
siRNA Nanoparticle Targeting PD-L1 Activates Tumor Immunity and Abrogates Pancreatic Cancer Growth in Humanized Preclinical Model

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of siPD-L1@PLGA NPs
2.2. Derivation of Primary Pancreatic Cancer Cell and Humanized PDX Model
2.3. Cell Culture and FACS
2.4. Antibodies and Reagents
2.5. Cellular Uptake of siPD-L1@PLGA NPs
2.6. Cytotoxicity Study of Scrambled siPD-L1@PLGA NPs on Blue #96 Cells
2.7. Isolation of Splenocytes and CD8+ T cells
2.8. In Vitro Cytolytic Assay of Ovalbumin(OVA)-Specific Cytotoxic T Lymphocytes (CTLs)
2.9. In Vitro Proliferation Study of OVA-Specific CTLs
2.10. Production of IFN-γ in Tumor Antigen-Stimulated CTLs
2.11. siPD-L1@PLGA NPs Treatment and Analysis of Tumor-Infiltrated Immune Cells in Humanized NSG Model
2.12. Measurement of Lymphocyte-Mediated Cytotoxicity from Tumor-Bearing Mouse
2.13. Multiplexed Fluorescent Immunohistochemistry
2.14. Multispectral Imaging and Analysis
2.15. DLS Analysis of siRNA@PLGA NPs
2.16. Statistics
3. Results
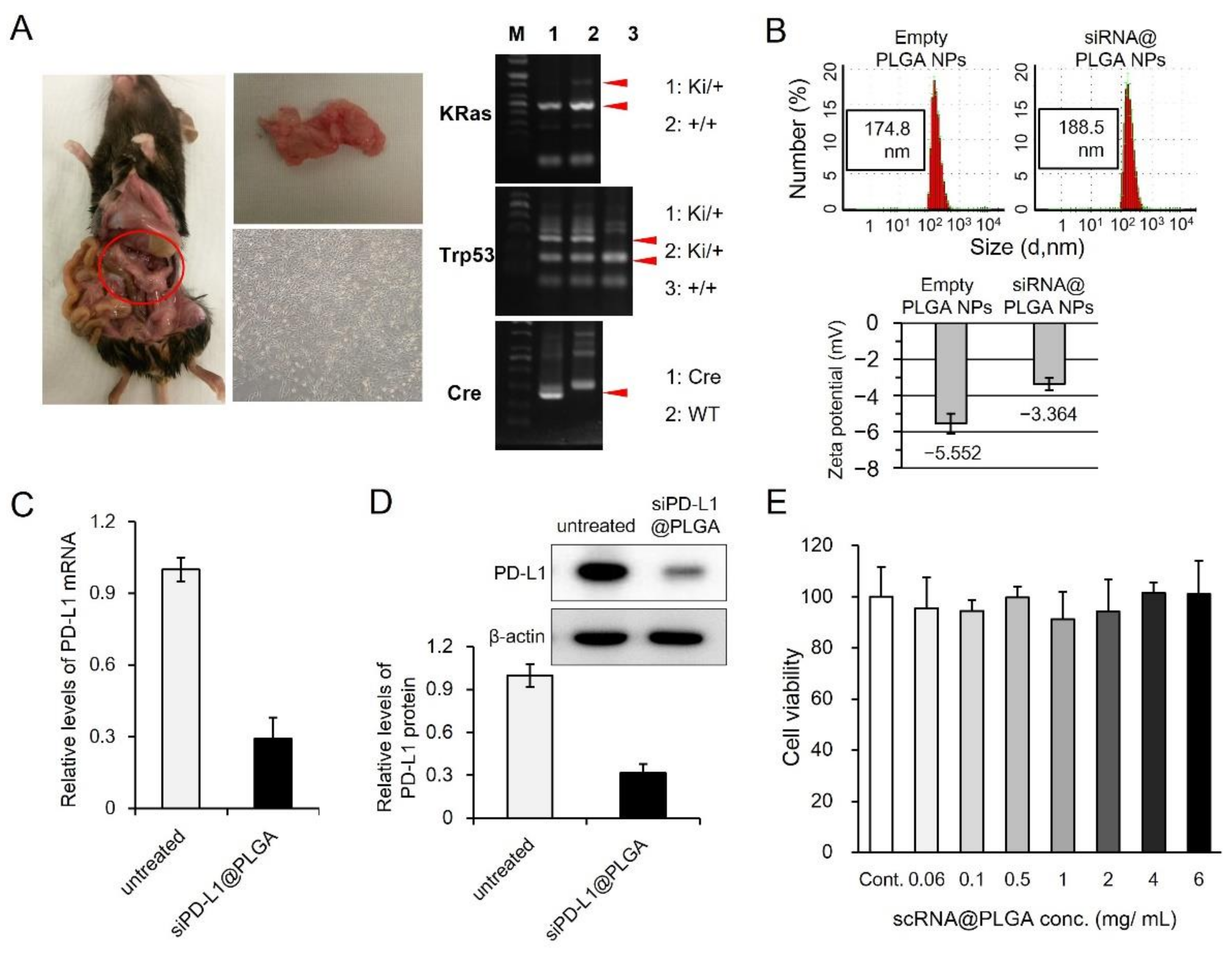
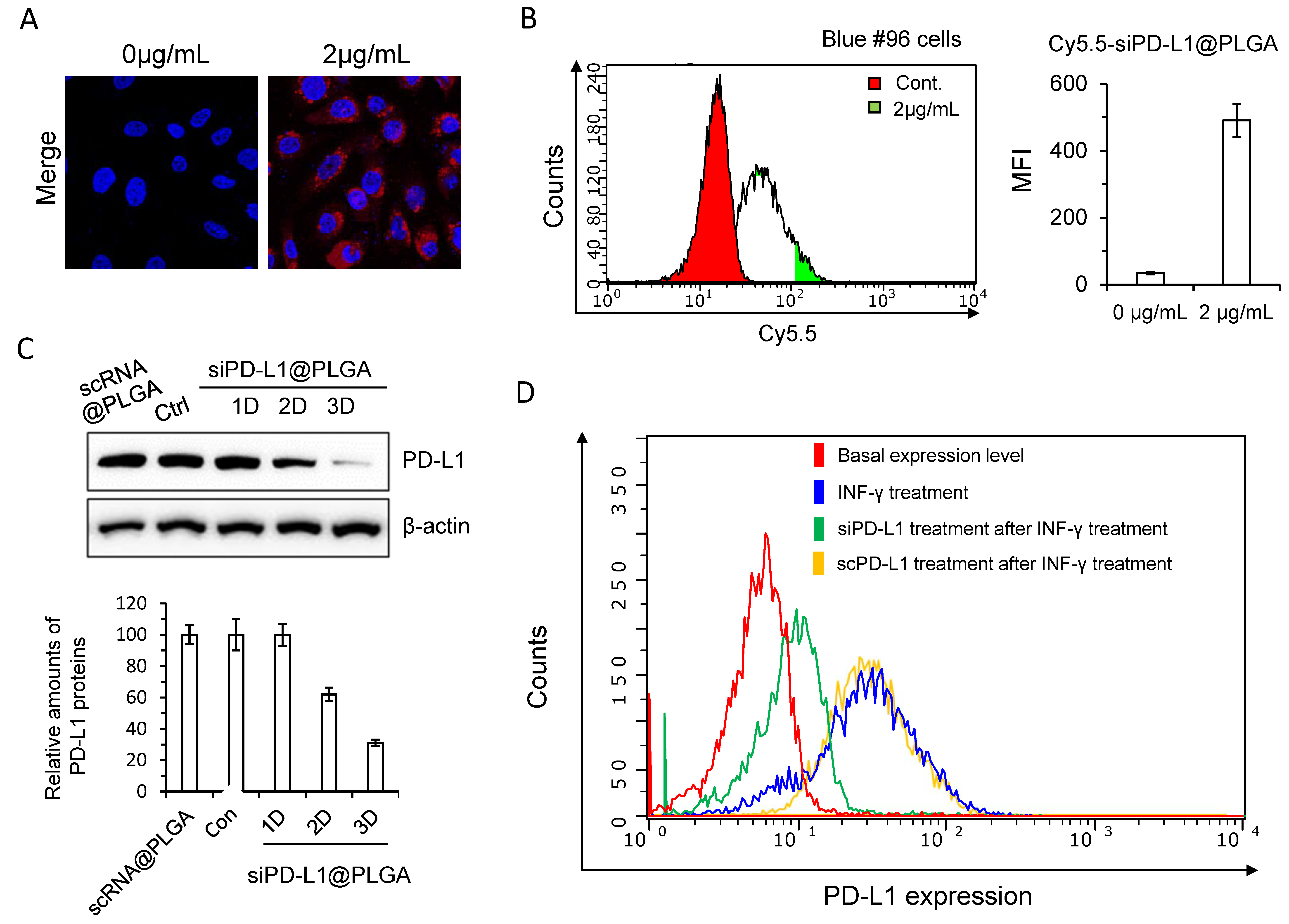
3.1. Synthesis of siRNA Nanoparticles Targeting PD-L1 and In Vitro Validation
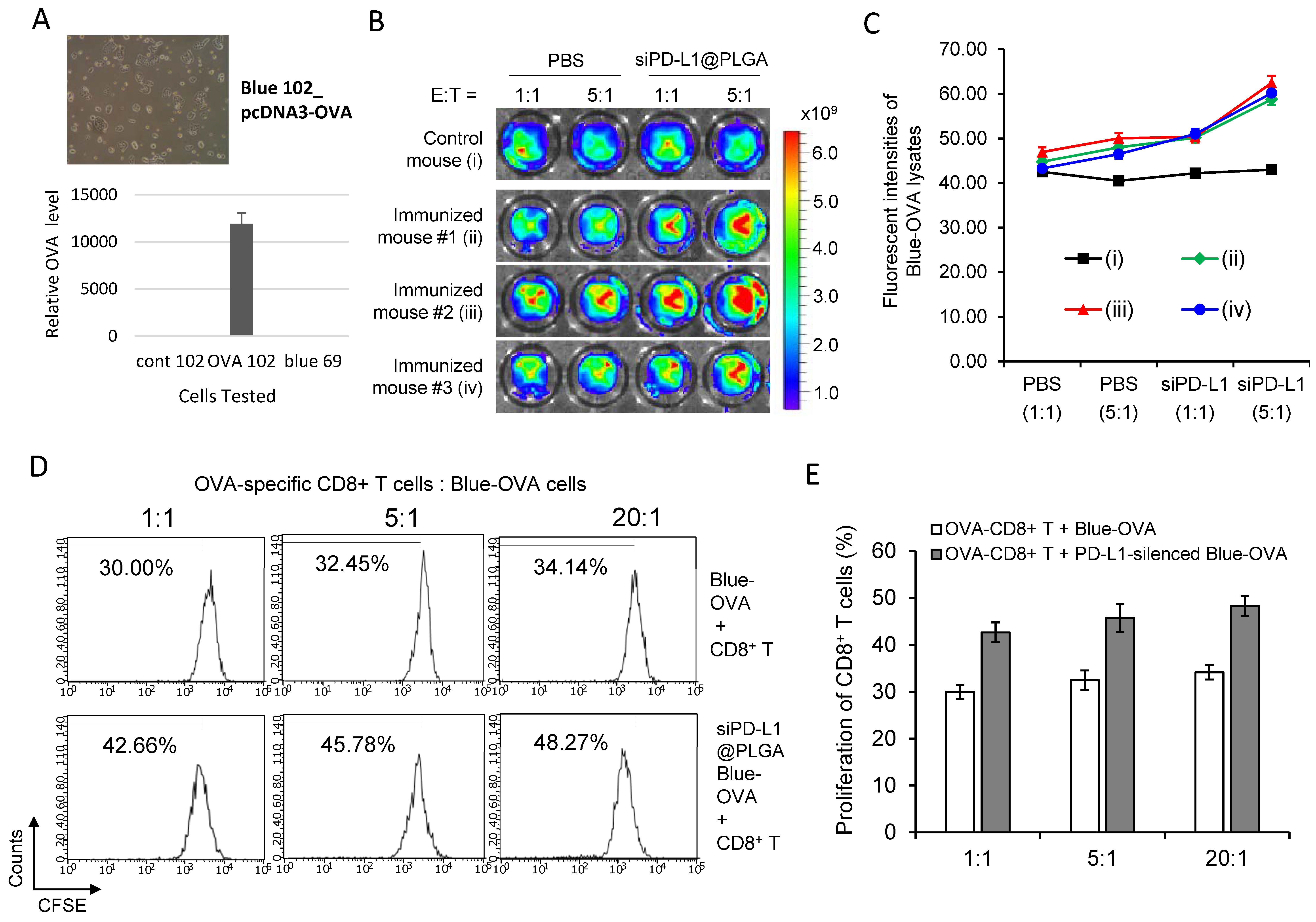
3.2. siPD-L1@PLGA Abrogates Immune Escape Function of Pancreatic Tumor Cells
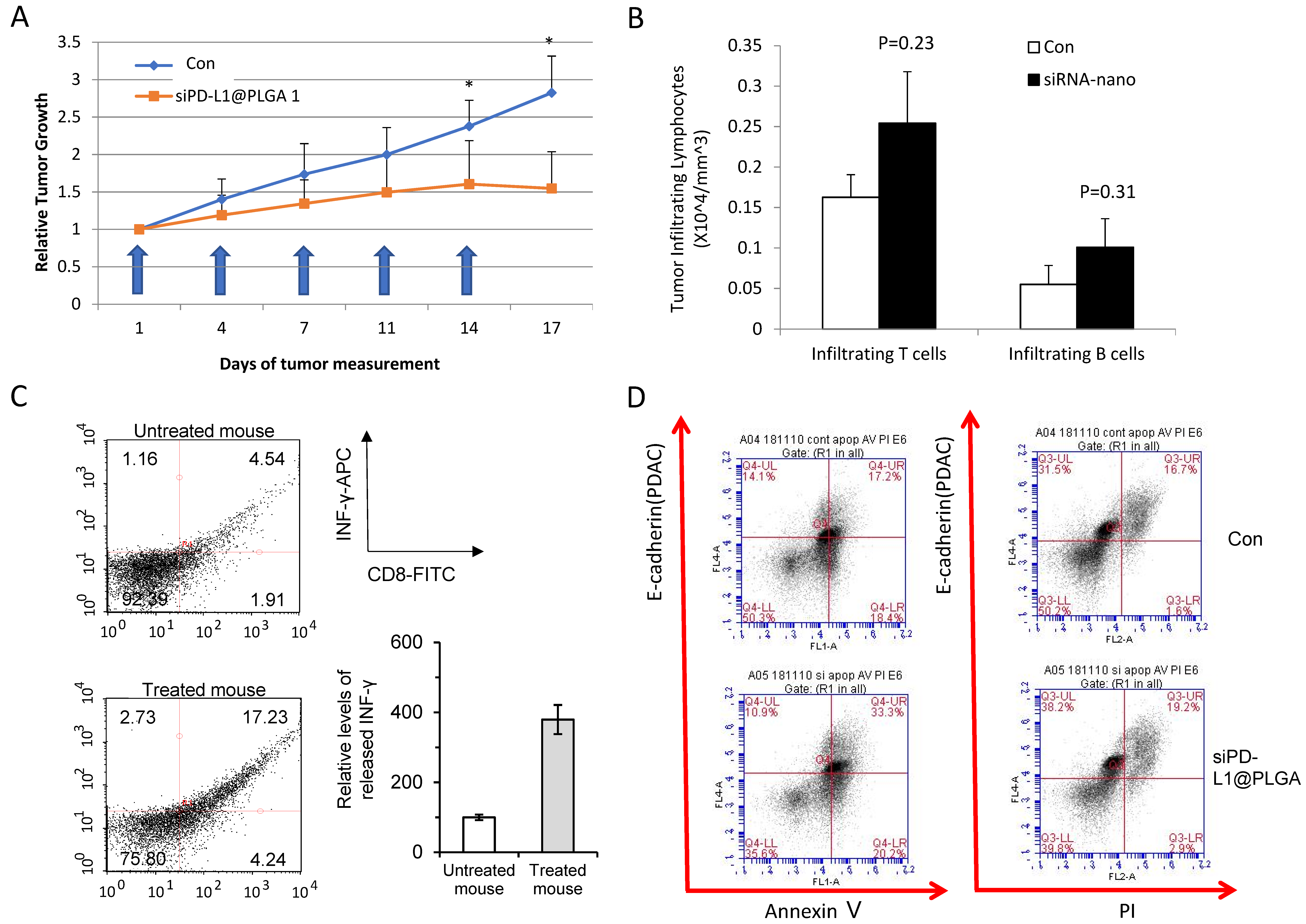
3.3. Efficacy Test in Humanized PDX Model Reveals Antitumor Effect of siPD-L1@PLGA via Immunomodulation
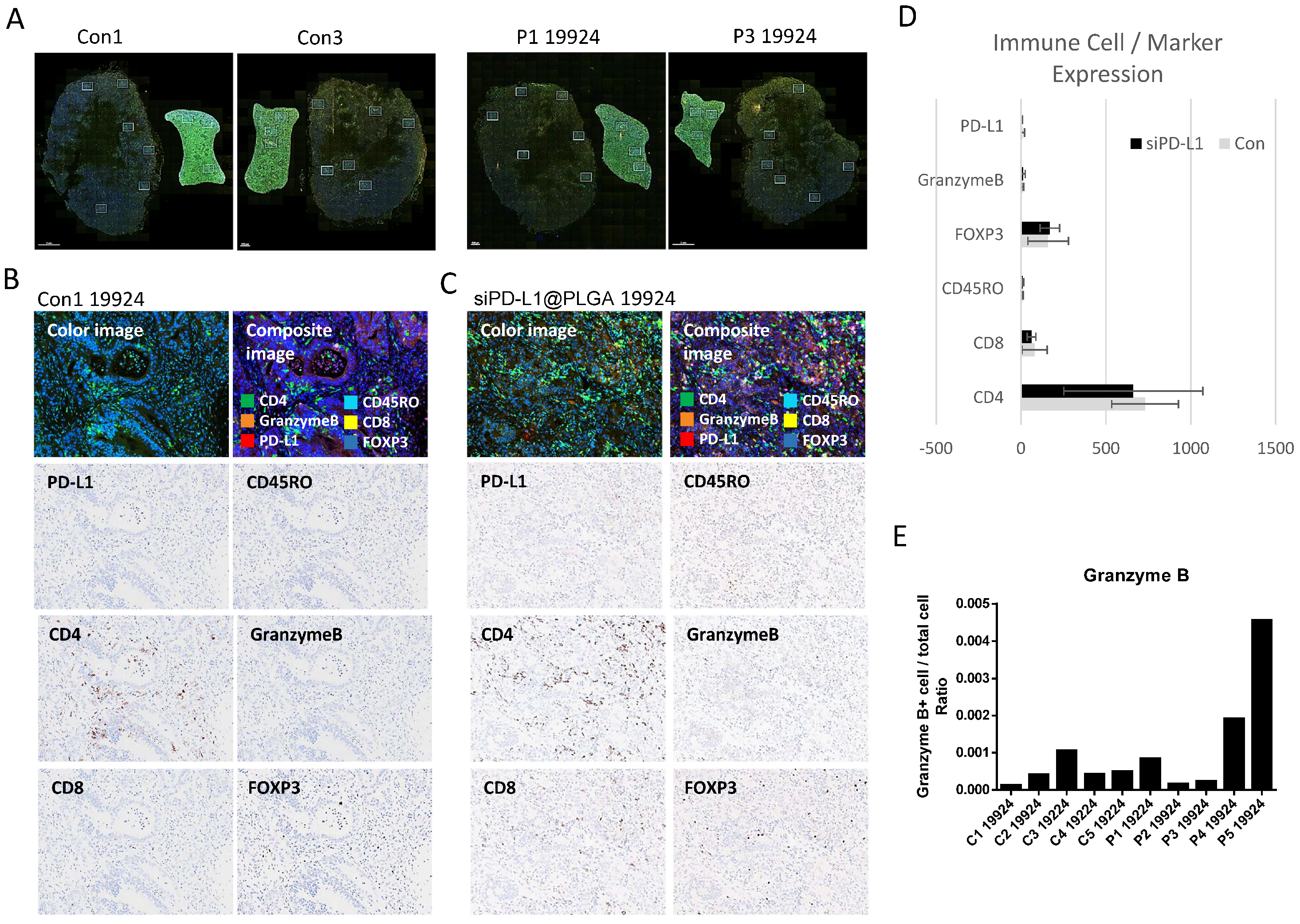
3.4. OPAL Multiplex IHC Analysis Reveals Granzyme B Upregulation in siPD-L1@PLGA-Treated Tumors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, F.; Jin, C.; Fu, D.L.; Warshaw, A.L. Modified FOLFIRINOX for resected pancreatic cancer: Opportunities and challenges. World J. Gastroenterol. 2019, 25, 2839–2845. [Google Scholar] [CrossRef]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.A. Immunosuppressive betaig-h3 links tumour stroma and dysfunctional T cells in pancreatic cancer. Gut 2019, 68, 581. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.K. The Pancreatic Cancer Microenvironment. Cancer J. 2017, 23, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Leinwand, J.; Miller, G. Regulation and modulation of antitumor immunity in pancreatic cancer. Nat. Immunol. 2020, 21, 1152–1159. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Wu, J.; Cai, J. Dilemma and Challenge of Immunotherapy for Pancreatic Cancer. Dig. Dis. Sci. 2021, 66, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug. Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Wang, C.; Shi, X.; Song, H.; Zhang, C.; Wang, X.; Huang, P.; Dong, A.; Zhang, Y.; Kong, D.; Wang, W. Polymer-lipid hybrid nanovesicle-enabled combination of immunogenic chemotherapy and RNAi-mediated PD-L1 knockdown elicits antitumor immunity against melanoma. Biomaterials 2021, 268, 120579. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Jordan, V.C.; Sheedy, P.; Billig, A.M.; Ross, A.; Pantazopoulos, P.; Medarova, Z. RNAi-Mediated PD-L1 Inhibition for Pancreatic Cancer Immunotherapy. Sci. Rep. 2019, 9, 4712. [Google Scholar] [CrossRef]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Panyam, J.; Zhou, W.Z.; Prabha, S.; Sahoo, S.K.; Labhasetwar, V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002, 16, 1217–1226. [Google Scholar] [CrossRef]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Perlstein, I. DNA delivery from an intravascular stent with a denatured collagen-polylactic-polyglycolic acid-controlled release coating: Mechanisms of enhanced transfection. Gene Ther. 2003, 10, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Okada, H. One- and three-month release injectable microspheres of the LH-RH superagonist leuprorelin ace tate. Adv. Drug Deliv. Rev. 1997, 28, 43–70. [Google Scholar] [CrossRef]
- Pacheco-Torres, J.; Penet, M.F.; Krishnamachary, B.; Mironchik, Y.; Chen, Z.; Bhujwalla, Z.M. PD-L1 siRNA Theranostics With a Dextran Nanoparticle Highlights the Importance of Nanoparticle Delivery for Effective Tumor PD-L1 Downregulation. Front. Oncol. 2020, 10, 614365. [Google Scholar] [CrossRef]
- Kwak, G.; Kim, D.; Nam, G.H.; Wang, S.Y.; Kim, I.S.; Kim, S.H.; Kwon, I.C.; Yeo, Y. Programmed Cell Death Protein Ligand-1 Silencing with Polyethylenimine-Dermatan Sulfate Complex for Dual Inhibition of Melanoma Growth. ACS Nano 2017, 11, 10135–10146. [Google Scholar] [CrossRef] [Green Version]
- Teo, P.Y.; Yang, C.; Whilding, L.M.; Parente-Pereira, A.C.; Maher, J.; George, A.J.; Hedrick, J.L.; Yang, Y.Y.; Ghaem-Maghami, S. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: Strategies to enhance T cell killing. Adv. Healthc. Mater. 2015, 4, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Jain, T.; Dudeja, V. The war against pancreatic cancer in 2020—Advances on all fronts. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 99–100. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, Z.; Du, X.; Chen, S.; Zhang, W.; Wang, J.; Li, H.; He, X.; Cao, J.; Wang, J. Co-inhibition of the TGF-beta pathway and the PD-L1 checkpoint by pH-responsive clustered nanoparticles for pancreatic cancer microenvironment regulation and anti-tumor immunotherapy. Biomater. Sci. 2020, 8, 5121–5132. [Google Scholar] [CrossRef]
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 67, 1845–1854. [Google Scholar] [CrossRef]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef] [Green Version]
- Risnayanti, C.; Jang, Y.S.; Lee, J.; Ahn, H.J. PLGA nanoparticles co-delivering MDR1 and BCL2 siRNA for overcoming resistance of paclitaxel and cisplatin in recurrent or advanced ovarian cancer. Sci. Rep. 2018, 8, 7498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Lee, C.H.; Seol, H.S.; Choi, Y.S.; Kim, E.; Lee, E.J.; Rhee, J.K.; Singh, S.R.; Jun, E.S.; Han, B.; et al. Generation and molecular characterization of pancreatic cancer patient-derived xenografts reveals their heterologous nature. Oncotarget 2016, 7, 62533–62546. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.T.; Wang, Z.; Kim, M.J.; Jiang, L.S.; Xu, S.J.; Jung, J.; Lee, E.; Park, J.H.; Bakheet, N.; Yoon, S.H.; et al. The Establishment of a Fast and Safe Orthotopic Colon Cancer Model Using a Tissue Adhesive Technique. Cancer Res. Treat. 2021, 53, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Somanchi, S.S.; McCulley, K.J.; Somanchi, A.; Chan, L.L.; Lee, D.A. A Novel Method for Assessment of Natural Killer Cell Cytotoxicity Using Image Cytometry. PLoS ONE 2015, 10, e0141074. [Google Scholar] [CrossRef] [PubMed]
- Delgiorno, K.E.; Hall, J.C.; Takeuchi, K.K.; Pan, F.C.; Halbrook, C.J.; Washington, M.K.; Olive, K.P.; Spence, J.R.; Sipos, B.; Wright, C.V.; et al. Identification and manipulation of biliary metaplasia in pancreatic tumors. Gastroenterology 2014, 146, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, M.; Krebbers, G.; Bekkenk, M.W.; Teunissen, M.B.M.; Luiten, R.M. Improvement of Opal Multiplex Immunofluorescence Workflow for Human Tissue Sections. J. Histochem. Cytochem. 2021, 69, 339–346. [Google Scholar] [CrossRef] [PubMed]
- King, M.A.; Covassin, L.; Brehm, M.A.; Racki, W.; Pearson, T.; Leif, J.; Laning, J.; Fodor, W.; Foreman, O.; Burzenski, L.; et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin. Exp. Immunol. 2009, 157, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Morillon, Y.M.I.; Smalley Rumfield, C.; Pellom, S.T.; Sabzevari, A.; Roller, N.T.; Horn, L.A.; Jochems, C.; Palena, C.; Greiner, J.W.; Schlom, J. The Use of a Humanized NSG-beta2m(-/-) Model for Investigation of Immune and Anti-tumor Effects Mediated by the Bifunctional Immunotherapeutic Bintrafusp Alfa. Front. Oncol. 2020, 10, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, R.; Bridle, K.R.; Cao, L.; Crawford, D.H.G.; Jayachandran, A. Dual Targeting of Sorafenib-Resistant HCC-Derived Cancer Stem Cells. Curr. Oncol. 2021, 28, 200. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Guo, Q.; Fu, H.; Yu, J.; Wang, L.; Sun, Y.; Zhang, J.; Duan, Y. Asynchronous blockade of PD-L1 and CD155 by polymeric nanoparticles inhibits triple-negative breast cancer progression and metastasis. Biomaterials 2021, 275, 120988. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, J.Y.; Ryu, H.J.; Lee, S.-H.; Kim, D.-Y.; Kim, M.J.; Lee, E.J.; Ryu, Y.-M.; Kim, S.-Y.; Kim, K.-P.; Choi, E.Y.; et al. siRNA Nanoparticle Targeting PD-L1 Activates Tumor Immunity and Abrogates Pancreatic Cancer Growth in Humanized Preclinical Model. Cells 2021, 10, 2734. https://doi.org/10.3390/cells10102734
Jung JY, Ryu HJ, Lee S-H, Kim D-Y, Kim MJ, Lee EJ, Ryu Y-M, Kim S-Y, Kim K-P, Choi EY, et al. siRNA Nanoparticle Targeting PD-L1 Activates Tumor Immunity and Abrogates Pancreatic Cancer Growth in Humanized Preclinical Model. Cells. 2021; 10(10):2734. https://doi.org/10.3390/cells10102734
Chicago/Turabian StyleJung, Jae Yun, Hyun Jin Ryu, Seung-Hwan Lee, Dong-Young Kim, Myung Ji Kim, Eun Ji Lee, Yeon-Mi Ryu, Sang-Yeob Kim, Kyu-Pyo Kim, Eun Young Choi, and et al. 2021. "siRNA Nanoparticle Targeting PD-L1 Activates Tumor Immunity and Abrogates Pancreatic Cancer Growth in Humanized Preclinical Model" Cells 10, no. 10: 2734. https://doi.org/10.3390/cells10102734