
A Hands-on Guide to the Synthesis of High-Purity and High-Surface-Area Magnesium Oxide
by
 , , ,
, , ,
Marek Gliński
1,*,
Agnieszka Czajka
1,
Urszula Ulkowska
1,
Ewa M. Iwanek (nee Wilczkowska)
1,
Dariusz Łomot
2 and
Zbigniew Kaszkur
2
1
Faculty of Chemistry, Warsaw University of Technology, 00-664 Warsaw, Poland
2
Institute of Physical Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Poland
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(12), 1595; https://doi.org/10.3390/catal12121595
Submission received: 30 October 2022
/
Revised: 24 November 2022
/
Accepted: 2 December 2022
/
Published: 6 December 2022
(This article belongs to the Special Issue Metal Oxide Catalysts: Synthesis and Applications)
Abstract
:In this study, magnesium nitrate, chloride or sulphate were used in the synthesis of Mg(OH)2, the precursor of MgO. It was found that the counter ion strongly influences the purity of the Mg(OH)2, as well as the specific surface area of the obtained MgO. The latter is also strongly influenced by the calcination temperature. The choice of the precipitating agent can lead to the introduction of K+ or Na+ ions and hence NH3 (aq) is the best choice. A multistep precipitation procedure of Mg(OH)2 was proposed to lower the concentration of typical impurities (Fe, Ni and Mn) found in commercial p.a. purity Mg(NO3)2. The effect of the number of portions of water used for washing of Mg(OH)2 on the purity of the final product has also been investigated in detail. The stages of formation of grains of Mg(OH)2 and their subsequent thermal decomposition was described together with determination of the introduction of new impurities into the material. Large scale (1500 g) preparation of Mg(OH)2 with an improved purity was performed and described. Therefore, this study explains what measures should be taken to obtain pure magnesia catalysts and is a valuable resource for catalytic research in which magnesia is used.
1. Introduction
Due to its unique properties, MgO has found its way into various studies in different scientific fields [1,2,3,4,5]. It belongs to the group of oxides of main elements (Al2O3, SiO2 and MgO) that are frequently used in heterogeneous catalysis. They play a role of support for various metallic phases [6,7], such as noble and transition metals, and also oxides of transition metals, as a part of a more complex support [1,3] or as a catalyst promoter [2,8]. Moreover, they act alone as heterogeneous catalysts [4,9,10,11,12,13]. The main advantage of these oxides is the high specific surface area, which enables obtaining high dispersions of metallic or oxide active phases of the obtained catalysts. Another extremely advantageous feature of this group of oxides is the presence of very different acid-base properties of their surfaces, from the dominant strongly acidic properties for the Al2O3·SiO2 and Al2O3 systems, through slightly acidic, slightly basic for SiO2, to strongly basic properties observed for the MgO surface [14]. Due to the high basicity of the MgO surface, this oxide has been used as a catalyst in a number of technological processes, such as the synthesis of higher olefins from ethylene (SHOP process) [15,16], in the synthesis of 2,6-dimethylphenol in the reaction of phenol alkylation with methanol [17], biodiesel production [12,13,18] or transesterification of fats with methanol to Fatty Acid Methyl Esters (FAME) [10,19,20].
An interesting recently published study on the application of MgO nano particles for FAME has shown that the synthesis conditions can lead to either a very high activity or none at all [19]. Similarly, in a study by Matsuda et al. [21], four different specimens of MgO were prepared by the hydrolysis of magnesium nitrate, sulphate, chloride and acetate with ammonia water and subsequent calcination, and the next two specimens were obtained by thermal decomposition of basic carbonate and oxalate. All MgO specimens were calcined at 823 K for 20 h in air, and their activity in isomerization of 1-butene was measured. It was found that only MgO from the nitrate, oxalate and acetate catalysts are active in the isomerization. It was shown that the activity is related to high surface basicity of the above-mentioned catalysts. According to the authors’ statement, the lower basicity of the MgO obtained from the sulphate, chloride and carbonate is the reason for the lack of their activity [21]. It was demonstrated by Tanabe et al. [22] that doping MgO with metal cations showed differentiated activity in aldol condensation of acetone. The following cations were tested as modifiers: Na+, K+, Rb+, Cs+, Al3+, Cr3+, Mn4+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+ and Zr4+. In the presence of Al3+, Cr3+, Mn4+, Fe3+, Co2+, Ni2+ and Cu2+ ions deposited onto MgO, a decrease in the activity of the catalysts was observed. On the other hand, the addition of Na+, Zn2+ and Zr4+ ions resulted in an increase of the catalysts’ activity [22].
Magnesium oxide is obtained by thermal decomposition of magnesium salts such as carbonate [21], oxalate [23] or nitrate [24]. The most commonly used MgO precursor is magnesium hydroxide, the thermal decomposition of which leads to the oxide. The influence of the type of magnesium oxide precursor and its decomposition conditions on the specific surface area of the obtained MgO, acid-base properties of the oxide surface and its catalytic activity was the subject of several studies [7,25,26,27]. It has been found that the surface area and strength of basic sites of MgO obtained from Mg(OH)2 strongly depend on the type of magnesium precursor, the type of base used for the precipitation of the hydroxide, the mode of mixing of reactants and the thermal conditions of the decomposition of Mg(OH)2 [27]. However, there have been no systematic studies on this topic thus far. This paper aims to fill this gap.
The research presented in this paper was conducted in order to achieve the three following goals:
- Developing a procedure for the synthesis of Mg(OH)2 by a wet method on a laboratory scale, the formation of the obtained hydroxide dust grains and the thermal decomposition of hydroxide grains to MgO;
- Determining the effect of the operation of obtaining magnesium hydroxide, its formation into grains and its subsequent calcination to the oxide on the final purity of the MgO catalyst;
- Performing a large-scale (of the order of 1500 g of the product) synthesis of very pure Mg(OH)2 as a MgO precursor on the basis of the conditions optimized in point 1.
In our opinion, the research contained in point 2 introduces an element of scientific novelty, as the research literature lacks the impact of the sequence of operations leading from the precursor (Mg(OH)2) to the catalyst (MgO) on its final purity. The rest of the research helps to compile and systemize the knowledge regarding MgO synthesis by performing the tests in a controlled manner with the constant monitoring of the key parameters and variables.
2. Results
The synthesis of magnesium hydroxide was performed from two starting materials, namely commercial MgO (Scheme 1a) and magnesium nitrate (Scheme 1b). Scheme 1 depicts the overview of the synthesis paths. The first path leads to obtaining two fractions of Batch II. In this path, the starting material is commercial MgO that is digested and purified by a two-step precipitation procedure. The results of the obtained solids are presented in Section 2.1. The second synthesis path (Scheme 1b) begins with magnesium nitrate and consists of a stepwise precipitation of magnesium hydroxide. The results of the characterization of the three fractions of Batch III obtained via this path are presented in Section 2.2. Next, a part of the purest fraction, fraction III, was treated with three different acids, i.e., nitric acid, sulphuric acid and hydrochloric acid, in order to investigate the impact of the counter ion on the purity of the precipitated magnesium hydroxide (see Results Section 2.3). For all of the batches of Mg(OH)2, the final step of synthesis was pellet pressing and grain formation, followed by the calcination of Mg(OH)2.
2.1. Properties of Mg(OH)2 Obtained from Commercial MgO
2.1.1. Composition Determination
The commercial powdered MgO had an assay (MgO) ≥ 97%. In order to increase its purity, as well as to obtain grains of MgO calcined at a specific temperature, the MgO was dissolved in nitric acid, and fractional precipitation was used to prepare magnesium hydroxide with a higher purity than the starting oxide. The resulting batch II fraction II of Mg(OH)2 was used to obtain grains of MgO that could be crushed to obtain a narrow range of particle sizes. The grains of MgO were characterized using numerous techniques. The spectral analysis of a powder of MgO-873, taken as representative, revealed the presence of the typical impurities (in ppm, in parentheses maximum concentration of selected impurities declared by the producer): Si 410 (500), B 85, Fe 35 (50), Ca 20 (200), Mn 15, Al 12 and Cu 5. The presence of the following anions was also found in classic chemical analysis of the MgO-873 sample (concentration in ppm): Cl− 80 (100), SO42− 7 (50) and PO43− 4 (20). The presence of silicon and boron are attributed to the use of borosilicate glass during the preparation of Mg(OH)2.
DTA-TGA-MS studies were carried out to determine two parameters, namely the amount of water (m/z = 18 signal was monitored) lost by the sample and the maximum temperature of Mg(OH)2 decomposition. Figure 1 shows the TG and DTA curves obtained for four of the samples, namely MgO-473, MgO-573, MgO-673 and MgO-873. The DTA-TGA-MS analysis revealed that the MgO-473 sample lost 30.39% of its weight (Figure 1a). The calculated weight loss for anhydrous Mg(OH)2 is 30.89%, which is within the determination error of the measurement. The Tmax for the decomposition of Mg(OH)2 is 692.9 K (Figure 1a). For the MgO-573 sample (Figure 1b), the weight loss (28.05%) is lower than the theoretical value, indicating that a partial dehydration of Mg(OH)2 took place during heating a sample at 573 K. For this sample, the highest rate of the hydroxide dehydration was observed at 691.7 K. A small weight loss (2.43%) observed for the MgO-673 sample (Figure 1c) indicates that the process of dehydration of Mg(OH)2 has already finished. This is also true for MgO-873 (Figure 1d). It is noteworthy that the Tmax for MgO-473, as well as for MgO-573 are distinctly higher than those reported in literature, e.g., for Mg(OH)2 obtained from the hydration of a commercial MgO of reagent purity the Tmax of 651 K has been reported [7]. However, it is commonly known that TG results are extremely sensitive to heating rates. A typical effect of an increase of the heating rate by 5 deg min−1 is to shift the maximum rate to a temperature about 20–40 deg higher than found at 1 deg min−1 [28]. During TG measurement when sample of Mg(OH)2 is heated with a heating rate of 1 deg min−1 the dehydration process commences at 558 K and ceases at 653 K [28]. Indeed, it was shown by us that Tmax for MgO-473 samples are equal 692.9 and 657.5 K for heating rates 10 and 2 deg min−1, respectively. It gives a 35.4 deg difference which falls in line with literature data.
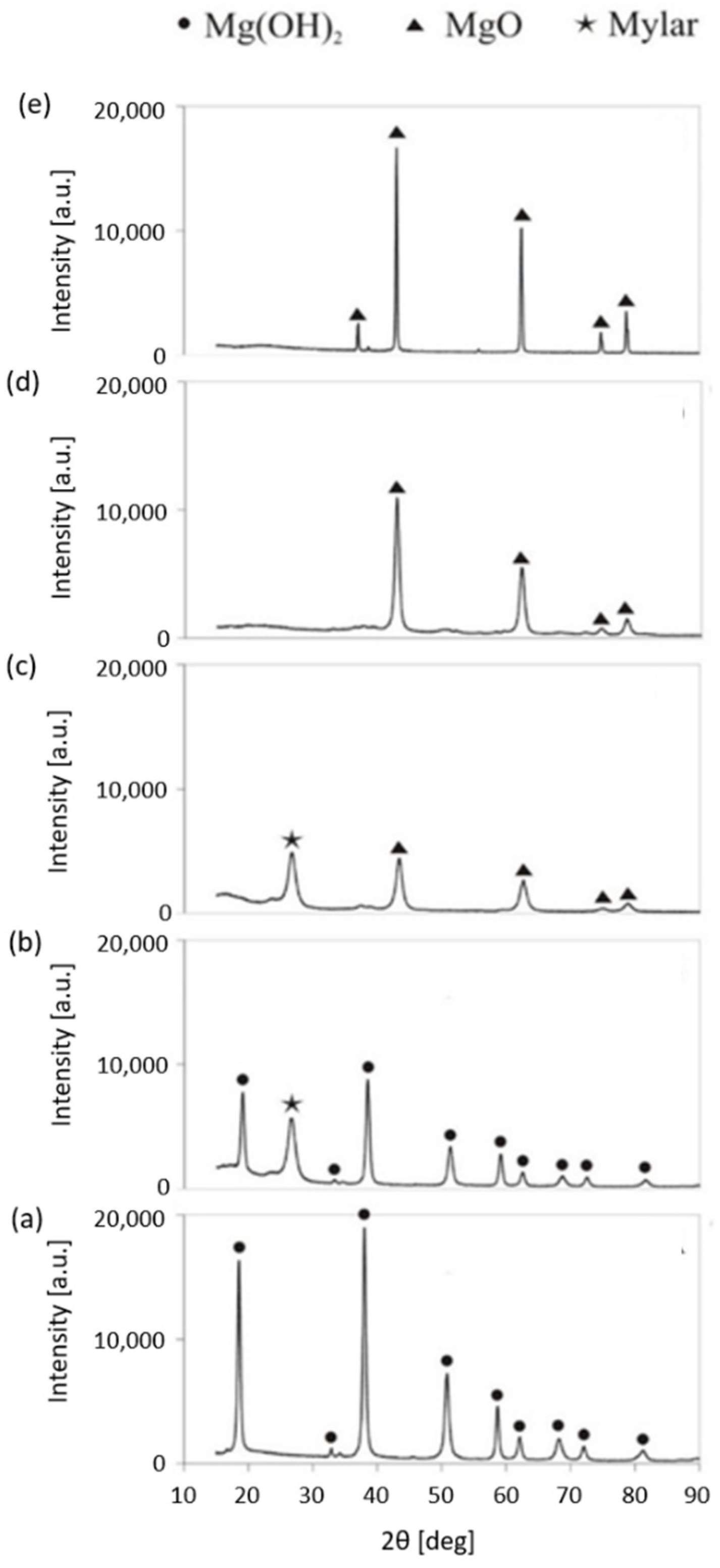
2.1.2. MgO-T Samples: XRD Measurements Results and Textural Properties
The XRD spectra of MgO-T samples calcined in the temperature range 473–1873 K are depicted in Figure 2a–e. Samples of MgO-473 and MgO-573 consist solely of the brucite phase, the latter sample is less crystalline when comparing the intensities of the appropriate signals in the spectra. Samples heated to 673 K and above showed the pure periclase phase only. The conclusions drawn based on both thermogravimetric and XRD studies are in good agreement as to the effect of the calcination temperature on the composition of the MgO-T sample.
In contrast, the SEM images of different MgO-T samples (Figure 3) do not reveal a substantial difference between the texture of Mg(OH)2 and MgO samples. All of the studied samples exhibit a flaky appearance.
The textural properties of the MgO-T samples have been summarized in Table 1. It can be seen that the properties of the first two samples substantially differ from those of the others and should be analysed separately. This is because the temperature is insufficient to decompose the magnesium hydroxide. Their specific surface areas, SBET, are 32 ± 5 m2·g−1, and the pore volume was small, though larger for samples calcined at 673 K and 873 K. The average crystallite size, D, in the samples calcined at the lowest temperature, i.e., magnesium hydroxide, is 12.4 ± 0.4 nm.
The values obtained for the rest of the samples, i.e., those that consist solely of MgO, show expected trends with temperature that are typical for the sintering of a solid, namely: increase in the average crystallite size, D, and decrease in specific surface area and pore volume with the increase in the calcination temperature of the oxide (Table 1). It can be seen that both the crystallite size and average pore diameter are lower for the freshly formed MgO phase than for Mg(OH)2 (the first two samples). The adsorption/desorption hysteresis for the sample calcined at 873 K is shown in Figure S1. It can be seen that it is a Type H3 loop, the same as that reported in [29].
2.1.3. Temperature-Programmed Desorption (TPD) Measurements of MgO-T Samples
Temperature-programmed desorption measurements were performed with five probe molecules: (CH3)3CCN, CH3CN, n-C4H9NH2, t-C4H9NH2 and (C2H5)3N. Two of them, namely t-BuCN and Et3N can only act as bases, whereas the other three also exhibit acidic properties and hence are able to probe basic sites of MgO. The pKa values of MeCN, n-BuNH2 and t-BuNH2 are 25 [30] and >33 for both amines (estimated by authors on the basis of value for ammonia). The pKBH+ values for t-BuCN and MeCN are −10.1 [31] and −10.4 [32]. The pKBH+ values that correspond to n-BuNH2, t-BuNH2 and Et3N are very similar according to the literature, namely 10.64 [32], 10.68 [33] and 10.75 [32]. All of the TPD profiles are compiled in Figure 4.
Figure 4a compiles three curves obtained for MgO-873 when n-butylamine, t-butylamine or triethylamine are desorbed from its surface. Despite a very similar basic strength of the three amines, their desorption profiles are very different from one another in terms of both the signal intensity and the maximum desorption temperature. The intensity of the desorption signal decreases in the following order: n-BuNH2 > t-BuNH2 >> Et3N. The very low relative intensity of the desorption signal of the third amine is particularly noteworthy. According to literature reports, the size of the bulky tertiary amine hinders the lone electron pair of the nitrogen atom from coming into the proximity of and reacting with a Lewis acidic site. This is why it preferentially reacts with Brønsted acidic sites, i.e., protons, rather than Lewis acidic sites, i.e., low coordinated Mg2+ ions. Apart from reacting with both Brønsted and Lewis acidic sites, the other two amines also exhibit acidic properties due to the presence of an amino group that can be deprotonated in a reaction with a very strong base. Their acid strength can be derived from the acid strength of ammonia, for which the pKa is 33 [32]. Therefore, it can be expected that this strength, due to the electron donating effect of alkyl groups, will be described by pKa > 33. Very strong solid bases, such as CaO, are able to react with ammonia as an N-H acid, which in the past has resulted in a false interpretation of results obtained in the temperature-programmed desorption of ammonia from their surface and consequently the conclusion that CaO is a stronger acid than some dealuminized zeolites [34,35]. Coluccia et al. and Borello et al. that on the MgO surface some of the adsorbed ammonia undergoes dissociative chemisorption to form surface amide groups [36,37]. It is also postulated that Mg2+(3c)O2−(3c) sites are the most reactive and should be able to dissociate molecules with pKa values higher than 30, such as ammonia, ethene, and benzene [14], indicating that the said reaction with amines can take place.
As known from the literature reports [21,38], as well as the authors’ own papers [39], there are basic sites that react with 4-chloroaniline as the indicator (H− > 26.5) on the surface of pure magnesium oxide. According to Tanabe’s definition, substances with sites whose basic strength exceeds 26.5 can be regarded as solid superbases [40]. This means that these strong sites seem to be able to deprotonate the studied primary amines. In other words, the desorption peak of n-butylamine (Figure 4a) mainly corresponds to amine molecules which desorb from Lewis acidic as well as from basic sites of extremely high basicity, with only a minor contribution from Brønsted acidic sites.
Figure 4b contains the curves obtained during the desorption of Et3N from the surface of MgO-473, MgO-873 and MgO-1873. As concluded from the weight loss balance, the MgO-473 sample represents anhydrous Mg(OH)2, which can be accomplished by heating magnesium hydroxide to temperatures above 453 K. The surface of MgO-473 is covered with a monolayer of hydroxyl groups and hence it has a larger variety of OH− surface groups than MgO-873. That is why the signal stretches across a broader range of temperatures. Since MgO-1873 has only residual surface hydroxyls, the signal was magnified five times in order to compare the temperature range of the desorption peak with those attained for the other two catalysts. It can be seen that the relative ratios of different types of hydroxyl groups vary with the calcination temperature (Figure 4b).
The desorption of pivalonitrile and acetonitrile from MgO-473, MgO-873 and MgO-1873 are depicted in Figure 4c,d, respectively. Both of these probe molecules possess a lone electron pair that can react with low coordinated magnesium cations (Lewis acidic sites), but only MeCN can also probe basic sites due to the acidity of the protons of the methyl group caused by the electron withdrawing properties of the CN group. The acidic strength of MeCN is comparable with that of acetylene, and for both compounds, the pKa value equals 25 [32]. Both nitriles can also exhibit basic properties because they can react with very strong Brønsted acidic sites. However, it does not happen in this case due to very weak acidic properties shown by all MgO samples, as evidenced in the reaction with triethylamine. It can be seen that MgO-473 has only one small desorption peak of t-BuCN whose maximum is observed at 352 K. This corresponds to the desorption of weakly bound, physisorbed molecules. In contrast, the sample calcined at 873 K exhibited two desorption peaks: one at 345 K, which again is attributed to the desorption of physisorbed molecules, and a much larger one at 527 K. This result indicates that the catalyst surface possesses a substantial number of Lewis acidic sites. A peak of pivalonitrile desorption at a similar temperature, 533 K, but much less intensive than that noted for MgO-873 is also observed in the TPD profile of MgO-1873. The loss of intensity is a consequence of the additional sintering and loss of Mg2+ cations with a small coordination number on the surface. The TPR profiles obtained for MgO-473 and MgO-873 using MeCN as the probe molecule (Figure 4d) are very similar as those obtained using t-BuCN. However, in the case of MgO-873 a narrow peak can be seen with a maximum at 416 K, which is attributed to the desorption of MeCN molecules from the strongest basic sites of MgO which are able to deprotonate such a weak acid as MeCN. The large peak that is present in the TPD curve with its maximum at 539 K is associated with Lewis acidic sites. The comparison of this peak in the TPR curves obtained for these two probe molecules indicates that most of the acidic sites on the surface of MgO-873 are Lewis type, i.e., magnesium cations with a low coordination number.
2.1.4. Surface Acidity and Basicity of MgO-T Samples
The concentrations of acidic and basic sites quantified using n-BuNH2, Et3N and PhCOOH are collected in Table 2. The numbers for n-BuNH2 correlate with the specific surface area of the catalysts to give approx. 1 μmol·m−2, whereas those noted for Et3N are substantially smaller. These results confirm that the acidity of the surface of MgO is closely related to the Lewis type acidity. In the literature, the concentration of hydroxyl groups in a monolayer referred to the surface unit of MgO is equal to the concentration of the <Mg-O> moieties on the surface and is 37.5–38.2 μmol·m−2 [41,42]. Only 3% of the total surface MgO moieties possess acidic properties according to the values of the titration with n-BuNH2. Their concentration depends only slightly on the thermal treatment of the sample. On the other hand, benzoic acid reacts with nearly half of the basic sites in the case of MgO-473 (47.5%) and MgO-873 (46.7%) and with practically all of the basic sites (99.5%) present on the surface of MgO-1873.
The impact of the calcination temperature on the strength of the acidic and basic sites on the surface of the series of MgO-T samples was established using nine Hammett indicators. All of the catalysts calcined at a temperature up to 1073 K possess only basic sites, whereas the sample calcined at 1873 K also exhibits the presence of acidic sites of the weakest strength (0.8 < H0 < 4.8) (Table 3). It has been observed that the range of strength of the basic active sites increases when the calcination temperature increases from 473 K to 573 K, and again increases when the calcination temperature changes from 573 K to 673 K. This increase in the strength of the basic sites is closely related to the transformation of magnesium hydroxide into magnesium oxide around the temperature of 673 K. The surface of magnesium oxide obtained as a result of thermal decomposition of the hydroxide at temperatures of 673 and 873 K reaches the highest basic strength among the studied samples. A further increase in calcination temperature leads to its gradual decline.
2.2. Magnesium Nitrate as a Starting Material
In order to obtain a substantial amount of pure magnesia large enough to carry out a wide range of reactions for several years and have enough for all kinds of characterisation studies, the precipitation of magnesium hydroxide was carried out from 10 kg of the magnesium nitrate precursor in three steps. The first precipitate contains the majority of the impurities. Figure 5 shows the three consecutive precipitates obtained from the first, second, and third precipitations. It can be seen that the first precipitate has a different colour than the other two, which indicates the presence of noticeable amounts of impurities. In order to quantify the impurities in each sample, they were subjected to different techniques, such as SEM-EDX, XPS, XRD and ICP-OES.
Figure 6 depicts the SEM and EDX results for MgO obtained from the three precipitates. It can be clearly seen that the topography of the samples significantly differs. The most regular shapes can be seen in MgO obtained from precipitate II (Figure 6b). EDX measurements were conducted on no fewer than three points of each sample, which is why the numbers are given as a range. The only three components detected with EDX are oxygen and magnesium, as well as a small amount of carbon, between 3 and 6 at.% (Figure 6d). The ratio of the Mg K to the total of Mg K and O K signals is approx. 50%, which means that there is a 1:1 stoichiometry of oxygen to magnesium within experimental error. This means that in each case, the precipitate is magnesium oxide. It can be seen in the obtained spectrum that no other elements are present. This means that in each case, the precipitate is magnesium oxide with all impurities below the detection limit. This technique, however, is not a very sensitive one, such as XPS. However, the XPS results of all of the samples show that the surface contains only three elements: magnesium, oxygen and carbon.
The fitting of the Mg 2p and O 1s detailed regions (Figure 7a) shows that the chemical environment of these elements is homogeneous, which indicates the lack of other compounds containing these elements. All other elements (except adventitious carbon) are below the detection limit. In order to probe deeper into the sample, depth analysis was performed with 15 and 30 s of etching. However, no contaminants were seen in the spectra (Figure 7b), even upon etching. This is because the contamination of the solid did not occur after synthesis and is not found in the top layers of the solids, but it comes from the precursor and is present throughout the solid. Moreover, the concentration of the impurities must be below the detection limit of the technique. Therefore, it can be concluded that to determine the concentration of the contaminants, a bulk method, such as ICP-OES of solutions of the digested samples, would be better.
Typical heavy metallic impurities of magnesium nitrate are iron, nickel and manganese. The first two are present in the commercial product probably due to leaching of these elements from the stainless-steel reactor in which the synthesis was carried out, whereas the third is an element similar in nature to magnesium and hence naturally present in magnesium-containing ores. Table 4 contains the compilation of results obtained with spectrophotometric methods for these three elements. It can be seen that the concentrations of the impurities were much higher in the first fraction and below the detection limits of these methods in the case of the third fraction. This is why the third fraction was tested using CPI-MPI-OES.
These results are presented in Table 5. The results indicate that the most abundant foreign element is silicon, present in the amount of 40 ppm. The other impurities are present in very minute amounts, i.e., less than half of the value noted for Si.
2.3. Impact of Post-Synthesis Treatment on Properties of Mg(OH)2 and MgO
2.3.1. Impact of Counter Ion and Precipitating Agent
Three acids were used to obtain magnesium salts of the same purity, but with different anions, namely NO3−, SO42− and Cl−. These salts were used to precipitate Mg(OH)2 with the same precipitating agent, NH3(aq), and the properties of thus obtained magnesium hydroxide, as well as the MgO resulting from a subsequent calcination, were determined. The maximum temperature of decomposition of the magnesium hydroxide washed with water in five portions is only different for the magnesium hydroxide obtained from the chloride (Table 6). As mentioned earlier, the lower temperature indicates that the hydroxide is impure. Moreover, washing the hydroxide with 10 more portions of water leads to only a slight increase of the Tmax and a further 10 portions lead to a temperature almost as high as that noted for the hydroxide from the other two salts.
The three precipitating agents used were aqueous ammonia, potassium hydroxide and sodium hydroxide. In the case of ammonia, the freshly precipitated magnesium hydroxide quickly settled out of the suspension. In contrast, using either potassium or sodium hydroxide as the precipitating agent resulted in a colloidal suspension of magnesium hydroxide (Table 6).
Magnesium hydroxide obtained from magnesium nitrate when the precipitating agent was aqueous ammonia was washed with equal portions of water repeatedly and after every fifth portion, a part of the solid was dried and used for characterisation, whereas another part was calcined so that the resulting MgO could also be characterized. The influence of the volume used for washing magnesium hydroxide and the surface area of both the hydroxide and the oxide was determined. The results are compiled in Figure 8. It can be seen that the surface area of the oxide is always substantially higher than that of the hydroxide.
In both cases, the lowest value was observed for the unwashed magnesium hydroxide. The area of these two compounds was 15 and 52 m2·g−1, respectively. The biggest increase of the surface area is noted between the unwashed sample and that washed with five portions of water. It is noteworthy that although the consecutive portions of water have only a small effect on the area of the hydroxide, they do impact the values attained for the resulting magnesium oxide, which continuously rises and equals 129 m2·g−1 for 25 portions of washing. (Figure 8).
The effect of the calcination atmosphere on the specific surface area of the obtained MgO was also studied. The results are presented in Table 7. It was shown that magnesium hydroxide calcined at 573 K in a stream of nitrogen for 6 h did not undergo thermal decomposition to magnesium oxide. The decomposition performed at the same temperature under vacuum led to the product comprised of 46% of MgO. In contrast, the calcination of Mg(OH)2 at 673 K for 6 h led to 100% of MgO, irrespective of the type of the atmosphere. It was also stated that the removal of the gaseous products (H2O) during the thermal decomposition of Mg(OH)2 exerts a beneficial effect on the specific surface area of the formed oxide. This is due to the fact that the presence of steam in the reaction zone led to sintering of the newly formed product and hence resulted in drastic lowering of its specific surface area, as it was observed at 873 K when Mg(OH)2 was decomposed under static air.
One factor that influences the surface area of the obtained MgO is the number of times applied in the washing of the hydroxide. This effect was different for the different batches (Table 8). When magnesium chloride was used as the precursor and the hydroxide was precipitated with an ammonia solution, the specific surface area was low, i.e., 32.1 and 38.6 after 15 and 25 portions of water were used for washing. This result is in line with the reports in the literature of using chlorides as the staring materials [7]. Slightly higher values were observed for the sulphate, and the difference upon further washing was also slightly larger (40.3 and 54.2 m2∙g−1). Measurements with three different precipitating agents used for precipitation of magnesium hydroxide from magnesium nitrate (sodium hydroxide, potassium hydroxide and ammonia solution) clearly show that the best way of obtaining pure magnesia with a high surface area is using an ammonia solution. The resulting surface areas are in the range of 60–70 m2∙g−1 for the hydroxide-precipitated solids, whereas the surface area of the final product obtained using an ammonia solution was almost twice as high (120–130 m2∙g−1). This shows that the synthesis method selection can result in substantial changes of the surface area of the obtained MgO.
The maximum temperature of decomposition of magnesium hydroxides for the three fractions of batch III are compiled in Table 9. As can be seen in Table 4, the sample of Mg(OH)2 from batch III fraction I contains the highest concentration of heavy metallic impurities (Ni, Fe, Mn). The impurities occur in the form of hydroxides that are more prone to thermal decomposition than Mg(OH)2 itself, e.g., Fe(OH)3 and Ni(OH)2 decompose at 493 and 563 K, respectively [43,44]. Therefore, the magnesium hydroxide from the first fraction has the lowest maximum decomposition rate temperature, i.e., 635 K, whereas the highest was noted for the third fraction (692 K). It is noteworthy that for all three fractions, this temperature was different, which indicates decreasing concentrations of impurities.
The surface area of the magnesia obtained from each of these fractions is also shown in Table 9. The changes in this parameter are substantial. The more impurities are present, the lower the final surface area. In our studies, the first fraction of MgO had a surface area of only 12.8 m2·g−1, whereas the second and third fractions had 62.9 and 129.2 m2·g−1, respectively. In other words, a tenfold decrease of the surface area due to the presence of impurities is observed despite the same calcination procedure.
2.3.2. Influence of the Formation of Mg(OH)2 Grains and Their Thermal Decomposition on the Final Purity of MgO
The next step on the way from the (Mg(OH)2) precursor to the catalyst is the formation of hydroxide grains and the thermal decomposition to MgO. This consists of the following steps:
- Pressing pure magnesium hydroxide dust into pellets;
- Crushing the obtained pellets;
- Separating the proper fraction of magnesium hydroxide using sieves;
- The thermal decomposition of the separated hydroxide fraction to magnesium oxide.
In order to determine the effect of the above-described steps on the final purity of the obtained oxide, changes in the concentration of elements in contact with the highest-purity magnesium hydroxide sample were investigated (batch III fraction III) before and after the forming Mg (OH)2 grains and the thermal decomposition to the oxide. These elements are: Fe (steel pellet press), Si and Al (agate and porcelain mortars) and Cu (sieves for fractionating hydroxide grains). The change in Si concentration after thermal decomposition of the hydroxide (quartz tube reactor) was also measured. The results of these studies are summarized in Table 10.
It was found that pressing the hydroxide powder increased the Fe content in the sample from the initial value of 17 ppm to 19 ppm. Grinding the hydroxide in a porcelain mortar increases the concentration of both Si and Al from 40 to 51 ppm and from 5 to 25 ppm, respectively. The reason for this is the contamination of the sample with the mortar material, the main component of which is mullite (3Al2O3·2SiO2). When an agate mortar is used instead, there is only a slight increase in Si concentration, i.e., to 45 ppm. This is definitely more beneficial than the introduction of Si and Al from the porcelain mortar to the sample because the contamination of the sample with Al2O3 will affect the acidic properties of the surface of the MgO obtained in the next step. The thermal decomposition of magnesium hydroxide carried out in a vertical tubular quartz reactor, in which the hydroxide bed is placed on a piece of glass wool, increases the Si concentration from 51 to 59 ppm.
3. Materials and Methods
3.1. Synthesis of Mg(OH)2
The synthesis of magnesium hydroxide was performed from two starting materials, namely commercial MgO and magnesium nitrate. Scheme 1 depicts the overview of the synthesis paths. Eight batches of Mg(OH)2 were obtained. For all of the batches of Mg(OH)2, the final step of synthesis was calcination of Mg(OH)2.
In order to obtain a large amount of pure magnesia suitable as a catalyst (batch III, details provided in Section 3.1.1), magnesium hydroxide was obtained in three subsequent precipitations so that the impurities would be precipitated mainly in the first two fractions.
The following aspects were investigated:
- Starting materials: There were two starting materials for the large-scale synthesis of MgO: commercial MgO for only batch II (Scheme 1a), details provided in Section 3.1.2, and commercial magnesium nitrate for all other batches (Scheme 1b).
- Precipitating agents: Three precipitating agents were used to obtain Mg(OH)2: aqueous ammonia (Scheme 1a,b), sodium hydroxide and potassium hydroxide (details provided in Section 3.1.3).
- Counter ions: There were three precursors of magnesium hydroxide used to test how much residual counter ions from the salt used was present in the obtained precipitate and hence for establishing the proper washing procedure (details provided in Section 3.1.4).
3.1.1. Large-Scale Preparation of Mg(OH)2 from Commercial Mg(NO3)2
In a polypropylene barrel of 150 dm3 capacity equipped with a mechanical stirrer, 10 kg (39.0 moles) of Mg(NO3)2·6H2O (puriss, p.a., Fluka Chemie GmbH, Buchs, Switzerland) was dissolved in 50 dm3 of redistilled water. To this solution 1.0 dm3 of ammonia solution in water (25%, p.a., POCh, Gliwice, Poland) was added with stirring. The resulting suspension of precipitated magnesium hydroxide was stirred slowly (30 rpm) for three days and filtered (Mg(OH)2 fraction I). To the purified solution of Mg(NO3)2, 8.0 dm3 of ammonia solution was added, and the procedure was repeated (Mg(OH)2, fraction II). Finally, to the purified solution of magnesium nitrate 10.0 dm3 of ammonia solution was slowly added, total time: 10 h, and the precipitate of the hydroxide left for ageing for a week in a closed barrel (Mg(OH)2, fraction III). Next, the precipitate was washed by decantation with redistilled water with 25 portions of water. Each portion of water was 10 dm3. The first five portions contained some of the ammonia solution (1.0 dm3 of 25% solution of ammonia was added to 9.0 dm3 of water). After filtration the cake of Mg(OH)2 was dried under normal pressure at 313, 353 and 393 K for 24 h at each temperature.
The first five portions of solution decanted after the third precipitation were collected and treated with an excess of 10% water solution of NaOH (p.a., POCh, Gliwice, Poland) to precipitate practically all Mg2+ ions still present in the solution (Mg(OH)2 IV fraction). Due to very slow sedimentation of the hydroxide from this fraction (gel-like) the washing procedure was changed to filtration of the hydroxide under reduced pressure and successive washing of the cake (10 times, 200 cm3 of water in each portion of wash water).
The following amounts of magnesium hydroxide were obtained in each fraction:
Mg(OH)2 I fraction—29.0 g, 1.27%;
Mg(OH)2 II fraction—275.4 g, 12.11%;
Mg(OH)2 III fraction—1538.4 g, 67.62%;
Mg(OH)2 IV fraction—304.7 g, 13.39%;
∑ (I—IV fraction)—2147.5 g, 94.40%;
Loss—97.4 g, 5.60%.
Fractions I–III were characterized (SBET, TG, content of impurities), but only fraction III was used in further experiments.
The lumps of magnesium hydroxide from fraction III were ground in an agate mortar. Powdered Mg(OH)2 was pressed in a hydraulic press (10 MPa), and the pellets were crushed using an agate mortar and pestle by a weak striking. A sieved fraction with a 0.16–0.40 mm grain diameter was used for further characterization of both Mg(OH)2 and the MgO obtained from it through calcination (3.0 g) at 873 K in a stream of air (1 h, V(air) = 3 dm3 h−1) and dry deoxygenated nitrogen (5 h, V(N2) = 3 dm3 h−1) in a vertical tubular quartz reactor. After cooling in a stream of N2, the MgO sample was transferred to a Schlenk-type container and stored under nitrogen.
3.1.2. Large Scale Preparation of Mg(OH)2 from Commercial MgO
In a glass reactor of 5 dm3 capacity equipped with a mechanical stirrer, 180 g of commercial MgO (purum p.a., Reachim, Chișinău, Moldova) were mixed with 2 dm3 of redistilled water. The content of the reactor was stirred and 540 cm3 of nitric acid (68%, p.a., POCh, Gliwice, Poland) was added in small portions (20 cm3) to control the reaction temperature. After cooling to room temperature, the slightly turbid solution was filtered, and the filtrate was transferred back to the reactor. The obtained solution of magnesium nitrate was treated with 130 cm3 of ammonia solution in water (25%, p.a., POCh, Gliwice, Poland). The resulting suspension of precipitated magnesium hydroxide was stirred slowly (30 rpm) for three days in aim to transfer metallic impurities in the form of their hydroxides into the sediment of Mg(OH)2 which was filtered off and discarded (batch II fraction I). To the purified clear solution of Mg(NO3)2, an excess of 25% ammonia solution (1.2 dm3) was added over 8 h with stirring. The precipitate of the hydroxide was left for ageing for a week in the closed reactor. Next, the precipitate was washed by decantation with redistilled water (25 times, 1 dm3 portion of water each time). The obtained slurry was filtered, and the cake was dried under normal pressure at 313, 353 and 393 K for 24 h at each temperature. The yield of Mg(OH)2 was 165 g (63.3% based on MgO) (batch II fraction II). The soft lumps of the hydroxide were ground in a porcelain mortar. Powdered Mg(OH)2 was pressed in a hydraulic press (Langzauner GmbH, Lambrechten, Austria) using 10 MPa of pressure and the pellets were crushed with a porcelain pestle in a mortar by striking. A sieved fraction of 0.16–0.40 mm grains was further used in the preparation of samples with the symbol MgO-T, e.g., MgO-473 in which the number after the dash indicates the temperature in Kelvins at which it was calcined.
MgO-T samples. Samples of Mg(OH)2 (5 g) were calcined in a tubular quartz reactor at elevated temperatures (T = 473–1073 K) for 1 h in a stream of dry air (20 dm3 h−1) and for 5 h in a stream of dry deoxygenated nitrogen (3.0 dm3 h−1). The sample MgO-1873 was prepared by calcination of the sample MgO-873 in an HTC Carbolite 18/8 electric oven (Carbolite Gerp, Hope, UK) at 1873 K for 5 h in nitrogen in an Al2O3 crucible. After cooling in a stream of N2 the MgO-T samples were transferred to Schlenk-type containers and stored under nitrogen.
3.1.3. Synthesis of Mg(OH)2 from Mg(NO3)2 and Alkali Metal Hydroxides
The solution of magnesium nitrate was prepared by treating 7.29 g (0.125 mol) of Mg(OH)2 (batch III, fraction III) in 100 cm3 of redistilled water with HNO3 (65%, p.a., POCh, Gliwice, Poland) diluted with water (1:1 v/v) in a polypropylene beaker of 800 cm3 capacity. The resulting solution was diluted with water to the volume of 250 cm3. To this solution a 1 M solution of alkali metal hydroxide in water was slowly added under stirring until all of the magnesium precipitated in the form of Mg(OH)2. Due to very slow sedimentation of the product (10 mm in 24 h at 80 mm total height of the suspension), it was impossible to wash the precipitate using sedimentation/decantation. In this case, the filtration/washing method was applied. The cake of Mg(OH)2 obtained by filtration on Buchner funnel with a double layer of a filter paper was washed with 25 portions of 100 cm3 of redistilled water. After every fifth portion of water, a part of the precipitate was taken for analysis (SBET, TG).
3.1.4. Synthesis of Mg(OH)2 from Mg(NO3)2, MgSO4 and MgCl2, and NH3 (aq)
In order to eliminate the impact of various impurities on the final purity of the product, the synthesis of magnesium salts (chloride, sulfate, nitrate) was performed with high-purity Mg(OH)2 (batch III, fraction III) as the starting material (Scheme 1).
General procedure. A suspension containing 7.29 g (0.125 mol) of Mg(OH)2 in 100 cm3 of redistilled water was prepared in a polypropylene beaker of 800 cm3 capacity. The appropriate inorganic acid of p.a. purity (HCl, H2SO4 or HNO3) diluted with water (1:1 v/v) was slowly added to the magnetically stirred suspension until the dissolution of the hydroxide occurred. The resulting solution was diluted with water to the volume of 250 cm3. To the stirred solution a concentrated solution of ammonia in water (25%, p. a., POCh, Gliwice, Poland) was slowly added at room temperature until the precipitation of the hydroxide was complete (pH ≈ 11). For all samples of Mg(OH)2 precipitated with the solution of ammonia a very fast sedimentation occurs (70 mm in 2 h at 80 mm total height of a suspension) that enables their purification by decantation. Each time, the same volume was applied, i.e., 300 cm3. The first ten portions contained ammonia (20 cm3 of 25% ammonia solution + 280 cm3 of water) to diminish the occurrence of the reverse reaction expressed by the following equation (Equation (1)):
Mg(OH)2 + 2 NH4NO3 → Mg(NO3)2 + 2 H2O + 2 NH3
The remaining portions (15) were pure redistilled water. After every fifth portion of water a part of the precipitate was taken for analysis (SBET, TG).
3.2. Formation of Grains of Mg(OH)2
Apart from purity, it is important that the catalyst and/or support have the proper dimension of grains. Formation of the grains of Mg(OH)2 consists of three stages:
- Pressing Mg(OH)2 powder in a steel die (20 mm i.d.) under pressure of 10 MPa into thin wafers (weight 317 ± 15 mg, the average of 10 weight measurements);
- Crushing the wafers with a pestle in a mortar (porcelain or agate) by weak striking;
- Sieving the grains of Mg(OH)2 through a set of sieves made of phosphorus bronze (94% Cu, 5% Sn and 0.4% P); the 0.4–0.5 mm fraction was selected for further investigations.
3.3. Preparation of MgO by Calcination of Mg(OH)2
After the grains of Mg(OH)2 were formed, they were calcined to obtain MgO, which was performed in accordance with the following procedures:
- At 873 or 1873 K (6 h) in static air (muffle furnace);
- At selected temperatures (473–1073 K) in a flow of air (3 dm3·h−1) for 1 h and flow of nitrogen (3 dm3·h−1) for 5 h (vertical tubular quartz reactor with a glass-wool plug), heating rate 10 deg·min−1;
- Under vacuum (pressure 0.013 hPa (rotary pump) or 27 hPa (water pump) in horizontal tubular quartz reactor, heating rate 2 and 10 deg min−1.
3.4. Reagents
Toluene (analytical reagent, POCh, Gliwice, Poland) was dried over metallic potassium with benzophenone as the indicator and distilled in dry nitrogen atmosphere.
n-Butylamine (99%), t-butylamine (98%) and triethylamine (98%), all from Aldrich (Poznań, Poland), were dried over KOH pellets for 2 weeks and distilled in the presence of metallic potassium. Benzoic acid (99%, Aldrich) was sublimed twice under normal pressure.
Acetonitrile (99.8%, anhydrous) and pivalonitrile (98%), both from Aldrich (Poznań, Poland), were dried over P2O5 and distilled in the presence of small amount of P2O5.
Water used as the solvent was prepared as follows: tap water was distilled in all metal laboratory apparatus. The distillate was filtered through two columns filled with cationic and anionic ion exchange resins and the obtained water was distilled twice in Pyrex glass apparatus. The distillate was kept in polypropylene containers.
3.5. Characterization of Mg(OH)2 and MgO
3.5.1. Composition Determination
Continuous Powder Introduction Microwave Induced Plasma Optical Emission Spectrometry. CPI-MIP-OES (Perkin Elmer Inc., Waltham, MA, USA), was applied for direct determination of trace elements in batch II fraction II, as well as batch III fraction III of MgO samples. The measurements were performed using an MIP-OES spectrometer (MIP 750MV, Analab, Warsaw, Poland) with a TEM cavity. Continuous introduction of the powdered MgO (c.a. 0.5 g, grain size 32–80 μm) was achieved by passing a flow of argon (up to 250 cm3 min−1) through the sample chamber of the CPI device (Analab, Warsaw, Poland). The analytical system was calibrated using mixtures of spectroscopic grade MgO with R.U. powder. The following elements were determined: Ni (231.6 nm), Fe (248.3 nm), B (249.8 nm), Si (251.6 nm), Cu (324.8 nm), Ca (393.4 nm), Al (396.2 nm) and Mn (403.1 nm).
Na+, K+ cations. The content of alkali metal ions in the Mg(OH)2 samples prepared using NaOH or KOH as reagents was measured using AES method at wavelength 766.5 and 589.0 nm for K+ and Na+, respectively.
Cl− anions. A weighed sample of Mg(OH)2 was digested with HNO3, diluted with water and titrated potentiometrically with 0.01047 M AgNO3 in the presence of 0.5 M KNO3 solution using an automatic titrator (Mettler Toledo, Greifensee, Switzerland).
SO42− anions. A weighed sample of Mg(OH)2 prepared by precipitation from MgSO4 solution was dissolved in HNO3, 5 wt.% BaCl2 solution in water was added and the whole solution was diluted with 50% ethanol. Absorbance of the solution was measured at the wavelength of 420 nm.
Mn+ ions (M = Ni, Mn and Fe). The content of these metal ions in Mg(OH)2 from batch III fractions I and II was determined using spectrophotometric methods after dissolving the samples in nitric acid. The following reagents were used for the measurements of the concentration of the above metals (in parentheses the wavelengths are given), dimethylglyoxime for Ni (λ = 380 nm), formaldoxime for Mn (λ = 480 nm) and 1,10-phenantroline for Fe (λ = 513 nm).
X-ray photoelectron spectroscopy. The XPS spectra were obtained on a Thermo Scientific K-Alpha instrument (Thermo Scientific, Waltham, MA, USA). The radiation source Al Kα operated at 1486.6 eV. The survey spectra were collected with pass energy of 200 eV, with a 1.0 eV step. Pass energy of 50 eV was used to obtain spectra of the following regions: C 1s, Mg 2p, and O 1s. The analysis was performed with Avantage 4.78 Surface Chemical Analysis (Thermo Scientific, Waltham, MA, USA). The analyzed area was approximately 50 × 35 μm. A Shirley-type background was subtracted, and the scale was calibrated to the C 1s signal. The peaks were fitted with functions with a 30:70 ratio of Gaussian-Lorentzian curves.
X-ray Diffraction Studies: Powder diffraction data were collected on a D-5000 diffractometer (Siemens, Dallas, TX, USA) equipped with a scintillation counter and Ni-filtered CuKα radiation with a sealed Cu tube operating at 40 kV and 40 mA. The measurements were performed in Bragg-Brentano focusing geometry using continuous scan mode with 0.01° collection step and counting time 1 s per step. Before measurements, samples of MgO-T were immobilized in a high-vacuum silicon grease or placed in envelopes made of Mylar because magnesium oxide reacts with an air humidity and in lesser extent also with atmospheric carbon dioxide. This is why freshly calcined samples of MgO for XRD measurements were placed into Mylar envelopes, made of thin polyester foil, to prevent the formation of Mg(OH)2.
3.5.2. Specific Surface Area
The specific surface areas of the samples were measured using a Micromeritics ASAP 2020 instrument (Micromeritics Instrument Corp., Norcross, GA, USA). During analysis, the pore size distribution and total pore volume were also measured and the average pore diameter (4V/S) was calculated.
3.5.3. Thermogravimetric Analysis
The DTA-TG-MS measurements were performed using a NETZSCH STA 449C thermobalance (NETZSCH, Selb, Germany). The experiments were conducted using samples of approximately 50 mg of catalyst powder heated up to 873 K at the constant rate of 10 K∙min−1 in a flow of Ar (10 cm3·min−1). The data was processed with NETZSCH Proteus Thermal Analysis software v.2 (NETZSCH, Selb, Germany). The m/z = 18 signal was monitored throughout the measurement. The mass of water released during each experiment was noted. The reference run was an experiment using 50 mg of Al2O3.
3.5.4. Temperature Programmed Desorption Studies
The TPD measurements were carried out in a stream of helium (25 cm3 min−1). A weighed sample of MgO-T (200 mg) was placed into the tubular quartz reactor and heated to 473 K for MgO-473 or 873 K for MgO-873 and MgO-1873 samples for 1h in a stream of He. After cooling to room temperature each sample was treated with vapors of n-BuNH2, Et3N, MeCN or t-BuCN in He for 1h and purged with He at 373 K for 1h. The outlet of the reactor was connected to the sampling valve of a Dycor Ametek MA 200 quadrupole mass spectrometer (Ametek Process Instruments, Pittsburg, PA, USA). The ionizing voltage was 70 V. The temperature of the reactor was ramped at a 10 K min−1 (up to 973 K) with an Omega CN 2011 temperature controller (Omega, Stamford, CT, USA). In order to separate contributions of different species, the obtained mass spectrum was analyzed using standard mass spectra of the respective compounds.
3.5.5. Secondary Emission Microscopy—Energy Dispersive X-ray Spectroscopy
The SEM images were taken using an FEI Quanta FEG 250 instrument (Field Electron and Ion Company, FEI, Hillsboro, OR, USA). The magnifications were 250, 5000, and 50,000. The EDS analysis was performed on three areas of each of the samples with the accelerating voltage of 10 keV (WD = 10 mm). The morphologies of samples were characterized by scanning electron microscope LEO 1530 (ZEISS, Oberkochen, Germany).
3.5.6. Basic/Acidic Site Determination and Quantification
The strength of the surface acid-base sites of MgO was determined by the Hammett method using a sequence of solutions (0.1%) of the following indicators (in parentheses, values of pKa or pKBH of indicators are given): crystal violet (0.8), methyl red (4.8), bromothymol blue (7.2), phenolphthalein (9.3), 2,4-dinitroaniline (15.0), 4-nitroaniline (18.4), diphenylamine (22.3), 4-chloroaniline (26.5), triphenylmethane (33.0) and diphenylmethane (35.0) in anhydrous toluene [40]. The measurements were performed under dry nitrogen atmosphere at room temperature with reading after 24 h [39].
The concentrations of acidic and basic sites of the MgO-T samples were determined according to the procedure described elsewhere [45]. The dilute solutions (0.01 mol dm−3) of benzoic acid, n-butylamine and triethylamine in anhydrous toluene were used as titers. The titrations were performed at room temperature in grease-less glass reactors under dry nitrogen atmosphere. Each sample of MgO-T (c.a. 250 mg) was treated with a known volume of the solution for 24 h. As was found in the preliminary experiments, an equilibrium is attained after 16–18 h of the reaction with occasional shaking only that excludes breaking up the grains. Aliquots of the solutions were taken, diluted with distilled water and titrated with 0.01 mol dm−3 solutions of hydrochloric acid or potassium hydroxide in the presence of phenolphthalein as the indicator.
4. Conclusions
The necessary conditions for the preparation of high-purity magnesium hydroxide from water-soluble magnesium salts by reaction with aqueous alkaline solutions were determined. It was shown that the type of magnesium salt used (nitrate, sulfate or chloride) and the type of base (NH3 (aq), NaOH or KOH) used for hydroxide precipitation have a significant impact on the final purity of the Mg(OH)2 precipitate, and, more importantly, on the specific surface area of magnesium oxide obtained by thermal decomposition. The specific surface areas of the obtained MgO are as follows (SBET [m2·g−1]/starting material): 129.2/nitrate > 54.2/sulfate > 38.6/chloride. It was also found that the Mg(NO3)2 + NH3 (aq) system provides the highest purity hydroxide. In the case of the MgCl2 + NH3 (aq) system, the obtained magnesium hydroxide contains chloride ions, which cause the sintering of MgO, while in the hydroxide precipitate formed in the MgSO4 + NH3 (aq) reaction, there are sulfate anions that cannot be removed, the presence of which will increase the acidic properties of the MgO surface.
The use of an aqueous ammonia solution as a magnesium hydroxide precipitating reagent does not introduce any impurities. In contrast, the presence of Na+ and K+ ions has been detected in the case of NaOH and KOH. Moreover, the use of these bases causes the Mg(OH)2 precipitate to sediment very slowly, and its washing is troublesome.
It has been shown that a multistep precipitation of Mg(OH)2, even in the case of analytically pure commercial precursors, results in a very efficient removal of metal ion (Fe, Ni, Mn, etc.) contamination from the product, which leads to a significant increase in the purity of the obtained hydroxide and, consequently, MgO. Studies of the effect of the following sequence of operations were carried out:
- Compacting pure magnesium hydroxide dust into pellets;
- Crushing the obtained pellets;
- Separation of the proper fraction of magnesium hydroxide on sieves;
- Thermal decomposition of the hydroxide to magnesium oxide
on the final purity of the obtained oxide. Changes in the concentration of elements (Fe, Si, Al and Cu) in contact with the highest-purity magnesium hydroxide sample (batch III fraction III) were investigated before and after the operation of forming Mg (OH)2 grains and its thermal decomposition to oxide. It was found that the performance of each of the above-mentioned operations causes a significant increase in the concentration of the indicated elements in the sample of magnesium hydroxide.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12121595/s1, Figure S1: Example of hysteresis loop of purified magnesia.
Author Contributions
Conceptualization, M.G.; formal analysis, M.G. and E.M.I.; investigation, M.G., A.C., U.U., E.M.I., D.Ł. and Z.K.; resources, M.G.; data curation, M.G.; writing—original draft preparation, M.G. and E.M.I.; writing—review and editing, M.G. and E.M.I.; visualization, U.U. and E.M.I.; project administration, M.G.; funding acquisition, M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Scientific Discipline Board (RDN) for Chemical Engineering of the Warsaw University of Technology, grant number: 2022 I-CHEM.3-1.2.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Ilya Gourevich from the Centre for Nanostructure Imaging (CNI) at the University of Toronto for performing the SEM-EDX analysis, and Peter Brodersen from the Ontario Centre for the Characterization of Advanced Materials (OCCAM) for the X-ray Photoelectron Spectroscopy measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ramlee, N.N.; Md Illias, R.; Rahman, R.A.; Toemen, S.; Selvasembian, R.; Ahmad, R.A.; Abdul Manas, N.H.; Wan Azelee, N.I. Biochemical and Physical Characterization of Immobilized Candida rugosa Lipase on Metal Oxide Hybrid Support. Catalysts 2022, 12, 854. [Google Scholar] [CrossRef]
- Zigla, A.A.; Kox, T.; Mevoa, D.; Assaouka, H.T.; Nsangou, I.N.; Daawe, D.M.; Kenmoe, S.; Kouotou, P.M. Magnesium-Modified Co3O4 Catalyst with Remarkable Performance for Toluene Low Temperature Deep Oxidation. Catalysts 2022, 12, 411. [Google Scholar] [CrossRef]
- Bagabas, A.; Al-Fatesh, A.S.; Kasim, S.O.; Arasheed, R.; Ibrahim, A.A.; Ashamari, R.; Anojaidi, K.; Fakeeha, A.H.; Abu-Dahrieh, J.K.; Abasaeed, A.E. Optimizing MgO Content for Boosting γ-Al2O3-Supported Ni Catalyst in Dry Reforming of Methane. Catalysts 2021, 11, 1233. [Google Scholar] [CrossRef]
- Saied, E.; Eid, A.M.; Hassan, S.E.-D.; Salem, S.S.; Radwan, A.A.; Halawa, M.; Saleh, F.M.; Saad, H.A.; Saied, E.M.; Fouda, A. The Catalytic Activity of Biosynthesized Magnesium Oxide Nanoparticles (MgO-NPs) for Inhibiting the Growth of Pathogenic Microbes, Tanning Effluent Treatment, and Chromium Ion Removal. Catalysts 2021, 11, 821. [Google Scholar] [CrossRef]
- Chowdhury, A.H.; Bhanja, P.; Salam, N.; Bhaumik, A.; Islam, S.M. Magnesium oxide as an efficient catalyst for CO2 fixation and N-formylation reactions under ambient conditions. Mol. Catal. 2018, 450, 46–54. [Google Scholar] [CrossRef]
- Julkapli, N.M.; Bagheri, S. Magnesium oxide as a heterogeneous catalyst support. Rev. Inorg. Chem. 2016, 36, 1–41. [Google Scholar] [CrossRef]
- Leofanti, G.; Solari, M.; Tauszik, G.; Garbassi, F.; Galvagno, S.; Schwank, J. Magnesium oxide as a catalyst support: The influence of chlorine. Appl. Catal. 1982, 3, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Al-Fatesh, A.S.; Kumar, R.; Fakeeha, A.H.; Kasim, S.O.; Khatri, J.; Ibrahim, A.A.; Arasheed, R.; Alabdulsalam, M.; Lanre, M.S.; Osman, A.; et al. Promotional effect of magnesium oxide for a stable nickel-based catalyst in dry reforming of methane. Sci. Rep. 2020, 10, 13861. [Google Scholar] [CrossRef] [PubMed]
- Di Cosimo, J.I.; Díez, V.K.; Ferretti, C.; Apesteguía, C.R. Chapter 1. Basic catalysis on MgO: Generation, characterization and catalytic properties of active sites. Catalysis 2014, 26, 1–28. [Google Scholar] [CrossRef]
- Montero, J.; Isaacs, M.; Lee, A.; Lynam, J.; Wilson, K. The surface chemistry of nanocrystalline MgO catalysts for FAME production: An in situ XPS study of H2O, CH3OH and CH3OAc adsorption. Surf. Sci. 2015, 646, 170–178. [Google Scholar] [CrossRef]
- Mohammadi, L.; Rahdar, A.; Bazrafshan, E.; Dahmardeh, H.; Thysiadou, A.; Kyzas, G.Z. Benzene Removal from Aqueous Solutions by Heterogeneous Catalytic Ozonation Process with Magnesium Oxide Nanoparticles. J. Int. Ozone Assoc. 2020, 43, 147–162. [Google Scholar] [CrossRef]
- Du, L.; Lia, Z.; Ding, S.; Chen, C.; Qu, S.; Yi, W.; Lu, J.; Ding, J. Synthesis and characterization of carbon-based MgO catalysts for biodiesel production from castor oil. Fuel 2019, 258, 116122. [Google Scholar] [CrossRef]
- Yousefi, S.; Haghighi, M.; Vahid, B.R. Facile and efficient microwave combustion fabrication of Mg-spinel as support for MgO nanocatalyst used in biodiesel production from sunflower oil: Fuel type approach. Chem. Eng. Res. Des. 2018, 138, 506–518. [Google Scholar] [CrossRef]
- Corma, A.; Iborra, S. Optimization of Alkaline Earth Metal Oxide and Hydroxide Catalysts for Base-Catalyzed Reactions. Adv. Catal. 2006, 49, 239–302. [Google Scholar] [CrossRef]
- Baird, M.J.; Lunsford, J.H. Catalytic sites for the isomerization of 1-butene over magnesium oxide. J. Catal. 1972, 26, 440–450. [Google Scholar] [CrossRef]
- Xiang, W.; Moa, X.; Feng, S.; Xu, F.; Zhou, G.; Zhou, H.; Xua, C.; Chen, B. Effect of MgO on WO3/SiO2-catalyzed light olefin metathesis using different feedstocks. Mol. Catal. 2017, 442, 49–56. [Google Scholar]
- Tanabe, K.; Holderich, W.F. ChemInform Abstract: Industrial Application of Solid Acid-base Catalysts. Appl. Catal. A Gen. 2010, 30, 199938249. [Google Scholar] [CrossRef]
- Margellou, A.; Koutsouki, A.; Petrakis, D.; Vaimakis, T.; Manos, G.; Kontominas, M.; Pomonis, P. Enhanced production of biodiesel over MgO catalysts synthesized in the presence of Poly-Vinyl-Alcohol (PVA). Ind. Crop. Prod. 2018, 114, 146–153. [Google Scholar] [CrossRef]
- Kampars, V.; Kampare, R.; Krumina, A. MgO Catalysts for FAME Synthesis Prepared Using PEG Surfactant during Precipitation and Calcination. Catalysts 2022, 12, 226. [Google Scholar] [CrossRef]
- Hung, C.-H.; Chen, C.-S.; Sheu, H.-S.; Chang, J.-R. Deactivation and rejuvenation of pellet MgO/SiO2 catalysts for trans-esterification of soybean oil with methanol to bio-diesel: Roles of MgO morphology change in catalysis. Ind. Eng. Chem. Res. 2018, 57, 456–469. [Google Scholar] [CrossRef]
- Matsuda, T.; Tanabe, J.; Hayashi, N.; Sasaki, Y.; Miura, H.; Sugiyama, K. Properties of magnesium oxides prepared from various salts and their catalytic activity in 1-butene isomerisation. Bull. Chem. Soc. Jpn. 1982, 55, 990–994. [Google Scholar] [CrossRef]
- Tanabe, K.; Zhang, G.; Hattori, H. Addition of metal cations to magnesium oxide catalysts for the aldol condensation of ace-tone. Appl. Catal. 1989, 48, 63–70. [Google Scholar] [CrossRef]
- Matsuda, T.; Sugimoto, M. High activity of MgO catalyst prepared from magnesium oxalate for the hydrogenation of butadiene. React. Kinet. Catal. Lett. 1991, 44, 69–73. [Google Scholar] [CrossRef]
- Ardizzone, S.; Bianchi, C.L.; Fadoni, M.; Vercelli, B. Magnesium salts and oxide: An XPS overview. Appl. Surf. Sci. 1997, 119, 253–259. [Google Scholar] [CrossRef]
- Holt, T.E.; Logan, A.D.; Chakraborti, S.; Datye, A.K. The effect of catalyst preparation conditions on the morphology of MgO catalyst supports. Appl. Catal. 1987, 34, 199–213. [Google Scholar] [CrossRef]
- Wanke, S.; Fiedorow, R. The Influence of Preparation Methods on Surface Area, Porosity and Crystallinity of Magnesium Oxide. Stud. Surf. Sci. Catal. 1987, 39, 601–609. [Google Scholar] [CrossRef]
- Choudhary, V.; Pandit, M. Surface properties of magnesium oxide obtained from magnesium hydroxide: Influence on preparation and calcination conditions of magnesium hydroxide. Appl. Catal. 1991, 71, 265–274. [Google Scholar] [CrossRef]
- Mu, J.; Pearlmutter, D.D. Thermal decomposition of carbonates, carboxylates, oxalates, acetates, formates, and hydroxides. Thermochim. Acta 1981, 49, 207–218. [Google Scholar] [CrossRef]
- Vahid, B.R.; Haghighi, M. Biodiesel production from sunflower oil over MgO/MgAl2O4 nanocatalyst: Effect of fuel type on catalyst nanostructure and performance. Energy Convers. Manag. 2017, 134, 290–300. [Google Scholar] [CrossRef]
- Pearson, R.G. Ionization potentials and electron affinities in aqueous solution. J. Am. Chem. Soc. 1986, 108, 6109–6114. [Google Scholar] [CrossRef]
- Martin, D.; Brause, W.; Radeglia, R. Struktur-Reaktivitätsuntersuchungen mit heterosubstituierten Nitrilen. II. H-Brückenwechselwirkungen zwischen OH-Protonendonatoren und Cyanverbindungen Korrelationen. J. Prakt. Chem. 1970, 312, 797–811. [Google Scholar] [CrossRef]
- Busca, G. Bases and Basic Materials in Chemical and Environmental Processes. Liquid versus Solid Basicity. Chem. Rev. 2010, 110, 2217–2249. [Google Scholar] [CrossRef] [PubMed]
- Bryantsev, V.S.; Diallo, M.S.; Goddard, W.A. pKa calculations of aliphatic amines, diamines, and aminoamides via Density Functional Theory with a Poisson−Boltzmann Continuum Solvent Model. J. Phys. Chem. A 2007, 111, 4422–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juskelis, M.V.; Slanga, J.P.; Roberie, T.G.; Peters, A.W. A comparison of CaO, beta, and a dealuminated Y by ammonia TPD and by temperature programmed 2-propylamine cracking. J. Catal. 1992, 138, 391–394. [Google Scholar] [CrossRef]
- Gorte, R.J.; Crossley, S.P. A perspective on catalysis in solid acids. J. Catal. 2019, 375, 524–530. [Google Scholar] [CrossRef]
- Coluccia, S.; Garrone, E.; Borello, E. Infrared spectroscopic study of molecular and dissociative adsorption of ammonia on magnesium oxide, calcium oxide and strontium oxide. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1983, 79, 607–613. [Google Scholar] [CrossRef]
- Borello, E.; Coluccia, S.; Zecchina, A. Infrared emission study of the reaction of CO with ammonia preadsorbed on MgO. J. Catal. 1985, 93, 331–339. [Google Scholar] [CrossRef]
- Kijeński, J.; Malinowski, S. Influence of sodium on the physico-chemical and catalytic properties of magnesium oxide. JCS Faraday Trans. I 1978, 74, 250–261. [Google Scholar] [CrossRef]
- Gliński, M.; Ulkowska, U. Reactivity of Alcohols in Chemoselective Transfer Hydrogenation of Acrolein over Magnesium Oxide as the Catalyst. Catal. Lett. 2010, 141, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Misono, M.; Ono, Y.; Hattori, H. Acid and base centers: Structure and acid-base property. Stud. Surf. Sci. Catal. 1989, 51, 27–213. [Google Scholar]
- Gregg, S.J.; Ramsay, J.D. Adsorption of carbon dioxide by magnesia studied by use of infrared and isotherm measurements. J. Chem. Soc. A 1970, 2784–2787. [Google Scholar] [CrossRef]
- Coluccia, S.; Lavagnino, S.; Marchese, L. The hydroxylated surface of MgO powders and the formation of surface sites. Mater. Chem. Phys. 1988, 18, 445–464. [Google Scholar] [CrossRef]
- Pinto, P.; Lanza, G.; Ardisson, J.; Lago, R. Controlled Dehydration of Fe(OH)3 to Fe2O3: Developing Mesopores with Complexing Iron Species for the Adsorption of β-Lactam Antibiotics. J. Braz. Chem. Soc. 2019, 30, 310–317. [Google Scholar] [CrossRef]
- Sato, T.; Nakamura, T.; Ozawa, F. Thermal decomposition of nickel hydroxide. J. Appl. Chem. Biotechnol. 1975, 25, 583–590. [Google Scholar] [CrossRef]
- Tamanaka, T.; Tanabe, K. New determination of acid-base strength distribution of a common scale on solid surfaces. J. Phys. Chem. 1975, 75, 2409–2411. [Google Scholar] [CrossRef]
Scheme 1.
Reaction pathways: starting materials, dissolving solutions and precipitating agents used. (a) commercial MgO as a starting material, (b) magnesium nitrate as a starting material.
Scheme 1.
Reaction pathways: starting materials, dissolving solutions and precipitating agents used. (a) commercial MgO as a starting material, (b) magnesium nitrate as a starting material.

Figure 1.
TG and DTA curves obtained for (a) MgO-473, (b) MgO-573, (c) MgO-673 and (d) MgO-873.

Figure 2.
XRD results obtained for (a) MgO-473, (b) MgO-573, (c) MgO-673; (d) MgO-873 and (e) MgO-1873.
Figure 2.
XRD results obtained for (a) MgO-473, (b) MgO-573, (c) MgO-673; (d) MgO-873 and (e) MgO-1873.

Figure 3.
SEM images of (a) MgO-473; (b) MgO-573; (c) MgO-673; (d) MgO-873 and (e) MgO-1873.

Figure 4.
TPD results: profiles obtained for (a) MgO-873 using n-butylamine, t-butylamine and triethylamine as probe molecules, (b) triethylamine for different MgO-T samples, (c) pivalonitrile, and (d) acetonitrile; MgO calcined at 473 K (black), 873 K (blue), 1873 K (red).
Figure 4.
TPD results: profiles obtained for (a) MgO-873 using n-butylamine, t-butylamine and triethylamine as probe molecules, (b) triethylamine for different MgO-T samples, (c) pivalonitrile, and (d) acetonitrile; MgO calcined at 473 K (black), 873 K (blue), 1873 K (red).

Figure 5.
Samples of magnesium oxide obtained by calcination at 873 K of the three precipitates of magnesium hydroxide; 1st, 2nd and 3rd precipitation in sequence from left to right.
Figure 5.
Samples of magnesium oxide obtained by calcination at 873 K of the three precipitates of magnesium hydroxide; 1st, 2nd and 3rd precipitation in sequence from left to right.

Figure 6.
SEM and EDX results: images with 50,000 times magnification of MgO obtained from the (a) first, (b) second, and (c) third fraction, and (d) typical EDX result; inset: numerical data for the precipitates.
Figure 6.
SEM and EDX results: images with 50,000 times magnification of MgO obtained from the (a) first, (b) second, and (c) third fraction, and (d) typical EDX result; inset: numerical data for the precipitates.

Figure 7.
XPS results collected from a sample of MgO obtained from batch III fraction I: (a) detailed regions: O 1s and Mg 2p (without prior etching) and (b) survey spectra at three different etch levels: 0 s, 15 s and 30 s.
Figure 7.
XPS results collected from a sample of MgO obtained from batch III fraction I: (a) detailed regions: O 1s and Mg 2p (without prior etching) and (b) survey spectra at three different etch levels: 0 s, 15 s and 30 s.

Figure 8.
Influence of number of portions of wash water used for washing Mg(OH)2 obtained from Mg(NO3)2 and NH3(aq) on the specific surface area of magnesium hydroxide and final product.
Figure 8.
Influence of number of portions of wash water used for washing Mg(OH)2 obtained from Mg(NO3)2 and NH3(aq) on the specific surface area of magnesium hydroxide and final product.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Textural properties of MgO-T samples calcined at various temperatures.
| T [K] | SBET [m2·g−1] | Pore Volume [cm3·g−1] | Av. Pore Diameter [nm] | D 2 [nm] | Lattice Parameter [nm] |
|---|---|---|---|---|---|
| 473 | 27 1 | 0.200 | 24.1 | 12.7 | a = 0.3153 c = 0.4744 |
| 573 | 36 | - | - | 12.0 | a = 0.3132 c = 0.4664 |
| 673 | 222 | 0.510 | 11.0 | 6.6 | a = 0.4222 |
| 773 | 134 | - | - | 8.7 | a = 0.4197 |
| 873 | 100 | 0.529 | 17.2 | 9.9 | a = 0.4191 |
| 973 | 69 | - | - | 11.4 | a = 0.4190 |
| 1073 | 47 | - | - | 15.5 | a = 0.4188 |
| 1873 | 5 | 0.059 | 42.1 | 38.5 | a = 0.4175 |
1 SBET = 31 m2·g−1 for unpressed powder of MgO-473; 2 crystallite dimensions based on integral-width of the most intensive XRD line.
Table 2.
The concentrations of acidic and basic sites on the surfaces of MgO-T samples (batch II fraction II).
Table 2.
The concentrations of acidic and basic sites on the surfaces of MgO-T samples (batch II fraction II).
| MgO-T | SBET [m2·g−1] | Conc. of Acidic Sites [μmol·g−1] | Conc. of Basic Sites [μmol·g−1] | |
|---|---|---|---|---|
| n-BuNH2 | Et3N | PhCOOH | ||
| MgO-473 | 27 | 30 1 | 0 2 | 490 3 |
| MgO-873 | 100 | 115 4 | 5 2 | 1785 5 |
| MgO-1873 | 5 | 5 2 | 2 2 | 190 4 |
1 ±5; 2 ±2; 3 ±15; 4 ±10; 5 ±40.
Table 3.
Distribution of the strength of acidic and basic sites on the surface of MgO-T samples determined using the Hammett indicator method.
Table 3.
Distribution of the strength of acidic and basic sites on the surface of MgO-T samples determined using the Hammett indicator method.
| MgO-T 1 | H0 | H- | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.8 | 4.8 | 7.2 | 9.3 | 15.0 | 18.4 | 22.3 | 26.5 | 33.0 | |
| MgO-473 | + | + | + | ||||||
| MgO-573 | + | + | + | + | |||||
| MgO-673 | + | + | + | + | + | + | |||
| MgO-873 | + | + | + | + | + | + | |||
| MgO-1073 | + | + | + | + | + | ||||
| MgO-1873 | + | + | + | + | + | ||||
1 The plus sign indicates that a color change occurred.
Table 4.
Results of determination of Fe, Ni and Mn impurities in batch III fractions I, II and III.
| Sample | Fe [%] | Ni [%] | Mn [%] |
|---|---|---|---|
| Mg(OH)2 (I) | 0.1683 | 0.3804 | 0.082 |
| Mg(OH)2 (II) | 0.0127 | 0.0296 | 0.0107 |
| Mg(OH)2 (III) | 0.00171 | 0.0012 1 | 0.00111 |
1 ±3%.
Table 5.
CPI-MIP-OES results of impurities determined in batch III fraction III.
| Element | [E] [%] 1 | Element | [E] [%] 1 |
|---|---|---|---|
| Si | 0.0040 | Ca | 0.0010 |
| B | 0.0019 | Na | 0.0008 |
| Fe | 0.0017 | Zn | 0.0007 |
| Ni | 0.0012 | Al | 0.0005 |
| Mn | 0.0011 | Cu | 0.0003 |
1 the method error of the determined values was ±3%.
Table 6.
Maximum temperature of decomposition of Mg(OH)2 samples obtained using three different counterions and three precipitating agents.
Table 6.
Maximum temperature of decomposition of Mg(OH)2 samples obtained using three different counterions and three precipitating agents.
| No. of Wash Portions | T max [K] 1 | ||||
|---|---|---|---|---|---|
| NO3− | SO42− | Cl− | NO3− | NO3− | |
| NH3 | NH3 | NH3 | KOH | NaOH | |
| 5 | 689 | 687 | 676 | 674 | 675 |
| 15 | 690 | 688 | 681 | 674 | 673 |
| 25 | 692 | 688 | 686 | 673 | 673 |
1 Tmax ± 2 deg.
Table 7.
Effect of Mg(OH)2 decomposition conditions on the specific surface area of the obtained MgO. Calcination for 6 h at final temperature, heating rate 10 deg·min−1.
Table 7.
Effect of Mg(OH)2 decomposition conditions on the specific surface area of the obtained MgO. Calcination for 6 h at final temperature, heating rate 10 deg·min−1.
| T [K] | Conditions | SBET [m2∙g−1] |
|---|---|---|
| 473 | Stream of N2 1 | 29 2 |
| 573 | Stream of N2 1 | 27 2 |
| Vacuum 3 | 143 4 | |
| 673 | Stream of N2 1 | 222 5 |
| Vacuum 2 | 326 | |
| Vacuum 3,6 | 337 | |
| 873 | Static air | 84 |
| Stream of N2 1 | 129 | |
| Vacuum 3 | 280 7 |
1 V(N2) = 3 dm3 h−1; 2 Mg(OH)2 is the solely product according to XRD measurements; 3 p = 0.013 hPa, rotary pump; 4 after measurement the sample contains 46.3% of MgO (TG); 5 MgO is the solely product according to XRD measurements; 6 heating rate 2 deg∙min−1; 7 S = 157 m2∙g−1 at p = 27 hPa (water pump).
Table 8.
Effect of the number of times applied in washing Mg(OH)2 on the specific surface area of MgO obtained from different precursors by calcination of Mg(OH)2 at 873 K for 6 h.
Table 8.
Effect of the number of times applied in washing Mg(OH)2 on the specific surface area of MgO obtained from different precursors by calcination of Mg(OH)2 at 873 K for 6 h.
| Origin of Mg(OH)2 | SBET [m2∙g−1] | |
|---|---|---|
| After 15 Washings | After 25 Washings | |
| MgCl2 + NH3 ∙ aq | 32.1 | 38.6 |
| MgSO4 + NH3 ∙ aq | 40.3 | 54.2 |
| Mg(NO3)2 + NaOH | 65.0 | 70.2 |
| Mg(NO3)2 + KOH | 60.3 | 66.1 |
| Mg(NO3)2 + NH3 ∙ aq | 118.4 | 129.2 |
Table 9.
Thermal analysis results of magnesium hydroxide samples (batch III) and specific surface area of MgO obtained from the decomposition at the temperature of 873 K (1 h in air and 5 h in nitrogen).
Table 9.
Thermal analysis results of magnesium hydroxide samples (batch III) and specific surface area of MgO obtained from the decomposition at the temperature of 873 K (1 h in air and 5 h in nitrogen).
| Sample | Tmax [K] | SBET of MgO [m2 g−1] |
|---|---|---|
| Mg(OH)2 (I) | 635 | 12.8 |
| Mg(OH)2 (II) | 681 | 62.9 |
| Mg(OH)2 (III) | 692 | 129.2 |
Table 10.
Determination of concentration of Fe, Si, Al and Cu in Mg(OH)2 and MgO (batch III fraction III) after formation of the grains of the hydroxide and subsequent thermal decomposition into MgO.
Table 10.
Determination of concentration of Fe, Si, Al and Cu in Mg(OH)2 and MgO (batch III fraction III) after formation of the grains of the hydroxide and subsequent thermal decomposition into MgO.
| Element | Concentration of Impurities 1 [μg·g−1] | |||
|---|---|---|---|---|
| Mg(OH)2 | Mg(OH)2 2 | Mg(OH)2 3 | MgO 4 | |
| Fe | 17 | 19 | - | - |
| Si | 40 | 51 5 | - | 59 |
| Al | 5 | 25 | - | - |
| Cu | 3 | 3 | 10 | - |
1 the average of the three measurements; 2 after pressing of a powder of Mg(OH)2 in a steel die and crushing of the wafers in a porcelain mortar; 3 after sieving the grains of Mg(OH)2 on a set of sifts made of a copper alloy (phosphor bronze containing c.a. 94% of Cu); 4 after calcination of the Mg(OH)2 bed at 873 K for 6 h placed on glass wool in vertical tubular quartz reactor; 5 after crushing the wafers of Mg(OH)2 in agate mortar a final Si concentration equalled to 45 μg∙g−1 was determined.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gliński, M.; Czajka, A.; Ulkowska, U.; Iwanek, E.M.; Łomot, D.; Kaszkur, Z. A Hands-on Guide to the Synthesis of High-Purity and High-Surface-Area Magnesium Oxide. Catalysts 2022, 12, 1595. https://doi.org/10.3390/catal12121595
AMA Style
Gliński M, Czajka A, Ulkowska U, Iwanek EM, Łomot D, Kaszkur Z. A Hands-on Guide to the Synthesis of High-Purity and High-Surface-Area Magnesium Oxide. Catalysts. 2022; 12(12):1595. https://doi.org/10.3390/catal12121595
Chicago/Turabian StyleGliński, Marek, Agnieszka Czajka, Urszula Ulkowska, Ewa M. Iwanek (nee Wilczkowska), Dariusz Łomot, and Zbigniew Kaszkur. 2022. "A Hands-on Guide to the Synthesis of High-Purity and High-Surface-Area Magnesium Oxide" Catalysts 12, no. 12: 1595. https://doi.org/10.3390/catal12121595
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.