Inter-Tumor Heterogeneity—Melanomas Respond Differently to GM-CSF-Mediated Activation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Melanoma- or Brain Cell-Conditioned Medium
2.3. ELISA Assay
2.4. Downregulation of GM-CSF Expression
2.5. Adhesion to Brain Endothelial Cells
2.6. Transendothelial Migration Through a Blood-Brain Barrier Model
2.7. Immunodetection of Proteins by Western Blot
2.8. Animals
2.9. Orthotopic Inoculation of Tumor Cells
2.10. Intracardiac Inoculation of Tumor Cells
2.11. RNA Preparation and Reverse Transcription Droplet Digital PCR (RT-ddPCR)
2.12. mRNA Sequencing Analysis
2.13. Biostatistical Analysis
3. Results
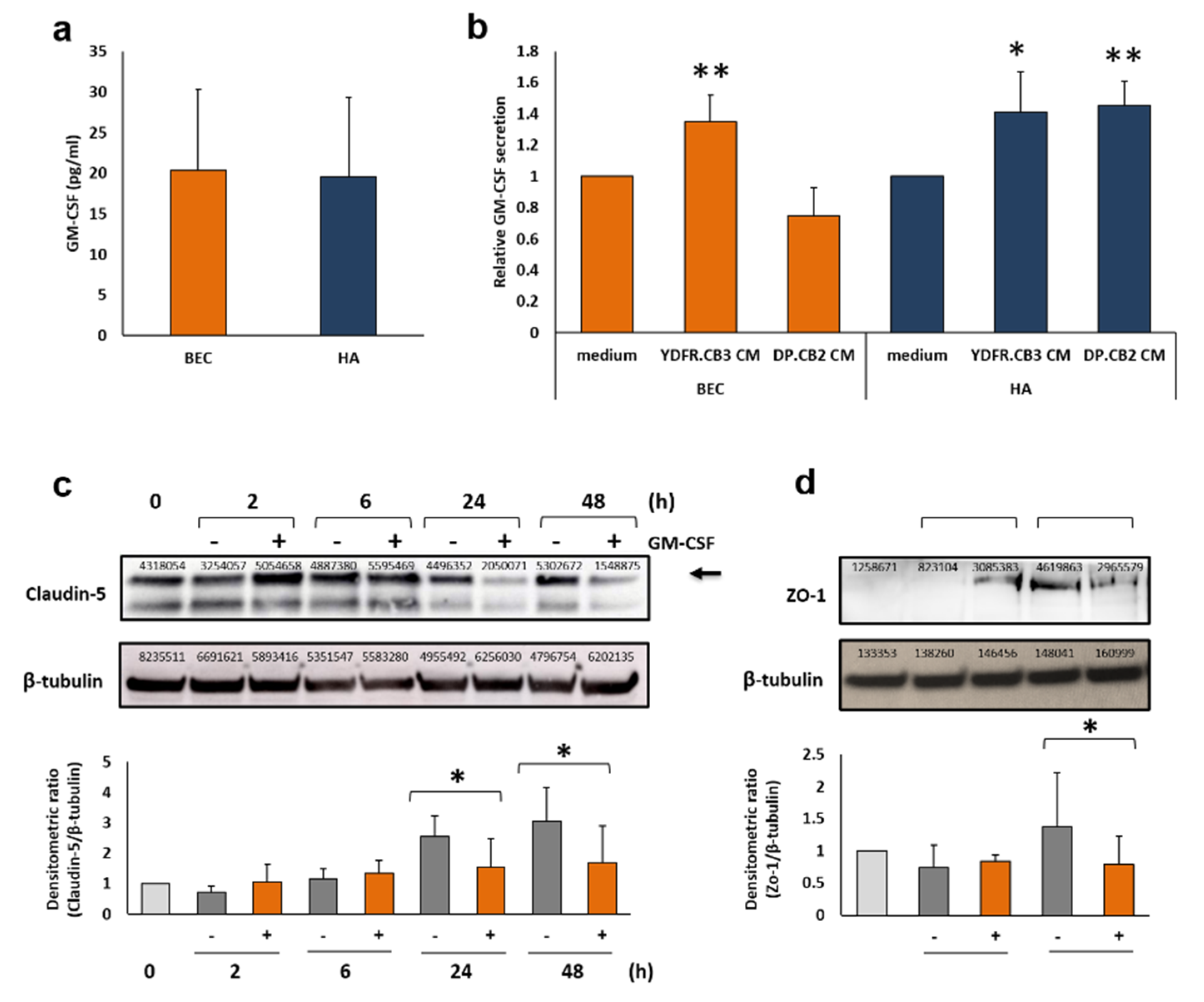
3.1. Melanoma-Derived Soluble Factors (MCM) Enhance GM-CSF Secretion from Brain Microenvironmental Cells
3.2. The Effects of GM-CSF on Brain Endothelial Cells
3.2.1. GM-CSF Alters the Gene Expression Profile of BEC
- Transendothelial migration related genes
- The downregulated gene interleukin-37 (IL-37, FC = −2.36) inhibits inflammatory response by suppressing the TLR2-NF-κB-ICAM-1 pathway in coronary artery endothelial cells and is possibly involved in the adhesion and transmigration of neutrophils through such endothelial cells [25]. Additionally, IL-37 promotes endothelial activation and angiogenesis [26].
- Angiogenesis related genes
- The upregulated gene carbonic anhydrase 9 (CA9, FC = 4.18) induces endothelial migration and angiogenesis in tumors [27].
- The upregulated gene serum/glucocorticoid regulated kinase 2 (SGK1, FC = 2.84) is required for endothelial cell migration and angiogenesis [28].
- The downregulated gene ephrin B3 (EFNB3, FC = −3.99) supports endothelial cell survival and its silencing decreases tumor vascularization and growth in a glioblastoma xenograft model [29].
- The downregulated gene RUNX family transcription factor 3 (RUNX3, FC = −2.33) contributes to endothelial-to-mesenchymal transition and endothelial cell dysfunction. RUNX3 downregulation reduced endothelial cell migration and promoted angiogenesis [30].
3.2.2. GM-CSF Down-Regulates the Expression of the Endothelial Tight Junction Proteins Claudin-5 and Zonula Occludens-1
3.3. GM-CSF is Secreted from Human Melanoma Cells
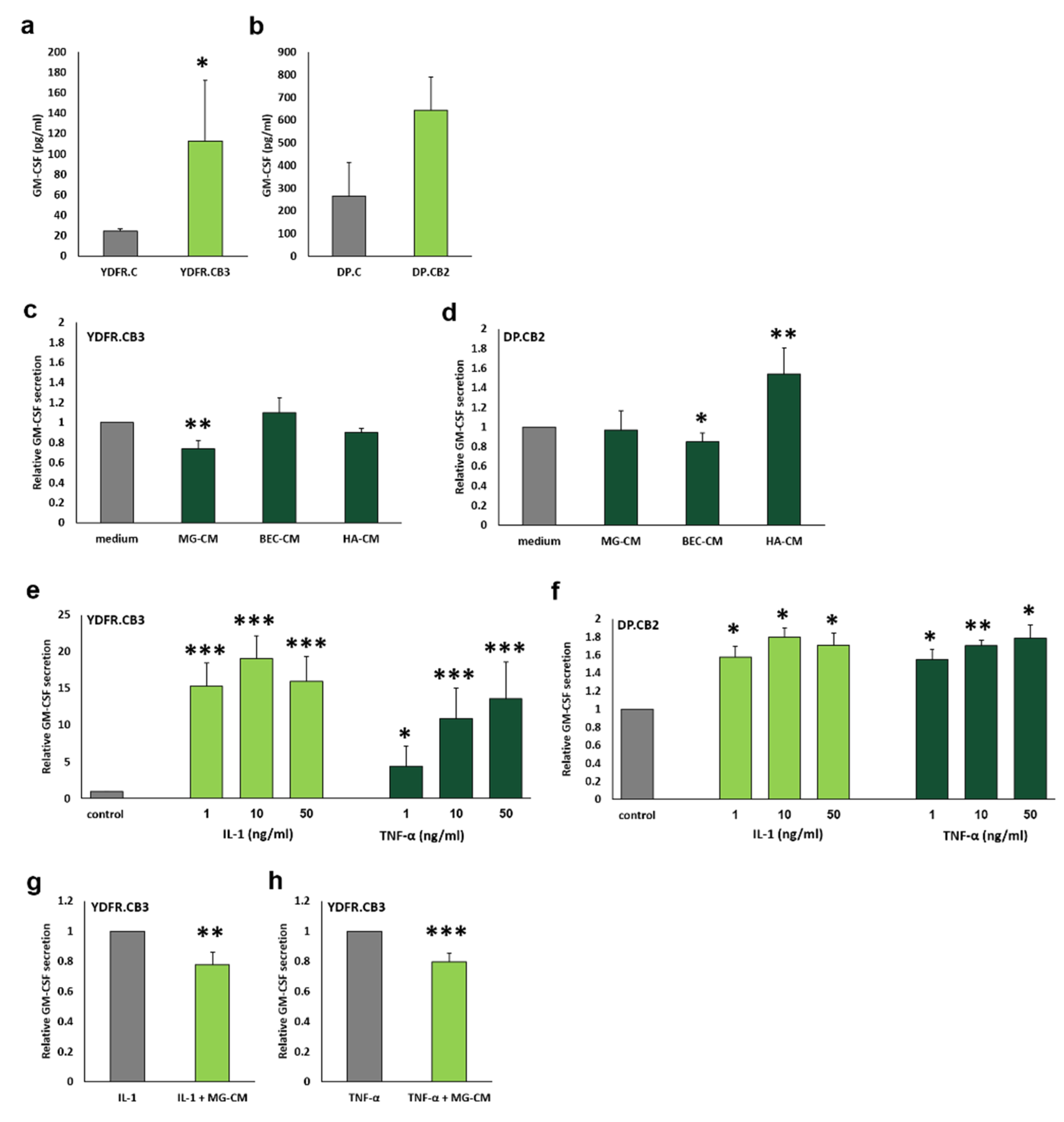
3.4. Brain-Derived Soluble Factors Alter GM-CSF Secretion from Brain-Metastasizing Melanoma Cells
3.5. IL-1α and TNF-α Differentially Influence GM-CSF Secretion from Melanoma Cells
3.6. Varied Responses of YDFR and DP Cells to GM-CSF-Mediated Activation
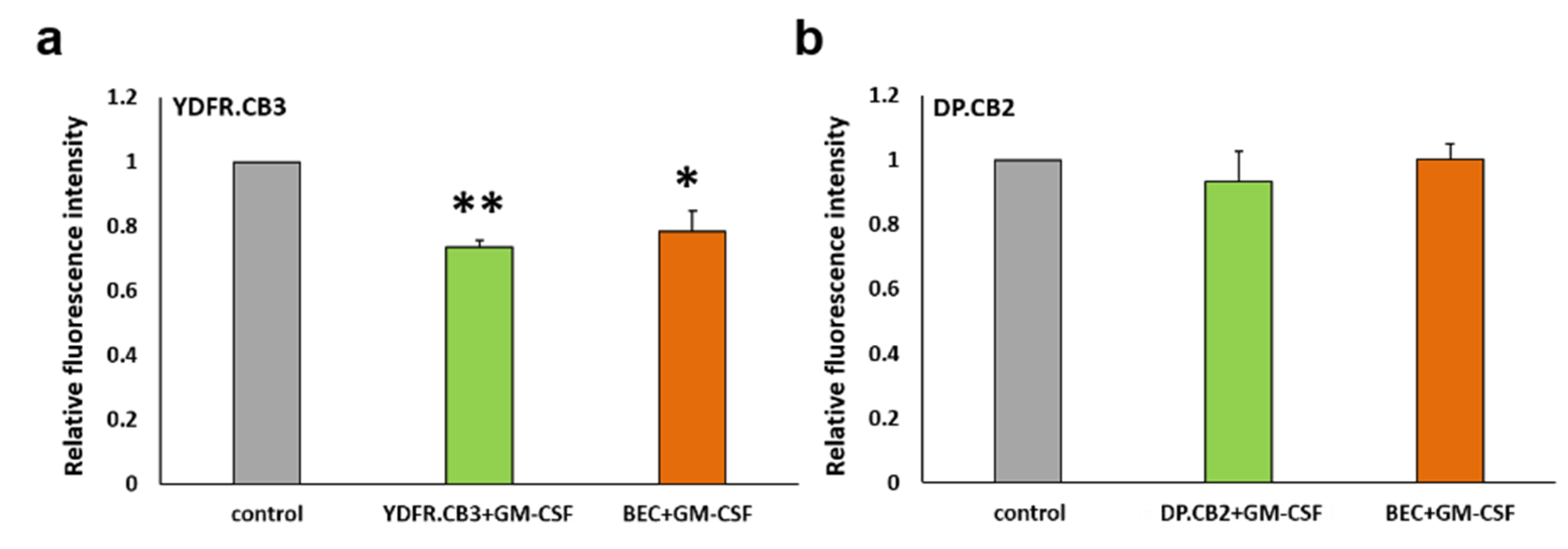
3.6.1. GM-CSF Differentially Affects the Interaction of Melanoma Cells with BEC
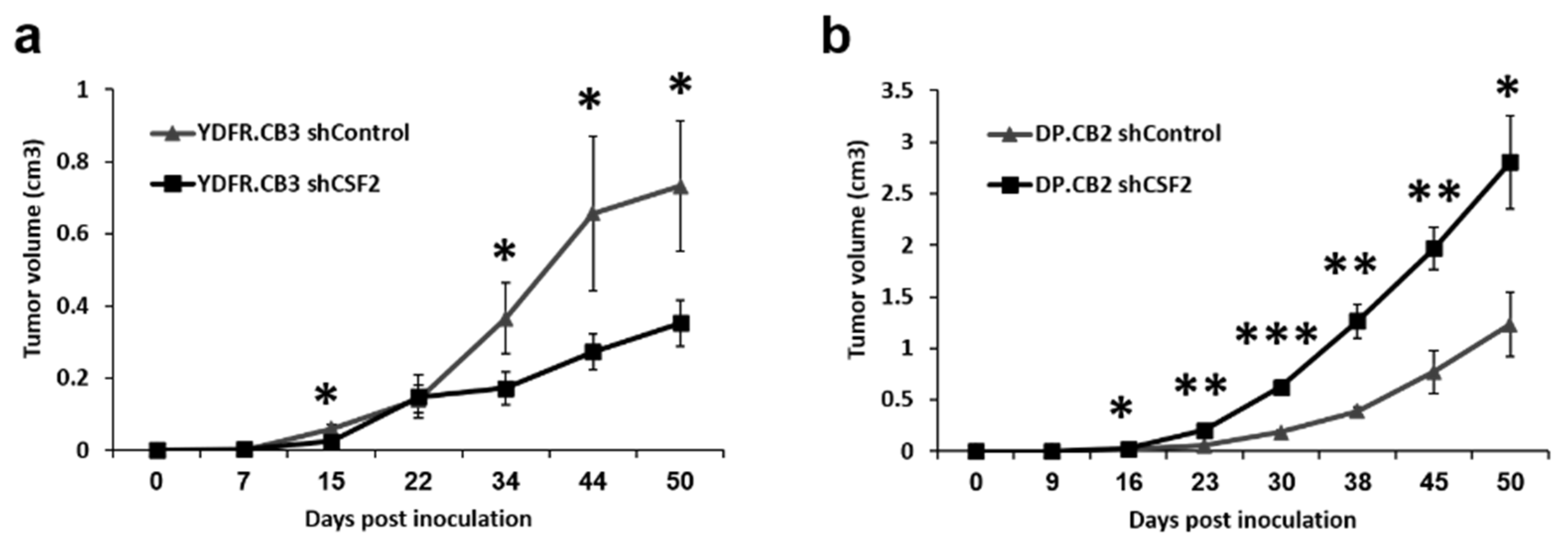
3.6.2. GM-CSF Either Promotes or Inhibits Local Tumor Formation by Melanoma Cells
3.6.3. GM-CSF May Impact Melanoma Brain Metastasis
- GM-CSF exerts regulatory functions on the metastatic microenvironment of the brain.
- GM-CSF impacts differently the malignant phenotype of melanoma cells from different patients; augmenting the malignancy of one melanoma while restraining the malignancy of another.
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Boire, A.; Brastianos, P.K.; Garzia, L.; Valiente, M. Brain metastasis. Nat. Rev. Cancer 2019, 20, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Oliva, I.G.; Tawbi, H.; Davies, M.A. Melanoma Brain Metastases. Cancer J. 2017, 23, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Boil. 2020, 60, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Margolin, K. What is new on the horizon in melanoma brain metastasis? ESMO Open 2019, 4, e000579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, I.J. The role of the organ microenvironment in brain metastasis. Semin. Cancer Boil. 2011, 21, 107–112. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.; Schwartz, H.; Sagi-Assif, O.; Meshel, T.; Izraely, S.; Ben Menachem, S.; Bengaiev, R.; Ben-Shmuel, A.; Nahmias, C.; Couraud, P.-O.; et al. Astrocytes facilitate melanoma brain metastasis via secretion of IL-23. J. Pathol. 2015, 236, 116–127. [Google Scholar] [CrossRef]
- Moshe, A.; Izraely, S.; Sagi-Assif, O.; Prakash, R.; Telerman, A.; Meshel, T.; Carmichael, T.; Witz, I.P. Cystatin C takes part in melanoma-microglia cross-talk: Possible implications for brain metastasis. Clin. Exp. Metastasis 2018, 35, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Izraely, S.; Ben-Menachem, S.; Sagi-Assif, O.; Telerman, A.; Zubrilov, I.; Ashkenazi, O.; Meshel, T.; Maman, S.; Orozco, J.I.J.; Salomon, M.P.; et al. The metastatic microenvironment: Melanoma-microglia cross-talk promotes the malignant phenotype of melanoma cells. Int. J. Cancer 2018, 144, 802–817. [Google Scholar] [CrossRef] [Green Version]
- Izraely, S.; Sagi-Assif, O.; Klein, A.; Meshel, T.; Tsarfaty, G.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.-O.; Ateh, E.; Bryant, J.L.; et al. The metastatic microenvironment: Brain-residing melanoma metastasis and dormant micrometastasis. Int. J. Cancer 2011, 131, 1071–1082. [Google Scholar] [CrossRef]
- Neuditschko, B.; Janker, L.; Niederstaetter, L.; Brunmair, J.; Krivanek, K.; Izraely, S.; Sagi-Assif, O.; Meshel, T.; Keppler, B.K.; Del Del Favero, G.; et al. The Challenge of Classifying Metastatic Cell Properties by Molecular Profiling Exemplified with Cutaneous Melanoma Cells and Their Cerebral Metastasis from Patient Derived Mouse Xenografts. Mol. Cell. Proteom. 2019, 19, 478–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzese, D.M.; Witz, I.P.; Kelly, D.F.; Hoon, D.S. Epigenomic landscape of melanoma progression to brain metastasis: Unexplored therapeutic alternatives. Epigenomics 2015, 7, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Izraely, S.; Ben-Menachem, S.; Sagi-Assif, O.; Meshel, T.; Marzese, D.M.; Ohe, S.; Zubrilov, I.; Pasmanik-Chor, M.; Hoon, D.S.; Witz, I.P. ANGPTL4 promotes the progression of cutaneous melanoma to brain metastasis. Oncotarget 2017, 8, 75778–75796. [Google Scholar] [CrossRef]
- Klein, A.; Sagi-Assif, O.; Meshel, T.; Telerman, A.; Izraely, S.; Ben-Menachem, S.; Bayry, J.; Marzese, D.M.; Ohe, S.; Hoon, D.S.; et al. CCR4 is a determinant of melanoma brain metastasis. Oncotarget 2017, 8, 31079–31091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, R.; Izraely, S.; Thareja, N.S.; Lee, R.; Rappaport, M.; Kawaguchi, R.; Sagi-Assif, O.; Ben-Menachem, S.; Meshel, T.; Machnicki, M.; et al. Regeneration Enhances Metastasis: A Novel Role for Neurovascular Signaling in Promoting Melanoma Brain Metastasis. Front. Mol. Neurosci. 2019, 13, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izraely, S.; Sagi-Assif, O.; Klein, A.; Meshel, T.; Ben-Menachem, S.; Zaritsky, A.; Ehrlich, M.; Prieto, V.G.; Bar-Eli, M.; Pirker, C.; et al. The metastatic microenvironment: Claudin-1 suppresses the malignant phenotype of melanoma brain metastasis. Int. J. Cancer 2014, 136, 1296–1307. [Google Scholar] [CrossRef]
- Izraely, S.; Klein, A.; Sagi-Assif, O.; Meshel, T.; Tsarfaty, G.; Hoon, D.S.; Witz, I.P. Chemokine–chemokine receptor axes in melanoma brain metastasis. Immunol. Lett. 2010, 130, 107–114. [Google Scholar] [CrossRef]
- Wang, J.; Hirose, H.; Du, G.-H.; Chong, K.; Kiyohara, E.; Witz, I.P.; Hoon, D.S. P-REX1 amplification promotes progression of cutaneous melanoma via the PAK1/P38/MMP-2 pathway. Cancer Lett. 2017, 407, 66–75. [Google Scholar] [CrossRef]
- Weksler, B.B.; Subileau, E.A.; Perriere, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinform. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinform. 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pozdniakova, S.; Ladilov, Y. Functional Significance of the Adcy10-Dependent Intracellular cAMP Compartments. J. Cardiovasc. Dev. Dis. 2018, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.L.; Buck, J.; Levin, L.R.; Winger, R.C.; Wang, J.; Arase, H.; Muller, W.A. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J. Exp. Med. 2015, 212, 1021–1041. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Xie, B.; Wu, G.; Hu, J.; Wang, D.; Cai, X.; Li, J. Interleukin-37: The Effect of Anti-Inflammatory Response in Human Coronary Artery Endothelial Cells. Mediat. Inflamm. 2019, 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Hu, Y.; Jin, J.; Yu, Y.; Zhang, S.; Cao, J.; Zhai, Y.; Wei, R.; Shou, J.; Cai, W.; et al. Interleukin 37 promotes angiogenesis through TGF-β signaling. Sci. Rep. 2017, 7, 6113. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.-R.; Shin, H.-J.; Kim, J.-Y.; Byun, H.-J.; Lee, J.H.; Sung, Y.K.; Rho, S.B. Dickkopf-1 (DKK-1) interrupts FAK/PI3K/mTOR pathway by interaction of carbonic anhydrase IX (CA9) in tumorigenesis. Cell. Signal. 2012, 24, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpashneh, E.; Poggioli, T.; Sarathchandra, P.; Lexow, J.; Monassier, L.; Terracciano, C.; Lang, F.; Damilano, F.; Zhou, J.Q.; Rosenzweig, A.; et al. Ablation of SGK1 Impairs Endothelial Cell Migration and Tube Formation Leading to Decreased Neo-Angiogenesis Following Myocardial Infarction. PLoS ONE 2013, 8, e80268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royet, A.; Broutier, L.; Coissieux, M.-M.; Malleval, C.; Gadot, N.; Maillet, D.; Gratadou-Hupon, L.; Bernet, A.; Nony, P.; Treilleux, I.; et al. Ephrin-B3 supports glioblastoma growth by inhibiting apoptosis induced by the dependence receptor EphA4. Oncotarget 2017, 8, 23750–23759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zou, J.; Li, B.; Wang, Y.; Wang, D.; Hao, Y.; Ke, X.; Li, X. RUNX3 modulates hypoxia-induced endothelial-to-mesenchymal transition of human cardiac microvascular endothelial cells. Int. J. Mol. Med. 2017, 40, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane proteins of the tight junctions at the blood–brain barrier: Structural and functional aspects. Semin. Cell Dev. Boil. 2015, 38, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.S.; Yang, Y.M.; Zhang, H.; Tian, L.; Jiang, J.S.; Dong, Y.B.; Zhang, K.; Li, B.; Zhao, W.D.; Fang, W.G.; et al. Intracerebral GM-CSF contributes to transendothelial monocyte migration in APP/PS1 Alzheimer’s disease mice. Br. J. Pharmacol. 2016, 36, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, S.; Zhang, J.; Liu, D.; Wei, J.; Fang, W.; Zhao, W.; Chen, Y.; Shang, D. ZO-1 expression is suppressed by GM-CSF via miR-96/ERG in brain microvascular endothelial cells. Br. J. Pharmacol. 2017, 38, 809–822. [Google Scholar] [CrossRef]
- Moynagh, P. The interleukin-1 signalling pathway in astrocytes: A key contributor to inflammation in the brain. J. Anat. 2005, 207, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Mierke, C.T. Role of the Endothelium during Tumor Cell Metastasis: Is the Endothelium a Barrier or a Promoter for Cell Invasion and Metastasis? J. Biophys. 2009, 2008, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Francisco-Cruz, A.; Aguilar-Santelises, M.; Ramos-Espinosa, O.; Mata-Espinosa, D.; Marquina-Castillo, B.; Barrios-Payan, J.; Hernández-Pando, R. Granulocyte–macrophage colony-stimulating factor: Not just another haematopoietic growth factor. Med. Oncol. 2013, 31, 774. [Google Scholar] [CrossRef]
- Aliper, A.M.; Frieden-Korovkina, V.P.; Buzdin, A.; Roumiantsev, S.; Zhavoronkov, A. A role for G-CSF and GM-CSF in nonmyeloid cancers. Cancer Med. 2014, 3, 737–746. [Google Scholar] [CrossRef]
- Belardelli, F. Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunol. 2002, 23, 201–208. [Google Scholar] [CrossRef]
- Hong, I.-S. Stimulatory versus suppressive effects of GM-CSF on tumor progression in multiple cancer types. Exp. Mol. Med. 2016, 48, e242. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Olson, O.C.; Bhardwaj, P.; Walsh, L.; Akkari, L.; Quick, M.L.; Chen, I.-C.; Wendel, N.; Ben-Chetrit, N.; Walker, J.; et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat. Cell Biol. 2017, 19, 974–987. [Google Scholar] [CrossRef]
- Hoeller, C.; Michielin, O.; Ascierto, P.A.; Szabo, Z.; Blank, C.U. Systematic review of the use of granulocyte-macrophage colony-stimulating factor in patients with advanced melanoma. Cancer Immunol. Immunother. 2016, 65, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Klein-Goldberg, A.; Maman, S.; Witz, I.P. The role played by the microenvironment in site-specific metastasis. Cancer Lett. 2014, 352, 54–58. [Google Scholar] [CrossRef]
- Cusnir, M.; Cavalcante, L. Inter-tumor heterogeneity. Hum. Vaccines Immunother. 2012, 8, 1143–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilkovitch, D.; Handel-Fernandez, M.E.; Herbert, L.M.; Lopez, D.M. Antitumor Effects of Mucin 1/sec Involves the Modulation of Urokinase-Type Plasminogen Activator and Signal Transducer and Activator of Transcription 1 Expression in Tumor Cells. Cancer Res. 2008, 68, 2427–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, J. Melanoma exosome induction of endothelial cell GM-CSF in pre-metastatic lymph nodes may result in different M1 and M2 macrophage mediated angiogenic processes. Med. Hypotheses 2016, 94, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | FC | p-Value |
|---|---|---|---|
| Upregulated | |||
| LYPD3 | LY6/PLAUR domain containing 3 | 7.62 | 0.000944 |
| HPD | 4-hydroxyphenylpyruvate dioxygenase | 5.26 | 0.000679 |
| CA9 | carbonic anhydrase 9 | 4.18 | 7.42 × 10−5 |
| MIR3124 | microRNA 3124 | 4.02 | 5.36 × 10−6 |
| PNPLA1 | patatin like phospholipase domain containing 1 | 3.84 | 7.94 × 10−6 |
| CGB7 | chorionic gonadotropin subunit beta 7 | 3.71 | 0.000861 |
| PCDHGA7 | protocadherin gamma subfamily A, 7 | 3.66 | 0.000385 |
| LINC02310 | long intergenic non-protein coding RNA 2310 | 3.63 | 0.000158 |
| ADCY10 | adenylate cyclase 10 | 3.01 | 1.1 × 10−5 |
| DIPK2B | divergent protein kinase domain 2B | 2.94 | 2.01 × 10−5 |
| SGK2 | serum/glucocorticoid regulated kinase 2 | 2.84 | 0.000594 |
| GLS2 | glutaminase 2 | 2.82 | 0.000387 |
| CASKIN1 | CASK interacting protein 1 | 2.62 | 0.000489 |
| LY6G5C | lymphocyte antigen 6 family member G5C | 2.42 | 0.000438 |
| PRSS27 | serine protease 27 | 2.40 | 0.000651 |
| ARHGAP40 | Rho GTPase activating protein 40 | 2.38 | 0.00044 |
| ARHGAP9 | Rho GTPase activating protein 9 | 2.15 | 0.000554 |
| SLC17A7 | solute carrier family 17 member 7 | 2.12 | 0.00088 |
| peptide deformylase, mitochondrial | 2.07 | 0.000363 | |
| RBM44 | RNA binding motif protein 44 | 2.02 | 0.000415 |
| ALLC | allantoicase | 2.01 | 5.31 × 10−6 |
| Downregulated | |||
| RAB11FIP4 | RAB11 family interacting protein 4 | −6.13 | 0.000349 |
| CES4A | carboxylesterase 4A | −4.98 | 6.78 × 10−5 |
| KHDRBS3 | KH RNA binding domain containing, signal transduction associated 3 | −4.41 | 0.000557 |
| EFNB3 | ephrin B3 | −3.99 | 8.79 × 10−5 |
| RPL21P28 | ribosomal protein L21 pseudogene 28 | −2.80 | 0.000986 |
| LINC00954 | long intergenic non-protein coding RNA 954 | −2.71 | 0.000508 |
| IRS2 | insulin receptor substrate 2 | −2.53 | 0.000306 |
| TSHZ3 | teashirt zinc finger homeobox 3 | −2.51 | 0.000497 |
| CSGALNACT1 | chondroitin sulfate N-acetylgalactosaminyltransferase 1 | −2.41 | 7.75 × 10−5 |
| KREMEN1 | kringle containing transmembrane protein 1 | −2.37 | 0.000685 |
| IL37 | interleukin 37 | −2.36 | 2 × 10−6 |
| RUNX3 | RUNX family transcription factor 3 | −2.33 | 0.000937 |
| ZFP37 | ZFP37 zinc finger protein | −2.06 | 0.000857 |
| YDFR.CB3 | DP.CB2 | ||
|---|---|---|---|
| GM-CSF expression | Expression in cutaneous vs. MBM 1 variants | Higher in the metastatic variant | Higher in the metastatic variant (NS 2) |
| Expression in MG-CM 3-treated MBM | Down-regulation | Not altered | |
| Expression in BEC-CM 4-treated MBM | Not altered | Down-regulation | |
| Expression in HA-CM 5-treated MBM | Not altered | Up-regulation | |
| IL-1α 6 treatment | Up-regulation | Up-regulation | |
| TNF-α 7 treatment | Up-regulation | Up-regulation | |
| IL-1α + MG-CM treatment | Down-regulation | NA 8 | |
| TNF-α + MG-CM treatment | Down-regulation | NA | |
| in-vitro function of GM-CSF | Adhesion to BEC | Decreased | Not altered |
| TEM 9 in the presence of rhGM-CSF 10 | Increased | Not altered | |
| TEM effect of shCSF2 11 | Decreased | Increased | |
| TEM MBM + rhGM-CSF | Not altered | Not altered | |
| TEM BEC + rhGM-CSF | Increased | Increased | |
| in-vivo function of GM-CSF | Tumorigenesis | Increased | Decreased |
| Brain metastasis | Increased | Decreased |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moshe, A.; Izraely, S.; Sagi-Assif, O.; Malka, S.; Ben-Menachem, S.; Meshel, T.; Pasmanik-Chor, M.; Hoon, D.S.B.; Witz, I.P. Inter-Tumor Heterogeneity—Melanomas Respond Differently to GM-CSF-Mediated Activation. Cells 2020, 9, 1683. https://doi.org/10.3390/cells9071683
Moshe A, Izraely S, Sagi-Assif O, Malka S, Ben-Menachem S, Meshel T, Pasmanik-Chor M, Hoon DSB, Witz IP. Inter-Tumor Heterogeneity—Melanomas Respond Differently to GM-CSF-Mediated Activation. Cells. 2020; 9(7):1683. https://doi.org/10.3390/cells9071683
Chicago/Turabian StyleMoshe, Adi, Sivan Izraely, Orit Sagi-Assif, Sapir Malka, Shlomit Ben-Menachem, Tsipi Meshel, Metsada Pasmanik-Chor, Dave S.B. Hoon, and Isaac P. Witz. 2020. "Inter-Tumor Heterogeneity—Melanomas Respond Differently to GM-CSF-Mediated Activation" Cells 9, no. 7: 1683. https://doi.org/10.3390/cells9071683