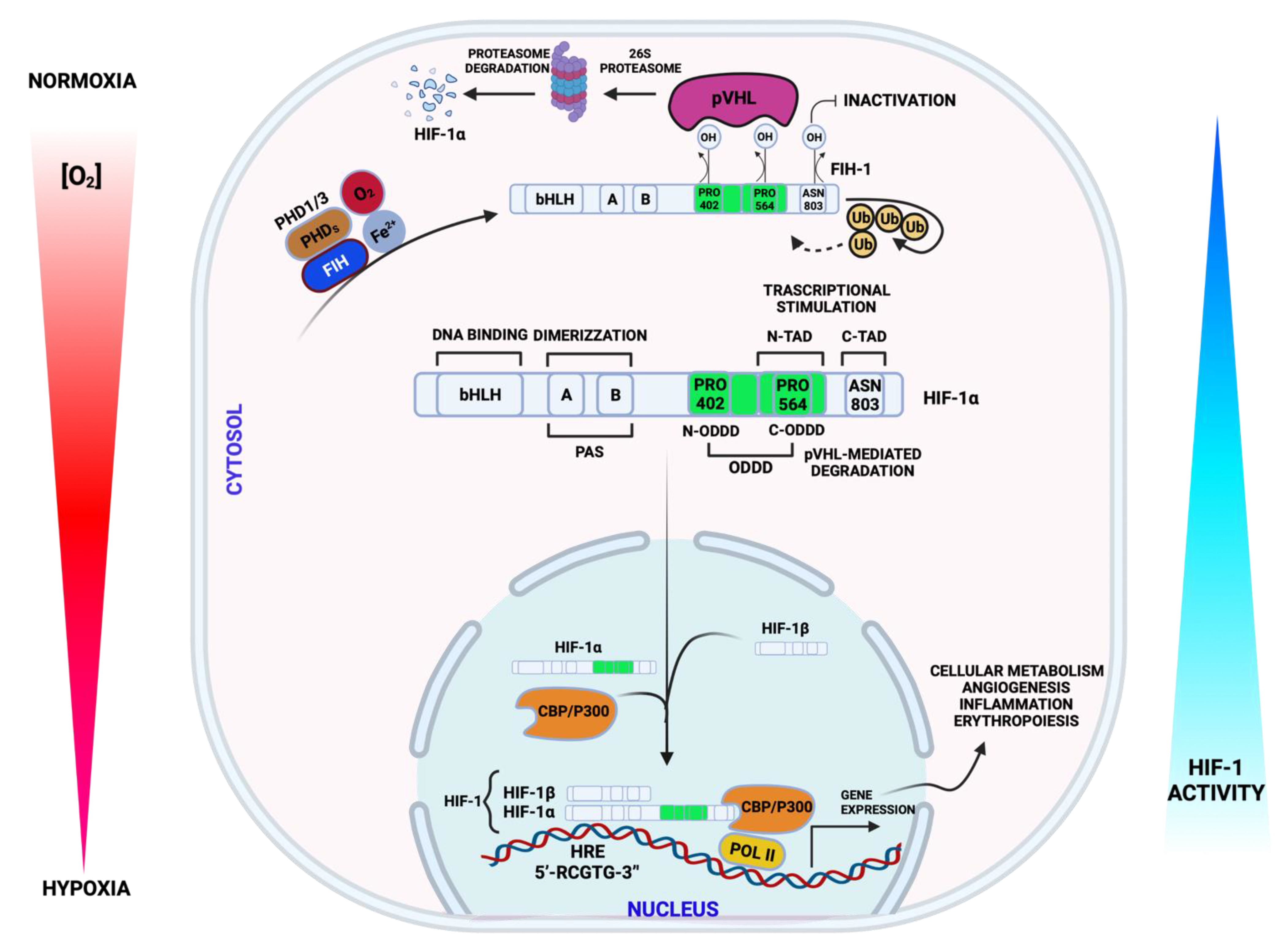
3.1. Hypoxia Signaling and Mitochondria in CVDs
3.2. Hypoxia Signaling and Mitochondria in Cancer
3.2.1. Hypoxia-Induced Modulation of Krebs Cycle and Oxidative Respiration
3.2.2. Hypoxia-Induced Mitochondrial ROS Production and Suppression
3.2.3. Hypoxia-Induced Mitochondrial Distribution and Dynamics
4. Mitochondrial Dysfunction and Inflammation in CVDs and Cancer
4.1. Oxidative Stress and Mitochondria
4.2. Inflammation and Mitochondria
4.3. Inflammation, Oxidative Stress, and Mitochondrial Dysfunction following Hypoxia in CVDs
4.4. Hypoxia-Mediating Signaling Pathways and Cell Death in CVDs
4.5. Inflammation, Oxidative Stress, and Mitochondrial Dysfunction following Hypoxia in Cancer Disease
4.6. Hypoxia-Mediated Signaling Pathways and Cell Death in Cancer
5. Novel Mechanisms and Therapeutic Targets in CVD and Cancer Disorders
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-HG | 2-hydroxyglutarate |
| 2-OG | 2-oxoglutarate |
| 4-HNE | 4-hydroxynonenal |
| AKAP121 | A-kinase anchor protein 1 |
| APC | Adenomatosis Polyposis Coli |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| ATP | Adenosine triphosphate |
| BAP1 | BRCA1-associated protein 1 |
| bHLH | Basic helix–loop–helix |
| BNIP3 | BCL2 and adenovirus E1B 19-kD-interacting protein 3 |
| C26 | Colon cancer |
| Ca2+ | Calcium |
| CAD | Coronary artery disease |
| CBP/p300 | CREB-binding protein |
| CC | Cancer cachexia |
| CCHD | Cyanotic congenital heart disease |
| CCL-39 | Chinese hamster Lung fibroblasts |
| CH | Chronic hypoxia |
| CHD | Coronary heart disease |
| CKD | Chronic kidney disease |
| CPT1A | Carnitine palmitoyltransferase 1A |
| CVD | Cardiovascular disease |
| DFO | Deferoxamine |
| DRP1 | Dynamin-related protein 1 |
| Ecs | Endothelial cells |
| EPO | Erythropoietin |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated kinase ½ |
| FAO | Fatty acid oxidation |
| FIH | Inhibiting HIF |
| Fis 1 | Fission 1 mitochondrial protein |
| FTH | Ferritin Heavy Chain |
| FUNDC1 | FUN14 Domain Containing 1 |
| GCLM | Glutamate–cysteine ligase |
| H2O2 | Hydrogen peroxide |
| HCC | Hepatocellular carcinoma |
| HCT 116 | Human colorectal carcinoma |
| HF | Heart failure |
| HIF-1 | Hypoxic-inducible factor-1 |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HMGB1 | High mobility group box 1 |
| HREs | Hypoxic-responsive elements |
| HSP70 | Heat shock protein 70 |
| HSP90 | Heat shock protein 90 |
| HUVEC | Human umbilical vein endothelial |
| IHD | Ischemic heart disease |
| IMM | Inner mitochondrial membrane |
| IMS | Mitochondrial intermembrane space |
| iNOS | Inducible nitric oxide synthase |
| ISCU | Iron-Sulfur Cluster Assembly Enzyme |
| JAK2/STAT3 | Janus kinase/signal transducer and activator of transcription |
| KRAS | GTPase Kras |
| LDH-A | Lactate dehydrogenase-A |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| LS174 | Human colonic adenocarcinoma cells |
| LV | Left ventricle |
| MAMs | Mitochondria-associated membranes |
| MAOs | Monoamine oxidases |
| MCF-7 | Human breast cancer cells |
| MCU | Mitochondrial calcium uniporter |
| MDH | Malate dehydrogenase |
| MECs | mammary epithelial cells |
| mETC | Mitochondrial electron respiratory chain |
| MFN1 | Mitofusin 1 |
| MI | Myocardial infarction |
| miRs | MicroRNAs |
| MLKL | Mixed lineage kinase domain-like |
| MMP-2 | Matrix metalloproteinase |
| MMP | Mitochondrial membrane potential |
| MMTV-PyMT | Mouse mammary tumor virus-polyoma middle tumor-antigen |
| mPTP | Mitochondrial permeability transition pore |
| MTOR | Rapamycin inhibitor rapamycin |
| mtROS | Mitochondrial ROS |
| MXI1 | MAX-interactor 1 |
| NLRs | Leucine-rich repeat-containing receptors |
| NOXs | NADPH oxidases |
| NRFs | Nuclear respiratory factors |
| O2− | Superoxide |
| O2 | Oxygen |
| ODDD | Oxygen-dependent degradation domain |
| OMM | Outer mitochondrial membrane |
| OSCC | Oral squamous cell carcinoma |
| OXPHOS | Oxidative phosphorylation |
| p70S6K | Phospho-p70 S6 Kinase |
| PAD | Peripheral arterial diseases |
| PASMCs | Pulmonary arterial smooth muscle cells |
| PCR | Polymerase chain reaction |
| PH | Pulmonary hypertension |
| PHDs | Propyl-hydroxylases |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K/AKT | Phosphoinositide-3-kinase/Akt |
| PKA | Protein kinase A |
| PKM2 | M2 isoform of pyruvate kinase |
| PRX/Trx | peroxiredoxin/thioredoxin |
| pVHL | Von Hippel–Lindau |
| RIP1, 3 | Receptor-interacting protein 1, 3 |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SLC7A11 | Solute carrier family 7-member 11 |
| SOD | Superoxide dismutase |
| SUM-149 | Triple-negative breast cancer cells |
| TCA | Tricarboxylic acid |
| TFR1 | transferrin receptor 1 |
| TGF-β | Transforming growth factor beta |
| TNBC | Triple negative breast cancer |
| TNF | Tumor necrosis factor |
| TRAF6 | Receptor-associated factor 6 |
| Tregs | regulatory T cells |
| VDAC1 | Voltage-dependent anion-selective channel 1 |
| VGF-A | vascular endothelial growth factor-A |
| WDR26 | WD Repeat Domain 26 |
| WNT | Wingless-related integration site |
| ΔΨm | Mitochondrial membrane potential |
References
- Masoudkabir, F.; Sarrafzadegan, N.; Gotay, C.; Ignaszewski, A.; Krahn, A.D.; Davis, M.K.; Franco, C.; Mani, A. Cardiovascular disease and cancer: Evidence for shared disease pathways and pharmacologic prevention. Atherosclerosis 2017, 263, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Gernaat, S.A.M.; Boer, J.M.A.; Bongard, D.V.D.; Maas, A.H.E.M.; Van Der Pol, C.C.; Bijlsma, R.M.; Grobbee, D.E.; Verkooijen, H.M.; Peeters, P.H. The risk of cardiovascular disease following breast cancer by Framingham risk score. Breast Cancer Res. Treat. 2018, 170, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coviello, J.S. Cardiovascular and Cancer Risk: The Role of Cardio-oncology. J. Adv. Pr. Oncol. 2018, 9, 160–176. [Google Scholar]
- De Boer, R.A.; Meijers, W.C.; Van Der Meer, P.; Van Veldhuisen, D.J. Cancer and heart disease: Associations and relations. Eur. J. Hear. Fail. 2019, 21, 1515–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Asp. Med. 2016, 47–48, 76–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Galli, G.; Wang, Y.; Fan, Q.; Wang, Z.; Wang, X.; Xiao, W. Novel Therapeutic Targets for Hypoxia-Related Cardiovascular Diseases: The Role of HIF-1. Front. Physiol. 2020, 11, 774. [Google Scholar] [CrossRef]
- Bousquet, P.A.; Meltzer, S.; Sønstevold, L.; Esbensen, Y.; Dueland, S.; Flatmark, K.; Sitter, B.; Bathen, T.F.; Seierstad, T.; Redalen, K.R.; et al. Markers of Mitochondrial Metabolism in Tumor Hypoxia, Systemic Inflammation, and Adverse Outcome of Rectal Cancer. Transl. Oncol. 2019, 12, 76–83. [Google Scholar] [CrossRef]
- Chen, P.-S.; Chiu, W.-T.; Hsu, P.-L.; Lin, S.-C.; Peng, I.-C.; Wang, C.-Y.; Tsai, S.-J. Pathophysiological implications of hypoxia in human diseases. J. Biomed. Sci. 2020, 27, 1–19. [Google Scholar] [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- McClelland, G.B.; Brooks, G.A. Changes in MCT 1, MCT 4, and LDH expression are tissue specific in rats after long-term hypobaric hypoxia. J. Appl. Physiol. 2002, 92, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; E Flook, B.; Voelkel, N.F. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J. Clin. Investig. 1995, 95, 1798–1807. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef]
- Melillo, G.; Musso, T.; Sica, A.; Taylor, L.S.; Cox, G.W.; Varesio, L. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J. Exp. Med. 1995, 182, 1683–1693. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factor 1 (HIF-1) Pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Nakayama, K.; Kataoka, N. Regulation of Gene Expression under Hypoxic Conditions. Int. J. Mol. Sci. 2019, 20, 3278. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. Purification and Characterization of Hypoxia-inducible Factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.C.; Sohn, H.A.; Park, Z.-Y.; Oh, S.; Kang, Y.K.; Lee, K.-M.; Kang, M.; Jang, Y.J.; Yang, S.-J.; Hong, Y.K.; et al. A Lactate-Induced Response to Hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF-1α in myocardial ischemia-reperfusion injury (Review). Mol. Med. Rep. 2021, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Costa, M. Hypoxia-Inducible Factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.-H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA Binding, and Transactivation Properties of Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruick, R.K.; McKnight, S.L. A Conserved Family of Prolyl-4-Hydroxylases That Modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zhou, N.; Yuan, J.; Lu, L.; Zhang, Q.; Wei, M.; Zou, Y.; Yuan, L. Heat shock transcription factor 1 regulates exercise-induced myocardial angiogenesis after pressure overload via HIF-1α/VEGF pathway. J. Cell. Mol. Med. 2020, 24, 2178–2188. [Google Scholar] [CrossRef]
- Hölscher, M.; Schäfer, K.; Krull, S.; Farhat, K.; Hesse, A.; Silter, M.; Lin, Y.; Pichler, B.J.; Thistlethwaite, P.; El-Armouche, A.; et al. Unfavourable consequences of chronic cardiac HIF-1α stabilization. Cardiovasc. Res. 2012, 94, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, Y.; Zhou, K.; Kao, G.; Yan, M.; Xiao, J. Hypoxia-inducible transcription factor-1α inhibition by topotecan protects against lipopolysaccharide-induced inflammation and apoptosis of cardiomyocytes. BioMed. Eng. OnLine 2021, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Pi, X.; Wang, Z.; He, J.; Willis, M.S.; Patterson, C. Depletion of PHD3 protects heart from ischemia/reperfusion injury by inhibiting cardiomyocyte apoptosis. J. Mol. Cell. Cardiol. 2015, 80, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Neckar, J.; Hsu, A.; Khan, A.H.; Gross, G.J.; Nithipatikom, K.; Cyprova, M.; Benak, D.; Hlavackova, M.; Sotáková-Kašparová, D.; Falck, J.R.; et al. Infarct size-limiting effect of epoxyeicosatrienoic acid analog EET-B is mediated by hypoxia-inducible factor-1α via downregulation of prolyl hydroxylase 3. Am. J. Physiol. Circ. Physiol. 2018, 315, H1148–H1158. [Google Scholar] [CrossRef] [Green Version]
- Date, T.; Mochizuki, S.; Belanger, A.J.; Yamakawa, M.; Luo, Z.; Vincent, K.A.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Expression of constitutively stable hybrid hypoxia-inducible factor-1α protects cultured rat cardiomyocytes against simulated ischemia-reperfusion injury. Am. J. Physiol. Physiol. 2005, 288, C314–C320. [Google Scholar] [CrossRef] [Green Version]
- Morand, J.; Arnaud, C.; Pépin, J.L.; Godin-Ribuot, D. Chronic intermittent hypoxia promotes myocardial ischemia-related ventricular arrhythmias and sudden cardiac death. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Meerson, F.Z.; Beloshitskiĭ, P.V.; Vorontsova, E.I.; Ustinova, E.E.; Rozhitskaia, I.I. Effect of adaptation to continuous and intermittent hypoxia on heart resistance to ischemic and reperfusion arrhythmias. Patol. Fiziol. Eksp Ter. 1989, 48–50. (In Russian) [Google Scholar]
- Belaidi, E.; Béguin, P.C.; Lévy, P.; Ribuot, C.; Godin-Ribuot, D. Prevention of HIF-1 activation and iNOS gene targeting by low-dose cadmium results in loss of myocardial hypoxic preconditioning in the rat. Am. J. Physiol. Circ. Physiol. 2008, 294, H901–H908. [Google Scholar] [CrossRef] [Green Version]
- Xi, L.; Tekin, D.; Gursoy, E.; Salloum, F.; Levasseur, J.E.; Kukreja, R.C. Evidence that NOS2 acts as a trigger and mediator of late preconditioning induced by acute systemic hypoxia. Am. J. Physiol. Circ. Physiol. 2002, 283, H5–H12. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.-L.; Zhu, H.-F.; Dong, J.-W.; Zhu, W.-Z.; Yang, W.-W.; Yang, H.-T.; Zhou, Z.-N. Inducible nitric oxide synthase contributes to intermittent hypoxia against ischemia/reperfusion injury1. Acta Pharmacol. Sin. 2005, 26, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamishima, Y.A.; Moslehi, J.; Bardeesy, N.; Cullen, D.; Bronson, R.T.; Kaelin, W.G. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 2008, 111, 3236–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, D.; Hu, H.; Zhang, P.; Xie, R.; Cui, W. HIF-1α/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2019, 120, 109464. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Pandey, S.; Day, C.H.; Chen, Y.-F.; Jiang, A.-Z.; Ho, T.-J.; Chen, R.-J.; Padma, V.V.; Kuo, W.-W.; Huang, C.-Y. Synergistic effect of HIF-1α and FoxO3a trigger cardiomyocyte apoptosis under hyperglycemic ischemia condition. J. Cell. Physiol. 2018, 233, 3660–3671. [Google Scholar] [CrossRef]
- Dorn, G.W.; Kirshenbaum, L.A. Cardiac reanimation: Targeting cardiomyocyte death by BNIP3 and NIX/BNIP3L. Oncogene 2008, 27, S158–S167. [Google Scholar] [CrossRef] [Green Version]
- Xin, T.; Lv, W.; Liu, D.; Jing, Y.; Hu, F. Opa1 Reduces Hypoxia-Induced Cardiomyocyte Death by Improving Mitochondrial Quality Control. Front. Cell Dev. Biol. 2020, 8, 853. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhu, P.; Guo, J.; Hu, N.; Wang, S.; Li, D.; Hu, S.; Ren, J.; Cao, F.; Chen, Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017, 13, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhao, J.; Hou, H.; Zhang, H.; Jiao, Y.; Wang, J.; Wang, Y.; Sun, Y. WDR26 promotes mitophagy of cardiomyocytes induced by hypoxia through Parkin translocation. Acta Biochim. Biophys. Sin. 2016, 48, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.-F.; Rodger, C.E.; Allen, G.F.G.; Weidlich, S.; Ganley, I.G. HIF1α-dependent mitophagy facilitates cardiomyoblast differentiation. Cell Stress 2020, 4, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Aich, A.; Jain, G.; Wozny, K.; Lüchtenborg, C.; Hartmann, M.; Bernhard, O.; Balleiniger, M.; Alfar, E.A.; Zieseniss, A.; et al. Defective Mitochondrial Cardiolipin Remodeling Dampens HIF-1α Expression in Hypoxia. Cell Rep. 2018, 25, 561–570.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, D.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Koutnikova, H.; Campuzano, V.; Foury, F.; Dollé, P.; Cazzalini, O.; Koenig, M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 1997, 16, 345–351. [Google Scholar] [CrossRef]
- Nanayakkara, G.; Alasmari, A.; Mouli, S.; Eldoumani, H.; Quindry, J.C.; McGinnis, G.; Fu, X.; Berlin, A.; Peters, B.; Zhong, J.; et al. Cardioprotective HIF-1α-frataxin signaling against ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2015, 309, H867–H879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of MiR-199a Derepresses Hypoxia-Inducible Factor-1α and Sirtuin 1 and Recapitulates Hypoxia Preconditioning in Cardiac Myocytes. Circ. Res. 2009, 104, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Briston, T.; Yang, J.; Ashcroft, M. HIF-1α localization with mitochondria. Cell Cycle 2011, 10, 4170–4171. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1α to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.-S.; Zhou, Y.-N.; Li, L.; Li, S.-F.; Long, D.; Chen, X.-L.; Zhang, J.-B.; Feng, L.; Li, Y.-P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Du, Y.; Ge, Y.; Xu, Z.-H.; Aa, N.; Gu, X.; Meng, H.; Lin, Z.; Zhu, N.; Shi, J.; Zhuang, R.; et al. Hypoxia-Inducible Factor 1 alpha (HIF-1α)/Vascular Endothelial Growth Factor (VEGF) Pathway Participates in Angiogenesis of Myocardial Infarction in Muscone-Treated Mice: Preliminary Study. Med. Sci. Monit. 2018, 24, 8870–8877. [Google Scholar] [CrossRef]
- Ikeda, M.; Ide, T.; Tadokoro, T.; Miyamoto, H.D.; Ikeda, S.; Okabe, K.; Ishikita, A.; Sato, M.; Abe, K.; Furusawa, S.; et al. Excessive Hypoxia-Inducible Factor-1α Expression Induces Cardiac Rupture via p53-Dependent Apoptosis After Myocardial Infarction. J. Am. Hear. Assoc. 2021, 10, 020895. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, L.; Yang, Y.; Chen, C.; Wang, D.; Wang, H. Acyl-CoA thioesterase 1 prevents cardiomyocytes from Doxorubicin-induced ferroptosis via shaping the lipid composition. Cell Death Dis. 2020, 11, 756. [Google Scholar] [CrossRef]
- Wei, H.; Bedja, D.; Koitabashi, N.; Xing, D.; Chen, J.; Fox-Talbot, K.; Rouf, R.; Chen, S.; Steenbergen, C.; Harmon, J.W.; et al. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF- signaling. Proc. Natl. Acad. Sci. 2012, 109, E841–E850. [Google Scholar] [CrossRef] [Green Version]
- Stamati, K.; Mudera, V.; Cheema, U. Evolution of oxygen utilization in multicellular organisms and implications for cell signalling in tissue engineering. J. Tissue Eng. 2011, 2, 2041731411432365. [Google Scholar] [CrossRef]
- Wolff, M.; Kosyna, F.K.; Dunst, J.; Jelkmann, W.; Depping, R. Impact of hypoxia inducible factors on estrogen receptor expression in breast cancer cells. Arch. Biochem. Biophys. 2017, 613, 23–30. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Schafer, X.; Fu, D.; Nehrke, K.; Munger, J.; Brookes, P.S. Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J. Biol. Chem. 2016, 291, 20188–20197. [Google Scholar] [CrossRef] [Green Version]
- Miska, J.; Lee-Chang, C.; Rashidi, A.; Muroski, M.E.; Chang, A.L.; Lopez-Rosas, A.; Zhang, P.; Panek, W.K.; Cordero, A.; Han, Y.; et al. HIF-1α Is a Metabolic Switch between Glycolytic-Driven Migration and Oxidative Phosphorylation-Driven Immunosuppression of Tregs in Glioblastoma. Cell Rep. 2019, 27, 226–237.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.-W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 Regulates Cytochrome Oxidase Subunits to Optimize Efficiency of Respiration in Hypoxic Cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Enríquez, S.; Carreño-Fuentes, L.; Gallardo-Pérez, J.C.; Saavedra, E.; Quezada, H.; Vega, A.; Marín-Hernández, A.; Olín-Sandoval, V.; Torres-Márquez, M.E.; Moreno-Sánchez, R. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int. J. Biochem. Cell Biol. 2010, 42, 1744–1751. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Zhang, Y.-Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 Controls Mitochondrial Metabolism during Hypoxia by Repressing the Iron-Sulfur Cluster Assembly Proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Guarino, F.; Zinghirino, F.; Mela, L.; Pappalardo, X.G.; Ichas, F.; De Pinto, V.; Messina, A. NRF-1 and HIF-1α contribute to modulation of human VDAC1 gene promoter during starvation and hypoxia in HeLa cells. Biochim. Biophys. Acta 2020, 1861, 148289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.; Semenza, G.L. HIF-1 Inhibits Mitochondrial Biogenesis and Cellular Respiration in VHL-Deficient Renal Cell Carcinoma by Repression of C-MYC Activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohme, S.; Yazdani, H.O.; Liu, Y.; Loughran, P.; Van Der Windt, D.J.; Huang, H.; Simmons, R.L.; Shiva, S.; Tai, S.; Tsung, A. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatol. 2017, 66, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Qiu, S.; Wang, P.; Liang, X.; Huang, F.; Wu, H.; Zhang, B.; Zhang, W.; Tian, X.; Xu, R.; et al. Cardamonin inhibits breast cancer growth by repressing HIF-1α-dependent metabolic reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1–dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. 2015, 112, E4600–E4609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.; E Drake, L.; Sachleben, J.R.; Asara, J.M.; Locasale, J.; Karczmar, G.S.; et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, S.; Ciccarone, F.; Tavian, D.; Ciriolo, M.R. ROS-dependent HIF1α activation under forced lipid catabolism entails glycolysis and mitophagy as mediators of higher proliferation rate in cervical cancer cells. J. Exp. Clin. Cancer Res. 2021, 40, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zuo, M.; Zeng, L.; Cui, K.; Liu, B.; Yan, C.; Chen, L.; Dong, J.; Shangguan, F.; Hu, W.; et al. OMA1 reprograms metabolism under hypoxia to promote colorectal cancer development. EMBO Rep. 2021, 22, e50827. [Google Scholar] [CrossRef]
- Chiche, J.; Rouleau, M.; Gounon, P.; Brahimi-Horn, M.C.; Pouyssã©Gur, J.; Mazure, N.M. Hypoxic enlarged mitochondria protect cancer cells from apoptotic stimuli. J. Cell. Physiol. 2009, 222, 648–657. [Google Scholar] [CrossRef]
- Devine, R.; Bicer, S.; Reiser, P.J.; Wold, L.E. Increased hypoxia-inducible factor-1α in striated muscle of tumor-bearing mice. Am. J. Physiol. Circ. Physiol. 2017, 312, H1154–H1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-S.; Chang, I.Y.-F.; Hung, J.-L.; Liao, W.-C.; Lai, Y.-R.; Chang, K.-P.; Li, H.-P.; Chang, Y.-S. ASC modulates HIF-1α stability and induces cell mobility in OSCC. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Coimbra-Costa, D.; Alva, N.; Duran, M.; Carbonell, T.; Rama, R. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biol. 2017, 12, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Akkol, E.; Tatlı, I.; Karatoprak, G.; Ağar, O.; Yücel, Ç.; Sobarzo-Sánchez, E.; Capasso, R. Is Emodin with Anticancer Effects Completely Innocent? Two Sides of the Coin. Cancers 2021, 13, 2733. [Google Scholar] [CrossRef]
- Hu, L.; Cui, R.; Liu, H.; Wang, F. Emodin and rhein decrease levels of hypoxia-inducible factor-1α in human pancreatic cancer cells and attenuate cancer cachexia in athymic mice carrying these cells. Oncotarget 2017, 8, 88008–88020. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.-P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St.-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.-H.; Li, C.-X.; Shen, S.-M.; Li, H.; Chen, G.-Q.; Wei, Q.; Wang, L.-S. Hypoxia-inducible factor 1α mediates the down-regulation of superoxide dismutase 2 in von Hippel–Lindau deficient renal clear cell carcinoma. Biochem. Biophys. Res. Commun. 2013, 435, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Missiroli, S.; Morciano, G.; Perrone, M.; Mantovani, C.M.; Anania, G.; Fiorica, F.; Pinton, P.; Giorgi, C. Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers. Cancers 2021, 13, 5622. [Google Scholar] [CrossRef]
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. 2021, 118, 2020078118. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872–883.e5. [Google Scholar] [CrossRef] [Green Version]
- Torrano, V.; Valcarcel-Jimenez, L.; Cortazar, A.R.; Liu, X.; Urosevic, J.; Castillo-Martin, M.; Fernández-Ruiz, S.; Morciano, G.; Caro-Maldonado, A.; Guiu, M.; et al. The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nat. Cell Biol. 2016, 18, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Scimia, M.C.; Wilkinson, D.; Trelles, R.D.; Wood, M.R.; Bowtell, D.; Dillin, A.; Mercola, M.; Ronai, Z.A. Fine-Tuning of Drp1/Fis1 Availability by AKAP121/Siah2 Regulates Mitochondrial Adaptation to Hypoxia. Mol. Cell 2011, 44, 532–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.G.; Wikman, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2014, 14, 177–185. [Google Scholar] [CrossRef]
- Araki, K.; Kawauchi, K.; Sugimoto, W.; Tsuda, D.; Oda, H.; Yoshida, R.; Ohtani, K. Mitochondrial protein E2F3d, a distinctive E2F3 product, mediates hypoxia-induced mitophagy in cancer cells. Commun. Biol. 2019, 2, 3. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-Induced Production of Mitochondrial Reactive Oxygen Species: Potential Mechanisms and Relevance for Cardiovascular Disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Lenaz, G. The Mitochondrial Production of Reactive Oxygen Species: Mechanisms and Implications in Human Pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, R.H.; Hirabara, S.M.; Silveira, L.D.R.; Levada-Pires, A.C.; Curi, R.; Pithon-Curi, T.C. Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J. Cell. Physiol. 2008, 216, 796–804. [Google Scholar] [CrossRef]
- Di Lisa, F.; Kaludercic, N.; Carpi, A.; Menabò, R.; Giorgio, M. Mitochondrial pathways for ROS formation and myocardial injury: The relevance of p66Shc and monoamine oxidase. Basic Res. Cardiol. 2009, 104, 131–139. [Google Scholar] [CrossRef]
- Starkov, A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial -Ketoglutarate Dehydrogenase Complex Generates Reactive Oxygen Species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial Aconitase Is a Source of Hydroxyl Radical. J. Biol. Chem. 2000, 275, 14064–14069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyemi, U.; Dupuy, C. The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutat. Res. Mutat. Res. 2012, 751, 77–81. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron Transfer between Cytochrome c and p66Shc Generates Reactive Oxygen Species that Trigger Mitochondrial Apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Simoes, I.C.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaiz, S.L.; Coronel, M.F.; Boveris, A. Nitric Oxide, Superoxide, and Hydrogen Peroxide Production in Brain Mitochondria after Haloperidol Treatment. Nitric Oxide 1999, 3, 235–243. [Google Scholar] [CrossRef]
- Tewari, S.; Santos, J.M.; Kowluru, R.A. Damaged Mitochondrial DNA Replication System and the Development of Diabetic Retinopathy. Antioxid. Redox Signal. 2012, 17, 492–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, S.; Zhong, Q.; Santos, J.M.; A Kowluru, R. Mitochondria DNA Replication and DNA Methylation in the Metabolic Memory Associated with Continued Progression of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4881–4888. [Google Scholar] [CrossRef]
- Morciano, G.; Naumova, N.; Koprowski, P.; Valente, S.; Sardão, V.A.; Potes, Y.; Rimessi, A.; Wieckowski, M.R.; Oliveira, P.J. The mitochondrial permeability transition pore: An evolving concept critical for cell life and death. Biol. Rev. 2021, 96, 2489–2521. [Google Scholar] [CrossRef]
- Radi, R.; Turrens, J.; Chang, L.; Bush, K.; Crapo, J.; Freeman, B. Detection of catalase in rat heart mitochondria. J. Biol. Chem. 1991, 266, 22028–22034. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.; Sinclair, D.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Hammad, H.; Van Nimwegen, M.; Kool, M.; Willart, M.A.M.; Muskens, F.; Hoogsteden, H.C.; Luttmann, W.; Ferrari, D.; Di Virgilio, F.; et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat. Med. 2007, 13, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chaung, W.W.; Wu, R.; Ji, Y.; Dong, W. Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int. J. Mol. Med. 2012, 30, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuominen, A.; Miller, Y.I.; Hansen, L.F.; Kesäniemi, Y.A.; Witztum, J.L.; Hörkkö, S. A Natural Antibody to Oxidized Cardiolipin Binds to Oxidized Low-Density Lipoprotein, Apoptotic Cells, and Atherosclerotic Lesions. Arter. Thromb. Vasc. Biol. 2006, 26, 2096–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Vitto, V.; Bouhamida, E.; Giorgi, C.; Pinton, P. Mitochondrial Bioenergetics and Dynamism in the Failing Heart. Life 2021, 11, 436. [Google Scholar] [CrossRef]
- Parathath, S.; Mick, S.L.; Feig, J.E.; Joaquin, V.; Grauer, L.; Habiel, D.M.; Gassmann, M.; Gardner, L.; Fisher, E.A. Hypoxia Is Present in Murine Atherosclerotic Plaques and Has Multiple Adverse Effects on Macrophage Lipid Metabolism. Circ. Res. 2011, 109, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Fu, J. Novel Insights Into the NLRP3 Inflammasome in Atherosclerosis. J. Am. Hear. Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification In Vitro. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Bin Goh, W.W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.M.; Pompilius, M.; Pinkerton, K.E.; Ballinger, S.W. Mitochondrial Oxidative Stress Significantly Influences Atherogenic Risk and Cytokine-Induced Oxidant Production. Environ. Heal. Perspect. 2011, 119, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The Life Span Determinant p66Shc Localizes to Mitochondria Where It Associates with Mitochondrial Heat Shock Protein 70 and Regulates Trans-membrane Potential. J. Biol. Chem. 2004, 279, 25689–25695. [Google Scholar] [CrossRef] [Green Version]
- Graiani, G.; Lagrasta, C.; Migliaccio, E.; Spillmann, F.; Meloni, M.; Madeddu, P.; Quaini, F.; Padura, I.M.; Lanfrancone, L.; Pelicci, P.; et al. Genetic Deletion of the p66 Shc Adaptor Protein Protects from Angiotensin II–Induced Myocardial Damage. Hypertension 2005, 46, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes Promotes Cardiac Stem Cell Aging and Heart Failure, Which Are Prevented by Deletion of the p66 shc Gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Lai, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine Oxidase A–Mediated Enhanced Catabolism of Norepinephrine Contributes to Adverse Remodeling and Pump Failure in Hearts with Pressure Overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef]
- Kaludercic, N.; Carpi, A.; Nagayama, T.; Sivakumaran, V.; Zhu, G.; Lai, E.W.; Bedja, D.; De Mario, A.; Chen, K.; Gabrielson, K.L.; et al. Monoamine Oxidase B Prompts Mitochondrial and Cardiac Dysfunction in Pressure Overloaded Hearts. Antioxid. Redox Signal. 2014, 20, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, T.; Suzuki, Y.-I.; Shimizu, T.; Isono, K.; Koseki, H.; Shirasawa, T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochem. Biophys. Res. Commun. 2002, 296, 729–736. [Google Scholar] [CrossRef]
- Sharma, S.; Bhattarai, S.; Ara, H.; Sun, G.; Clair, D.K.S.; Bhuiyan, S.; Kevil, C.; Watts, M.N.; Dominic, P.; Shimizu, T.; et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol. 2020, 37, 101740. [Google Scholar] [CrossRef] [PubMed]
- Souiden, Y.; Mallouli, H.; Meskhi, S.; Chaabouni, Y.; Rebai, A.; Chéour, F.; Mahdouani, K. MnSOD and GPx1 polymorphism relationship with coronary heart disease risk and severity. Biol. Res. 2016, 49, 22. [Google Scholar] [CrossRef] [Green Version]
- Faraci, F.M.; Didion, S.P. Vascular Protection. Arter. Thromb. Vasc. Biol. 2004, 24, 1367–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardanaz, N.; Yang, X.-P.; Cifuentes, M.E.; Haurani, M.J.; Jackson, K.W.; Liao, T.-D.; Carretero, O.A.; Pagano, P.J. Lack of Glutathione Peroxidase 1 Accelerates Cardiac-Specific Hypertrophy and Dysfunction in Angiotensin II Hypertension. Hypertension 2010, 55, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Chua, C.C.; Gao, J.; Chua, K.-W.; Ho, Y.-S.; Hamdy, R.C.; Chua, B.H.L. Prevention of ischemia/reperfusion-induced cardiac apoptosis and injury by melatonin is independent of glutathione peroxdiase 1. J. Pineal Res. 2009, 46, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, S.; Ide, T.; Yamato, M.; Matsusaka, H.; Hattori, F.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Hasegawa, Y.; Kurihara, T.; et al. Overexpression of Mitochondrial Peroxiredoxin-3 Prevents Left Ventricular Remodeling and Failure After Myocardial Infarction in Mice. Circulation 2006, 113, 1779–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 Inhibits Mitochondrial Reactive Oxygen Species Generation and Apoptosis Stress Kinase-1 Activity to Maintain Cardiac Function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [Green Version]
- Flores-Mateo, G.; Carrillo-Santisteve, P.; Elosua, R.; Guallar, E.; Marrugat, J.; Bleys, J.; Covas, M.-I. Antioxidant Enzyme Activity and Coronary Heart Disease: Meta-analyses of Observational Studies. Am. J. Epidemiol. 2009, 170, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Tonet, E.; Bernucci, D.; Morciano, G.; Campo, G. Pharmacological protection of reperfusion injury in ST-segment elevation myocardial infarction. Gone with the wind? Postepy Kardiol Interwencyjnej 2018, 14, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Calbet, J.A. Chronic hypoxia increases blood pressure and noradrenaline spillover in healthy humans. J. Physiol. 2003, 551, 379–386. [Google Scholar] [CrossRef]
- Lee, S.H.; Wolf, P.L.; Escudero, R.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Early Expression of Angiogenesis Factors in Acute Myocardial Ischemia and Infarction. New Engl. J. Med. 2000, 342, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, Q.; Su, Z.; Xing, J.; Wu, J.; Xiang, L.; Huang, Y.; Pan, H.; Wu, X.; Zhang, X.; et al. Suppression of Myocardial Hypoxia-Inducible Factor-1α Compromises Metabolic Adaptation and Impairs Cardiac Function in Patients with Cyanotic Congenital Heart Disease During Puberty. Circulation 2021, 143, 2254–2272. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2021, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Modesti, L.; Danese, A.; Vitto, V.A.M.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.; Lambert, J.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Lewis, T.S.; Powers, C.; Khuchua, Z.; Baines, C.P.; Wipf, P.; Javadov, S. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia–Reperfusion. Antioxid. Redox Signal. 2017, 27, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. 2011, 108, 19725–19730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia–reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, R.; Tyson, D.W.; Rosevear, H.M.; Brosius, F.C. Hypoxia-inducible factor-1alpha is a critical mediator of hypoxia induced apoptosis in cardiac H9c2 and kidney epithelial HK-2 cells. BMC Cardiovasc. Disord. 2008, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Moslehi, J.; Minamishima, Y.A.; Shi, J.; Neuberg, D.; Charytan, D.; Padera, R.F.; Signoretti, S.; Liao, R.; Kaelin, W.G. Loss of Hypoxia-Inducible Factor Prolyl Hydroxylase Activity in Cardiomyocytes Phenocopies Ischemic Cardiomyopathy. Circulation 2010, 122, 1004–1016. [Google Scholar] [CrossRef] [Green Version]
- Loor, G.; Schumacker, P.T. Role of hypoxia-inducible factor in cell survival during myocardial ischemia–reperfusion. Cell Death Differ. 2008, 15, 686–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Li, M.; Li, X.; Li, H.; Lai, Y.; Huang, S.; He, X.; Si, X.; Zheng, H.; Liao, W.; et al. Sirt1-inducible deacetylation of p21 promotes cardiomyocyte proliferation. Aging 2019, 11, 12546–12567. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Jian, Z.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 2019, 43, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Robador, P.A.; José, G.S.; Rodríguez, C.; Guadall, A.; Moreno, M.U.; Beaumont, J.; Fortuño, A.; Díez, J.; Martínez-González, J.; Zalba, G. HIF-1-mediated up-regulation of cardiotrophin-1 is involved in the survival response of cardiomyocytes to hypoxia. Cardiovasc. Res. 2011, 92, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Tennant, D.; Howell, N.J. The role of HIFs in ischemia-reperfusion injury. Hypoxia 2014, 2, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Wan, D.-Y.; Zhang, Z.; Yang, H.-H. Cardioprotective effect of miR-214 in myocardial ischemic postconditioning by down-regulation of hypoxia inducible factor 1, alpha subunit inhibitor. Cell. Mol. Biol. 2015, 61, 1–6. [Google Scholar]
- Stanley, M.; Crowcroft, N.; Quigley, J.; Parkinson, E. Responses of human cervical keratinocytes in vitro to tumour promoters and diethylstilboestrol. Carcinogenesis 1985, 6, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.D.; Banik, A.; Mandal, B.; Sarkar, S. Cardiac-specific overexpression of HIF-1α during acute myocardial infarction ameliorates cardiomyocyte apoptosis via differential regulation of hypoxia-inducible pro-apoptotic and anti-oxidative genes. Biochem. Biophys. Res. Commun. 2021, 537, 100–108. [Google Scholar] [CrossRef]
- Yang, B.; He, K.; Zheng, F.; Wan, L.; Yu, X.; Wang, X.; Zhao, D.; Bai, Y.; Chu, W.; Sun, Y.; et al. Over-expression of hypoxia-inducible factor-1 alpha in vitro protects the cardiac fibroblasts from hypoxia-induced apoptosis. J. Cardiovasc. Med. 2014, 15, 579–586. [Google Scholar] [CrossRef]
- Zhu, H.; Sun, A. Programmed necrosis in heart disease: Molecular mechanisms and clinical implications. J. Mol. Cell. Cardiol. 2018, 116, 125–134. [Google Scholar] [CrossRef]
- Piamsiri, C.; Maneechote, C.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Targeting necroptosis as therapeutic potential in chronic myocardial infarction. J. Biomed. Sci. 2021, 28, 1–13. [Google Scholar] [CrossRef]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress–induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Karshovska, E.; Wei, Y.; Subramanian, P.; Mohibullah, R.; Geißler, C.; Baatsch, I.; Popal, A.; Campos, J.C.; Exner, N.; Schober, A. HIF-1α (Hypoxia-Inducible Factor-1α) Promotes Macrophage Necroptosis by Regulating miR-210 and miR-383. Arter. Thromb. Vasc. Biol. 2020, 40, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhao, C.; Li, H.; Chen, X.; Ding, Y.; Xu, S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem. Biophys. Res. Commun. 2018, 497, 233–240. [Google Scholar] [CrossRef]
- Chen, X.; Xu, S.; Zhao, C.; Liu, B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 2019, 516, 37–43. [Google Scholar] [CrossRef]
- Yoshimura, C.; Nagasaka, A.; Kurose, H.; Nakaya, M. Efferocytosis during myocardial infarction. J. Biochem. 2020, 168, 1–6. [Google Scholar] [CrossRef]
- Davis, C.K.; Jain, S.A.; Bae, O.-N.; Majid, A.; Rajanikant, G.K. Hypoxia Mimetic Agents for Ischemic Stroke. Front. Cell Dev. Biol. 2019, 6, 175. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, W.; Leng, Y.; Xiong, Y.; Xia, Z. Ferroptosis Is Involved in Diabetes Myocardial Ischemia/Reperfusion Injury Through Endoplasmic Reticulum Stress. DNA Cell Biol. 2020, 39, 210–225. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.-J.; Luo, X.-J.; Tu, H.; Chen, H.; Xiong, X.-M.; Li, N.-S.; Peng, J. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn-Schmiedebergs Arch. fur Exp. Pathol. Pharmakol. 2021, 394, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; Fang, Y.-H.; Beh, S.T.; Liu, Y.-W.; Hsu, L.-W.; Yen, C.-J.; Liu, P.-Y. Programmed Cell Death-1: Programmed Cell Death-Ligand 1 Interaction Protects Human Cardiomyocytes Against T-Cell Mediated Inflammation and Apoptosis Response In Vitro. Int. J. Mol. Sci. 2020, 21, 2399. [Google Scholar] [CrossRef] [Green Version]
- Van der Windt, G.J.W.; O’Sullivan, D.; Everts, B.; Huang, S.C.-C.; Buck, M.; Curtis, J.D.; Chang, C.-H.; Smith, A.M.; Ai, T.; Faubert, B.; et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA 2013, 110, 14336–14341. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Carmen, F.C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualdoni, G.A.; Mayer, K.A.; Göschl, L.; Boucheron, N.; Ellmeier, W.; Zlabinger, G.J. The AMP analog AICAR modulates the T reg /T h 17 axis through enhancement of fatty acid oxidation. FASEB J. 2016, 30, 3800–3809. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Tian, T.; Gao, J.; Liu, X.; Hou, H.; Cao, R.; Li, B.; Quan, M.; Guo, L. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J. Neuroimmunol. 2016, 292, 58–67. [Google Scholar] [CrossRef]
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPKα1: A glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Putignani, L.; Raffa, S.; Pescosolido, R.; Aimati, L.; Signore, F.; Torrisi, M.R.; Grammatico, P. Alteration of expression levels of the oxidative phosphorylation system (OXPHOS) in breast cancer cell mitochondria. Breast Cancer Res. Treat. 2007, 110, 439–452. [Google Scholar] [CrossRef]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Beroud, C.; Demont, J.; Bouvier, R.; Schägger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinog. 2002, 23, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Bonora, E.; Porcelli, A.M.; Gasparre, G.; Biondi, A.; Ghelli, A.; Carelli, V.; Baracca, A.; Tallini, G.; Martinuzzi, A.; Lenaz, G.; et al. Defective Oxidative Phosphorylation in Thyroid Oncocytic Carcinoma Is Associated with Pathogenic Mitochondrial DNA Mutations Affecting Complexes I and III. Cancer Res. 2006, 66, 6087–6096. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease−Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dmitrieva-Posocco, O.; Dzutsev, A.; Posocco, D.F.; Hou, V.; Yuan, W.; Thovarai, V.; Mufazalov, I.; Gunzer, M.; Shilovskiy, I.; Khaitov, M.; et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 2019, 50, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., II; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Panchanathan, R.; Liu, H.; Choubey, D. Hypoxia primes human normal prostate epithelial cells and cancer cell lines for the NLRP3 and AIM2 inflammasome activation. Oncotarget 2016, 7, 28183–28194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, X.; Xia, J.; Zhang, L.; Chen, B.; Lian, G.; Yun, C.; Yang, J.; Yan, Y.; Wang, P.; et al. Macrophage HIF-2α suppresses NLRP3 inflammasome activation and alleviates insulin resistance. Cell Rep. 2021, 36, 109607. [Google Scholar] [CrossRef]
- Moon, J.-S.; Nakahira, K.; Chung, K.-P.; DeNicola, G.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.-H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Boncompagni, C.; Borghi, C.; Campagnaro, A.; Marchetti, F.; Anania, G.; Greco, P.; Fiorica, F.; Pinton, P.; et al. Targeting the NLRP3 Inflammasome as a New Therapeutic Option for Overcoming Cancer. Cancers 2021, 13, 2297. [Google Scholar] [CrossRef]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2018, 116, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kong, H.; Zeng, X.; Liu, W.; Wang, Z.; Yan, X.; Wang, H.; Xie, W. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol. Rep. 2016, 35, 2053–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively Active Inflammasome in Human Melanoma Cells Mediating Autoinflammation via Caspase-1 Processing and Secretion of Interleukin-1β. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Muneer, K.M.; Tamimi, I.A.; Chang, M.E.; Ata, M.O.; Yusuf, N. Thymoquinone suppresses metastasis of melanoma cells by inhibition of NLRP3 inflammasome. Toxicol. Appl. Pharmacol. 2013, 270, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Zaki, H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.-D. IL-18 Production Downstream of the Nlrp3 Inflammasome Confers Protection against Colorectal Tumor Formation. J. Immunol. 2010, 185, 4912–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, S.; Gagliani, N.; Zenewicz, L.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’connor, W.; Murphy, A.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2013, 94, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma. Cancers 2019, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-Mediated Suppression of Acyl-CoA Dehydrogenases and Fatty Acid Oxidation Is Critical for Cancer Progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Kato, H.; Kitajima, S.; Lee, K.L.; Gradin, K.; Okamoto, T.; Poellinger, L. Interaction between von Hippel-Lindau Protein and Fatty Acid Synthase Modulates Hypoxia Target Gene Expression. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.S.; Lee, J.H.; Kong, J.; Ji, Y.; Kim, J.; Choe, S.S.; Kim, J.B. Hypoxia Restrains Lipid Utilization via Protein Kinase A and Adipose Triglyceride Lipase Downregulation through Hypoxia-Inducible Factor. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation via HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Nejad, A.E.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Javanmard, S.H.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 1–26. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef] [Green Version]
- Akakura, N.; Kobayashi, M.; Horiuchi, I.; Suzuki, A.; Wang, J.; Chen, J.; Niizeki, H.; Ki, K.; Hosokawa, M.; Asaka, M. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001, 61, 6548–6554. [Google Scholar] [PubMed]
- Steinbach, J.P.; Wolburg, H.; Klumpp, A.; Probst, H.; Weller, M. Hypoxia-induced cell death in human malignant glioma cells: Energy deprivation promotes decoupling of mitochondrial cytochrome c release from caspase processing and necrotic cell death. Cell Death Differ. 2003, 10, 823–832. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, L.; Chew, S.H.; Hirayama, T.; Sekido, Y.; Toyokuni, S. Carbonic anhydrase 9 confers resistance to ferroptosis/apoptosis in malignant mesothelioma under hypoxia. Redox Biol. 2019, 26, 101297. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.-Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022, 22, 1–19. [Google Scholar] [CrossRef]
- Novelli, F.; Bononi, A.; Wang, Q.; Bai, F.; Patergnani, S.; Kricek, F.; Haglund, E.; Suarez, J.S.; Tanji, M.; Xu, R.; et al. BAP1 forms a trimer with HMGB1 and HDAC1 that modulates gene × environment interaction with asbestos. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, L.; Gan, B. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol. Cell. Oncol. 2019, 6, 1536845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronkhorst, I.H.G.; Ly, L.V.; Jordanova, E.S.; Vrolijk, J.; Versluis, M.; Luyten, G.P.M.; Jager, M.J. Detection of M2-Macrophages in Uveal Melanoma and Relation with Survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, N.J.; Wierenga, A.P.A.; Gezgin, G.; Marinkovic, M.; Luyten, G.P.M.; Kroes, W.G.M.; Versluis, M.; Van Der Velden, P.A.; Verdijk, R.M.; Jager, M.J. Ischemia Is Related to Tumour Genetics in Uveal Melanoma. Cancers 2019, 11, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wong, C.C.L.; Wei, H.; Gilkes, D.M.; Korangath, P.; Chaturvedi, P.; Schito, L.; Chen, J.; Krishnamachary, B.; Winnard, P.T.; et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012, 31, 1757–1770. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 Inhibitors for Cancer Therapy: From Gene Expression to Drug Discovery. Curr. Pharm. Des. 2009, 15, 3839–3843. [Google Scholar] [CrossRef]
- Fujiwara, S.; Nakagawa, K.; Harada, H.; Nagato, S.; Furukawa, K.; Teraoka, M.; Seno, T.; Oka, K.; Iwata, S.; Ohnishi, T. Silencing hypoxia-inducible factor-1α inhibits cell migration and invasion under hypoxic environment in malignant gliomas. Int. J. Oncol. 2007, 30, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Greenberger, L.M.; Horak, I.D.; Filpula, D.; Sapra, P.; Westergaard, M.; Frydenlund, H.F.; Albaek, C.; Schrøder, H.D.; Ørum, H. A RNA antagonist of hypoxia-inducible factor-1α, EZN-2968, inhibits tumor cell growth. Mol. Cancer Ther. 2008, 7, 3598–3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1α. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, J.S.; Jung, Y.-J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 Regulates a von Hippel Lindau-independent Hypoxia-inducible Factor-1α-degradative Pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef] [Green Version]
- Voit, R.A.; Sankaran, V.G. Stabilizing HIF to Ameliorate Anemia. Cell 2020, 180, 6. [Google Scholar] [CrossRef]
- Bai, W.-W.; Xing, Y.-F.; Wang, B.; Lu, X.-T.; Wang, Y.-B.; Sun, Y.-Y.; Liu, X.-Q.; Guo, T.; Zhao, Y.-X. Tongxinluo Improves Cardiac Function and Ameliorates Ventricular Remodeling in Mice Model of Myocardial Infarction through Enhancing Angiogenesis. Evidence-Based Complement. Altern. Med. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-W.; Lu, M.-K.; Zhong, H.-T.; Wang, L.-H.; Fu, Y.-P. Panax Notoginseng Saponins Attenuate Myocardial Ischemia-Reperfusion Injury Through the HIF-1α/BNIP3 Pathway of Autophagy. J. Cardiovasc. Pharmacol. 2019, 73, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Nikolopoulou, E.A.; Xu, Q.; Qu, A. Hypoxia inducible factor as a therapeutic target for atherosclerosis. Pharmacol. Ther. 2018, 183, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A.; Ahmad, S.; Malcolm, K.C.; Miller, S.M.; Hendry-Hofer, T.; Schaack, J.B.; White, C.W. Differential Regulation of Pulmonary Vascular Cell Growth by Hypoxia-Inducible Transcription Factor–1α and Hypoxia-Inducible Transcription Factor–2α. Am. J. Respir. Cell Mol. Biol. 2013, 49, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.; Machado, R.; Zhang, X.; Zhao, Y.-Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2α Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
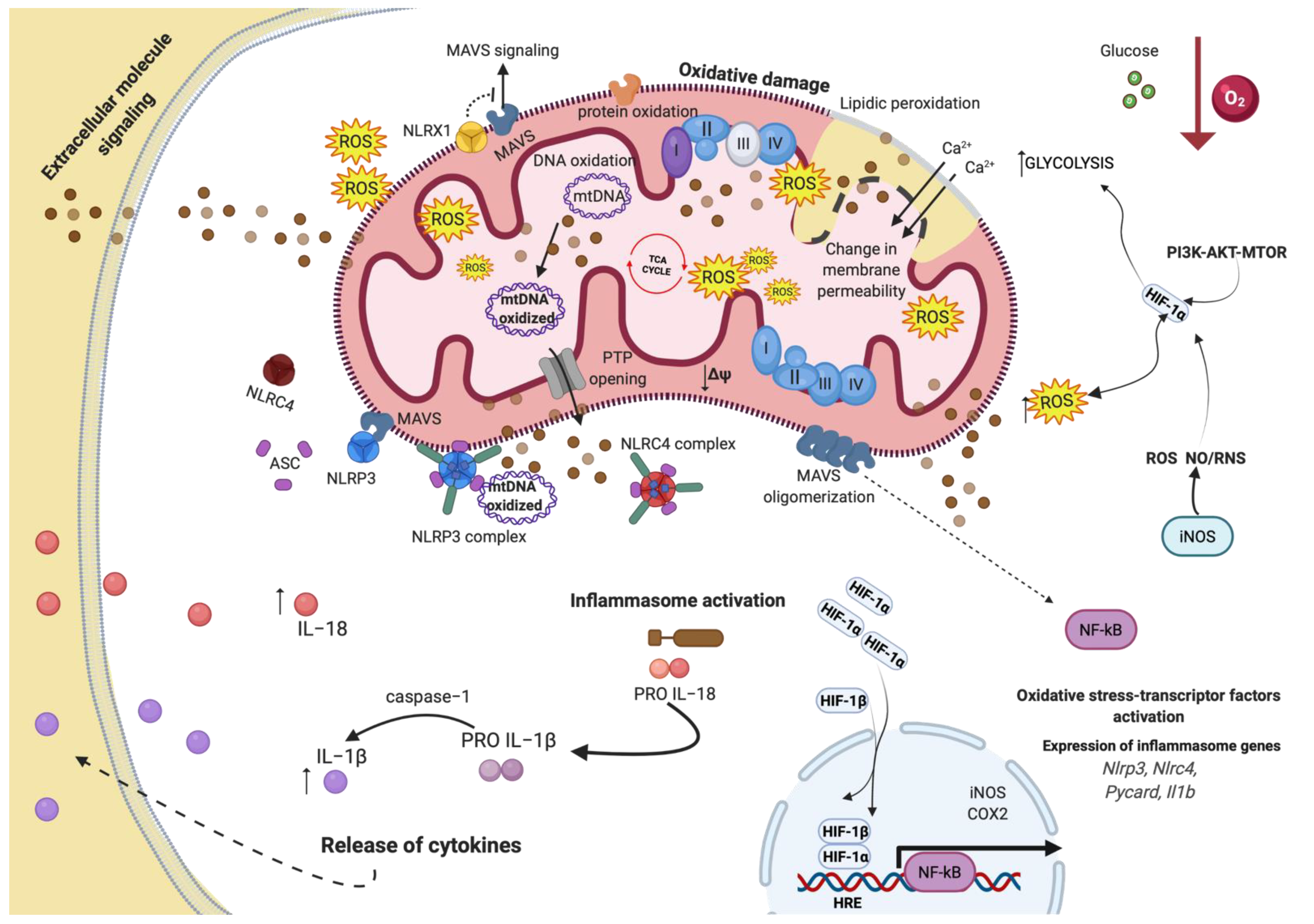{kind=link}
| Cardiovascular Disorders | In Vivo/In Vitro | Animal Models | Cell Lines | HIF-1α Effect | References |
|---|---|---|---|---|---|
| Ischemia-reperfusion injury (I/R injury) | In vitro | - | Rat neonatal ventricular cardiomyocytes cells | Cardioprotective effect, overexpression of HIF-1α elevated target genes (iNOS, VEGF, HSP70, and GLUT1-4) | [32] |
| In Vivo/in vitro | PHD3−/− mice | HL-1 cardiomyocytes | Cardioprotection, PHD3 deletion increased HIF-1α, resulted in cardiomyocytes death suppression | [30,31] | |
| In Vivo/in vitro | Sprague Dawley (SD)/rat model | H9C2 cardiomyoblasts | Cardioprotection, BNIP3-mediated autophagy modulation | [39] | |
| Myocardial infraction (MI) | In Vivo | Post-MI mice | - | Cardioprotection, upregulated angiogenesis | [57] |
| In vivo/in vitro | MI-mice | Rat neonatal cardiomyocytes | Detrimental, stimulated apoptosis through p53 following MI | [58] | |
| Heart failure (HF) | In vivo | HIF-1αtgmice | - | Detrimental, prolonged HIF-1α accumulation increased disease development | [28] |
| Myocarditis | In vitro | - | H9C2 cardiomyoblasts | Detrimental, repression of HIF-1α improved cardiomyocytes at odds with LPS-stimulated cell death | [29] |
| Dilated cardiomyopathy | In vivo | PHD2−/− mice | - | Detrimental, prolonged HIF-1α upregulation promoted dilated cardiomyopathy | [38] |
| Cyanotic congenital heart disease (CCHD) | In Vivo | CH rodent models | - | Cardioprotection, HIF-1α overexpression alleviated maladapted metabolic | [59] |
| Cardiac hypertrophy | In Vivo | HIF-1α KO mice | - | Cardioprotection by controlling negatively TGF-β | [60] |
| Cancer Disorders | In Vivo/In Vitro | Animal Models | Cell Lines | HIF-1α Effect | References |
|---|---|---|---|---|---|
| Human Breast ductal carcinoma | In vitro | - | MCF-7 cells | Inhibited ER Estrogen receptor expression | [62] |
| Renal carcinoma | In vitro | - | RCC4 and RCC10 | Increased mitochondrial biogenesis | [69] |
| Hepatocellular carcinoma (HCC) | In vivo | Mice and Diethylnitrosamine model of Murine HCC | HCC cell lines | Promoted mitochondrial biogenesis and reduced ATP | [70] |
| Triple negative breast cancer (TNBC) | In vitro/in vivo | Nude mice | MDA-MB-231 | Enhanced mitochondrial OXPHOS and elevated ROS generation | [71] |
| In vitro | - | MDA-MB-231 and SUM-149 cells | Increased intracellular glutathione levels | [72] | |
| In vivo | MMTV-PyMT mice | Primary MECs | Regulated mitochondrial mass | [73,74] | |
| Colorectal cancer | In vitro/in vivo | Oma1−/− mice | HCT116 cells | Increased mitochondrial ROS | [75] |
| Several human cancers | In vitro | - | A549, CCL39, HeLa, LS174, MCF7, PC3, ORL33, SKMel, and 786-O cells | Enlarged mitochondrial phenotype | [76] |
| Glioblastoma | In vitro/in vivo | Foxp3-YFP-CRE × HIF-1α -fl/fl mice | Murine glioma GL-261 | Promoted fatty acids oxidation for mitochondrial metabolism | [64] |
| Cancer cachexia (CC) | In Vitro/in vivo | C26 mice model | Colon-26 (C26) adenocarcinoma | Affected the metabolic changes | [77] |
| Oral cancer | In vitro | - | Oral squamous cell carcinoma (OSCC) | Stimulated migration and invasion in the indicated cells | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouhamida, E.; Morciano, G.; Perrone, M.; Kahsay, A.E.; Della Sala, M.; Wieckowski, M.R.; Fiorica, F.; Pinton, P.; Giorgi, C.; Patergnani, S. The Interplay of Hypoxia Signaling on Mitochondrial Dysfunction and Inflammation in Cardiovascular Diseases and Cancer: From Molecular Mechanisms to Therapeutic Approaches. Biology 2022, 11, 300. https://doi.org/10.3390/biology11020300
Bouhamida E, Morciano G, Perrone M, Kahsay AE, Della Sala M, Wieckowski MR, Fiorica F, Pinton P, Giorgi C, Patergnani S. The Interplay of Hypoxia Signaling on Mitochondrial Dysfunction and Inflammation in Cardiovascular Diseases and Cancer: From Molecular Mechanisms to Therapeutic Approaches. Biology. 2022; 11(2):300. https://doi.org/10.3390/biology11020300
Chicago/Turabian StyleBouhamida, Esmaa, Giampaolo Morciano, Mariasole Perrone, Asrat E. Kahsay, Mario Della Sala, Mariusz R. Wieckowski, Francesco Fiorica, Paolo Pinton, Carlotta Giorgi, and Simone Patergnani. 2022. "The Interplay of Hypoxia Signaling on Mitochondrial Dysfunction and Inflammation in Cardiovascular Diseases and Cancer: From Molecular Mechanisms to Therapeutic Approaches" Biology 11, no. 2: 300. https://doi.org/10.3390/biology11020300