Adding Size Exclusion Chromatography (SEC) and Light Scattering (LS) Devices to Obtain High-Quality Small Angle X-Ray Scattering (SAXS) Data

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
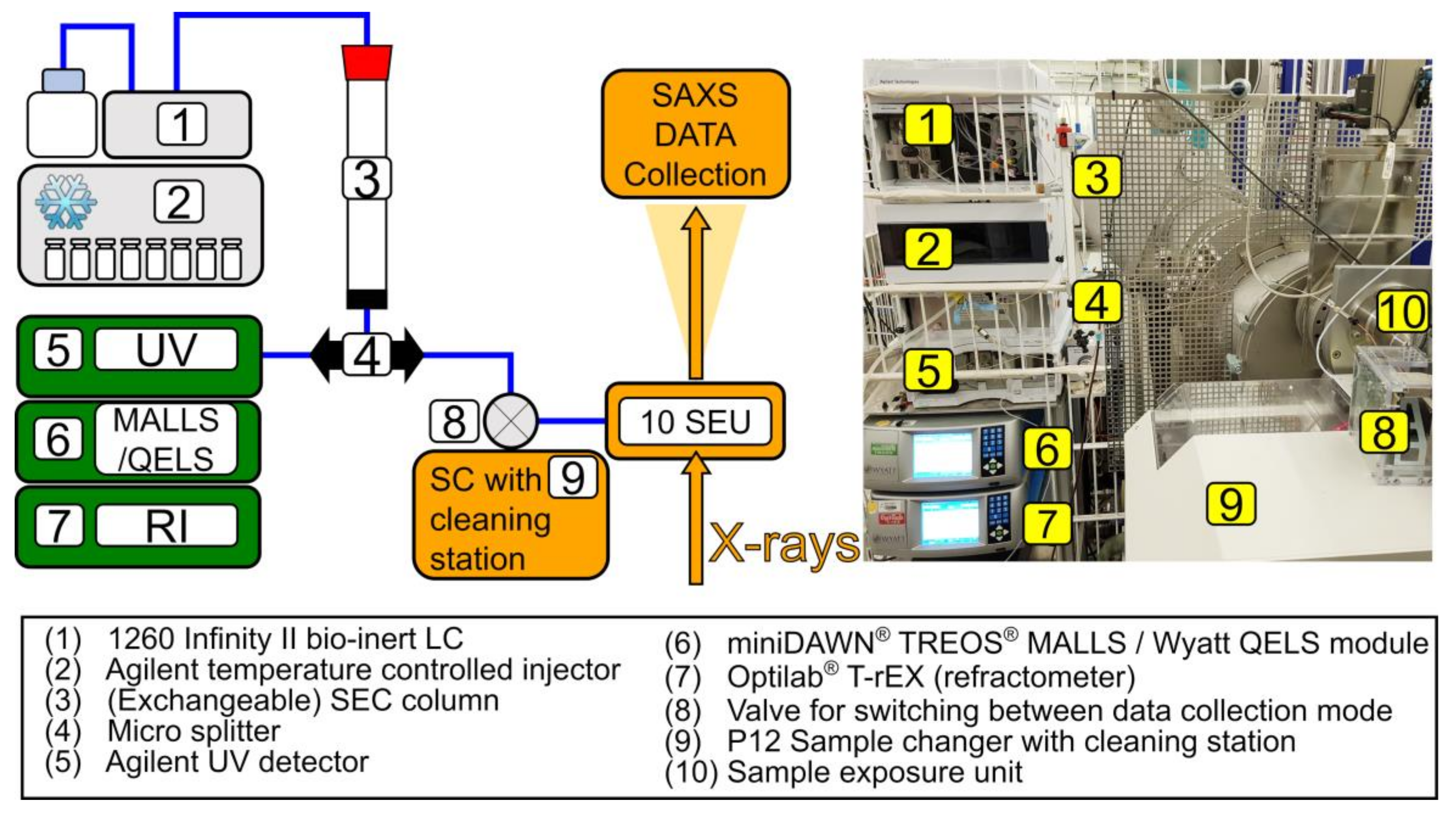
2.2. Experimental Set-Up
2.2.1. Chromatography System
2.2.2. UV, MALLS, and RI Data Collection and Analysis
2.3. SAXS Data Collection and Reduction
2.4. SAXS (Automated) Data Processing and Analysis
2.4.1. Background Subtraction of the SEC-SAXS Data
2.4.2. SAXS Data Analysis
2.4.3. Data Presentation
3. Results
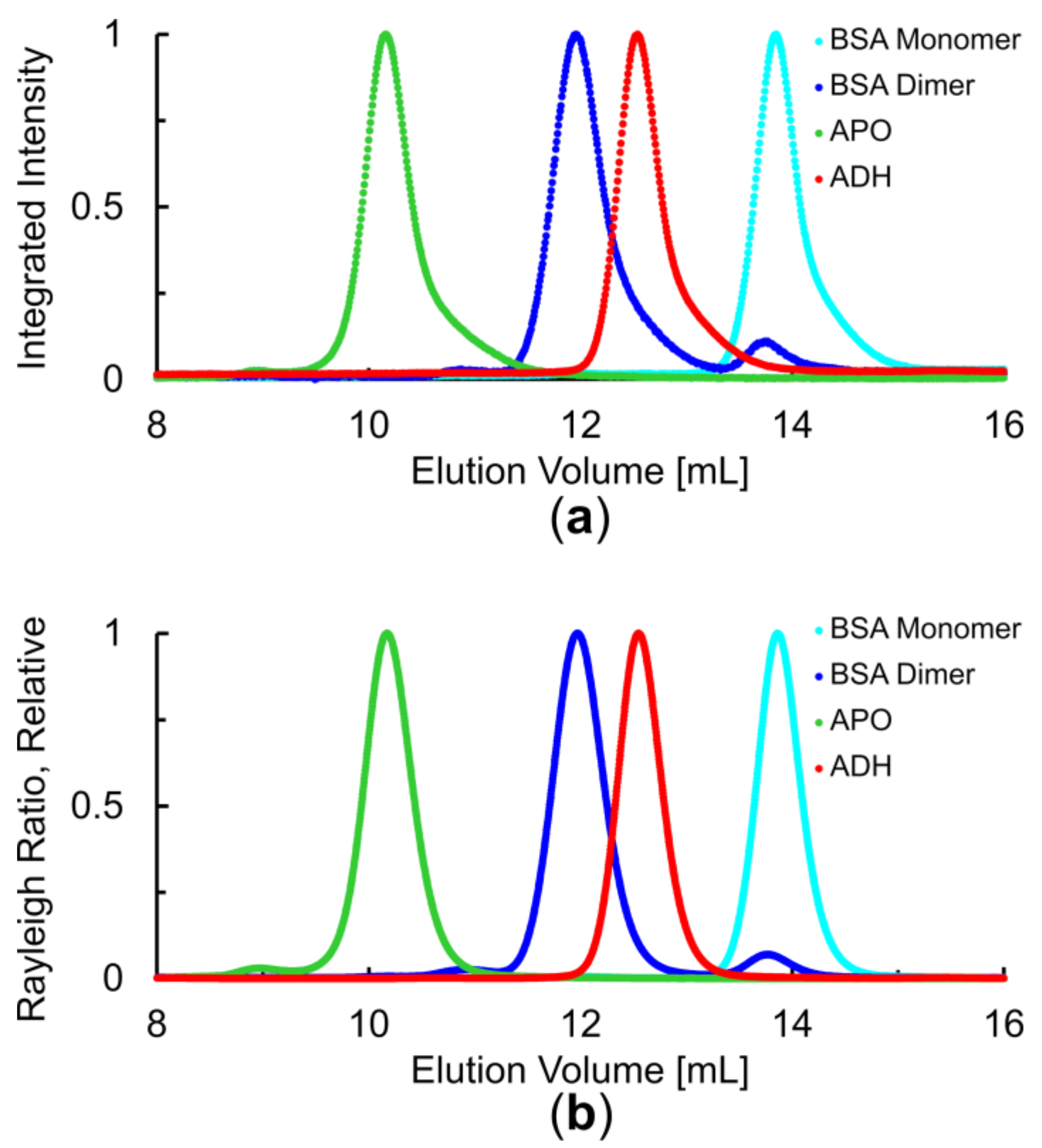
3.1. Overall Assessment of SEC Performance
3.2. Concentration Determinion via UV Absorption and/or dRI
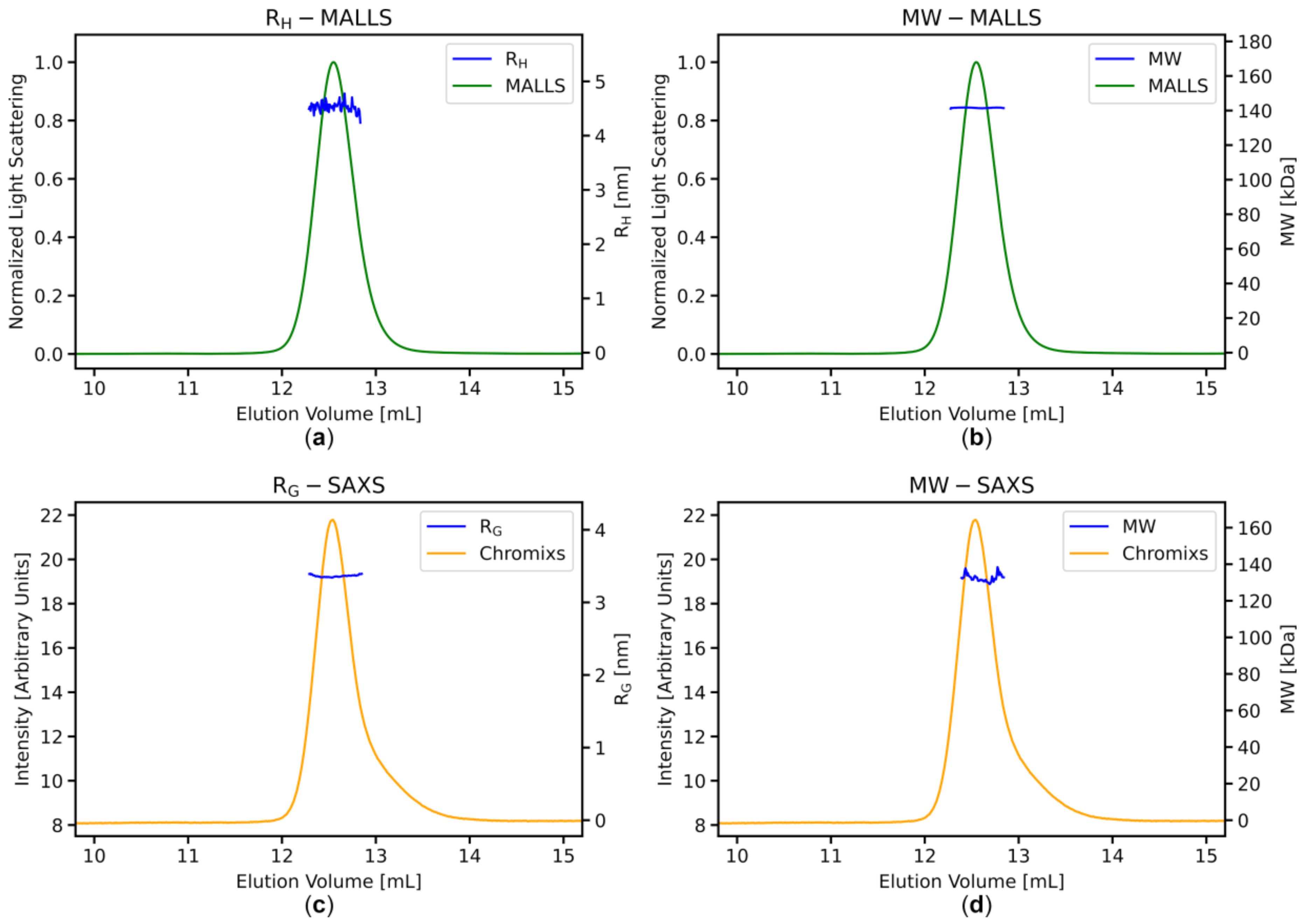
3.3. Stable MWMALLS and RH Estimates Across the Elution Peaks Indicate Homogeneous Sample Populations
3.4. SAXS Data Analysis: Stable RG and MWI(0) Estimates across the Elution Peaks
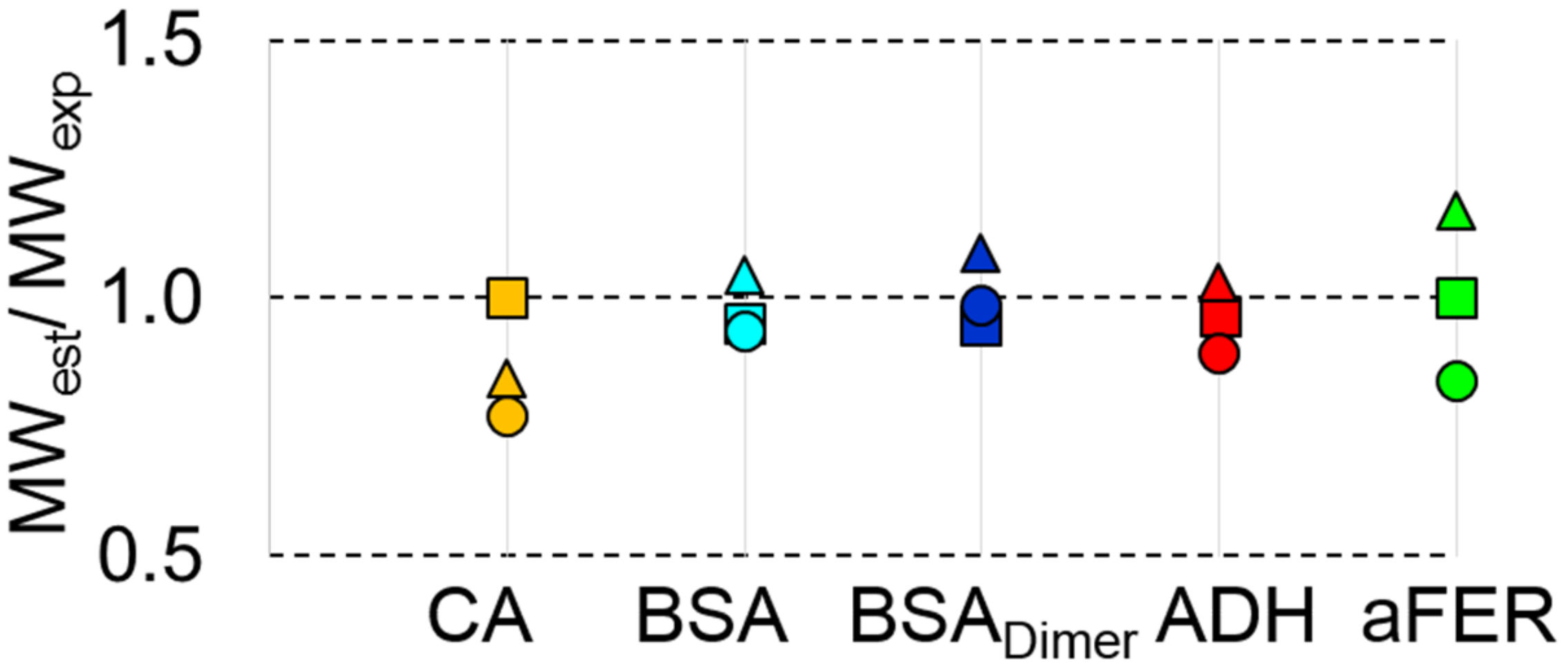
3.5. MWMALLS is the Most Robust Estimate for Determining the Molecular Weight for SEC-SAXS Applications
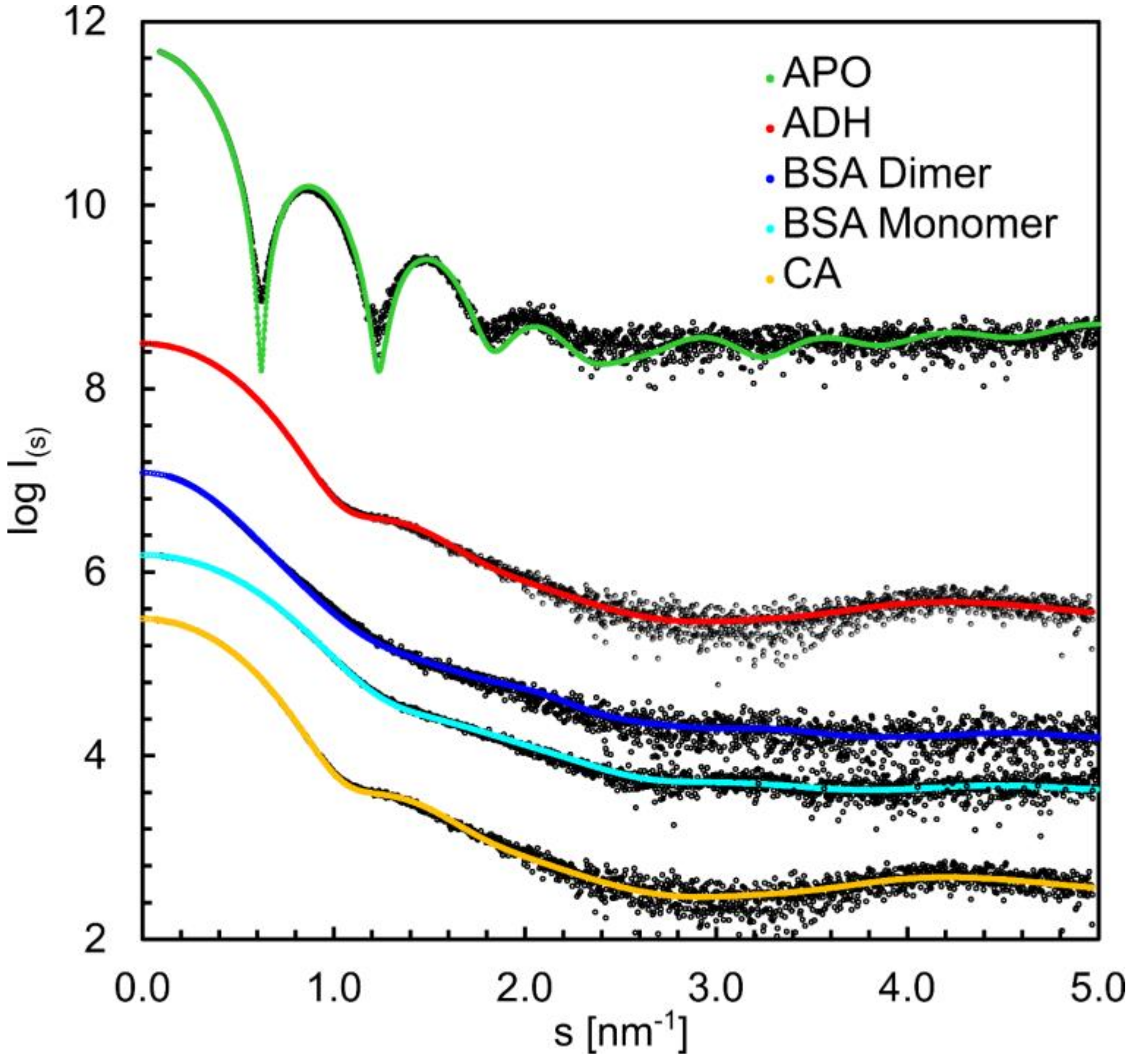
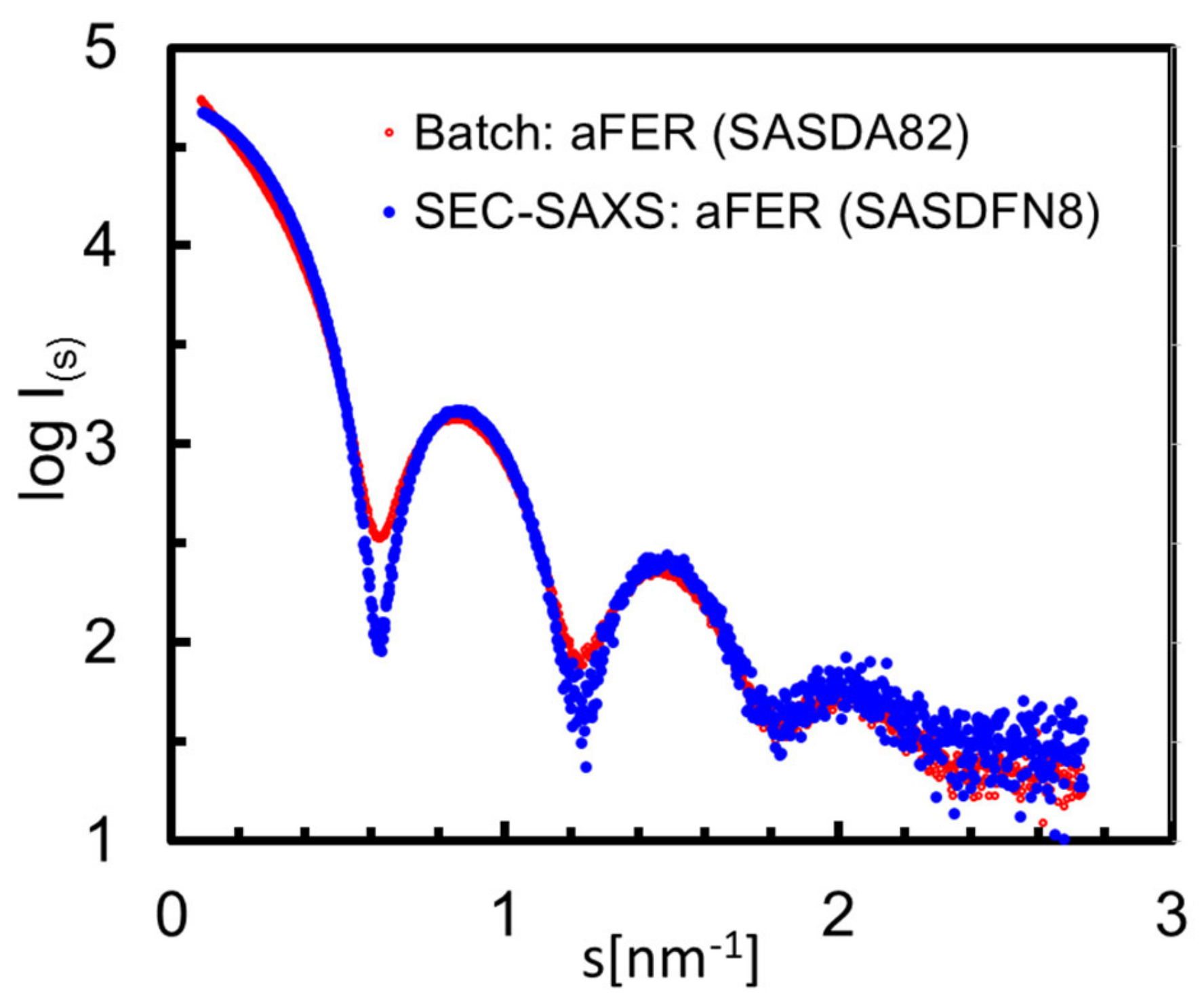
3.6. Structural Information Derived from the Buffer-Subtracted SAXS Scattering Curves
4. Discussion
- (i)
- Upgraded hardware modules such as the automated injector and switching valve for better use of allocated beamtime (the need to physically enter the experimental hutch by the operators is reduced, as several samples can be queued, and washing between samples can be initiated remotely).
- (ii)
- The robust HPLC pumps allow extensive use of columns (also at high pressures). Quaternary solvent blending offers increased flexibility in remote preparation of buffers e.g., varying in ionic strength or pH gradient.
- (iii)
- The additional acquisition of QELS data allows for further assessment of sample homogeneity (stability of RH across elution peak) as well as structural information through the RG/RH ratio.
- (iv)
- Collection of MALLS data at three angles provides more precise MWMALLS estimates.
- (v)
- A combination of software tools and a common beamline-control interface (BEQUEREL) for ease of use and the necessary communication between integrated devices during data collection [22] as well as for data analysis such as CHROMIXS [27] and the new QuickPlot tools described here. Evaluation of data quality “on the spot” can optimize beam time usage, as improving measures can be made for subsequent runs.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tuukkanen, A.T.; Spilotros, A.; Svergun, D.I. Progress in small-angle scattering from biological solutions at high-brilliance synchrotrons. IUCrJ 2017, 4, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Bernadó, P.; Shimizu, N.; Zaccai, G.; Kamikubo, H.; Sugiyama, M. Solution scattering approaches to dynamical ordering in biomolecular systems. Biochim. Biophys. Acta BBA Gen. Subj. 2018, 1862, 253–274. [Google Scholar] [CrossRef]
- Vallat, B.; Webb, B.; Westbrook, J.; Sali, A.; Berman, H.M. Development of a prototype system for archiving integrative/hybrid structure models of biological macromolecules. Structure 2018, 26, 894–904. [Google Scholar] [CrossRef] [Green Version]
- Brosey, C.A.; Tainer, J.A. Evolving SAXS versatility: Solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Curr. Opin. Struct. Biol. 2019, 58, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, C.M.; Graewert, M.A.; Blanchet, C.E.; Langley, D.B.; Whitten, A.E.; Svergun, D.I. Preparing monodisperse macromolecular samples for successful biological small-angle X-ray and neutron-scattering experiments. Nat. Protoc. 2016, 11, 2122–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graewert, M.A.; Jeffries, C.M. Sample and Buffer Preparation for SAXS. Adv. Exp. Med. Biol. 2017, 1009, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Mathew, E.; Mirza, A.; Menhart, N. Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. J. Synchrotron Radiat. 2004, 11, 314–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Inoko, Y. Size-exclusion chromatography combined with small-angle X-ray scattering optics. J. Chromatogr. A 2009, 1216, 7461–7465. [Google Scholar] [CrossRef]
- Pérez, J.; Vachette, P. A successful combination: Coupling SE-HPLC with SAXS. Adv. Exp. Med. Biol. 2017, 1009, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Johansen, N.T.; Pedersen, M.C.; Porcar, L.; Martel, A.; Arleth, L. Introducing SEC–SANS for studies of complex self-organized biological systems. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 1178–1191. [Google Scholar] [CrossRef]
- Bucciarelli, S.; Midtgaard, S.R.; Pedersen, M.N.; Skou, S.; Arleth, L.; Vestergaard, B. Size-exclusion chromatography small-angle X-ray scattering of water soluble proteins on a laboratory instrument. J. Appl. Crystallogr. 2018, 51, 1623–1632. [Google Scholar] [CrossRef]
- Inoue, R.; Nakagawa, T.; Morishima, K.; Sato, N.; Okuda, A.; Urade, R.; Yogo, R.; Yanaka, S.; Yagi-Utsumi, M.; Kato, K.; et al. Newly developed laboratory-based size exclusion chromatography small-angle X-ray scattering system (La-SSS). Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graewert, M.A.; Franke, D.; Jeffries, C.M.; Blanchet, C.E.; Ruskule, D.; Kuhle, K.; Flieger, A.; Schäfer, B.; Tartsch, B.; Meijers, R.; et al. Automated pipeline for purification, biophysical and X-ray analysis of biomacromolecular solutions. Sci. Rep. 2015, 5, 10734. [Google Scholar] [CrossRef]
- Jacques, D.A.; Guss, J.M.; Svergun, D.I.; Trewhella, J. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 620–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trewhella, J.; Duff, A.P.; Durand, D.; Gabel, F.; Guss, J.M.; Hendrickson, W.A.; Hura, G.L.; Jacques, D.A.; Kirby, N.M.; Kwan, A.H.; et al. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Valentini, E.; Kikhney, A.G.; Previtali, G.; Jeffries, C.M.; Svergun, D.I. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2014, 43, D357–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikhney, A.G.; Borges, C.R.; Molodenskiy, D.S.; Jeffries, C.M.; Svergun, D.I. SASBDB: Towards an automatically curated and validated repository for biological scattering data. Protein Sci. 2019, 29, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.-C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, C.E.; Spilotros, A.; Schwemmer, F.; Graewert, M.A.; Kikhney, A.; Jeffries, C.M.; Frank, S.; Mark, D.; Zengerle, R.; Cipriani, F.; et al. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystallogr. 2015, 48, 431–443. [Google Scholar] [CrossRef]
- Round, A.; Felisaz, F.; Fodinger, L.; Gobbo, A.; Huet, J.; Villard, C.; Blanchet, C.E.; Pernot, P.; McSweeney, S.; Roessle, M.; et al. BioSAXS Sample Changer: A robotic sample changer for rapid and reliable high-throughput X-ray solution scattering experiments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajizadeh, N.R.; Franke, D.; Svergun, D.I. Integrated beamline control and data acquisition for small-angle X-ray scattering at the P12 BioSAXS beamline at PETRAIII storage ring DESY. J. Synchrotron Radiat. 2018, 25, 906–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinias, J.P.; Bunner, B.; Gilar, M.; Jorgenson, J.W. Measurement and Modeling of Extra-Column Effects Due to Injection and Connections in Capillary Liquid Chromatography. Chromatography 2015, 2, 669–690. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Brown, P.H.; Schuck, P. On the distribution of protein refractive index increments. Biophys. J. 2011, 100, 2309–2317. [Google Scholar] [CrossRef] [Green Version]
- Volk, A.; Kähler, C.J. Density model for aqueous glycerol solutions. Exp. Fluids 2018, 59, 75. [Google Scholar] [CrossRef] [Green Version]
- Schroer, M.A.; Blanchet, C.E.; Gruzinov, A.Y.; Gräwert, M.A.; Brennich, M.E.; Hajizadeh, N.R.; Jeffries, C.M.; Svergun, D.I. Smaller capillaries improve the small-angle X-ray scattering signal and sample consumption for biomacromolecular solutions. J. Synchrotron Radiat. 2018, 25, 1113–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.C.; Toraya, H.; Blanton, T.N.; Wu, Y. X-ray powder diffraction analysis of silver behenate, a possible low-angle diffraction standard. J. Appl. Crystallogr. 1993, 26, 180–184. [Google Scholar] [CrossRef] [Green Version]
- Panjkovich, A.; Svergun, D.I. CHROMIXS: Automatic and interactive analysis of chromatography-coupled small-angle X-ray scattering data. Bioinformatics 2017, 34, 1944–1946. [Google Scholar] [CrossRef]
- Franke, D.; Kikhney, A.G.; Svergun, D.I. Automated acquisition and analysis of small angle X-ray scattering data. Nucl. Inst. Meth. A 2012, 689, 52–59. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, H.; Neto, M.D.O.; Napolitano, H.B.; Polikarpov, I.; Craievich, A.F. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 2009, 43, 101–109. [Google Scholar] [CrossRef]
- Hajizadeh, N.R.; Franke, D.; Jeffries, C.M.; Svergun, D.I. Consensus Bayesian assessment of protein molecular mass from solution X-ray scattering data. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nat. Cell Biol. 2013, 496, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Franke, D.; Jeffries, C.M.; Svergun, D.I. Machine learning methods for X-ray scattering data analysis from biomacromolecular solutions. Biophys. J. 2018, 114, 2485–2492. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, E.; Svergun, D.I. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 2007, 40, s245–s249. [Google Scholar] [CrossRef] [Green Version]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Kirby, N.; Cowieson, N.; Hawley, A.M.; Mudie, S.T.; McGillivray, D.J.; Kusel, M.; Samardzic-Boban, V.; Ryan, T.M. Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 1254–1266. [Google Scholar] [CrossRef]
- Jeffries, C.M.; Graewert, M.A.; Svergun, D.I.; Blanchet, C.E. Limiting radiation damage for high-brilliance biological solution scattering: Practical experience at the EMBL P12 beamline PETRAIII. J. Synchrotron Radiat. 2015, 22, 273–279. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Carbonic Anyhdrase 2 | Serum Albumin | Serum Albumin (dimer) | Alcohol Dehydrogenase | Apoferritin |
|---|---|---|---|---|---|
| Abbreviation | CA | BSA | BSA dimer | ADH | aFER |
| Source | Bos taurus–erythrocytes | Bos taurus | Bos taurus | Saccharomyces cerevisiae | Equus caballus |
| Oligomeric state | Monomer | Monomer | Dimer | Tetramer | Tetramer |
| Calculated MW * | 29 kDa | 66 kDa | 133 kDa | 147 kDa | 454 kDa |
| SEC-SAXS buffer | 50 mM HEPES, 150 mM NaCl, 2% v/v glycerol, pH 7 | ||||
| SEC-SAXS column | Superdex 75 inc. 10/300 | Superdex 200 increase, 10/300 | |||
| SEC-SAXS injection volume | 100 μL | 100 μL | 100 μL | 100 μL | 50 μL |
| SEC-SAXS injection concentration | 11.9 mg/mL | 8.8 mg/mL | 5.5 mg/mL | 9.2 mg/mL | 11 mg/mL |
| A280 nm Extinction coefficient, E0.1% ** | 1.732 | 0.646 | 0.646 | 1.326 | 0.729 |
| dn/dc (mL/g) *** | 0.1870 | 0.1852 | 0.1852 | 0.1842 | 0.1860 |
| UniProt entry (amino acid sequence range) | P00921 (1-260) | P02769 (25-607) | P02769 (25-607) | P00330 (1-348) | P02791 (1-175) |
| CA | BSA | BSA Dimer | ADH | aFER | |
|---|---|---|---|---|---|
| Data Collection Parameters | |||||
| Instrument | EMBLP12 (PETRAIII, DESY, Hamburg) | ||||
| Beam geometry (mm2) | 0.2 × 0.12 | ||||
| Wavelength (nm) | 0.124 | ||||
| s range (nm−1) | 0.03–6.0 | ||||
| Temperature (K) | 293 | ||||
| Exposure time (s) | 41 | 81 | 25 | 67 | 85 |
| Average conc. from dRI (mg/mL) | 1.85 | 1.3 | 0.5 | 1.2 | 0.56 |
| Dilution factor | 6.4 | 6.8 | 11.0 | 7.7 | 19.6 |
| Structural Parameters | |||||
| RG (nm) | 1.8 ± 0.1 | 2.8 ± 0.1 | 4.0 ± 0.2 | 3.3 ± 0.2 | 5.4 ± 0.3 |
| RH (nm) | 2.4 ± 0.1 | 3.5 ± 0.1 | 4.6 ± 0.2 | 4.5 ± 0.3 | 6.7 ± 0.4 |
| RG/RH | 0.75 | 0.80 | 0.87 | 0.73 | 0.81 |
| Dmax (nm) | 5.1 ± 0.5 | 8 ± 0.5 | 13.2 ± 1 | 9.3 ± 1 | 12.5 ± 1 |
| Molecular Weight Determination (kDa) | |||||
| MWexp (kDa) | 29 | 66.4 | 133 | 147 | 454 |
| MWMALLS (kDa) | 29 ± 1.5 | 63 ± 3 | 126 ± 5 | 142 ± 7 | 454 ± 20 |
| MWQP (kDa) | 20 ± 2 | 61 ± 5 | 135 ± 12 | 131 ± 12 | 518 ± 40 |
| MWMoW (kDa) | 23 ± 2 | 60 ± 5 | 128 ± 12 | 137 ± 12 | 419 ± 40 |
| MWVC (kDa) | 23 ± 2 | 61 ± 5 | 126 ± 12 | 122 ± 12 | 464 ± 40 |
| MWS&S (kDa) | 23 ± 2 | 60 ± 5 | 147 ± 12 | 135 ± 12 | 418 ± 40 |
| MWI(0) (kDa) | 24 ± 2 | 69 ± 5 | 144 ± 12 | 151 ± 12 | 530 ± 40 |
| MWBayesian (kDa) | 22.4 | 59.5 | 130.9 | 131 | 381 |
| Cred. interval, kDa range (%) | 20.9–24 98.99 | 56.2–60.2 (94.7) | 121.5–134.4 (91.6) | 116–134 (96.24) | 264–455 (96.3) |
| Software Employed | |||||
| Primary data reduction | SASFLOW/CHROMIXS | ||||
| Data processing | PRIMUS | ||||
| Ab initio modeling | DAMMIF | ||||
| Computation of model intensities | CRYSOL | ||||
| Final Models | |||||
| Crysol fit (χ2) | 1.1 | 1.3 | 4.3 | 1.4 | 8.8 |
| PDB ID | 5A25 | 4F5S | 4F5S | 4W6Z | 1IER |
| SASBDB ID | SASDFP8 | SASDFQ8 | SASDFR8 | SASDFS8 | SASDFN8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graewert, M.A.; Da Vela, S.; Gräwert, T.W.; Molodenskiy, D.S.; Blanchet, C.E.; Svergun, D.I.; Jeffries, C.M. Adding Size Exclusion Chromatography (SEC) and Light Scattering (LS) Devices to Obtain High-Quality Small Angle X-Ray Scattering (SAXS) Data. Crystals 2020, 10, 975. https://doi.org/10.3390/cryst10110975
Graewert MA, Da Vela S, Gräwert TW, Molodenskiy DS, Blanchet CE, Svergun DI, Jeffries CM. Adding Size Exclusion Chromatography (SEC) and Light Scattering (LS) Devices to Obtain High-Quality Small Angle X-Ray Scattering (SAXS) Data. Crystals. 2020; 10(11):975. https://doi.org/10.3390/cryst10110975
Chicago/Turabian StyleGraewert, Melissa A., Stefano Da Vela, Tobias W. Gräwert, Dmitry S. Molodenskiy, Clément E. Blanchet, Dmitri I. Svergun, and Cy M. Jeffries. 2020. "Adding Size Exclusion Chromatography (SEC) and Light Scattering (LS) Devices to Obtain High-Quality Small Angle X-Ray Scattering (SAXS) Data" Crystals 10, no. 11: 975. https://doi.org/10.3390/cryst10110975