
Knockdown of Antisense Noncoding Mitochondrial RNA Reduces Tumorigenicity of Patient-Derived Clear Cell Renal Carcinoma Cells in an Orthotopic Xenograft Mouse Model
 , , and
, , and 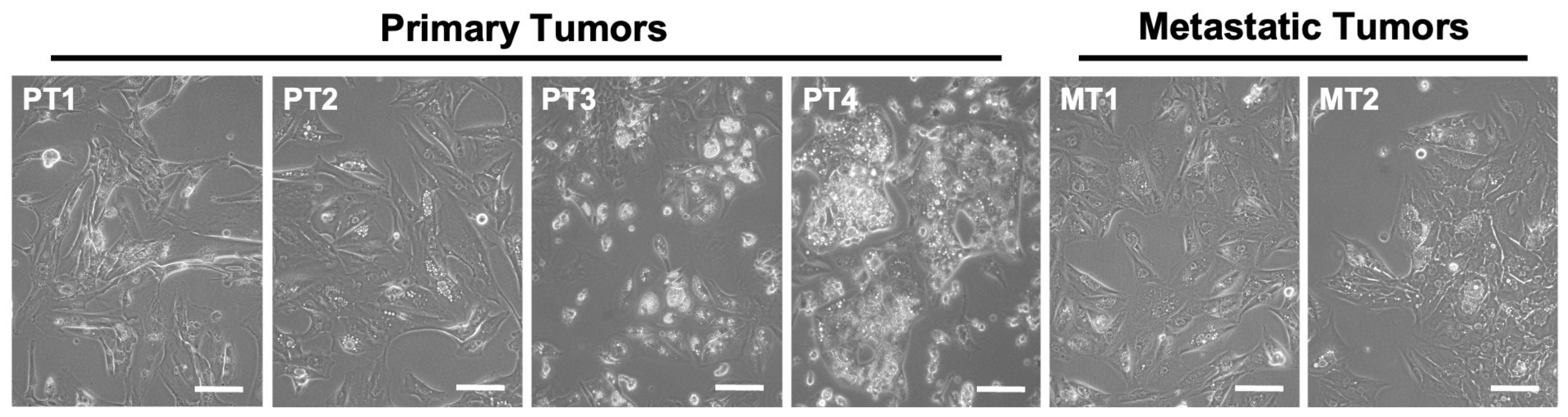{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Cultures
2.2. Transfection
2.3. Determination of Apoptosis
2.4. Western Blot
2.5. Spheroid Formation Assay
2.6. Orthotopic Xenograft Model
2.7. Subcutaneous Xenograft Model of MT1 Cells
2.8. Graphs and Statistical Analysis
3. Results
3.1. Primary Cultures of Human ccRCC
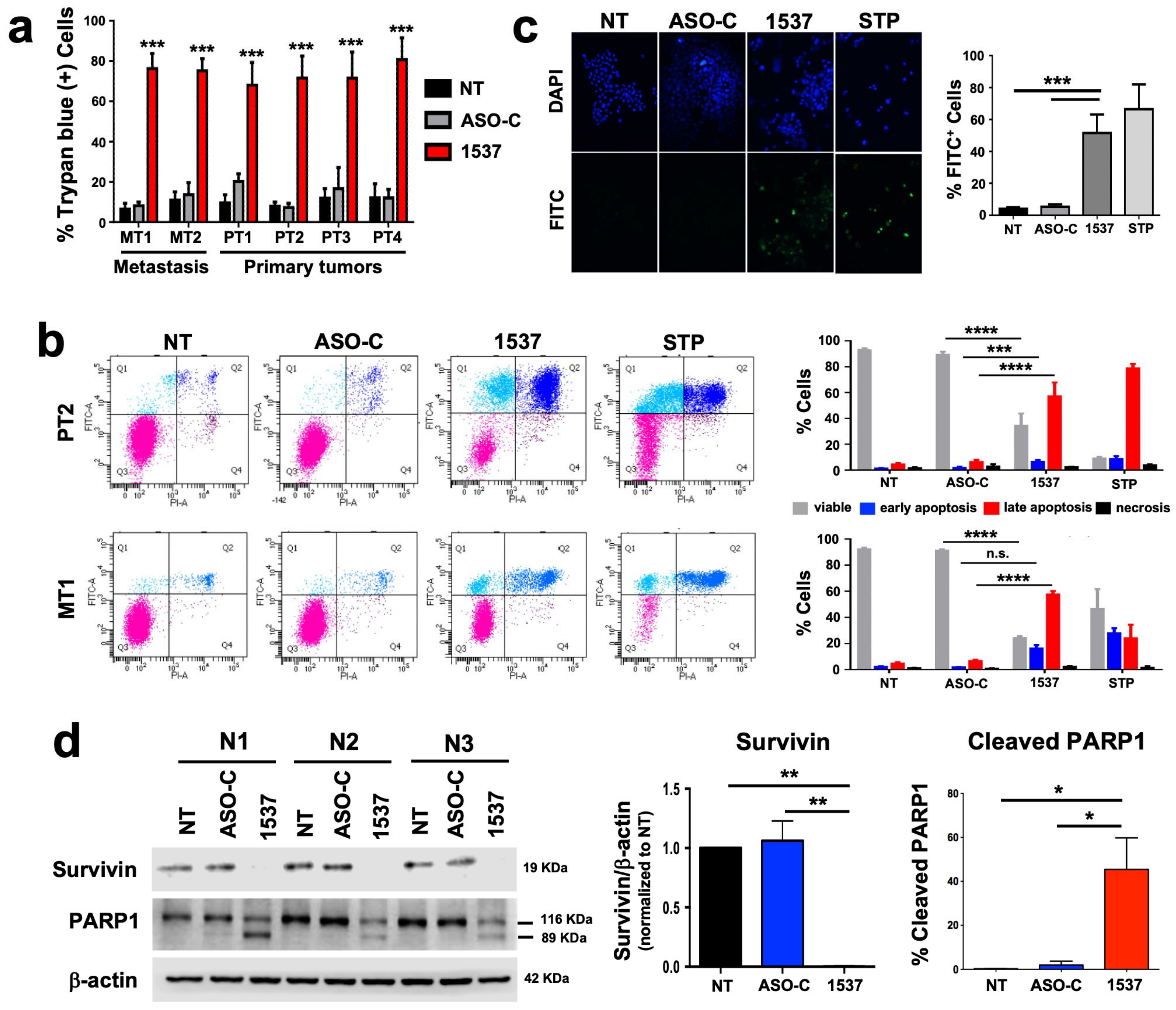
3.2. Knockdown of ASncmtRNA Induces Apoptotic Death of Primary ccRCC Cultures
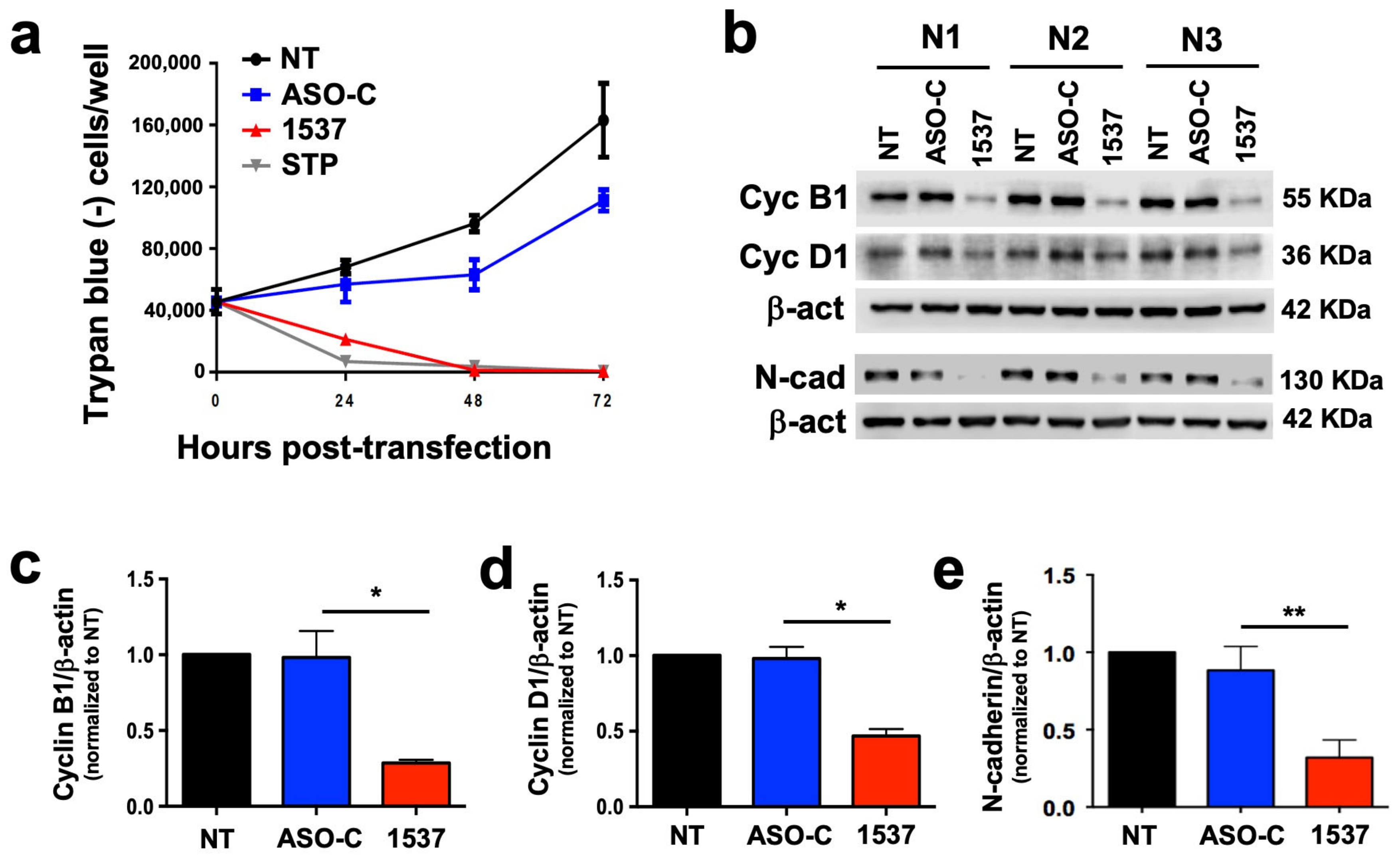
3.3. Knockdown of ASncmtRNA Reduces Proliferative Capacity of Primary ccRCC Cultures
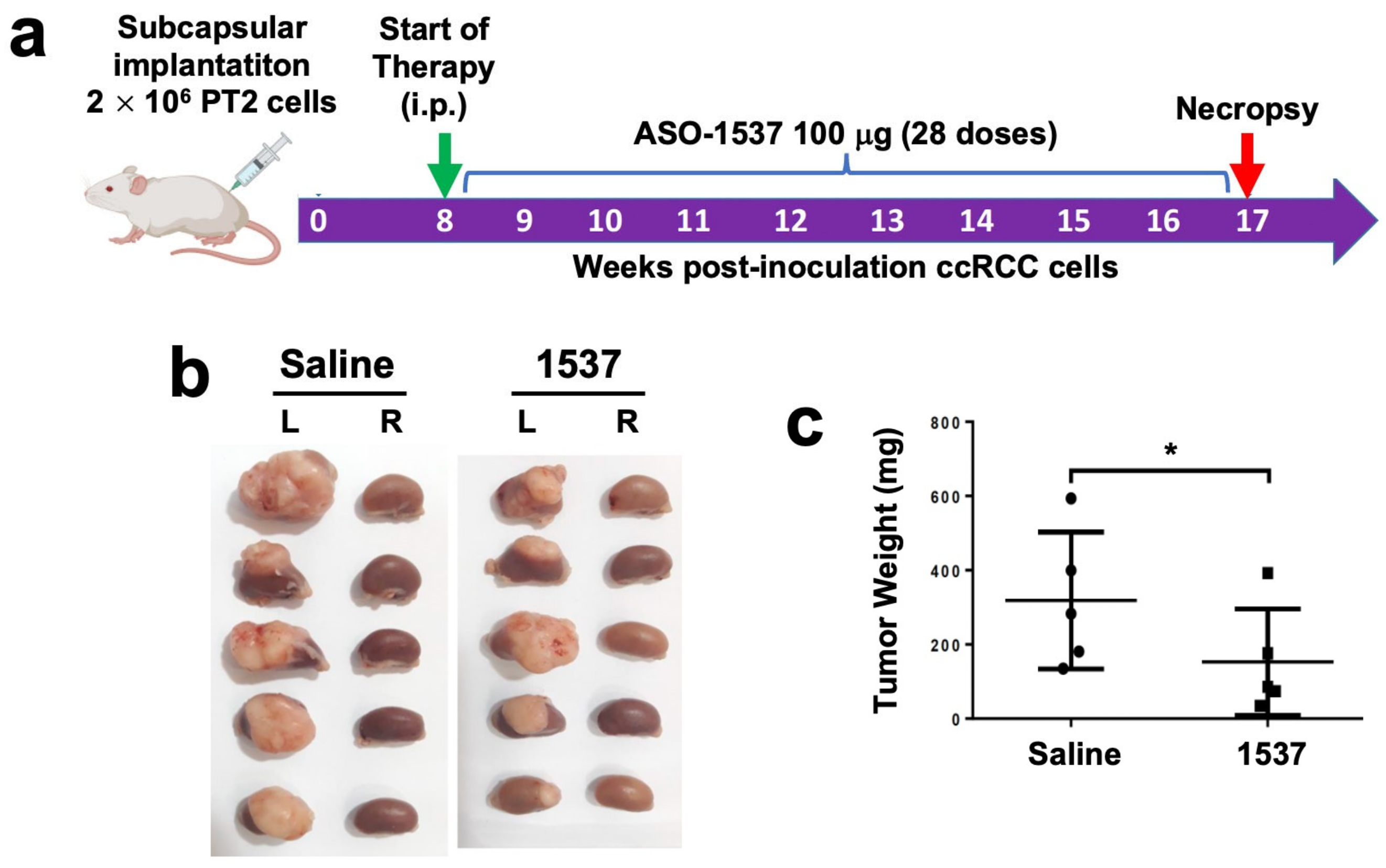
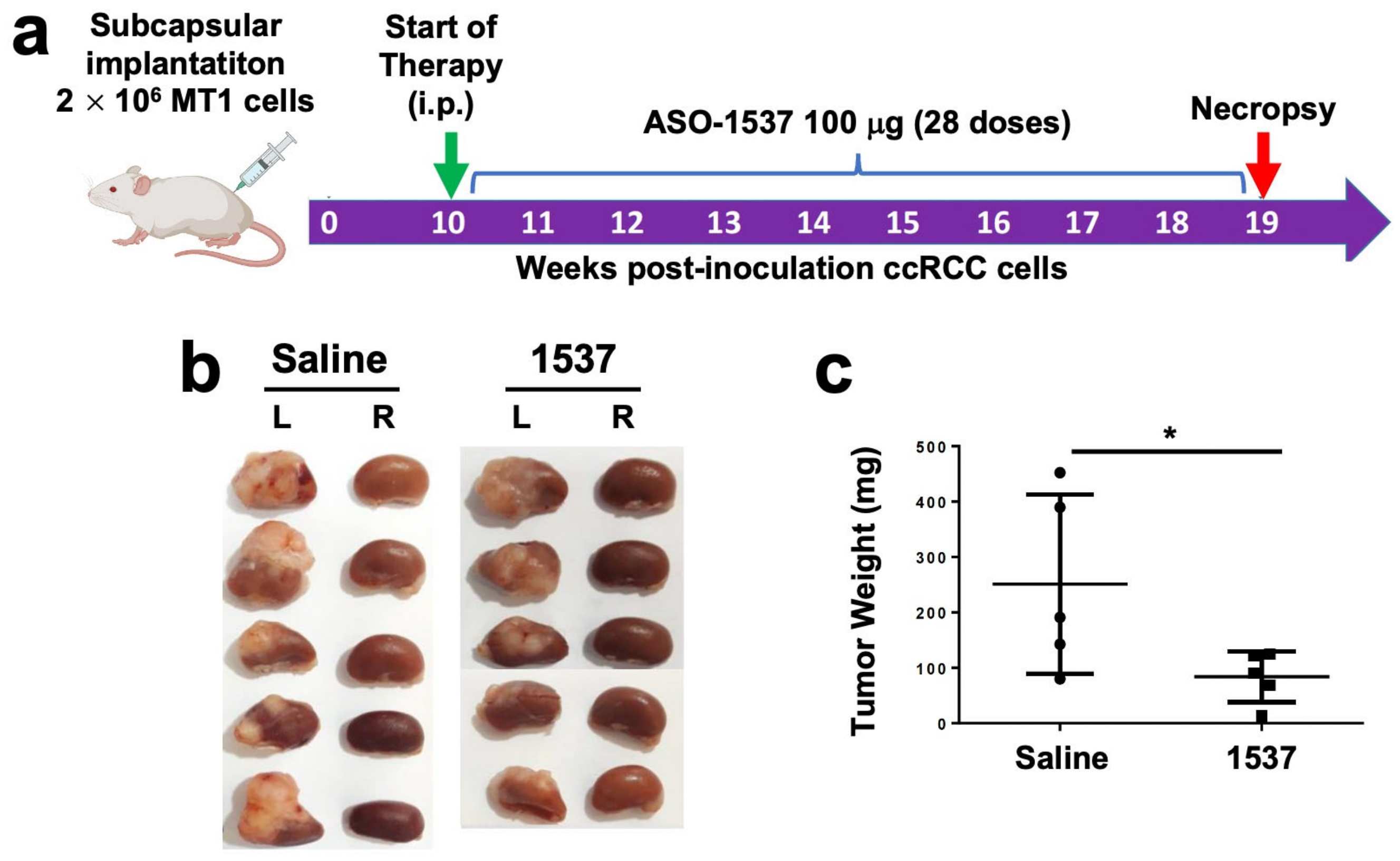
3.4. Knockdown of ASncmtRNA Reduces Growth of Orthotopic Tumors Generated with ccRCC Primary Cultures
3.5. Comparison of ASO-1537 and Sunitinib in a Subcutaneous Xenograft ccRCC Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Muglia, V.F.; Prando, A. Renal cell carcinoma: Histological classification and correlation with imaging findings. Radiol. Bras. 2015, 48, 166–174. [Google Scholar] [CrossRef]
- Yang, J.; Wang, K.; Yang, Z. Treatment strategies for clear cell renal cell carcinoma: Past, present and future. Front. Oncol. 2023, 13, 1133832. [Google Scholar] [CrossRef] [PubMed]
- Granados-Romero, J.J.; Valderrama-Treviño, A.I.; Mendoza-Barrera, G.-E.; Ceballos-Villalva, J.C.; Contreras-Flores, E.H.; Barrera-Mera, B.; Espejel-Deloiza, M.; Estrada-Mata, A.G.; Badillo-Martínez, K.D.; Rodal-Soler, Á. Open radical nephrectomy for early treatment of renal cell carcinoma: A case report and review. Int. J. Res. Med. Sci. 2016, 4, 1752–1759. [Google Scholar] [CrossRef]
- Astore, S.; Baciarello, G.; Cerbone, L.; Calabrò, F. Primary and acquired resistance to first-line therapy for clear cell renal cell carcinoma. Cancer Drug Resist. 2023, 6, 517–546. [Google Scholar] [CrossRef]
- Aweys, H.; Lewis, D.; Sheriff, M.; Rabbani, R.D.; Lapitan, P.; Sanchez, E.; Papadopoulos, V.; Ghose, A.; Boussios, S. Renal Cell Cancer—Insights in Drug Resistance Mechanisms. Anticancer Res. 2023, 43, 4781–4792. [Google Scholar] [CrossRef]
- Ljungberg, B.; Albiges, L.; Abu-Ghanem, Y.; Bedke, J.; Capitanio, U.; Dabestani, S.; Fernández-Pello, S.; Giles, R.H.; Hofmann, F.; Hora, M.; et al. European Association of Urology Guidelines on Renal Cell Carcinoma: The 2022 Update. Eur. Urol. 2022, 82, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Harshman, L.C.; Srinivas, S. The bevacizumab experience in advanced renal cell carcinoma. Onco Targets Ther. 2010, 3, 179–189. [Google Scholar] [CrossRef]
- Ljungberg, B.; Albiges, L.; Bensalah, K.; Bex, A.; Giles, R.H.; Hora, M.; Volpe, A. EAU Guidelines. Edn. presented at the EAU Annual Congress Amsterdam 2020. Eur. Urol. 2020, 67, 913–924. [Google Scholar] [CrossRef]
- Lasorsa, F.; di Meo, N.A.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; et al. Immune Checkpoint Inhibitors in Renal Cell Carcinoma: Molecular Basis and Rationale for Their Use in Clinical Practice. Biomedicines 2023, 11, 1071. [Google Scholar] [CrossRef]
- Villegas, J.; Burzio, V.; Villota, C.; Landerer, E.; Martinez, R.; Santander, M.; Martinez, R.; Pinto, R.; Vera, M.I.; Boccardo, E.; et al. Expression of a novel non-coding mitochondrial RNA in human proliferating cells. Nuc. Acids Res. 2007, 35, 7336–7347. [Google Scholar] [CrossRef]
- Lobos-González, L.; Silva, V.; Araya, M.; Restovic, F.; Echenique, J.; Oliveira-Cruz, L.; Fitzpatrick, C.; Briones, M.; Villegas, J.; Villota, C.; et al. Targeting antisense mitochondrial ncRNAs inhibits murine melanoma tumor growth and metastasis through reduction in survival and invasion factors. Oncotarget 2016, 7, 58331–58350. [Google Scholar] [CrossRef] [PubMed]
- Burzio, V.A.; Villota, C.; Villegas, J.; Landerer, E.; Boccardo, E.; Villa, L.L.; Martínez, R.; Lopez, C.; Gaete, F.; Toro, V.; et al. Expression of a family of noncoding mitochondrial RNAs distinguishes normal from cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9430–9434. [Google Scholar] [CrossRef] [PubMed]
- Borgna, V.; Villegas, J.; Burzio, V.A.; Belmar, S.; Araya, M.; Jeldes, E.; Lobos-González, L.; Silva, V.; Villota, C.; Oliveira-Cruz, L.; et al. Mitochondrial ASncmtRNA-1 and ASncmtRNA-2 as potent targets to inhibit tumor growth and metastasis in the RenCa murine renal adenocarcinoma model. Oncotarget 2017, 8, 43692–43708. [Google Scholar] [CrossRef]
- Vidaurre, S.; Fitzpatrick, C.; Burzio, V.A.; Briones, M.; Villota, C.; Villegas, J.; Echenique, J.; Oliveira-Cruz, L.; Araya, M.; Borgna, V.; et al. Down-regulation of the antisense mitochondrial non-coding RNAs (ncRNAs) is a unique vulnerability of cancer cells and a potential target for cancer therapy. J. Biol. Chem. 2014, 289, 27182–27198. [Google Scholar] [CrossRef] [PubMed]
- Borgna, V.; Lobos-González, L.; Guevara, F.; Landerer, E.; Bendek, M.; Ávila, R.; Silva, V.; Villota, C.; Araya, M.; Rivas, A.; et al. Targeting antisense mitochondrial noncoding RNAs induces bladder cancer cell death and inhibition of tumor growth through reduction of survival and invasion factors. J. Cancer 2020, 11, 1780–1791. [Google Scholar] [CrossRef]
- Fitzpatrick, C.; Bendek, M.F.; Briones, M.; Farfán, N.; Silva, V.A.; Nardocci, G.; Montecino, M.; Boland, A.; Deleuze, J.F.; Villegas, J.; et al. Mitochondrial ncRNA targeting induces cell cycle arrest and tumor growth inhibition of MDA-MB-231 breast cancer cells through reduction of key cell cycle progression factors. Cell Death Dis. 2019, 10, 423. [Google Scholar] [CrossRef]
- Chow, L.Q.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–896. [Google Scholar] [CrossRef]
- Babaei, G.; Aziz, S.G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, jcs223826. [Google Scholar] [CrossRef]
- Bendek, M.F.; Fitzpatrick, C.; Jeldes, E.; Boland, A.; Deleuze, J.F.; Farfán, N.; Villegas, J.; Nardocci, G.; Montecino, M.; Burzio, L.O.; et al. Inverse Modulation of Aurora Kinase A and Topoisomerase IIα in Normal and Tumor Breast Cells upon Knockdown of Mitochondrial ASncmtRNA. Non-Coding RNA 2023, 9, 59. [Google Scholar] [CrossRef]
- Ma, H.T.; Poon, R.Y.C. Aurora kinases and DNA damage response. Mutat. Res. 2020, 821, 111716. [Google Scholar] [CrossRef]
- Uusküla-Reimand, L.; Wilson, M.D. Untangling the roles of TOP2A and TOP2B in transcription and cancer. Sci. Adv. 2022, 8, eadd4920. [Google Scholar] [CrossRef]
- Lu, C.H.; Wu, C.H.; Hsieh, P.F.; Wu, C.Y.; Kuo, W.W.; Ou, C.H.; Lin, V.C.H. Small interfering RNA targeting N-cadherin regulates cell proliferation and migration in enzalutamide-resistant prostate cancer. Oncol. Lett. 2022, 23, 90. [Google Scholar] [CrossRef]
- Ciołczyk-Wierzbicka, D.; Gil, D.; Laidler, P. The inhibition of cell proliferation using silencing of N-cadherin gene by siRNA process in human melanoma cell lines. Curr. Med. Chem. 2012, 19, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Bianchessi, V.; Badi, I.; Bertolotti, M.; Nigro, P.; D’Alessandra, Y.; Capogrossi, M.C.; Zanobini, M.; Pompilio, G.; Raucci, A.; Lauri, A. The mitochondrial lncRNA ASncmtRNA-2 is induced in aging and replicative senescence in Endothelial Cells. J. Mol. Cell Cardiol. 2015, 81, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Powles, T.; McFaul, S.; Stebbing, J.; Wilson, P.; Oliver, T.; Tranter, N.; Shamash, J. The efficacy and safety of irinotecan cisplatin and mitomycin chemotherapy in sunitinib pre-treated metastatic clear cell renal cancer. Onco Targets Ther. 2008, 1, 35–39. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduc. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Zimmermann, K.; Schmittel, A.; Steiner, U.; Asemissen, A.M.; Knoedler, M.; Thiel, E.; Miller, K.; Keilholz, U. Sunitinib treatment for patients with advanced clear-cell renal-cell carcinoma after progression on sorafenib. Oncology 2009, 76, 350–354. [Google Scholar] [CrossRef]
- Morais, C. Sunitinib resistance in renal cell carcinoma. J. Kidney Cancer VHL 2014, 1, 1–11. [Google Scholar] [CrossRef]
- Dhawan, M.S.; Aggarwal, R.R.; Boyd, E.; Comerford, K.; Zhang, J.; Méndez, B.; Valenzuela, P.; Grabowsky, J.; Thomas, S.; Munster, P.N. Phase 1 study of ANDES-1537: A novel antisense oligonucleotide against non-coding mitochondrial DNA in advanced solid tumors. J. Clin. Oncol. 2018, 36, 2557. [Google Scholar] [CrossRef]
- Marona, P.; Górka, J.; Kwapisz, O.; Jura, J.; Rys, J.; Hoffman, R.M.; Miekus, K. Resistance to tyrosine kinase inhibitors promotes renal cancer progression through MCPIP1 tumor-suppressor downregulation and c-Met activation. Cell Death Dis. 2022, 13, 814. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
- Gassenmaier, M.; Chen, D.; Buchner, A.; Henkel, L.; Schiemann, M.; Mack, B.; Schendel, D.J.; Zimmermann, W.; Pohla, H. CXC chemokine receptor 4 is essential for maintenance of renal cell carcinoma-initiating cells and predicts metastasis. Stem Cells 2013, 31, 1467–1476. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araya, M.; Sepúlveda, F.; Villegas, J.; Alarcón, L.; Burzio, L.O.; Burzio, V.A.; Borgna, V. Knockdown of Antisense Noncoding Mitochondrial RNA Reduces Tumorigenicity of Patient-Derived Clear Cell Renal Carcinoma Cells in an Orthotopic Xenograft Mouse Model. Cancers 2024, 16, 830. https://doi.org/10.3390/cancers16040830
Araya M, Sepúlveda F, Villegas J, Alarcón L, Burzio LO, Burzio VA, Borgna V. Knockdown of Antisense Noncoding Mitochondrial RNA Reduces Tumorigenicity of Patient-Derived Clear Cell Renal Carcinoma Cells in an Orthotopic Xenograft Mouse Model. Cancers. 2024; 16(4):830. https://doi.org/10.3390/cancers16040830
Chicago/Turabian StyleAraya, Mariela, Francisca Sepúlveda, Jaime Villegas, Luis Alarcón, Luis O. Burzio, Verónica A. Burzio, and Vincenzo Borgna. 2024. "Knockdown of Antisense Noncoding Mitochondrial RNA Reduces Tumorigenicity of Patient-Derived Clear Cell Renal Carcinoma Cells in an Orthotopic Xenograft Mouse Model" Cancers 16, no. 4: 830. https://doi.org/10.3390/cancers16040830