
Understanding the Neurotrophic Virus Mechanisms and Their Potential Effect on Systemic Lupus Erythematosus Development
 ,
,
Abstract
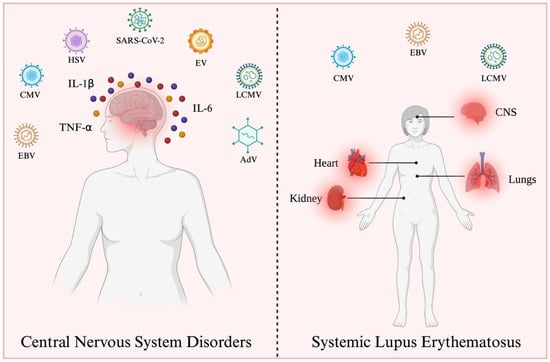
:
1. Introduction
2. Neurotrophic Viruses
2.1. Adenovirus
2.2. Cytomegalovirus
2.3. Enteroviruses
2.4. Epstein-Barr Virus
2.5. Herpes Simplex Virus
2.6. Lymphocytic Choriomeningitis Virus
2.7. Severe Acute Respiratory Syndrome Coronavirus-2
3. Viral Infections Could Trigger SLE Development
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.; Song, Y.; Kang, J.; Duan, S.; Li, Q.; Feng, F.; Duan, J. Epidemiology of Patients with Central Nervous System Infections, Mainly Neurosurgical Patients: A Retrospective Study from 2012 to 2019 in a Teaching Hospital in China. BMC Infect. Dis. 2021, 21, 826. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.G.L.; Boni, M.; Primo, C.C. The Invasive Behaviour of Shape Cryptococcus Neoformans: A Possibility of Direct Access to the Central Nervous System? Mycopathologia 1997, 140, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Medana, I.M.; Mai, N.T.H.; Day, N.P.J.; Hien, T.T.; Bethell, D.; Phu, N.H.; Farrar, J.; White, N.J.; Turner, G.D.H. Cellular Stress and Injury Responses in the Brains of Adult Vietnamese Patients with Fatal Plasmodium falciparum Malaria. Neuropathol. Appl. Neurobiol. 2001, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Itamura, S.; Nishimura, H.; Sato, Y.; Tashiro, M.; Hashikawa, T.; Kurata, T. Productive Infection in the Murine Central Nervous System with Avian Influenza Virus A (H5N1) after Intranasal Inoculation. Acta Neuropathol. 2004, 108, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Flexner, S. Contributions to the pathology of experimental virus encephalitis. J. Exp. Med. 1928, 47, 9–22. [Google Scholar] [CrossRef]
- McGill, F.; Griffiths, M.J.; Bonnett, L.J.; Geretti, A.M.; Michael, B.D.; Beeching, N.J.; McKee, D.; Scarlett, P.; Hart, I.J.; Mutton, K.J.; et al. Incidence, Aetiology, and Sequelae of Viral Meningitis in UK Adults: A Multicentre Prospective Observational Cohort Study. Lancet Infect. Dis. 2018, 18, 992–1003. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, S.; Wang, S.; Zheng, Y.; Wang, S.; Chen, H.; Pang, J.; Ma, J.; Yang, X.; Chen, Y. Global Magnitude of Encephalitis Burden and Its Evolving Pattern over the Past 30 Years. J. Infect. 2022, 84, 777–787. [Google Scholar] [CrossRef]
- Handley, G.; Pankow, S.; Bard, J.D.; Yee, R.; Nigo, M.; Hasbun, R. Distinguishing Cytomegalovirus Meningoencephalitis from Other Viral Central Nervous System Infections. J. Clin. Virol. 2021, 142, 104936. [Google Scholar] [CrossRef]
- Cheng, H.; Chen, D.; Peng, X.; Wu, P.; Jiang, L.; Hu, Y. Clinical Characteristics of Epstein–Barr Virus Infection in the Pediatric Nervous System. BMC Infect. Dis. 2020, 20, 886. [Google Scholar] [CrossRef]
- McBride, M.; Williman, J.; Best, E.; Walls, T.; Sadarangani, M.; Grant, C.C.; Martin, N.G. The Epidemiology of Aseptic Meningitis in New Zealand Children from 1991 to 2020. J Paediatr. Child Health 2022, 58, 1980–1989. [Google Scholar] [CrossRef]
- Mathew, S.; Al Khatib, H.A.; Al Ansari, K.; Nader, J.; Nasrallah, G.K.; Younes, N.N.; Coyle, P.V.; Al Thani, A.A.; Al Maslamani, M.A.; Yassine, H.M. Epidemiology Profile of Viral Meningitis Infections Among Patients in Qatar (2015–2018). Front. Med. 2021, 8, 663694. [Google Scholar] [CrossRef]
- Gonzalez-Quintial, R.; Mayeux, J.M.; Kono, D.H.; Theofilopoulos, A.N.; Pollard, K.M.; Baccala, R. Silica Exposure and Chronic Virus Infection Synergistically Promote Lupus-like Systemic Autoimmunity in Mice with Low Genetic Predisposition. Clin. Immunol. 2019, 205, 75–82. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.; Wu, C.; Zhang, Y. Epstein-Barr Virus and Human Herpesvirus 6 Infection in Patients with Systemic Lupus Erythematosus. Virol. J. 2023, 20, 29. [Google Scholar] [CrossRef]
- Pérez-Mercado, A.E.; Vilá-Pérez, S. Cytomegalovirus as a Trigger for Systemic Lupus Erythematosus. JCR J. Clin. Rheumatol. 2010, 16, 335–337. [Google Scholar] [CrossRef]
- Jiyad, Z.; Moriarty, B.; Creamer, D.; Higgins, E. Generalized Pustular Psoriasis Associated with Epstein-Barr Virus. Clin. Exp. Dermatol. 2015, 40, 146–148. [Google Scholar] [CrossRef]
- Laitinen, O.H.; Honkanen, H.; Pakkanen, O.; Oikarinen, S.; Hankaniemi, M.M.; Huhtala, H.; Ruokoranta, T.; Lecouturier, V.; André, P.; Harju, R.; et al. Coxsackievirus B1 Is Associated With Induction of β-Cell Autoimmunity That Portends Type 1 Diabetes. Diabetes 2014, 63, 446–455. [Google Scholar] [CrossRef]
- Masuoka, S.; Kusunoki, N.; Takamatsu, R.; Takahashi, H.; Tsuchiya, K.; Kawai, S.; Nanki, T. Epstein-Barr Virus Infection and Variants of Epstein-Barr Nuclear Antigen-1 in Synovial Tissues of Rheumatoid Arthritis. PLoS ONE 2018, 13, e0208957. [Google Scholar] [CrossRef]
- Rothe, K.; Quandt, D.; Schubert, K.; Rossol, M.; Klingner, M.; Jasinski-Bergner, S.; Scholz, R.; Seliger, B.; Pierer, M.; Baerwald, C.; et al. Latent Cytomegalovirus Infection in Rheumatoid Arthritis and Increased Frequencies of Cytolytic LIR-1+CD8+ T Cells. Arthritis Rheumatol. 2016, 68, 337–346. [Google Scholar] [CrossRef]
- Sanadgol, N.; Ramroodi, N.; Ahmadi, G.A.; Komijani, M.; Moghtaderi, A.; Bouzari, M.; Rezaei, M.; Kardi, M.T.; Dabiri, S.; Moradi, M.; et al. Prevalence of Cytomegalovirus Infection and Its Role in Total Immunoglobulin Pattern in Iranian Patients with Different Subtypes of Multiple Sclerosis. New Microbiol. 2011, 34, 263–274. [Google Scholar]
- Maple, P.A.C.; Tanasescu, R.; Gran, B.; Constantinescu, C.S. A Different Response to Cytomegalovirus (CMV) and Epstein–Barr Virus (EBV) Infection in UK People with Multiple Sclerosis (PwMS) Compared to Controls. J. Infect. 2020, 80, 320–325. [Google Scholar] [CrossRef]
- Zandman-Goddard, G.; Berkun, Y.; Barzilai, O.; Boaz, M.; Ram, M.; Anaya, J.; Shoenfeld, Y. Neuropsychiatric Lupus and Infectious Triggers. Lupus 2008, 17, 380–384. [Google Scholar] [CrossRef]
- Hammad, A.; Coles, A.; Gibson, M.D.J. 8. False Positive Mumps Serology in a Patient with Cerebral Systemic Lupus Erythematosus. Rheumatol. Adv. Pract. 2018, 2, rky033. [Google Scholar] [CrossRef]
- Turtzo, L.C.; Jikaria, N.; Cota, M.R.; Williford, J.P.; Uche, V.; Davis, T.; MacLaren, J.; Moses, A.D.; Parikh, G.; Castro, M.A.; et al. Meningeal Blood–Brain Barrier Disruption in Acute Traumatic Brain Injury. Brain Commun. 2020, 2, fcaa143. [Google Scholar] [CrossRef]
- Tumani, H.; Huss, A.; Bachhuber, F. The Cerebrospinal Fluid and Barriers—Anatomic and Physiologic Considerations. Handb. Clin. Neurol. 2018, 146, 21–32. [Google Scholar]
- Khan, M.; Clijsters, M.; Choi, S.; Backaert, W.; Claerhout, M.; Couvreur, F.; Van Breda, L.; Bourgeois, F.; Speleman, K.; Klein, S.; et al. Anatomical Barriers against SARS-CoV-2 Neuroinvasion at Vulnerable Interfaces Visualized in Deceased COVID-19 Patients. Neuron 2022, 110, 3919–3935.e6. [Google Scholar] [CrossRef]
- Moseman, E.A.; Blanchard, A.C.; Nayak, D.; McGavern, D.B. T Cell Engagement of Cross-Presenting Microglia Protects the Brain from a Nasal Virus Infection. Sci. Immunol. 2020, 5, eabb1817. [Google Scholar] [CrossRef]
- Schrauwen, E.J.A.; Herfst, S.; Leijten, L.M.; van Run, P.; Bestebroer, T.M.; Linster, M.; Bodewes, R.; Kreijtz, J.H.C.M.; Rimmelzwaan, G.F.; Osterhaus, A.D.M.E.; et al. The Multibasic Cleavage Site in H5N1 Virus Is Critical for Systemic Spread along the Olfactory and Hematogenous Routes in Ferrets. J. Virol. 2012, 86, 3975–3984. [Google Scholar] [CrossRef]
- Delbue, S.; Franciotta, D.; Giannella, S.; Dolci, M.; Signorini, L.; Ticozzi, R.; D’Alessandro, S.; Campisciano, G.; Comar, M.; Ferrante, P.; et al. Human Polyomaviruses in the Cerebrospinal Fluid of Neurological Patients. Microorganisms 2019, 8, 16. [Google Scholar] [CrossRef]
- Chacko, A.; Delbaz, A.; Choudhury, I.N.; Eindorf, T.; Shah, M.; Godfrey, C.; Sullivan, M.J.; St John, J.A.; Ulett, G.C.; Ekberg, J.A.K. Streptococcus Agalactiae Infects Glial Cells and Invades the Central Nervous System via the Olfactory and Trigeminal Nerves. Front. Cell Infect. Microbiol. 2022, 12, 188. [Google Scholar] [CrossRef]
- Bhat, N.R.; Fan, F. Adenovirus Infection Induces Microglial Activation: Involvement of Mitogen-Activated Protein Kinase Pathways. Brain Res. 2002, 948, 93–101. [Google Scholar] [CrossRef]
- Patra, P.; Rani, A.; Sharma, N.; Mukherjee, C.; Jha, H.C. Unraveling the Connection of Epstein–Barr Virus and Its Glycoprotein M 146–157 Peptide with Neurological Ailments. ACS Chem. Neurosci. 2023, 14, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Malik, J.R.; Acharya, A.; Avedissian, S.N.; Byrareddy, S.N.; Fletcher, C.V.; Podany, A.T.; Dyavar, S.R. ACE-2, TMPRSS2, and Neuropilin-1 Receptor Expression on Human Brain Astrocytes and Pericytes and SARS-CoV-2 Infection Kinetics. Int. J. Mol. Sci. 2023, 24, 8622. [Google Scholar] [CrossRef] [PubMed]
- Afrasiabi, A.; Keane, J.T.; Ong, L.T.C.; Alinejad-Rokny, H.; Fewings, N.L.; Booth, D.R.; Parnell, G.P.; Swaminathan, S. Genetic and Transcriptomic Analyses Support a Switch to Lytic Phase in Epstein Barr Virus Infection as an Important Driver in Developing Systemic Lupus Erythematosus. J. Autoimmun. 2022, 127, 102781. [Google Scholar] [CrossRef] [PubMed]
- Luethy, L.N.; Erickson, A.K.; Jesudhasan, P.R.; Ikizler, M.; Dermody, T.S.; Pfeiffer, J.K. Comparison of Three Neurotropic Viruses Reveals Differences in Viral Dissemination to the Central Nervous System. Virology 2016, 487, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tabor-Godwin, J.M.; Ruller, C.M.; Bagalso, N.; An, N.; Pagarigan, R.R.; Harkins, S.; Gilbert, P.E.; Kiosses, W.B.; Gude, N.A.; Cornell, C.T.; et al. A Novel Population of Myeloid Cells Responding to Coxsackievirus Infection Assists in the Dissemination of Virus within the Neonatal CNS. J. Neurosci. 2010, 30, 8676–8691. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus Infections in the Nervous System. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Girvin, A.M.; Miller, S.D. Direct Activation of Innate and Antigen-Presenting Functions of Microglia Following Infection with Theiler’s Virus. J. Virol. 2001, 75, 9780–9789. [Google Scholar] [CrossRef]
- Liu, P.; Wang, X.; Yang, Q.; Yan, X.; Fan, Y.; Zhang, S.; Wei, Y.; Huang, M.; Jiang, L.; Feng, L. Collaborative Action of Microglia and Astrocytes Mediates Neutrophil Recruitment to the CNS to Defend against Escherichia Coli K1 Infection. Int. J. Mol. Sci. 2022, 23, 6540. [Google Scholar] [CrossRef]
- Watanabe, M.; Nishikawaji, Y.; Kawakami, H.; Kosai, K. Adenovirus Biology, Recombinant Adenovirus, and Adenovirus Usage in Gene Therapy. Viruses 2021, 13, 2502. [Google Scholar] [CrossRef]
- Saban, S.D.; Silvestry, M.; Nemerow, G.R.; Stewart, P.L. Visualization of α-Helices in a 6-Ångstrom Resolution Cryoelectron Microscopy Structure of Adenovirus Allows Refinement of Capsid Protein Assignments. J. Virol. 2006, 80, 12049–12059. [Google Scholar] [CrossRef]
- Davison, A.J.; Benkő, M.; Harrach, B. Genetic Content and Evolution of Adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Wiethoff, C.M.; Stewart, P.L.; Nemerow, G.R. Adenovirus. In Current topics in microbiology and immunology; Springer: New York, NY, USA, 2010; Volume 343, pp. 195–224. [Google Scholar] [CrossRef]
- Rosa-Calatrava, M.; Grave, L.; Puvion-Dutilleul, F.; Chatton, B.; Kedinger, C. Functional Analysis of Adenovirus Protein IX Identifies Domains Involved in Capsid Stability, Transcriptional Activity, and Nuclear Reorganization. J. Virol. 2001, 75, 7131–7141. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.L.; Besser, E.S.; Nemerow, G.R. A Single Maturation Cleavage Site in Adenovirus Impacts Cell Entry and Capsid Assembly. J. Virol. 2016, 90, 521–532. [Google Scholar] [CrossRef] [PubMed]
- San Martín, C. Latest Insights on Adenovirus Structure and Assembly. Viruses 2012, 4, 847–877. [Google Scholar] [CrossRef] [PubMed]
- van Riel, D.; Verdijk, R.; Kuiken, T. The Olfactory Nerve: A Shortcut for Influenza and Other Viral Diseases into the Central Nervous System. J. Pathol. 2015, 235, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Baker, L.A.; Zhou, L.; Klein, R.S. Viral Interactions with the Blood-Brain Barrier: Old Dog, New Tricks. Tissue Barriers 2016, 4, e1142492. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-C.; Huang, S.-L.; Chen, S.-P.; Huang, Y.-L.; Huang, C.-G.; Tsao, K.-C.; Lin, T.-Y. Adenovirus Infection Associated with Central Nervous System Dysfunction in Children. J. Clin. Virol. 2013, 57, 300–304. [Google Scholar] [CrossRef]
- Fonseca, G.J.; Thillainadesan, G.; Yousef, A.F.; Ablack, J.N.; Mossman, K.L.; Torchia, J.; Mymryk, J.S. Adenovirus Evasion of Interferon-Mediated Innate Immunity by Direct Antagonism of a Cellular Histone Posttranslational Modification. Cell Host Microbe 2012, 11, 597–606. [Google Scholar] [CrossRef]
- Sohn, S.-Y.; Hearing, P. Adenovirus Sequesters Phosphorylated STAT1 at Viral Replication Centers and Inhibits STAT Dephosphorylation. J. Virol. 2011, 85, 7555–7562. [Google Scholar] [CrossRef]
- Lu, Q.; Yu, D.-H.; Fang, C.; Liu, F.; Ye, X.; Zhao, Y.; Qin, J.; Guo, X.-K.; Liang, M.; Hu, F.; et al. Influence of E3 Region on Conditionally Replicative Adenovirus Mediated Cytotoxicity in Hepatocellular Carcinoma Cells. Cancer Biol. Ther. 2009, 8, 1125–1132. [Google Scholar] [CrossRef]
- Sester, M.; Koebernick, K.; Owen, D.; Ao, M.; Bromberg, Y.; May, E.; Stock, E.; Andrews, L.; Groh, V.; Spies, T.; et al. Conserved Amino Acids within the Adenovirus 2 E3/19K Protein Differentially Affect Downregulation of MHC Class I and MICA/B Proteins. J. Immunol. 2010, 184, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.; Van Loock, M. Functional Annotation of Human Cytomegalovirus Gene Products: An Update. Front. Microbiol. 2014, 5, 218. [Google Scholar] [CrossRef]
- Zeng, J.; Cao, D.; Yang, S.; Jaijyan, D.K.; Liu, X.; Wu, S.; Cruz-Cosme, R.; Tang, Q.; Zhu, H. Insights into the Transcriptome of Human Cytomegalovirus: A Comprehensive Review. Viruses 2023, 15, 1703. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Kosugi, I.; Sakao-Suzuki, M.; Meguro, S.; Arai, Y.; Tsutsui, Y.; Iwashita, T. Cytomegalovirus Initiates Infection Selectively from High-Level Β1 Integrin–Expressing Cells in the Brain. Am. J. Pathol. 2015, 185, 1304–1323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Savitz, J. Effect of Cytomegalovirus Infection on the Central Nervous System: Implications for Psychiatric Disorders. In Microorganisms and Mental Health; Springer International Publishing: Cham, Switzerland, 2022; pp. 215–241. [Google Scholar]
- Sharma, S.; Mohan, A.; Smith-Rohrberg, D.; Sethu, M. Cytomegalovirus Polyradiculopathy: A Rare Neurological Manifestation of Acquired Immunodeficiency Syndrome. Neurol. India 2008, 56, 493. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, I.; Shinmura, Y.; Kawasaki, H.; Arai, Y.; Li, R.-Y.; Baba, S.; Tsutsui, Y. Cytomegalovirus Infection of the Central Nervous System Stem Cells from Mouse Embryo: A Model for Developmental Brain Disorders Induced by Cytomegalovirus. Lab. Investig. 2000, 80, 1373–1383. [Google Scholar] [CrossRef]
- Grigoleit, U.; Riegler, S.; Einsele, H.; Laib Sampaio, K.; Jahn, G.; Hebart, H.; Brossart, P.; Frank, F.; Sinzger, C. Human Cytomegalovirus Induces a Direct Inhibitory Effect on Antigen Presentation by Monocyte-Derived Immature Dendritic Cells. Br. J. Haematol. 2002, 119, 189–198. [Google Scholar] [CrossRef]
- Baasch, S.; Giansanti, P.; Kolter, J.; Riedl, A.; Forde, A.J.; Runge, S.; Zenke, S.; Elling, R.; Halenius, A.; Brabletz, S.; et al. Cytomegalovirus Subverts Macrophage Identity. Cell 2021, 184, 3774–3793.e25. [Google Scholar] [CrossRef]
- LoPiccolo, D.M.; Gold, M.C.; Kavanagh, D.G.; Wagner, M.; Koszinowski, U.H.; Hill, A.B. Effective Inhibition of Kb- and Db-Restricted Antigen Presentation in Primary Macrophages by Murine Cytomegalovirus. J. Virol. 2003, 77, 301–308. [Google Scholar] [CrossRef]
- Baicus, A.; Baicus, C. Neuronal Infections; Singh, S.K., Ruzek, D., Eds.; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C Protein of Enterovirus 71 Inhibits Retinoid Acid-Inducible Gene I-Mediated Interferon Regulatory Factor 3 Activation and Type I Interferon Responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef]
- Coyne, C.B.; Bozym, R.; Morosky, S.A.; Hanna, S.L.; Mukherjee, A.; Tudor, M.; Kim, K.S.; Cherry, S. Comparative RNAi Screening Reveals Host Factors Involved in Enterovirus Infection of Polarized Endothelial Monolayers. Cell Host Microbe 2011, 9, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, J.; Wang, N.; Sun, Z.; Ma, Q.; Li, J.; Zhang, M.; Xu, J. Enterovirus A71 Capsid Protein VP1 Increases Blood–Brain Barrier Permeability and Virus Receptor Vimentin on the Brain Endothelial Cells. J. Neurovirol. 2020, 26, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.Q.; Ng, Q.Y.; Oo, Y.; Chu, J.J.H.; Ng, S.Y.; Sze, S.K.; Alonso, S. Enterovirus-A71 Exploits Peripherin and Rac1 to Invade the Central Nervous System. EMBO Rep. 2021, 22, e51777. [Google Scholar] [CrossRef] [PubMed]
- Piralla, A.; Pellegrinelli, L.; Giardina, F.; Galli, C.; Binda, S.; Pariani, E.; Baldanti, F. Contribution of Enteroviruses to Acute Central Nervous System or Systemic Infections in Northern Italy (2015–2017): Is It Time to Establish a National Laboratory-Based Surveillance System? BioMed Res. Int. 2020, 2020, 9393264. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.-H.; Chow, Y.-H.; Chung, N.-H.; Chen, T.-C.; Shie, F.-S.; Juang, J.-L. Neurotropic EV71 Causes Encephalitis by Engaging Intracellular TLR9 to Elicit Neurotoxic IL12-P40-INOS Signaling. Cell Death Dis. 2022, 13, 328. [Google Scholar] [CrossRef]
- de Ceano-Vivas, M.; García, M.L.; Velázquez, A.; Martín del Valle, F.; Menasalvas, A.; Cilla, A.; Epalza, C.; Romero, M.P.; Cabrerizo, M.; Calvo, C. Neurodevelopmental Outcomes of Infants Younger Than 90 Days Old Following Enterovirus and Parechovirus Infections of the Central Nervous System. Front. Pediatr. 2021, 9, 719119. [Google Scholar] [CrossRef]
- Chang, L.-Y.; Huang, L.-M.; Gau, S.S.-F.; Wu, Y.-Y.; Hsia, S.-H.; Fan, T.-Y.; Lin, K.-L.; Huang, Y.-C.; Lu, C.-Y.; Lin, T.-Y. Neurodevelopment and Cognition in Children after Enterovirus 71 Infection. N. Engl. J. Med. 2007, 356, 1226–1234. [Google Scholar] [CrossRef]
- Tseng, J.-J.; Lin, C.-H.; Lin, M.-C. Long-Term Outcomes of Pediatric Enterovirus Infection in Taiwan: A Population-Based Cohort Study. Front. Pediatr. 2020, 8, 285. [Google Scholar] [CrossRef]
- Wang, H.; Lei, X.; Xiao, X.; Yang, C.; Lu, W.; Huang, Z.; Leng, Q.; Jin, Q.; He, B.; Meng, G.; et al. Reciprocal Regulation between Enterovirus 71 and the NLRP3 Inflammasome. Cell Rep. 2015, 12, 42–48. [Google Scholar] [CrossRef]
- Xiang, Z.; Liu, L.; Lei, X.; Zhou, Z.; He, B.; Wang, J. 3C Protease of Enterovirus D68 Inhibits Cellular Defense Mediated by Interferon Regulatory Factor 7. J. Virol. 2016, 90, 1613–1621. [Google Scholar] [CrossRef]
- Kang, J.; Pang, Z.; Zhou, Z.; Li, X.; Liu, S.; Cheng, J.; Liu, P.; Tan, W.; Wang, Z.; Wang, T. Enterovirus D68 Protease 2A pro Targets TRAF3 To Subvert Host Innate Immune Responses. J. Virol. 2021, 95, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S. Epstein–Barr Virus: General Features. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 148–157. [Google Scholar]
- Indari, O.; Chandramohanadas, R.; Jha, H.C. Epstein–Barr Virus Infection Modulates Blood–Brain Barrier Cells and Its Co-Infection with Plasmodium falciparum Induces RBC Adhesion. Pathog. Dis. 2021, 79, ftaa080. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.V.; Cox, B.; Lafuse, W.P.; Ariza, M.E. Epstein-Barr Virus DUTPase Induces Neuroinflammatory Mediators: Implications for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Clin. Ther. 2019, 41, 848–863. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.A.; English, S.W.; Fugate, J.E.; Tosh, P.K. Acute Epstein-Barr Virus Infection Presenting as Guillain-Barre Syndrome. IDCases 2021, 25, e01196. [Google Scholar] [CrossRef] [PubMed]
- Singhi, P.; Sharma, J.P.; Gautam, R.; Indra, R.M.; Rafli, A. Extensive Longitudinal Transverse Myelitis Associated with CSF Epstein-Barr Virus Infection: A Case Report. Child. Neurol. Open 2021, 8, 2329048X2110499. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, D.; Singh, V.K.; Baral, B.; Pathak, D.K.; Jayabalan, J.; Kumar, R.; Tapryal, S.; Jha, H.C. Indication of Neurodegenerative Cascade Initiation by Amyloid-like Aggregate-Forming EBV Proteins and Peptide in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 3957–3967. [Google Scholar] [CrossRef]
- Woulfe, J.M.; Gray, M.T.; Gray, D.A.; Munoz, D.G.; Middeldorp, J.M. Hypothesis: A Role for EBV-Induced Molecular Mimicry in Parkinson’s Disease. Park. Relat. Disord. 2014, 20, 685–694. [Google Scholar] [CrossRef]
- Yiu, S.P.T.; Zerbe, C.; Vanderwall, D.; Huttlin, E.L.; Weekes, M.P.; Gewurz, B.E. An Epstein-Barr Virus Protein Interaction Map Reveals NLRP3 Inflammasome Evasion via MAVS UFMylation. Mol. Cell 2023, 83, 2367–2386.e15. [Google Scholar] [CrossRef]
- Liu, X.; Sadaoka, T.; Krogmann, T.; Cohen, J.I. Epstein-Barr Virus (EBV) Tegument Protein BGLF2 Suppresses Type I Interferon Signaling To Promote EBV Reactivation. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Wang, P.; Deng, Y.; Guo, Y.; Xu, Z.; Li, Y.; Ou, X.; Xie, L.; Lu, M.; Zhong, J.; Li, B.; et al. Epstein-Barr Virus Early Protein BFRF1 Suppresses IFN-β Activity by Inhibiting the Activation of IRF3. Front. Immunol. 2020, 11, 513383. [Google Scholar] [CrossRef]
- Li, L.; Liu, D.; Hutt-Fletcher, L.; Morgan, A.; Masucci, M.G.; Levitsky, V. Epstein-Barr Virus Inhibits the Development of Dendritic Cells by Promoting Apoptosis of Their Monocyte Precursors in the Presence of Granulocyte Macrophage–Colony-Stimulating Factor and Interleukin-4. Blood 2002, 99, 3725–3734. [Google Scholar] [CrossRef]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein–Barr Virus Reprograms Human B Lymphocytes Immediately in the Prelatent Phase of Infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed]
- Megyola, C.; Ye, J.; Bhaduri-McIntosh, S. Identification of a Sub-Population of B Cells That Proliferates after Infection with Epstein-Barr Virus. Virol. J. 2011, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Lu, J.; Pei, Y.; Robertson, E.S. Transcriptome Reprogramming of Epstein-Barr Virus Infected Epithelial and B Cells Reveals Distinct Host-Virus Interaction Profiles. Cell Death Dis. 2022, 13, 894. [Google Scholar] [CrossRef] [PubMed]
- Lehman, I.R.; Boehmer, P.E. Replication of Herpes Simplex Virus DNA. J. Biol. Chem. 1999, 274, 28059–28062. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, J.M.; Lazear, H.M.; Awasthi, S.; Wang, F.; Friedman, H.M. The Herpes Simplex Virus 1 IgG Fc Receptor Blocks Antibody-Mediated Complement Activation and Antibody-Dependent Cellular Cytotoxicity In Vivo. J. Virol. 2011, 85, 3239–3249. [Google Scholar] [CrossRef]
- Komala Sari, T.; Gianopulos, K.A.; Nicola, A.V. Glycoprotein C of Herpes Simplex Virus 1 Shields Glycoprotein B from Antibody Neutralization. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Andreu, S.; López-Guerrero, J.A. The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 5026. [Google Scholar] [CrossRef]
- Kopp, S.J.; Ranaivo, H.R.; Wilcox, D.R.; Karaba, A.H.; Wainwright, M.S.; Muller, W.J. Herpes Simplex Virus Serotype and Entry Receptor Availability Alter CNS Disease in a Mouse Model of Neonatal HSV. Pediatr. Res. 2014, 76, 528–534. [Google Scholar] [CrossRef]
- He, Q.; Liu, H.; Huang, C.; Wang, R.; Luo, M.; Lu, W. Herpes Simplex Virus 1-Induced Blood-Brain Barrier Damage Involves Apoptosis Associated With GM130-Mediated Golgi Stress. Front. Mol. Neurosci. 2020, 13, 2. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, K.; He, Q.; Lei, Q.; Lu, W. Mechanisms of Blood-Brain Barrier Disruption in Herpes Simplex Encephalitis. J. Neuroimmune Pharmacol. 2019, 14, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Rozenberg, F. Herpes Simplex Virus and Central Nervous System Infections: Encephalitis, Meningitis, Myelitis. Virologie 2020, 24, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.J.; Fani, L.; Ikram, M.K.; Ghanbari, M.; Ikram, M.A. Herpes Simplex Virus 1 and the Risk of Dementia: A Population-Based Study. Sci. Rep. 2021, 11, 8691. [Google Scholar] [CrossRef]
- Ma, Y.; He, B. Recognition of Herpes Simplex Viruses: Toll-Like Receptors and Beyond. J. Mol. Biol. 2014, 426, 1133–1147. [Google Scholar] [CrossRef]
- Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes Simplex Virus and Toll-Like Receptors. Viral Immunol. 2005, 18, 457–465. [Google Scholar] [CrossRef]
- Stefanidou, M.; Ramos, I.; Mas Casullo, V.; Trépanier, J.B.; Rosenbaum, S.; Fernandez-Sesma, A.; Herold, B.C. Herpes Simplex Virus 2 (HSV-2) Prevents Dendritic Cell Maturation, Induces Apoptosis, and Triggers Release of Proinflammatory Cytokines: Potential Links to HSV-HIV Synergy. J. Virol. 2013, 87, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- Smiley, J.R. Herpes Simplex Virus Virion Host Shutoff Protein: Immune Evasion Mediated by a Viral RNase? J. Virol. 2004, 78, 1063–1068. [Google Scholar] [CrossRef]
- Koppers-Lalic, D.; Rijsewijk, F.A.M.; Verschuren, S.B.E.; van Gaans-van den Brink, J.A.M.; Neisig, A.; Ressing, M.E.; Neefjes, J.; Wiertz, E.J.H.J. The UL41-Encoded Virion Host Shutoff (Vhs) Protein and Vhs-Independent Mechanisms Are Responsible for down-Regulation of MHC Class I Molecules by Bovine Herpesvirus 1. J. Gen. Virol. 2001, 82, 2071–2081. [Google Scholar] [CrossRef]
- Tigges, M.A.; Leng, S.; Johnson, D.C.; Burke, R.L. Human Herpes Simplex Virus (HSV)-Specific CD8+ CTL Clones Recognize HSV-2-Infected Fibroblasts after Treatment with IFN-Gamma or When Virion Host Shutoff Functions Are Disabled. J. Immunol. 1996, 156, 3901–3910. [Google Scholar] [CrossRef]
- Koyanagi, N.; Kawaguchi, Y. Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses. Viruses 2020, 12, 1354. [Google Scholar] [CrossRef]
- Buchmeier, M.J.; de la Torre, J.C.; Peters, C.J. Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Kang, S.S.; McGavern, D.B. Lymphocytic Choriomeningitis Infection of the Central Nervous System. Front. Biosci. 2008, 4529, 4529. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.R.; Hsu, T.-H. Viral Disruption of the Blood–Brain Barrier. Trends Microbiol. 2012, 20, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D.B. Myelomonocytic Cell Recruitment Causes Fatal CNS Vascular Injury during Acute Viral Meningitis. Nature 2009, 457, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Matullo, C.M.; O’Regan, K.J.; Hensley, H.; Curtis, M.; Rall, G.F. Lymphocytic Choriomeningitis Virus-Induced Mortality in Mice Is Triggered by Edema and Brain Herniation. J. Virol. 2010, 84, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.L.; Mets, M.B.; Beauchamp, C.L. Lymphocytic Choriomeningitis Virus: Emerging Fetal Teratogen. Am. J. Obs. Gynecol. 2002, 187, 1715–1716. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Singhapakdi, K.; Maertens, P. Echoencephalography of Encephalopathy Due to Congenital Lymphocytic Choriomeningitis Virus. J. Neuroimaging 2022, 32, 412–419. [Google Scholar] [CrossRef]
- Wright, R.; Johnson, D.; Neumann, M.; Ksiazek, T.G.; Rollin, P.; Keech, R.V.; Bonthius, D.J.; Hitchon, P.; Grose, C.F.; Bell, W.E.; et al. Congenital Lymphocytic Choriomeningitis Virus Syndrome: A Disease That Mimics Congenital Toxoplasmosis or Cytomegalovirus Infection. Pediatrics 1997, 100, e9. [Google Scholar] [CrossRef]
- Pythoud, C.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Lymphocytic Choriomeningitis Virus Differentially Affects the Virus-Induced Type I Interferon Response and Mitochondrial Apoptosis Mediated by RIG-I/MAVS. J. Virol. 2015, 89, 6240–6250. [Google Scholar] [CrossRef]
- Pythoud, C.; Rodrigo, W.W.S.I.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus Nucleoprotein Targets Interferon Regulatory Factor-Activating Kinase IKKε. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Pellett, P.E.; Mitra, S.; Holland, T.C. Basics of Virology. In Handbook of Clinical Neurology; Academic Press: Cambridge, MA, USA, 2014; pp. 45–66. [Google Scholar]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z.; et al. SARS-CoV-2 Nsp12 Attenuates Type I Interferon Production by Inhibiting IRF3 Nuclear Translocation. Cell Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Arshad, N.; Laurent-Rolle, M.; Ahmed, W.S.; Hsu, J.C.-C.; Mitchell, S.M.; Pawlak, J.; Sengupta, D.; Biswas, K.H.; Cresswell, P. SARS-CoV-2 Accessory Proteins ORF7a and ORF3a Use Distinct Mechanisms to down-Regulate MHC-I Surface Expression. Proc. Natl. Acad. Sci. USA 2023, 120, e2208525120. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 Suppresses the Antiviral Innate Immune Response by Degrading MAVS through Mitophagy. Cell Mol. Immunol. 2022, 19, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q.; et al. SARS-CoV-2 Crosses the Blood–Brain Barrier Accompanied with Basement Membrane Disruption without Tight Junctions Alteration. Signal Transduct. Target. Ther. 2021, 6, 337. [Google Scholar] [CrossRef] [PubMed]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 Spike Protein Alters Barrier Function in 2D Static and 3D Microfluidic in-Vitro Models of the Human Blood–Brain Barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Vitale-Cross, L.; Szalayova, I.; Scoggins, A.; Palkovits, M.; Mezey, E. SARS-CoV-2 Entry Sites Are Present in All Structural Elements of the Human Glossopharyngeal and Vagal Nerves: Clinical Implications. EBioMedicine 2022, 78, 103981. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Mayilsamy, K.; McGill, A.R.; Martinez, T.E.; Chandran, B.; Blair, L.J.; Bickford, P.C.; Mohapatra, S.S.; Mohapatra, S. SARS-CoV-2 Infection Increases the Gene Expression Profile for Alzheimer’s Disease Risk. Mol. Ther. Methods Clin. Dev. 2022, 27, 217–229. [Google Scholar] [CrossRef]
- Vossler, D.G. Does SARS-CoV-2 Cause Seizures and Epilepsy in COVID-19 via Inflammation or by Direct Infection? Epilepsy Curr. 2023, 23, 153–155. [Google Scholar] [CrossRef]
- Zhang, P.-P.; He, Z.-C.; Yao, X.-H.; Tang, R.; Ma, J.; Luo, T.; Zhu, C.; Li, T.-R.; Liu, X.; Zhang, D.; et al. COVID-19-Associated Monocytic Encephalitis (CAME): Histological and Proteomic Evidence from Autopsy. Signal Transduct. Target. Ther. 2023, 8, 24. [Google Scholar] [CrossRef]
- Abenza Abildúa, M.J.; Atienza, S.; Carvalho Monteiro, G.; Erro Aguirre, M.E.; Imaz Aguayo, L.; Freire Álvarez, E.; García-Azorín, D.; Gil-Olarte Montesinos, I.; Lara Lezama, L.B.; Navarro Pérez, M.P.; et al. Encephalopathy and Encephalitis during Acute SARS-CoV-2 Infection. Spanish Society of Neurology’s COVID-19 Registry. Neurología 2021, 36, 127–134. [Google Scholar] [CrossRef]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.-W.; Xu, Y.; Zhang, J.; Nan, M.-L.; Xiao, Y.; Zhan, P.; Liu, X.; et al. SARS-CoV-2 NSP5 and N Protein Counteract the RIG-I Signaling Pathway by Suppressing the Formation of Stress Granules. Signal Transduct. Target. Ther. 2022, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV Nucleocapsid Protein Antagonizes IFN-β Response by Targeting Initial Step of IFN-β Induction Pathway, and Its C-Terminal Region Is Critical for the Antagonism. Virus Genes. 2011, 42, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Z.; Guo, J.; Xu, S.; Zhou, J.; Chen, Q.; Tong, X.; Wang, D.; Peng, G.; Fang, L.; et al. SARS-CoV-2 Nsp5 Exhibits Stronger Catalytic Activity and Interferon Antagonism than Its SARS-CoV Ortholog. J. Virol. 2022, 96, e00037-22. [Google Scholar] [CrossRef] [PubMed]
- López-Muñoz, A.D.; Kosik, I.; Holly, J.; Yewdell, J.W. Cell Surface SARS-CoV-2 Nucleocapsid Protein Modulates Innate and Adaptive Immunity. Sci. Adv. 2022, 8, eabp9770. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.; Kajon, A. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-F.; Tan, C.-B.; Yao, Z.-X.; Jiang, L.; Hong, S.-Q. Adenovirus Infection-Associated Central Nervous System Disease in Children. Pediatr. Infect. Dis. J. 2021, 40, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, K.L.; Richardson, S.E.; MacGregor, D.; Mahant, S.; Raghuram, K.; Bitnun, A. Adenovirus-Associated Central Nervous System Disease in Children. J. Pediatr. 2019, 205, 130–137. [Google Scholar] [CrossRef]
- Yamada, M.; Nakamura, K.; Yoshii, M.; Kaku, Y.; Narita, M. Brain Lesions Induced by Experimental Intranasal Infection of Japanese Encephalitis Virus in Piglets. J. Comp. Pathol. 2009, 141, 156–162. [Google Scholar] [CrossRef]
- Ginsberg, H.S.; Moldawer, L.L.; Sehgal, P.B.; Redington, M.; Kilian, P.L.; Chanock, R.M.; Prince, G.A. A Mouse Model for Investigating the Molecular Pathogenesis of Adenovirus Pneumonia. Proc. Natl. Acad. Sci. USA 1991, 88, 1651–1655. [Google Scholar] [CrossRef]
- Carlin, C.R. New Insights to Adenovirus-Directed Innate Immunity in Respiratory Epithelial Cells. Microorganisms 2019, 7, 216. [Google Scholar] [CrossRef]
- Kotha, P.L.N.; Sharma, P.; Kolawole, A.O.; Yan, R.; Alghamri, M.S.; Brockman, T.L.; Gomez-Cambronero, J.; Excoffon, K.J.D.A. Adenovirus Entry From the Apical Surface of Polarized Epithelia Is Facilitated by the Host Innate Immune Response. PLoS Pathog. 2015, 11, e1004696. [Google Scholar] [CrossRef] [PubMed]
- Zemke, N.R.; Berk, A.J. The Adenovirus E1A C Terminus Suppresses a Delayed Antiviral Response and Modulates RAS Signaling. Cell Host Microbe 2017, 22, 789–800.e5. [Google Scholar] [CrossRef] [PubMed]
- Yunis, J.; Farrell, H.E.; Bruce, K.; Lawler, C.; Wyer, O.; Davis-Poynter, N.; Brizić, I.; Jonjić, S.; Adler, B.; Stevenson, P.G. Murine Cytomegalovirus Glycoprotein O Promotes Epithelial Cell Infection In Vivo. J. Virol. 2019, 93, e01378-18. [Google Scholar] [CrossRef] [PubMed]
- Lopper, M.; Compton, T. Coiled-Coil Domains in Glycoproteins B and H Are Involved in Human Cytomegalovirus Membrane Fusion. J. Virol. 2004, 78, 8333–8341. [Google Scholar] [CrossRef] [PubMed]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, Novel MHC Class I–Related Molecules, Bind to CMV Glycoprotein UL16 and Stimulate NK Cytotoxicity through the NKG2D Receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; Pati, P.; Jensen, T.L.; Goll, J.B.; Gelber, C.E.; Singh, A.; McNeal, M.; Boppana, S.B.; Bernstein, D.I. Cytomegalovirus Genetic Diversity Following Primary Infection. J. Infect. Dis. 2020, 221, 715–720. [Google Scholar] [CrossRef]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional Profiling of a Human Cytomegalovirus Genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the Worldwide Seroprevalence of Cytomegalovirus: A Systematic Review and Meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef]
- Anduze-Faris, B.M.; Fillet, A.-M.; Gozlan, J.; Lancar, R.; Boukli, N.; Gasnault, J.; Caumes, E.; Livartowsky, J.; Matheron, S.; Leport, C.; et al. Induction and Maintenance Therapy of Cytomegalovirus Central Nervous System Infection in HIV-Infected Patients. AIDS 2000, 14, 517–524. [Google Scholar] [CrossRef]
- Krstanović, F.; Britt, W.J.; Jonjić, S.; Brizić, I. Cytomegalovirus Infection and Inflammation in Developing Brain. Viruses 2021, 13, 1078. [Google Scholar] [CrossRef]
- Slavuljica, I.; Kveštak, D.; Csaba Huszthy, P.; Kosmac, K.; Britt, W.J.; Jonjić, S. Immunobiology of Congenital Cytomegalovirus Infection of the Central Nervous System—The Murine Cytomegalovirus Model. Cell Mol. Immunol. 2015, 12, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Singh, J.; Perry, S.T.; Compton, T. Human Cytomegalovirus Elicits a Coordinated Cellular Antiviral Response via Envelope Glycoprotein B. J. Virol. 2004, 78, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Yurochko, A.D.; Hwang, E.S.; Rasmussen, L.; Keay, S.; Pereira, L.; Huang, E.S. The Human Cytomegalovirus UL55 (GB) and UL75 (GH) Glycoprotein Ligands Initiate the Rapid Activation of Sp1 and NF-KappaB during Infection. J. Virol. 1997, 71, 5051–5059. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.; Abendroth, A.; McSharry, B.; Slobedman, B. Interferon-Independent Upregulation of Interferon-Stimulated Genes during Human Cytomegalovirus Infection Is Dependent on IRF3 Expression. Viruses 2019, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Billstrom Schroeder, M.; Worthen, G.S. Viral Regulation of RANTES Expression during Human Cytomegalovirus Infection of Endothelial Cells. J. Virol. 2001, 75, 3383–3390. [Google Scholar] [CrossRef]
- Browne, E.P.; Wing, B.; Coleman, D.; Shenk, T. Altered Cellular MRNA Levels in Human Cytomegalovirus-Infected Fibroblasts: Viral Block to the Accumulation of Antiviral MRNAs. J. Virol. 2001, 75, 12319–12330. [Google Scholar] [CrossRef] [PubMed]
- Simmen, K.A.; Singh, J.; Luukkonen, B.G.M.; Lopper, M.; Bittner, A.; Miller, N.E.; Jackson, M.R.; Compton, T.; Früh, K. Global Modulation of Cellular Transcription by Human Cytomegalovirus Is Initiated by Viral Glycoprotein B. Proc. Natl. Acad. Sci. USA 2001, 98, 7140–7145. [Google Scholar] [CrossRef]
- Maxim C-J Cheeran, S.H.S. Cytomegalovirus Induces Cytokine and Chemokine Production Differentially in Microglia and Astrocytes: Antiviral Implications. J. Neurovirol 2001, 7, 135–147. [Google Scholar] [CrossRef]
- Füzik, T.; Moravcová, J.; Kalynych, S.; Plevka, P. Structure of Human Enterovirus 70 and Its Inhibition by Capsid-Binding Compounds. J. Virol. 2022, 96, e00604-22. [Google Scholar] [CrossRef]
- Lee, K.-M.; Wu, C.-C.; Wu, S.-E.; Lin, Y.-H.; Wang, L.-T.; Chang, C.-R.; Huang, P.-N.; Shih, S.-R.; Kuo, R.-L. The RNA-Dependent RNA Polymerase of Enterovirus A71 Associates with Ribosomal Proteins and Positively Regulates Protein Translation. RNA Biol. 2020, 17, 608–622. [Google Scholar] [CrossRef]
- Xiao, X.; Lei, X.; Zhang, Z.; Ma, Y.; Qi, J.; Wu, C.; Xiao, Y.; Li, L.; He, B.; Wang, J. Enterovirus 3A Facilitates Viral Replication by Promoting Phosphatidylinositol 4-Kinase IIIβ–ACBD3 Interaction. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Visser, L.J.; Langereis, M.A.; Rabouw, H.H.; Wahedi, M.; Muntjewerff, E.M.; de Groot, R.J.; van Kuppeveld, F.J.M. Essential Role of Enterovirus 2A Protease in Counteracting Stress Granule Formation and the Induction of Type I Interferon. J. Virol. 2019, 93, e00222-19. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, L.; Moreni, G.; Wolthers, K.C.; Pajkrt, D. World-Wide Prevalence and Genotype Distribution of Enteroviruses. Viruses 2021, 13, 434. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-S.; Lee, H.-C.; Lee, K.-M.; Gong, Y.-N.; Shih, S.-R. Enterovirus and Encephalitis. Front. Microbiol. 2020, 11, 261. [Google Scholar] [CrossRef]
- Feng, M.; Guo, S.; Fan, S.; Zeng, X.; Zhang, Y.; Liao, Y.; Wang, J.; Zhao, T.; Wang, L.; Che, Y.; et al. The Preferential Infection of Astrocytes by Enterovirus 71 Plays a Key Role in the Viral Neurogenic Pathogenesis. Front. Cell Infect. Microbiol. 2016, 6, 192. [Google Scholar] [CrossRef]
- Poelaert, K.C.K.; van Kleef, R.G.D.M.; Liu, M.; van Vliet, A.; Lyoo, H.; Gerber, L.-S.; Narimatsu, Y.; Büll, C.; Clausen, H.; de Vries, E.; et al. Enterovirus D-68 Infection of Primary Rat Cortical Neurons: Entry, Replication, and Functional Consequences. mBio 2023, 14, e00245-23. [Google Scholar] [CrossRef]
- Ye, N.; Gong, X.; Pang, L.; Gao, W.; Zhang, Y.; Li, X.; Liu, N.; Li, D.; Jin, Y.; Duan, Z. Cytokine Responses and Correlations Thereof with Clinical Profiles in Children with Enterovirus 71 Infections. BMC Infect. Dis. 2015, 15, 225. [Google Scholar] [CrossRef]
- Gong, X.; Zhou, J.; Zhu, W.; Liu, N.; Li, J.; Li, L.; Jin, Y.; Duan, Z. Excessive Proinflammatory Cytokine and Chemokine Responses of Human Monocyte-Derived Macrophages to Enterovirus 71 Infection. BMC Infect. Dis. 2012, 12, 224. [Google Scholar] [CrossRef]
- Chang, C.; Li, J.; Ou, Y.; Chen, W.; Liao, S.; Raung, S.; Hsiao, A.; Chen, C. Enterovirus 71 Infection Caused Neuronal Cell Death and Cytokine Expression in Cultured Rat Neural Cells. IUBMB Life 2015, 67, 789–800. [Google Scholar] [CrossRef]
- Sugimoto, A.; Yamashita, Y.; Kanda, T.; Murata, T.; Tsurumi, T. Epstein-Barr Virus Genome Packaging Factors Accumulate in BMRF1-Cores within Viral Replication Compartments. PLoS ONE 2019, 14, e0222519. [Google Scholar] [CrossRef]
- Henson, B.W.; Perkins, E.M.; Cothran, J.E.; Desai, P. Self-Assembly of Epstein-Barr Virus Capsids. J. Virol. 2009, 83, 3877–3890. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.H.J.; Rowe, M. The Epstein-Barr Virus G-Protein-Coupled Receptor Contributes to Immune Evasion by Targeting MHC Class I Molecules for Degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef] [PubMed]
- Walston, J.J.; Hayman, I.R.; Gore, M.; Ferguson, M.; Temple, R.M.; Liao, J.; Alam, S.; Meyers, C.; Tugizov, S.M.; Hutt-Fletcher, L.; et al. The Epstein-Barr Virus Glycoprotein BDLF2 Is Essential for Efficient Viral Spread in Stratified Epithelium. J. Virol. 2023, 97, e01528-22. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.; Jardetzky, T.S.; Longnecker, R. The Epstein-Barr Virus (EBV) Glycoprotein B Cytoplasmic C-Terminal Tail Domain Regulates the Energy Requirement for EBV-Induced Membrane Fusion. J. Virol. 2014, 88, 11686–11695. [Google Scholar] [CrossRef]
- Sathiyamoorthy, K.; Hu, Y.X.; Möhl, B.S.; Chen, J.; Longnecker, R.; Jardetzky, T.S. Structural Basis for Epstein–Barr Virus Host Cell Tropism Mediated by Gp42 and GHgL Entry Glycoproteins. Nat. Commun. 2016, 7, 13557. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Sakaida, K.; Yoshida, M.; Al Masud, H.M.A.; Sato, Y.; Goshima, F.; Kimura, H.; Murata, T. The C-Terminus of Epstein-Barr Virus BRRF2 Is Required for Its Proper Localization and Efficient Virus Production. Front. Microbiol. 2017, 8, 125. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Z.; Gong, W.; Xiao, X.; Feng, X.; Li, W.; Shan, S.; Xu, D.; Zhou, Z. Epstein-Barr Virus Protein BKRF4 Restricts Nucleosome Assembly to Suppress Host Antiviral Responses. Proc. Natl. Acad. Sci. USA 2022, 119, e2203782119. [Google Scholar] [CrossRef]
- Hung, C.-H.; Chiu, Y.-F.; Wang, W.-H.; Chen, L.-W.; Chang, P.-J.; Huang, T.-Y.; Lin, Y.-J.; Tsai, W.-J.; Yang, C.-C. Interaction Between BGLF2 and BBLF1 Is Required for the Efficient Production of Infectious Epstein–Barr Virus Particles. Front. Microbiol. 2020, 10, 3021. [Google Scholar] [CrossRef]
- Al Masud, H.M.A.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H.; Murata, T. Epstein-Barr Virus BBRF2 Is Required for Maximum Infectivity. Microorganisms 2019, 7, 705. [Google Scholar] [CrossRef]
- Uddin, M.K.; Watanabe, T.; Arata, M.; Sato, Y.; Kimura, H.; Murata, T. Epstein-Barr Virus BBLF1 Mediates Secretory Vesicle Transport to Facilitate Mature Virion Release. J. Virol. 2023, 97, e00437-23. [Google Scholar] [CrossRef]
- Balfour, H.H.; Sifakis, F.; Sliman, J.A.; Knight, J.A.; Schmeling, D.O.; Thomas, W. Age-Specific Prevalence of Epstein–Barr Virus Infection Among Individuals Aged 6–19 Years in the United States and Factors Affecting Its Acquisition. J. Infect. Dis. 2013, 208, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Sharifipour, S.; Davoodi Rad, K. Seroprevalence of Epstein–Barr Virus among Children and Adults in Tehran, Iran. New Microbes New Infect. 2020, 34, 100641. [Google Scholar] [CrossRef]
- Ye, Z.; Chen, L.; Zhong, H.; Cao, L.; Fu, P.; Xu, J. Epidemiology and Clinical Characteristics of Epstein-Barr Virus Infection among Children in Shanghai, China, 2017–2022. Front Cell Infect. Microbiol 2023, 13, 1139068. [Google Scholar] [CrossRef]
- Jakhmola, S.; Jha, H.C. Glial Cell Response to Epstein-Barr Virus Infection: A Plausible Contribution to Virus-Associated Inflammatory Reactions in the Brain. Virology 2021, 559, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Mehta, D.; Lu, J.; El-Naccache, D.; Shukla, S.K.; Kovacsics, C.; Kolson, D.; Robertson, E.S. Gammaherpesvirus Infection of Human Neuronal Cells. mBio 2015, 6, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; McDonnell, W.J.; Gonzalez-Ericsson, P.I.; Al-Rohil, R.N.; Mobley, B.C.; Salem, J.-E.; Wang, D.Y.; Sanchez, V.; Wang, Y.; Chastain, C.A.; et al. A Case Report of Clonal EBV-like Memory CD4+ T Cell Activation in Fatal Checkpoint Inhibitor-Induced Encephalitis. Nat. Med. 2019, 25, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, E.; Khalighinejad, F.; Ionete, C. Intravenous Immunoglobulins in an Adult Case of Post-EBV Cerebellitis. BMJ Case Rep. 2020, 13, e231661. [Google Scholar] [CrossRef]
- Wada, T.; Muraoka, M.; Yokoyama, T.; Toma, T.; Kanegane, H.; Yachie, A. Cytokine Profiles in Children with Primary Epstein-Barr Virus Infection. Pediatr. Blood Cancer 2013, 60, E46–E48. [Google Scholar] [CrossRef]
- Lui, W.-Y.; Bharti, A.; Wong, N.-H.M.; Jangra, S.; Botelho, M.G.; Yuen, K.-S.; Jin, D.-Y. Suppression of CGAS- and RIG-I-Mediated Innate Immune Signaling by Epstein-Barr Virus Deubiquitinase BPLF1. PLoS Pathog. 2023, 19, e1011186. [Google Scholar] [CrossRef]
- Hooykaas, M.J.G.; van Gent, M.; Soppe, J.A.; Kruse, E.; Boer, I.G.J.; van Leenen, D.; Groot Koerkamp, M.J.A.; Holstege, F.C.P.; Ressing, M.E.; Wiertz, E.J.H.J.; et al. EBV MicroRNA BART16 Suppresses Type I IFN Signaling. J. Immunol. 2017, 198, 4062–4073. [Google Scholar] [CrossRef]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr Virus Gp42 Protein Bound to the MHC Class II Receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Taylor, T.J. Herpes Simplex Virus. Front. Biosci. 2002, 7, d752. [Google Scholar] [CrossRef] [PubMed]
- Kamperschroer, C.; Quinn, D.G. The Role of Proinflammatory Cytokines in Wasting Disease During Lymphocytic Choriomeningitis Virus Infection. J. Immunol. 2002, 169, 340–349. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes Simplex Virus: Global Infection Prevalence and Incidence Estimates, 2016. Bull World Health Organ 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Feige, L.; Zaeck, L.M.; Sehl-Ewert, J.; Finke, S.; Bourhy, H. Innate Immune Signaling and Role of Glial Cells in Herpes Simplex Virus- and Rabies Virus-Induced Encephalitis. Viruses 2021, 13, 2364. [Google Scholar] [CrossRef] [PubMed]
- Babaei, A.; Shatizadeh Malekshahi, S.; Pirbonyeh, N.; Moattari, A. Prevalence and Clinical Manifestations of Herpes Simplex Virus Infection among Suspected Patients of Herpes Simplex Encephalitis in Shiraz, Iran. Virusdisease 2021, 32, 266–271. [Google Scholar] [CrossRef]
- Choi, R.; Kim, G.; Jo, I.J.; Sim, M.S.; Song, K.J.; Kim, B.J.; Na, D.L.; Huh, H.J.; Kim, J.; Ki, C.; et al. Incidence and Clinical Features of Herpes Simplex Viruses (1 and 2) and Varicella-zoster Virus Infections in an Adult Korean Population with Aseptic Meningitis or Encephalitis. J. Med. Virol. 2014, 86, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.S.; Fitzgerald, K.A. The CGAS-STING Pathway for DNA Sensing. Mol. Cell 2013, 51, 135–139. [Google Scholar] [CrossRef]
- Unterholzner, L. The Interferon Response to Intracellular DNA: Why so Many Receptors? Immunobiology 2013, 218, 1312–1321. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Smith, J.B.; Herbert, J.J.; Truong, N.R.; Cunningham, A.L. Cytokines and Chemokines: The Vital Role They Play in Herpes Simplex Virus Mucosal Immunology. Front. Immunol. 2022, 13, 936235. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Bowen, G.N.; Zhou, S.; Cerny, A.; Zacharia, A.; Knipe, D.M.; Finberg, R.W.; Kurt-Jones, E.A. Role of Specific Innate Immune Responses in Herpes Simplex Virus Infection of the Central Nervous System. J. Virol. 2012, 86, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.M.; Knipe, D.M.; Finberg, R.W. Herpes Simplex Virus 1 Interaction with Toll-like Receptor 2 Contributes to Lethal Encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N.; Hu, S.; Rowen, T.N.; Palmquist, J.M.; Lokensgard, J.R. Cutting Edge: TLR2-Mediated Proinflammatory Cytokine and Chemokine Production by Microglial Cells in Response to Herpes Simplex Virus. J. Immunol. 2005, 175, 4189–4193. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Chiu, Y.; Liang, X.; Xia, S.; Ayrapetyan, M.; Liu, S.; He, C.; Song, R.; Zeng, J.; Deng, X.; et al. Microglia Innate Immune Response Contributes to the Antiviral Defense and Blood–CSF Barrier Function in Human Choroid Plexus Organoids during HSV-1 Infection. J. Med. Virol. 2023, 95, e28472. [Google Scholar] [CrossRef]
- Michael, B.D.; Bricio-Moreno, L.; Sorensen, E.W.; Miyabe, Y.; Lian, J.; Solomon, T.; Kurt-Jones, E.A.; Luster, A.D. Astrocyte- and Neuron-Derived CXCL1 Drives Neutrophil Transmigration and Blood-Brain Barrier Permeability in Viral Encephalitis. Cell Rep. 2020, 32, 108150. [Google Scholar] [CrossRef]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR Activation by the Proline-Rich RNA Binding Domain of the Herpes Simplex Virus Type 1 Us11 Protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef]
- Gobeil, P.A.M.; Leib, D.A. Herpes Simplex Virus Γ34.5 Interferes with Autophagosome Maturation and Antigen Presentation in Dendritic Cells. mBio 2012, 3, 10-1128. [Google Scholar] [CrossRef]
- Trgovcich, J.; Johnson, D.; Roizman, B. Cell Surface Major Histocompatibility Complex Class II Proteins Are Regulated by the Products of the γ 1 34.5 and U L 41 Genes of Herpes Simplex Virus 1. J. Virol. 2002, 76, 6974–6986. [Google Scholar] [CrossRef]
- Riviere, Y.; Ahmed, R.; Southern, P.J.; Buchmeier, M.J.; Dutko, F.J.; Oldstone, M.B. The S RNA Segment of Lymphocytic Choriomeningitis Virus Codes for the Nucleoprotein and Glycoproteins 1 and 2. J. Virol. 1985, 53, 966–968. [Google Scholar] [CrossRef]
- Morin, B.; Coutard, B.; Lelke, M.; Ferron, F.; Kerber, R.; Jamal, S.; Frangeul, A.; Baronti, C.; Charrel, R.; de Lamballerie, X.; et al. The N-Terminal Domain of the Arenavirus L Protein Is an RNA Endonuclease Essential in MRNA Transcription. PLoS Pathog. 2010, 6, e1001038. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Fuller-Pace, F.V.; Buchmeier, M.J.; Southern, P.J. Analysis of the Genomic l Rna Segment from Lymphocytic Choriomeningitis Virus. Virology 1987, 161, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Charrel, R.N.; Coutard, B.; Baronti, C.; Canard, B.; Nougairede, A.; Frangeul, A.; Morin, B.; Jamal, S.; Schmidt, C.L.; Hilgenfeld, R.; et al. Arenaviruses and Hantaviruses: From Epidemiology and Genomics to Antivirals. Antivir. Res. 2011, 90, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Salvato, M.S.; Shimomaye, E.M. The Completed Sequence of Lymphocytic Choriomeningitis Virus Reveals a Unique RNA Structure and a Gene for a Zinc Finger Protein. Virology 1989, 173, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Riaño, E.; Cheng, B.Y.H.; de la Torre, J.C.; Martínez-Sobrido, L. The C-Terminal Region of Lymphocytic Choriomeningitis Virus Nucleoprotein Contains Distinct and Segregable Functional Domains Involved in NP-Z Interaction and Counteraction of the Type I Interferon Response. J. Virol. 2011, 85, 13038–13048. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; García-Sastre, A.; de la Torre, J.C. Inhibition of the Type I Interferon Response by the Nucleoprotein of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef] [PubMed]
- Pinschewer, D.D.; Perez, M.; de la Torre, J.C. Role of the Virus Nucleoprotein in the Regulation of Lymphocytic Choriomeningitis Virus Transcription and RNA Replication. J. Virol. 2003, 77, 3882–3887. [Google Scholar] [CrossRef] [PubMed]
- Labudova, M.; Tomaskova, J.; Skultety, L.; Pastorek, J.; Pastorekova, S. The Nucleoprotein of Lymphocytic Choriomeningitis Virus Facilitates Spread of Persistent Infection through Stabilization of the Keratin Network. J. Virol. 2009, 83, 7842–7849. [Google Scholar] [CrossRef]
- Hashizume, M.; Takashima, A.; Iwasaki, M. A Small Stem-Loop-Forming Region within the 3′-UTR of a Nonpolyadenylated LCMV MRNA Promotes Translation. J. Biol. Chem. 2022, 298, 101576. [Google Scholar] [CrossRef]
- Igonet, S.; Vaney, M.-C.; Vonrhein, C.; Bricogne, G.; Stura, E.A.; Hengartner, H.; Eschli, B.; Rey, F.A. X-Ray Structure of the Arenavirus Glycoprotein GP2 in Its Postfusion Hairpin Conformation. Proc. Natl. Acad. Sci. USA 2011, 108, 19967–19972. [Google Scholar] [CrossRef]
- Sullivan, B.M.; Emonet, S.F.; Welch, M.J.; Lee, A.M.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B. Point Mutation in the Glycoprotein of Lymphocytic Choriomeningitis Virus Is Necessary for Receptor Binding, Dendritic Cell Infection, and Long-Term Persistence. Proc. Natl. Acad. Sci. USA 2011, 108, 2969–2974. [Google Scholar] [CrossRef]
- Dykewicz, C.A. Lymphocytic Choriomeningitis Outbreak Associated With Nude Mice in a Research Institute. JAMA J. Am. Med. Assoc. 1992, 267, 1349. [Google Scholar] [CrossRef]
- Vilibic-Cavlek, T.; Savic, V.; Ferenc, T.; Mrzljak, A.; Barbic, L.; Bogdanic, M.; Stevanovic, V.; Tabain, I.; Ferencak, I.; Zidovec-Lepej, S. Lymphocytic Choriomeningitis—Emerging Trends of a Neglected Virus: A Narrative Review. Trop. Med. Infect. Dis. 2021, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; Mahoney, J.; Buchmeier, M.J.; Karacay, B.; Taggard, D. Critical Role for Glial Cells in the Propagation and Spread of Lymphocytic Choriomeningitis Virus in the Developing Rat Brain. J. Virol. 2002, 76, 6618–6635. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Otto, G.; Colby, L.A. Selected Zoonoses. In Laboratory Animal Medicine; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1313–1370. [Google Scholar]
- Puccini, J.M.; Ruller, C.M.; Robinson, S.M.; Knopp, K.A.; Buchmeier, M.J.; Doran, K.S.; Feuer, R. Distinct Neural Stem Cell Tropism, Early Immune Activation, and Choroid Plexus Pathology Following Coxsackievirus Infection in the Neonatal Central Nervous System. Lab. Investig. 2014, 94, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J. Lymphocytic Choriomeningitis Virus: An Underrecognized Cause of Neurologic Disease in the Fetus, Child, and Adult. Semin. Pediatr. Neurol. 2012, 19, 89–95. [Google Scholar] [CrossRef]
- Pearce, B.D.; Po, C.L.; Pisell, T.L.; Miller, A.H. Lymphocytic Responses and the Gradual Hippocampal Neuron Loss Following Infection with Lymphocytic Choriomeningitis Virus (LCMV). J. Neuroimmunol. 1999, 101, 137–147. [Google Scholar] [CrossRef]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. MyD88 Is Critical for the Development of Innate and Adaptive Immunity during Acute Lymphocytic Choriomeningitis Virus Infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar] [CrossRef]
- Pratumchai, I.; Zak, J.; Huang, Z.; Min, B.; Oldstone, M.B.A.; Teijaro, J.R. B Cell–Derived IL-27 Promotes Control of Persistent LCMV Infection. Proc. Natl. Acad. Sci. USA 2022, 119, e2116741119. [Google Scholar] [CrossRef]
- Lin, A.A.; Wojciechowski, S.E.; Hildeman, D.A. Androgens Suppress Antigen-Specific T Cell Responses and IFN-γ Production during Intracranial LCMV Infection. J. Neuroimmunol. 2010, 226, 8–19. [Google Scholar] [CrossRef]
- Che Mat, N.F.; Siddiqui, S.; Mehta, D.; Seaver, K.; Banete, A.; Alothaimeen, T.; Gee, K.; Basta, S. Lymphocytic Choriomeningitis Virus Infection of Dendritic Cells Interferes with TLR-Induced IL-12/IL-23 Cytokine Production in an IL-10 Independent Manner. Cytokine 2018, 108, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic Choriomeningitis Virus (LCMV) Infection of CNS Glial Cells Results in TLR2-MyD88/Mal-Dependent Inflammatory Responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic Characterization of a Novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Hardenbrook, N.J.; Zhang, P. A Structural View of the SARS-CoV-2 Virus and Its Assembly. Curr. Opin. Virol. 2022, 52, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.-L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 Envelope and Membrane Proteins Modulate Maturation and Retention of the Spike Protein, Allowing Assembly of Virus-like Particles. J. Biol. Chem. 2021, 296, 100111. [Google Scholar] [CrossRef]
- Zhang, Z.; Nomura, N.; Muramoto, Y.; Ekimoto, T.; Uemura, T.; Liu, K.; Yui, M.; Kono, N.; Aoki, J.; Ikeguchi, M.; et al. Structure of SARS-CoV-2 Membrane Protein Essential for Virus Assembly. Nat. Commun. 2022, 13, 4399. [Google Scholar] [CrossRef]
- Zaffagni, M.; Harris, J.M.; Patop, I.L.; Pamudurti, N.R.; Nguyen, S.; Kadener, S. SARS-CoV-2 Nsp14 Mediates the Effects of Viral Infection on the Host Cell Transcriptome. Elife 2022, 11, e71945. [Google Scholar] [CrossRef]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The Role of the Nsp2 and Nsp3 in Its Pathogenesis. J. Med. Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; Nakagawa, S.; Sato, K. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Itoh, Y.; Suzuki, T.; Tanaka, T.; Sakai, Y.; Koido, M.; Hata, C.; Wang, C.-X.; Otani, M.; Moriishi, K.; et al. SARS-CoV-2 ORF6 Disrupts Nucleocytoplasmic Trafficking to Advance Viral Replication. Commun. Biol. 2022, 5, 483. [Google Scholar] [CrossRef]
- Yang, R.; Zhao, Q.; Rao, J.; Zeng, F.; Yuan, S.; Ji, M.; Sun, X.; Li, J.; Yang, J.; Cui, J.; et al. SARS-CoV-2 Accessory Protein ORF7b Mediates Tumor Necrosis Factor-α-Induced Apoptosis in Cells. Front. Microbiol. 2021, 12, 654709. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhuang, M.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.; Zhang, X.; Gao, C.; Wang, P. SARS-CoV-2 ORF9b Antagonizes Type I and III Interferons by Targeting Multiple Components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and CGAS–STING Signaling Pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef] [PubMed]
- Mena, G.E.; Martinez, P.P.; Mahmud, A.S.; Marquet, P.A.; Buckee, C.O.; Santillana, M. Socioeconomic Status Determines COVID-19 Incidence and Related Mortality in Santiago, Chile. Science 2021, 372, eabg5298. [Google Scholar] [CrossRef] [PubMed]
- Balloff, C.; Bandlow, C.; Bernhard, M.; Brandenburger, T.; Bludau, P.; Elben, S.; Feldt, T.; Hartmann, C.J.; Heinen, E.; Ingwersen, J.; et al. Prevalence and Prognostic Value of Neurological Affections in Hospitalized Patients with Moderate to Severe COVID-19 Based on Objective Assessments. Sci. Rep. 2023, 13, 19619. [Google Scholar] [CrossRef] [PubMed]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.S.L.M.; Vendramini, P.H.; Valença, A.G.F.; Brandão-Teles, C.; Zuccoli, G.d.S.; et al. Morphological, Cellular, and Molecular Basis of Brain Infection in COVID-19 Patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2200960119. [Google Scholar] [CrossRef]
- Martínez-Mármol, R.; Giordano-Santini, R.; Kaulich, E.; Cho, A.-N.; Przybyla, M.; Riyadh, M.A.; Robinson, E.; Chew, K.Y.; Amor, R.; Meunier, F.A.; et al. SARS-CoV-2 Infection and Viral Fusogens Cause Neuronal and Glial Fusion That Compromises Neuronal Activity. Sci. Adv. 2023, 9, eadg2248. [Google Scholar] [CrossRef]
- Zhu, X.; Gebo, K.A.; Abraham, A.G.; Habtehyimer, F.; Patel, E.U.; Laeyendecker, O.; Gniadek, T.J.; Fernandez, R.E.; Baker, O.R.; Ram, M.; et al. Dynamics of Inflammatory Responses after SARS-CoV-2 Infection by Vaccination Status in the USA: A Prospective Cohort Study. Lancet Microbe 2023, 4, e692–e703. [Google Scholar] [CrossRef]
- Pereson, M.J.; Badano, M.N.; Aloisi, N.; Chuit, R.E.; de Bracco, M.M.; Bare, P. TNF-α Levels in Respiratory Samples Are Associated with SARS-CoV-2 Infection. Microbiol. Spectr. 2022, 10, e01411-21. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.-Y.; Plociennikowska, A.; Heigwer, F.; Prasad, V.; Joecks, S.; Burkart, S.S.; et al. SARS-CoV-2 Infection Induces a pro-Inflammatory Cytokine Response through CGAS-STING and NF-ΚB. Commun. Biol. 2022, 5, 45. [Google Scholar] [CrossRef]
- Planès, R.; Bert, J.-B.; Tairi, S.; BenMohamed, L.; Bahraoui, E. SARS-CoV-2 Envelope (E) Protein Binds and Activates TLR2 Pathway: A Novel Molecular Target for COVID-19 Interventions. Viruses 2022, 14, 999. [Google Scholar] [CrossRef]
- Vojdani, A.; Pollard, K.M.; Campbell, A.W. Environmental Triggers and Autoimmunity. Autoimmune Dis. 2014, 2014, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, B.; Shirafkan, F.; Ripperger, K.; Rattay, K. The Role of Viral Infections in the Onset of Autoimmune Diseases. Viruses 2023, 15, 782. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal Analysis Reveals High Prevalence of Epstein-Barr Virus Associated with Multiple Sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, S.; Sakuma, Y.; Yanagida, T.; Yoshio, T. Association of Cerebrospinal Fluid Anti-Sm Antibodies with Acute Confusional State in Systemic Lupus Erythematosus. Arthritis Res. Ther. 2014, 16, 450. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tu, Z.; Zhang, X.; Du, K.; Xie, Z.; Lin, Z. Pathogenesis and Treatment of Neuropsychiatric Systemic Lupus Erythematosus: A Review. Front. Cell Dev. Biol. 2022, 10, 998328. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.S.; Stocks, B.T.; Hoopes, E.M.; Rhoads, J.P.; McNew, K.L.; Major, A.S.; Moore, D.J. Metabolic Preconditioning in CD4+ T Cells Restores Inducible Immune Tolerance in Lupus-Prone Mice. JCI Insight 2021, 6, e143245. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, A.M.; Zhang, J.; Mackay, M.; Aranow, C.; Diamond, B. Phenotypic Characterization of Autoreactive B Cells—Checkpoints of B Cell Tolerance in Patients with Systemic Lupus Erythematosus. PLoS ONE 2009, 4, e5776. [Google Scholar] [CrossRef]
- Mihaylova, N.; Chipinski, P.; Bradyanova, S.; Velikova, T.; Ivanova-Todorova, E.; Chausheva, S.; Herbáth, M.; Kalinova, D.; Prechl, J.; Kyurkchiev, D.; et al. Suppression of Autoreactive T and B Lymphocytes by Anti-Annexin A1 Antibody in a Humanized NSG Murine Model of Systemic Lupus Erythematosus. Clin. Exp. Immunol. 2020, 199, 278–293. [Google Scholar] [CrossRef]
- Ding, J.; Su, S.; You, T.; Xia, T.; Lin, X.; Chen, Z.; Zhang, L. Serum Interleukin-6 Level Is Correlated with the Disease Activity of Systemic Lupus Erythematosus: A Meta-Analysis. Clinics 2020, 75, e1801. [Google Scholar] [CrossRef]
- Rafat, S.; Mohamed, Y.Y.; Ahmed, M.E.; Abdelaleem, E.A. Interleukin-23 Serum Level in Systemic Lupus Erythematosus Patients: Relation to Disease Activity and Different Disease Parameters. Egypt. Rheumatol. 2022, 44, 139–143. [Google Scholar] [CrossRef]
- Zucchi, D.; Elefante, E.; Schilirò, D.; Signorini, V.; Trentin, F.; Bortoluzzi, A.; Tani, C. One Year in Review 2022: Systemic Lupus Erythematosus. Clin. Exp. Rheumatol. 2022, 40, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bunting, K.A.; Kalsi, J.; Hinks, J.A.; Latchman, D.S.; Pearl, L.H.; Isenberg, D.A. Lupus Autoantibodies to Native DNA Preferentially Bind DNA Presented on PolIV. Immunology 2005, 114, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K. Inflammatory Cytokines in Systemic Lupus Erythematosus. J. Biomed. Biotechnol. 2011, 2011, 432595. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.R.W.; Falasinnu, T.; Ramsey-Goldman, R.; Clarke, A.E. The Global Epidemiology of SLE: Narrowing the Knowledge Gaps. Rheumatology 2023, 62, i4–i9. [Google Scholar] [CrossRef] [PubMed]
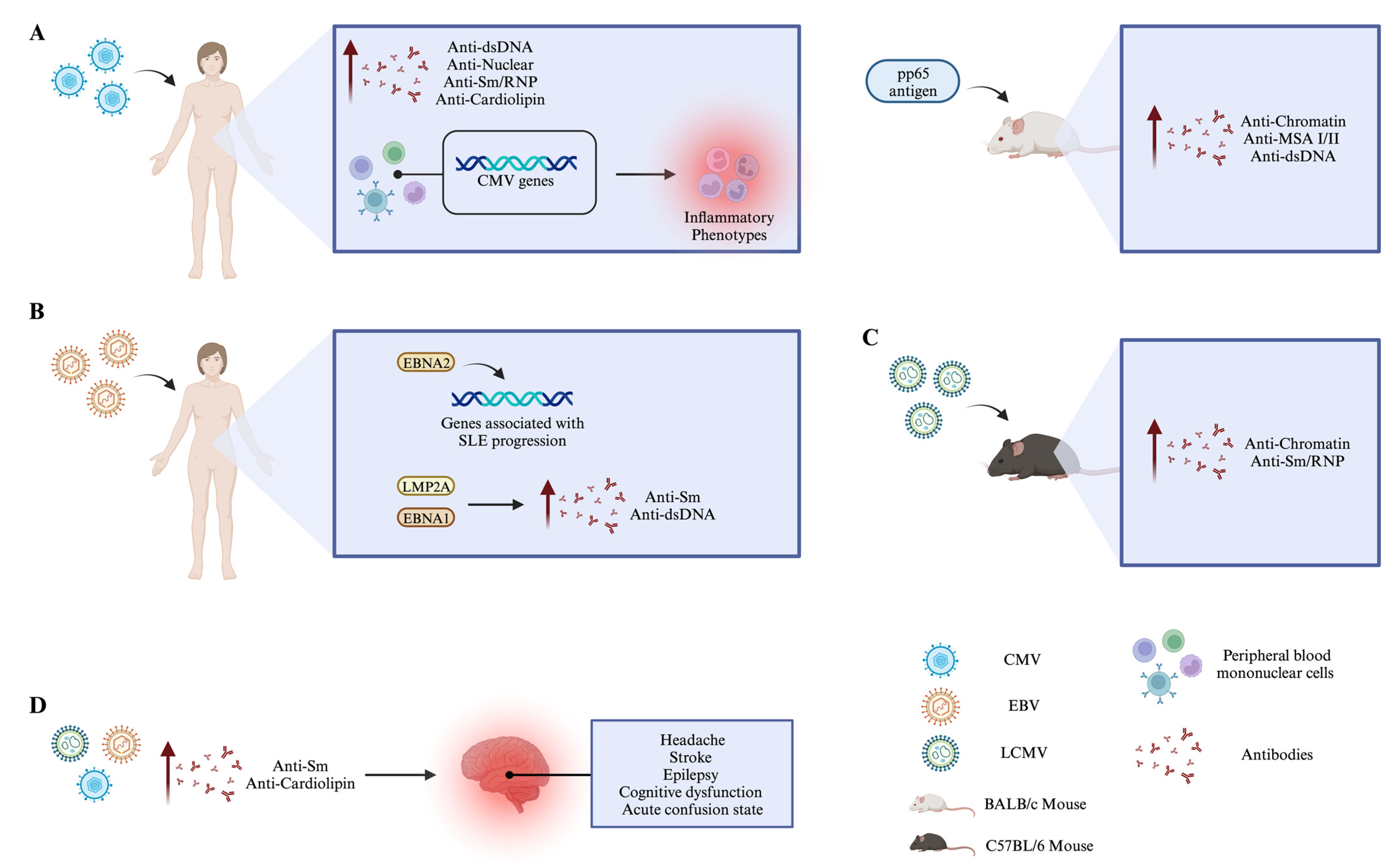
- Guo, G.; Ye, S.; Xie, S.; Ye, L.; Lin, C.; Yang, M.; Shi, X.; Wang, F.; Li, B.; Li, M.; et al. The Cytomegalovirus Protein US31 Induces Inflammation through Mono-Macrophages in Systemic Lupus Erythematosus by Promoting NF-ΚB2 Activation. Cell Death Dis. 2018, 9, 104. [Google Scholar] [CrossRef]
- Neo, J.Y.J.; Wee, S.Y.K.; Bonne, I.; Tay, S.H.; Raida, M.; Jovanovic, V.; Fairhurst, A.-M.; Lu, J.; Hanson, B.J.; MacAry, P.A. Characterisation of a Human Antibody That Potentially Links Cytomegalovirus Infection with Systemic Lupus Erythematosus. Sci. Rep. 2019, 9, 9998. [Google Scholar] [CrossRef]
- Hsieh, A.-H.; Jhou, Y.-J.; Liang, C.-T.; Chang, M.; Wang, S.-L. Fragment of Tegument Protein Pp65 of Human Cytomegalovirus Induces Autoantibodies in BALB/c Mice. Arthritis Res. Ther. 2011, 13, R162. [Google Scholar] [CrossRef]
- Richard, M.L.; Gilkeson, G. Mouse Models of Lupus: What They Tell Us and What They Don’t. Lupus Sci. Med. 2018, 5, e000199. [Google Scholar] [CrossRef]
- Moon, U.; Park, S.; Oh, S.; Kim, W.-U.; Park, S.-H.; Lee, S.-H.; Cho, C.-S.; Kim, H.-Y.; Lee, W.-K.; Lee, S. Patients with Systemic Lupus Erythematosus Have Abnormally Elevated Epstein–Barr Virus Load in Blood. Arthritis Res. Ther. 2004, 6, R295. [Google Scholar] [CrossRef]
- Wang, H.; Nicholas, M.W.; Conway, K.L.; Sen, P.; Diz, R.; Tisch, R.M.; Clarke, S.H. EBV Latent Membrane Protein 2A Induces Autoreactive B Cell Activation and TLR Hypersensitivity. J. Immunol. 2006, 177, 2793–2802. [Google Scholar] [CrossRef]
- Singh, D.; Oudit, O.; Hajtovic, S.; Sarbaugh, D.; Salis, R.; Adebowale, T.; James, J.; Spatz, L.A. Antibodies to an Epstein Barr Virus Protein That Cross-React with DsDNA Have Pathogenic Potential. Mol. Immunol. 2021, 132, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Quintial, R.; Nguyen, A.; Kono, D.H.; Oldstone, M.B.A.; Theofilopoulos, A.N.; Baccala, R. Lupus Acceleration by a MAVS-Activating RNA Virus Requires Endosomal TLR Signaling and Host Genetic Predisposition. PLoS ONE 2018, 13, e0203118. [Google Scholar] [CrossRef]
- Hanly, J.G.; Urowitz, M.B.; Su, L.; Bae, S.C.; Gordon, C.; Wallace, D.J.; Clarke, A.; Bernatsky, S.; Isenberg, D.; Rahman, A.; et al. Prospective Analysis of Neuropsychiatric Events in an International Disease Inception Cohort of Patients with Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2010, 69, 529–535. [Google Scholar] [CrossRef]
- Liang, M.H.; Corzillius, M.; Bae, S.C.; Lew, R.A.; Fortin, P.R.; Gordon, C.; Isenberg, D.; Alarcón, G.S.; Straaton, K.V.; Denburg, J.; et al. The American College of Rheumatology Nomenclature and Case Definitions for Neuropsychiatric Lupus Syndromes. Arthritis Rheum. 1999, 42, 599–608. [Google Scholar] [CrossRef]
- Sled, J.G.; Spring, S.; van Eede, M.; Lerch, J.P.; Ullal, S.; Sakic, B. Time Course and Nature of Brain Atrophy in the MRL Mouse Model of Central Nervous System Lupus. Arthritis Rheum. 2009, 60, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Foster, J.; Sakic, B. Distribution and Prevalence of Leukocyte Phenotypes in Brains of Lupus-Prone Mice. J. Neuroimmunol. 2006, 179, 26–36. [Google Scholar] [CrossRef] [PubMed]
- ElMelegy, T.T.H.; Radwan, A.H.; Herdan, O.M.; Rashed, H.G. Does Herpes Simplex Virus Have a Role in the Pathogenesis of Systemic Lupus Erythematosus in Egyptians? Egypt. J. Immunol. 2022, 29, 48–56. [Google Scholar] [CrossRef]
- Chang, M.; Liu, M.; Chen, M.; Li, N.; Zhao, Y.; Zhang, S.; He, P.; Yu, Q. Mendelian Randomization Analysis Suggests No Associations of Herpes Simplex Virus Infections with Systemic Lupus Erythematosus. J. Med. Virol. 2023, 95, e28649. [Google Scholar] [CrossRef]
- Kajon, A.E.; Dickson, L.M.; Fisher, B.T.; Hodinka, R.L. Fatal Disseminated Adenovirus Infection in a Young Adult with Systemic Lupus Erythematosus. J. Clin. Virol. 2011, 50, 80–83. [Google Scholar] [CrossRef]
- Terrasson, J.; De Haes, P.; De Langhe, E. Systemic Lupus Erythematosus Mimicry Caused by Viral Infection with Coxsackie B4. Rheumatology 2021, 60, e203–e204. [Google Scholar] [CrossRef]
- Perlejewski, K.; Bukowska-Ośko, I.; Rydzanicz, M.; Pawełczyk, A.; Caraballo Cortès, K.; Osuch, S.; Paciorek, M.; Dzieciątkowski, T.; Radkowski, M.; Laskus, T. Next-Generation Sequencing in the Diagnosis of Viral Encephalitis: Sensitivity and Clinical Limitations. Sci. Rep. 2020, 10, 16173. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, M.; Greber, U.F. Virus Infection Variability by Single-Cell Profiling. Viruses 2021, 13, 1568. [Google Scholar] [CrossRef]
- Ren, M.; Zhou, Y.; Tu, T.; Jiang, D.; Pang, M.; Li, Y.; Luo, Y.; Yao, X.; Yang, Z.; Wang, Y. RVG Peptide-Functionalized Favipiravir Nanoparticle Delivery System Facilitates Antiviral Therapy of Neurotropic Virus Infection in a Mouse Model. Int. J. Mol. Sci. 2023, 24, 5851. [Google Scholar] [CrossRef] [PubMed]
- Surnar, B.; Shah, A.S.; Park, M.; Kalathil, A.A.; Kamran, M.Z.; Ramirez Jaime, R.; Toborek, M.; Nair, M.; Kolishetti, N.; Dhar, S. Brain-Accumulating Nanoparticles for Assisting Astrocytes to Reduce Human Immunodeficiency Virus and Drug Abuse-Induced Neuroinflammation and Oxidative Stress. ACS Nano 2021, 15, 15741–15753. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Duggal, S.; Chugh, T. Das Cytomegalovirus Infection in Non-Immunosuppressed Critically Ill Patients. J. Infect. Dev. Ctries. 2011, 5, 571–579. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
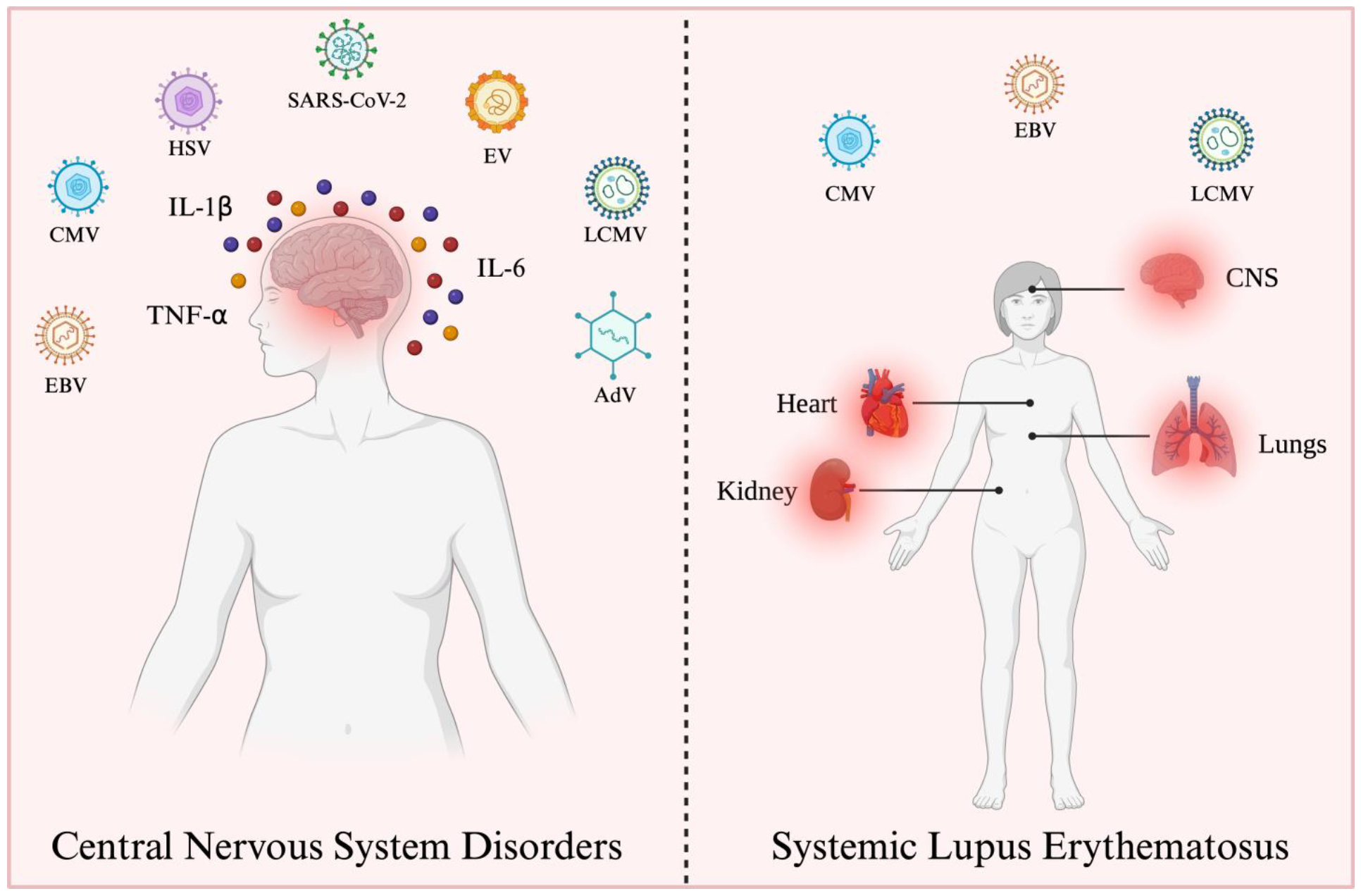
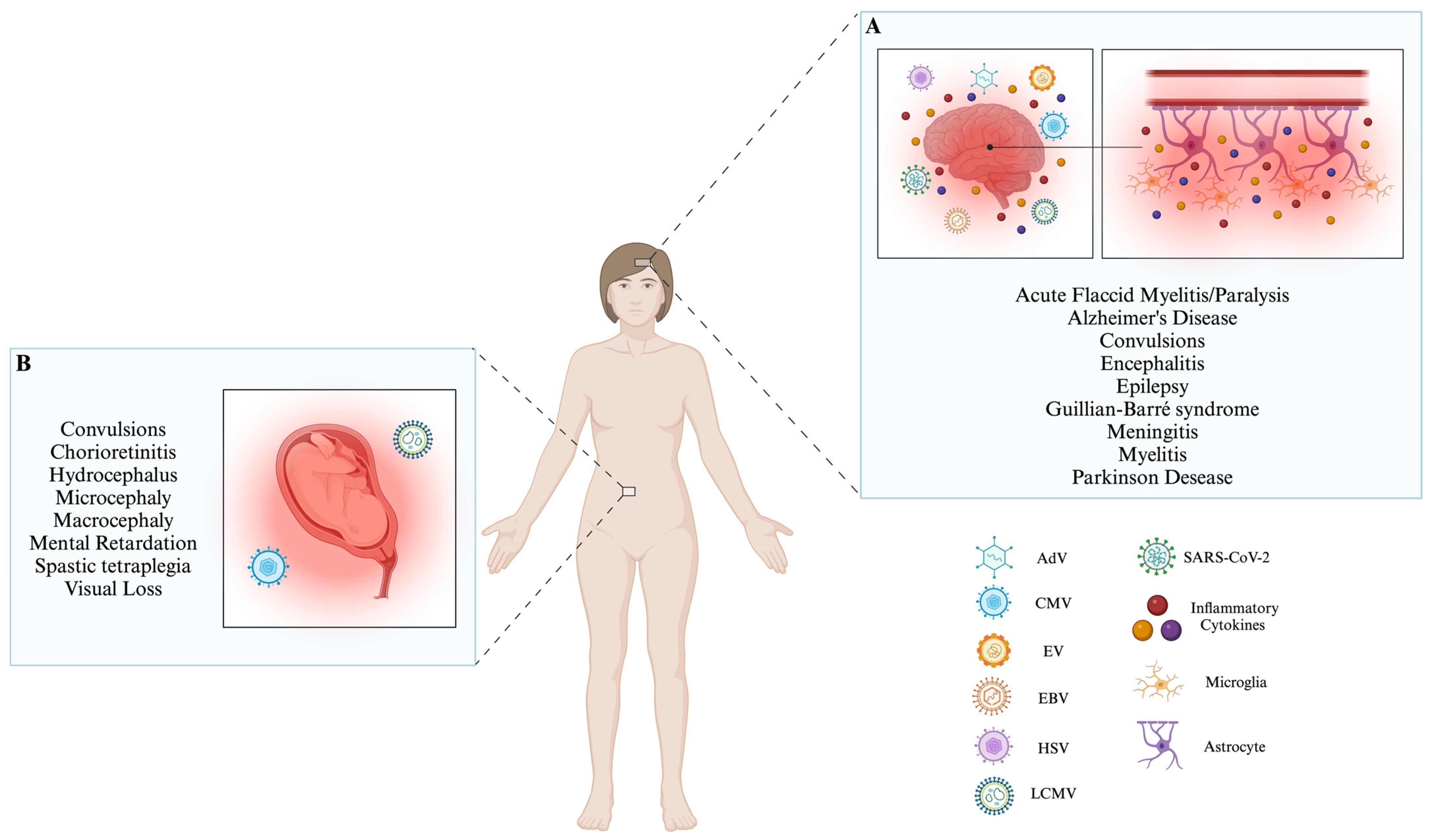
| Virus | Genome | Invasion to CNS | Immune Response Evasion | Clinical Signs | References |
|---|---|---|---|---|---|
| AdV | dsDNA | BBB, and Olfactory nerves | IFN-I pathway modulates the innate and adaptive immune response, and affects antigen presentation processes | Aseptic meningitis convulsions, encephalitis, and acute disseminated encephalomyelitis. | [10,11,39,46,47,48,49,50,51,52] |
| CMV | dsDNA | BBB | Affects antigen presentation processes module phenotypic and functional changes in macrophages. | Aseptic meningitis, polyradiculopathy, myelitis, ventriculitis, dementia, cranial nerve involvement, microcephaly, periventricular calcifications, convulsions, spastic tetraplegia, and hydrocephalus suppress the development of neural stem cells. | [10,11,53,54,55,56,57,58,59,60,61] |
| EV | ssRNA− | BBB, Myeloid cells infected, motoneurons. | IFN-I pathway regulates the cytosolic RIG-I pathway and NLRP3 inflammasome. | Aseptic meningitis, encephalitis, AFM, cognitive impairments in motor skills, problem-solving abilities, socialization, communication, neuronal development delay, ADHD, and epilepsy. | [10,11,35,62,63,64,65,66,67,68,69,70,71,72,73,74] |
| EBV | dsDNA | BBB | IFN-I pathway, MAVS, and NLRP3 inflammasome induce apoptosis and inhibit DC development, reprogramming B cells. | Aseptic meningitis, encephalitis, transverse myelitis, cerebellitis, ataxia, Guillian–Barré syndrome, Alzheimer’s Disease, and Parkinson’s disease. | [10,11,75,76,77,78,79,80,81,82,83,84,85,86,87,88] |
| HSV | dsDNA | BBB, trigeminal or olfactory nerves, latent HSV-1 in situ in the brain, and reactivation of initial infection in the periphery. | IFN-I pathway hinders the complement activation, hinders the ability of IgG antibodies, induces DCs apoptosis, and affects antigen presentation processes. | Aseptic meningitis, acute encephalitis, and cognitive impairment. | [10,11,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104] |
| LCMV | ssRNA− | BBB | IFN-I production. | Aseptic meningitis, hydrocephalus, chorioretinitis, macrocephaly, microcephaly, chorioretinopathy, spastic quadriparesis, seizures, visual loss, mental retardation, and neurodegenerative manifestations. | [10,11,105,106,107,108,109,110,111,112,113,114] |
| SARS-CoV-2 | ssRNA+ | BBB, and cranial nerves. | It affects dsRNA recognition via the RIG-I IFN pathway, induces MAVS autophagy, modules the immune chemotaxis, and affects antigen presentation processes. | Encephalitis, encephalopathy, seizures, epilepsy, and Alzheimer’s Disease. | [115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uribe, F.R.; González, V.P.I.; Kalergis, A.M.; Soto, J.A.; Bohmwald, K. Understanding the Neurotrophic Virus Mechanisms and Their Potential Effect on Systemic Lupus Erythematosus Development. Brain Sci. 2024, 14, 59. https://doi.org/10.3390/brainsci14010059
Uribe FR, González VPI, Kalergis AM, Soto JA, Bohmwald K. Understanding the Neurotrophic Virus Mechanisms and Their Potential Effect on Systemic Lupus Erythematosus Development. Brain Sciences. 2024; 14(1):59. https://doi.org/10.3390/brainsci14010059
Chicago/Turabian StyleUribe, Felipe R., Valentina P. I. González, Alexis M. Kalergis, Jorge A. Soto, and Karen Bohmwald. 2024. "Understanding the Neurotrophic Virus Mechanisms and Their Potential Effect on Systemic Lupus Erythematosus Development" Brain Sciences 14, no. 1: 59. https://doi.org/10.3390/brainsci14010059