
Exploiting the DNA Damage Response for Prostate Cancer Therapy

1
Radiation Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA
2
Molecular Imaging Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
Cancers 2024, 16(1), 83; https://doi.org/10.3390/cancers16010083
Submission received: 18 October 2023
/
Revised: 14 December 2023
/
Accepted: 20 December 2023
/
Published: 23 December 2023
(This article belongs to the Special Issue Recent Perspectives on Mechanisms of Radiation-Mediated DNA Damage Induction and Response in Cancers)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Localized prostate cancer has a favorable prognosis and can be effectively treated with radiotherapy, as well as therapies that target hormone production and signaling. However, if the disease progresses past these treatments, treatment options are limited, and the disease often becomes fatal. In this review, we discuss the changes in treatment as prostate cancer develops, as well as the genetic mutations that accompany advanced prostate cancer. We highlight how these mutations can provide clinical opportunities to manipulate the DNA damage response for therapeutic gain using cancer-specific alterations in the genome and new therapeutic agents that are under development or entering clinical trials.
Abstract
Prostate cancers that progress despite androgen deprivation develop into castration-resistant prostate cancer, a fatal disease with few treatment options. In this review, we discuss the current understanding of prostate cancer subtypes and alterations in the DNA damage response (DDR) that can predispose to the development of prostate cancer and affect its progression. We identify barriers to conventional treatments, such as radiotherapy, and discuss the development of new therapies, many of which target the DDR or take advantage of recurring genetic alterations in the DDR. We place this in the context of advances in understanding the genetic variation and immune landscape of CRPC that could help guide their use in future treatment strategies. Finally, we discuss several new and emerging agents that may advance the treatment of lethal disease, highlighting selected clinical trials.
Keywords:
prostate cancer; androgen; DNA damage response; radiotherapy; radiopharmaceutical therapy; PARP; ATR; ATM; DNA-PK; immunotherapy; hypoxia1. Introduction
Prostate cancer (PCa) is the second most common cancer in men and the second leading cause of cancer-related death amongst men worldwide [1]. The clinical diagnosis staging, and prognostic stratification of PCa relies on digital rectal examination, the histological analysis of tumor biopsies with grade grouping using the Gleason scoring system, and serum prostatic specific antigen (PSA) levels [2]. The majority of PCa patients are diagnosed with low-risk localized disease and require minimal intervention at diagnosis, with active surveillance considered a favored approach. Radical prostatectomy or radiotherapy (RT), in the form of either external beam RT (EBRT) or internal RT via brachytherapy, are the major treatment options for intermediate-risk localized PCa, with surveillance offered to patients with favorable risk factors and low-volume disease [3]. Patients diagnosed with high-risk localized disease are treated with RT with androgen deprivation therapy (ADT) or surgery to provide local control and reduce the risk of recurrence or metastasis [2]. In the setting of metastatic disease at presentation, systemic therapy with ADT and next-generation androgen receptor (AR) signaling inhibitors (ARSIs), such as enzalutamide, are a first-line therapy to prevent the reactivation of AR signaling [4].
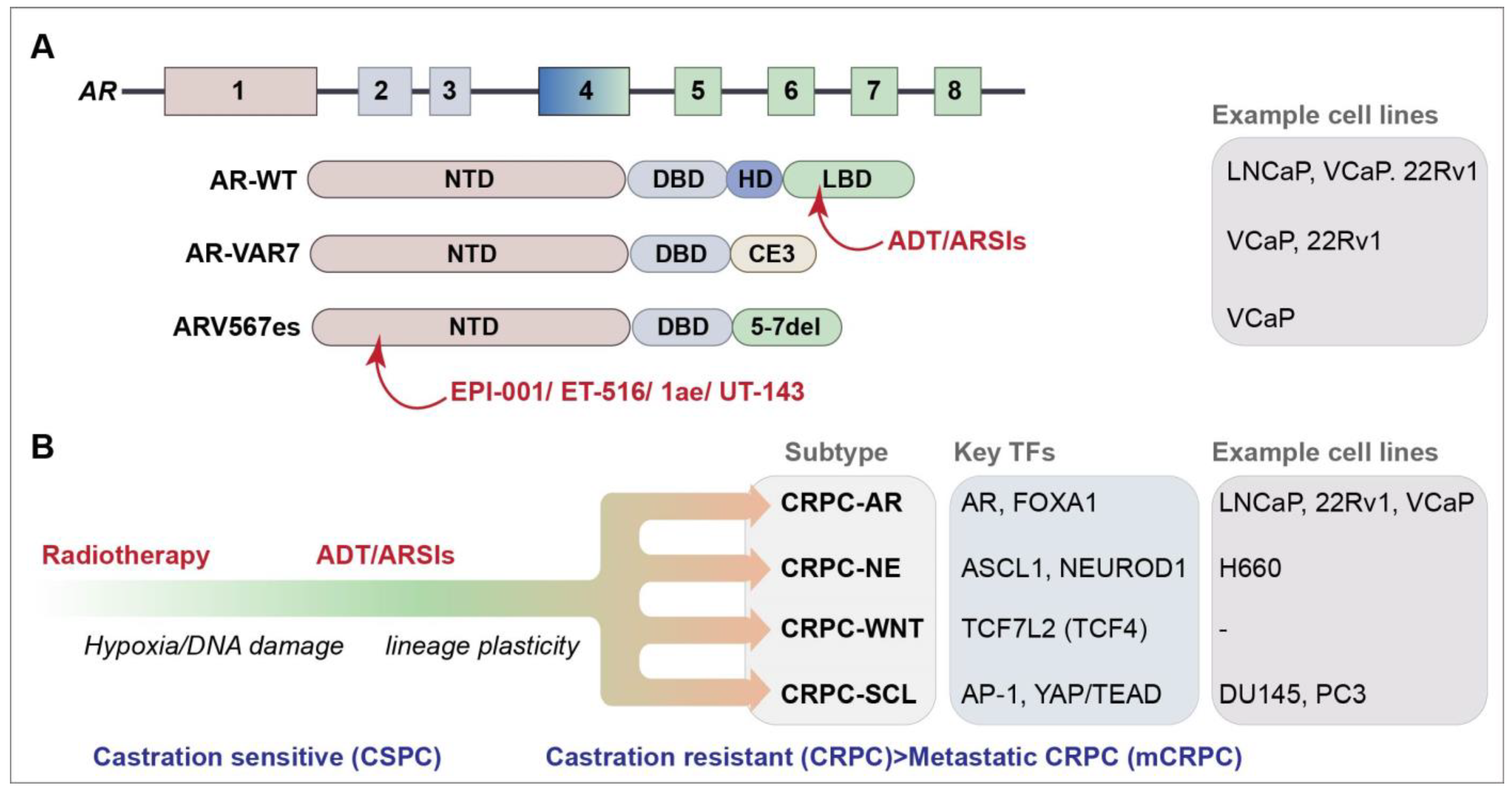
The AR is a master transcription factor (TF) that facilitates the effects of androgens on target tissues and is a major target in PCa therapy [5]. The AR is comprised of four domains: an intrinsically disordered N-terminal domain (NTD) that is essential for activating transcription, a DNA-binding domain (DBD) that recognizes androgen response elements (AREs) in target genes, a hinge domain (HD) and a ligand-binding domain (LBD) that binds androgen (Figure 1A). The LBD is the target of frontline ADT and ARSI therapies that have had a strong impact on the control of castration-sensitive PCa (Figure 1A). Extensive research efforts have focused on the role of AR-dependent resistance mechanisms in PCa, which play vital roles in disease progression and treatment resistance. These mechanisms include AR amplification and overexpression, the expression of AR mutants and overexpression of AR variants (AR-Vs) that often lack the LBD (Figure 1A) [6]. Over 20 AR-Vs have been identified, but the functional relevance of many of these to disease progression remains unclear (reviewed in references [7,8]). Some AR-Vs, including AR-V7 and AR-V567es, are produced at the protein level and have constitutive transcriptional activity that can drive proliferation in the absence of androgens. The presence of AR-V7 is a clinical biomarker of resistance to AR-targeted therapies; however, its direct clinical targeting is hampered by the lack of the LBD targeted by ARSIs (Figure 1A) [9,10,11,12,13,14,15].
While many tumors are initially responsive to RT, ADT, and ARSIs, around 20–30% of patients will progress within 2–3 years and eventually transition to a more aggressive, AR-independent form of PCa known as castration-resistant prostate cancer (CRPC) or metastatic CRPC (mCRPC), both of which are incurable and often lethal within 2 years [16,17].
Figure 1.
(A) Schematic of the AR gene and AR and selected AR-V protein domains. Protein domains are described in the text and color coded to the exon schematic. Selected agents that target protein domains are indicated in red (see text for further details) [18,19,20,21]. AR-VAR7 arises due to the splicing to cryptic exon 3 (CE3) and ARV567es from the deletion of exons 5–7; cell lines that express each of the selected genes endogenously are listed [6]. (B) Progression of PCa and identified CRPC subtypes are depicted [22]. Key transcription factors (TFs) and some commonly used cell lines representing the subtypes are shown.
Figure 1.
(A) Schematic of the AR gene and AR and selected AR-V protein domains. Protein domains are described in the text and color coded to the exon schematic. Selected agents that target protein domains are indicated in red (see text for further details) [18,19,20,21]. AR-VAR7 arises due to the splicing to cryptic exon 3 (CE3) and ARV567es from the deletion of exons 5–7; cell lines that express each of the selected genes endogenously are listed [6]. (B) Progression of PCa and identified CRPC subtypes are depicted [22]. Key transcription factors (TFs) and some commonly used cell lines representing the subtypes are shown.

The genomic landscape of CRPC, as well as the full extent of its heterogeneity remains unclear. Lineage plasticity, facilitated in part by defects in chromatin regulation, plays a significant role in treatment resistance in CRPC [23,24]. Recent work has revealed four CRPC subtypes based on the chromatin accessibility and gene expression profiles of patient-derived cell lines and organoids (Figure 1B) [22,24]. Two known subtypes were identified: the AR-dependent subtype (CRPC-AR), characterized by AR amplifications or mutations and the AR gene signature, and the neuroendocrine (NE) subtype (CRPC-NE), characterized by the expression of NE markers, such as SYP and CHGA [22]. Two new subtypes were defined: the WNT-dependent subtype (CRPC-WNT), which featured alterations in the WNT pathway genes, such as CTNNB1 (also referred to as beta-catenin), and the stem cell-like subtype (CRPC-SCL), defined by the enrichment of mammary stem cell signature genes and the high expression of the cancer stem cell markers CD44 and TACSTD2.
Hypoxia is a prominent feature of the tumor microenvironment (TME) proposed to promote lineage plasticity, increase DNA damage, and drive treatment resistance in cancers, including PCa [24,25,26]. Hypoxia is a major form of cellular stress, driving cells to adapt through transcriptional and molecular changes, including a decrease in proliferation, heightened glycolysis, the modulation of DNA repair factors, and angiogenesis [27]. Tumor hypoxia is characterized by low tissue oxygenation, ranging from 0.3% to 4.2% oxygen (2–32 mmHg) and PCa has been determined to have low oxygenation levels compared to tumors from other tissues [28,29,30,31]. In response to hypoxia, tumor cells regulate distinct physiological responses through hypoxia-inducible factors (HIFs) [27]. Tumor hypoxia can have several biological and clinical consequences in PCa, including elevated genomic instability, increased mutational burden, castration resistance, an increased likelihood of metastasis, chemoresistance, and radioresistance [31,32,33,34].
RT is a central and effective treatment modality for localized PCa. Hypoxic conditions reduce the efficacy of RT by preventing the oxygen-dependent “fixation” of damage caused by free radicals [35]. Hypoxic signaling can also contribute to the abnormal vascularization of tumors, limiting drug delivery and contributing to the reduced radiosensitivity and chemosensitivity of tumor cells [35]. Moreover, hypoxia contributes to an immunosuppressive TME and represents a challenge to immunotherapy [36]. Substantial efforts have been made to identify strategies to overcome hypoxia-induced therapeutic resistance (reviewed in reference [36]). These include agents that enhance oxygen delivery to radiosensitize hypoxic cells, electron-affinic radiosensitizers and hypoxia-activated prodrugs (HAPs). However, these approaches have yet to make a major clinical impact in PCa treatment.
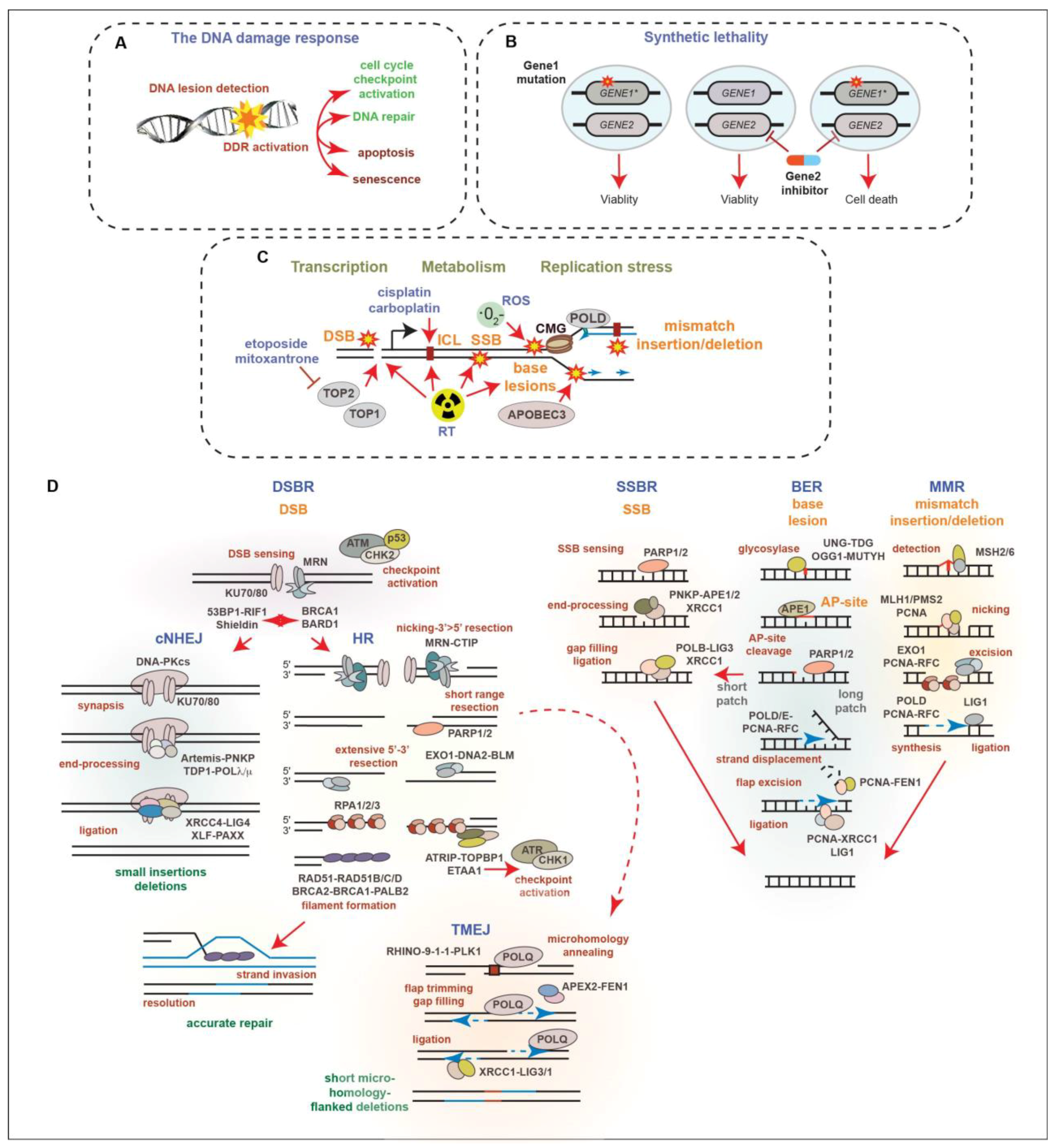
Ionizing radiation induces DNA damage in the form of a variety of base lesions, single-strand breaks (SSBs), double-strand breaks (DSBs), which are considered the most toxic DNA lesion, and DNA crosslinks [35]. The success of RT and many chemotherapies is strongly influenced by the status of the DNA damage response (DDR), a signal transduction network that identifies and responds to many different types of DNA lesions (Figure 2A) [37,38]. The DDR triggers changes in cellular proliferation, through the activation of cell cycle checkpoints, and regulates the DNA repair machinery to resolve DNA lesions and protect genome integrity. Depending on the cell type or the extent of DNA damage, the DDR can also activate cell death or senescence pathways to promote the removal of cells with potentially oncogenic DNA damage. The DDR acts as an inducible barrier to oncogenesis, creating a selective pressure to lose the key components of DDR signaling that allow for tumor growth through the evasion of tumor-suppressive mechanisms, including DNA repair, cell cycle checkpoint arrest, and cell fate signaling pathways [39]. Defects in the DDR result in genomic instability, a hallmark of cancer, in the forms of DNA amplifications and deletions, translocations, and mutations [40]. As the disease advances, the progressive loss of DDR genes can accelerate tumor growth and DNA damage tolerance. However, this can also enhance the efficacy of some DNA-damaging treatments, as recurrent gene losses can provide opportunities to take advantage of synthetic lethal interactions (Figure 2B) that can be targeted using specific inhibitors or RT [41].
There is an unmet critical need for new therapeutic modalities for CRPC and mCRPC and several promising strategies taking advantage of recurring genetic alterations, particularly those affecting the DNA damage response (DDR), have entered clinical trials. This review will focus on the current barriers to the treatment of CRPC and new developments in therapeutic approaches that act by causing DNA damage, taking advantage of mutations in the DDR or modulating the DDR to improve treatment responses.
2. The DNA Damage Response in Prostate Cancer
Several types of endogenous DNA damage arise in PCa due to cellular processes, including metabolism, transcription, and DNA replication (Figure 2C). These include mismatched bases, base insertions or deletions due to polymerase errors, as well as base lesions caused by reactive oxygen species (ROS), spontaneous deamination, or the activities of the Apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC) family of enzymes that deaminate cytosines [44,45,52]. SSBs can arise due to the processing of base lesions and DSBs can be caused by the activity of topoisomerase enzymes acting during transcription and replication, the processing of stalled replication forks during replication stress or after collisions with transcriptional machinery, and the mitotic breakage of under-replicated DNA regions [42,46,49]. RT introduces many types of base lesions, DNA crosslinks, and SSBs that can elevate replication stress levels, as well as DSBs that trigger cell death or senescence pathways. Compared to many other cancer types, few DNA-damaging agents aside from RT are used in the treatment of PCa, as localized PCa is well-controlled with RT and CRPC is generally poorly responsive to genotoxic compounds, potentially due to slow growth rates. Genotoxic agents, including topoisomerase-2 inhibitors, such as etoposide and mitoxantrone, as well as platinum DNA crosslinking agents, like cisplatin, are used in palliative care, as well as the treatment of CRPC-NE in accordance with the guidelines for small cell lung cancer [2]. The use of taxanes, which poison microtubules to prevent proliferation, has largely overtaken mitoxantrone used in selected settings in CRPC treatment, but acquired resistance remains a significant issue [53,54,55,56].
DNA double-strand breaks (DSBs) activate a number of kinases, including ATM, ATR, and DNA-PKcs, as well as additional effector kinases such as CHK1 and CHK2, which together, regulate cell cycle checkpoints, DNA repair, and cell fate decisions (Figure 2D) [37]. ATM is activated directly by the detection of DSBs by the MRE11-RAD50-NBS1 (MRN) complex, and subsequently activates CHK2 that in turn modifies many of the same targets as ATM. The accumulation of ssDNA, which can arise due to the nucleolytic resection of DSBs, replication stress (replication fork stalling and ssDNA accumulation), or by gaps that can form behind the replication fork, leads to the activation of ATR and CHK1 [57]. ATR activation is triggered by ssDNA coated with RPA and the activities of the ATRIP, TOPBP1, and ETAA1 proteins. Like ATM/CHK2, ATR/CHK1 plays a key role in regulating cell cycle checkpoints, as well as multiple DNA repair factors and cell fate pathways.
The two major pathways of DSB repair are classical nonhomologous end-joining (cNHEJ) and homologous recombination (HR) (Figure 2D) [42]. These pathways compete for DNA ends, with cNHEJ functioning throughout the cell cycle and HR operating in S and G2 due to the need for a sister chromatid template to guide accurate recombinational repair. DNA-PKcs, along with KU70 and KU80, form the DNA-dependent protein kinase that plays a central role in promoting cNHEJ of DSBs. KU70/80 recognizes DNA ends, promoting the activation of DNA-PKcs, as well as the recruitment of the Artemis nuclease, and other factors, which can clean up blocked ends, allowing for their ligation by LIG4 and additional factors, including XRCC4, XLF, and PAXX [42,43]. Short insertions and deletions at the breakpoint can occur as a result of cNHEJ repair. A protein complex that includes 53BP1, RIF1, and Shieldin (SHLD1/2/3 and REV7) is critical for preventing the access of BRCA1-BARD1 and multiple nucleases that resect DNA ends to promote HR-mediated repair. The MRN complex can introduce nicks near the break via its endonuclease activity and resect 3′-5′, generating ends that release KU70/80 and are incompatible with cNHEJ-mediated ligation. Subsequent 5′-3′ resection by multiple nucleases and helicases, including EXO1, DNA2, and BLM, generates the overhangs needed for stand invasion and the HR-mediated repair of DSBs. Overhangs are bound by the tripartite RPA (RPA1/2/3) that is exchanged for RAD51, as well as its paralogs, RAD51A/B/C/D, to facilitate HR, resulting in accurate repair. Additional alternative end-joining pathways can contribute to DSB repair and DNA damage tolerance [42,43]. These include polymerase theta-mediated end-joining (TMEJ) (also referred to as alternative end joining or microhomology-mediated end-joining). TMEJ uses microhomology exposed by limited resection to anneal and modify ends, fill gaps, and ligate ends through the activities of PARP1/2, POLQ (Polymerase theta), XRCC1, and LIG3, resulting in short microhomology-flanked deletions (Figure 2D) [37,41]. Recent work has demonstrated that TMEJ can operate during mitosis in homologous recombination-deficient (HRD) cells in conjunction with RHINO, the 9-1-1 complex, and PLK1, as well as the MDC1-CIP2A-TOPBP1 proteins that tether mitotic DNA breaks [58,59,60,61,62].
Base and nucleotide lesions can be processed by several excision mechanisms, which include single-strand base repair (SSBR) and base excision repair (BER) (Figure 2B). The Poly (ADP-ribose) polymerases, particularly PARP1 and PARP2, play important roles in regulating these pathways [48,49]. PARP1/2 activity promotes the recruitment of additional factors, including XRCC1. The processing of some base lesion types leads to SSBs and unrepaired SSBs can result in DSBs, through replication fork progression or replication–transcription collisions. In addition to SSBR and BER, MMR also processes base lesions that include base mismatches, insertions, and deletions that are generated by the replicative polymerases. MMR is critical to prevent mutagenesis and the loss of MMR is particularly relevant in many cancer types including PCa, as it increases microsatellite instability (MSI) and tumor mutational burden (TMB) [63]. The increased mutational load can lead to oncogene activation or tumor suppressor inactivation, for example through mutations in KRAS or TP53. Multiple single base pair mutational signatures for MMR deficiency (dMMR) have been identified, including SBS6 and SBS15, as well as numerous doublet-base substitution and indel signatures [64].
High TMB can also occur due to mutations in several polymerases that affect proofreading capabilities, including POLE and POLD1, as well as the APOBEC3 cytidine deaminases that can be activated by replication stress [52,65,66]. The APOBEC3-mediated deamination of cytosines to uracil causes mispairing during DNA replication or the subsequent processing of uracil by glycosylases, such as UNG, generates abasic sites (AP-sites) that can elevate TMB [52]. APOBEC3A and APOBEC3B are the prominent mutagenic drivers in several cancer types, giving rise to a distinct mutational signature (SBS2 and SBS13) that can be distinguished from dMMR and identified by cancer genome sequencing and analysis [67,68,69,70].
While only a single hypermutagenic polymerase mutation has been reported in CRPC to our knowledge, both dMMR and APOBEC signatures appear in CRPC, but occur infrequently compared to some other cancer types [71,72,73]. MSI-high/dMMR tumors were identified in some CRPC cohorts, constituting up to 5% of analyzed patients depending on the cohort analyzed [74,75]. Numerous prostate cancer samples exhibit APOBEC signature mutations that increase along the spectrum of disease progression from localized to mCRPC, although the overall frequency is low compared to several other tumor types [71,76]. Recent work has also suggested that APOBEC3B activity in CRPC may have an important role in mutagenesis that can facilitate lineage plasticity and ARSI resistance (see Section 6 for additional details) [77].
Germline mutations in DNA repair genes affecting multiple pathways have been implicated in PCa susceptibility. These are estimated to occur in ~5–15% of total PCa cases, with around 12% occurring in mCRPC, where several are associated with high-grade disease. Germline mutations in DDR genes primarily occur in genes involved in cell cycle checkpoint signaling (ATM~2%, CHEK2~2.8%, TP53~0.7%), HR (BRCA2~4.8%, BRCA1~1.3%, PALB2~0.6%, NBN~0.3%, RAD50~0.3%, RAD51~3%, RAD51C~0.2% and RAD51D < 0.2%), BER (MUTYH~2.4%), and MMR (MSH2~0.7%, PMS2~0.5%, MSH6~0.5%, MLH1~0.06%) [78,79]. Somatic mutations in the DDR genes occur in localized PCa (~11%) and increase in frequency as disease progresses, with DDR mutations estimated to occur in 20% or more of mCRPC cases [80,81,82,83,84]. These include ATM (~7%), BRCA2 (~10%), and BRCA1 (0.5%), accounting for around 20% of the DDR mutations, as well as ATR (~8%), RAD51 (~2%), PARP1 (~6%), MLH1 (~1.4%), and MSH2 (~3%). The high frequency of DDR mutations in CRPC represents a potential opportunity for exploiting synthetic lethalities or targeted therapies in patients that progress from RT and anti-androgen therapies.
3. New Approaches for Targeting AR-Variants
ARSIs, such as Enzalutamide, are effective at targeting AR function through the competitive binding of its LBD (Figure 1A). However, the emergence of AR-Vs that lack the LBD, through mutations or cryptic splicing, confer resistance to ADT and ARSIs in CRPC [9,85]. One prominent variant, AR-V7, enhances the expression of DNA repair genes and confers radioresistance in various PCa cell lines, indicating that targeting AR-Vs could not only address the resistance to ARSIs, but also augment RT and potentially other therapies targeting the DDR [86,87].
Screening for small molecules that inhibited the transcriptional activity of the intrinsically disordered AR NTD led to the identification of EPI-001, the first compound to directly target this domain (Figure 1A) [18]. Recent studies have described new molecules, some derived from EPI-001, which target the AR or AR-V NTD (Figure 1A) [18,20,21,88]. These new molecules disrupt the liquid–liquid phase separation (LLPS) propensity, protein–protein interactions and transcriptional activity of AR and AR-Vs and showed anti-tumor effects in animal models, including those expressing AR-Vs. While the additional development of these compounds will be required for clinical use, they further demonstrate that targeting the intrinsically disordered domain of the AR and AR-Vs is a feasible strategy for impairing its function in tumorigenesis. These agents will also allow for the testing of combination strategies with other agents being explored in CRPC, including PARP inhibitors that also impair AR and AR-V transcriptional activity, albeit indirectly [89,90]. The approach of targeting intrinsically disordered domains could be employed against other TFs in future work, potentially guided by the recent subtype classification that identified specific TFs associated with AR-negative/low CRPCs (Figure 1B) [22]. In addition, as many DNA repair proteins contain intrinsically disordered domains and undergo LLPS, these approaches could potentially be used to directly target a number of key DNA repair factors, including 53BP1 and TOPBP1, in future therapies [91].
4. PARP Inhibitors in CRPC
The seminal discovery that HRD was synthetically lethal with PARP inhibitors ignited interest in targeting PARP activity across many cancer types [92,93]. The low toxicity of PARP inhibitors, as well as the loss of heterozygosity in germline HRD mutations or somatic mutations in cancer cells, but not surrounding tissues, makes PARP inhibitor lethality with HRD attractive due to its tumor-selective effects. Several potent PARP inhibitors were developed and approved for clinical use and multiple mechanisms have been proposed to explain the synthetic lethal interaction between PARP inhibitors and HRD, which has been reviewed extensively elsewhere [37,94]. In brief, the generation of DSBs due to the combined impairment of BER and HR, toxic ssDNA gaps behind the replication fork, the incorporation of ribonucleotides that can trap PARP, and the degree of PARP1/2 trapping, have all been proposed to contribute to the toxicity of PARP inhibitors in different settings. Extensive CRISPR screening has identified many additional modifiers of PARP inhibitor toxicity and notable resistance mechanisms, which in some cases, appear to be specific to the particular HR mutation [94,95,96]. A prominent example is the loss of 53BP1, RIF1, or members of the Shieldin complex that rescues PARP inhibitor sensitivity resulting from the loss of BRCA1 (Figure 2B) [95].
PARP inhibitor clinical trials for prostate cancer were recently reviewed and analyzed extensively, so here, we will summarize several major trials for four PARP inhibitors that have been tested in mCRPC: Olaparib, Rucaparib, Niraparib, and Talazaporib [97,98,99]. The important takeaways overall are that PARP inhibitors appear to be safe, although anemia and fatigue are observed in some patients, and in biomarker-selected populations, PARP inhibitors are beneficial for patients. However, there remains a need for a more detailed understanding of how specific genomic alterations influence PARP inhibitor efficacy.
The TOPARP (NCT01682772), PROfound (NCT02987543), and TRITON2 (NCT02952534) trials tested the two currently FDA-approved PARP inhibitors Olaparib (Lynparza) and Rucaparib (Rubraca) as monotherapies in mCRPC with HRD [100,101,102,103,104]. The TOPARP trial tested Olaparib monotherapy on an unselected patient population that had progressed on ARSIs or taxanes and found a higher response rate in patients with DDR mutations, including all patients with BRCA2 mutations and the majority of those with ATM mutations [105]. The PROfound trial selected patients that had progressed following ARSI treatment and that had HRD, defined either by BRCA1, BRCA2, or ATM mutations, or a panel of another 12 DDR related genes. The trial showed that Olaparib monotherapy was more effective than ARSI treatment, measuring radiographic progression-free survival and overall survival [100,102]. The TRITON2 trial selected patients that progressed on previous ARSI or taxane treatment and had mutations in BRCA1/2, ATM, or additionally selected DDR genes [101,103,104]. Improved objective response rates (ORRs) to Rucaparib were observed, particularly for BRCA2-deficient patients, and differences in responses based on the specific DDR gene altered were also notable. The loss of PALB2, FANCA, BRIP1, and RAD51B were associated with improved responses, while responses were limited in ATM-, CHEK2-, and CDK12-deficient cancers [103].
The Phase II GALAHAD (NCT02854436) and TALAPRO-1 (NCT03148795) trials recently provided compelling evidence for the efficacy of the PARP inhibitors Niraparib and Talazaporib in mCRPC [106,107]. The GALAHAD trial assessed patients post-ARSI or -taxane treatment, using two cohorts that had either BRCA1/2 alterations or alterations in a panel of other DDR genes including ATM, BRIP1, CHEK2, FANCA, HDAC2, or PALB2, with improved ORRs observed in both, but with the BRCA1/2 cohort showing the most dramatic responses to Niraparib [106]. The TALAPRO-1 trial again assessed patients post-ARSI or -taxane treatment, using a panel of DDR-related genes for the selection of a single cohort [107]. The carriers of BRCA2 alterations showed the most dramatic responses, but tumors with alterations in BRCA1, PALB2, and ATM also showed partial or complete responses.
Despite its identification as a PARP inhibitor sensitizer in multiple settings, ATM loss in PCa does not appear to consistently confer a strong benefit to PARP inhibitor-treated patients, in contrast to the loss of BRCA2 that was consistently associated with the highest benefit to patients treated with multiple PARP inhibitors [98,108,109,110,111,112,113,114]. The extent of PARP trapping was demonstrated to correlate with PARP inhibitor toxicity, with Talazaparib being the most potent PARP inhibitor tested, followed by Niraparib and Rucaparib/ Olaparib [115,116]. The differing conclusions of the TRITON2 and TALAPRO-1 trials regarding ATM may suggest that the extent of PARP trapping is important for the synthetic interaction with ATM loss. Alternatively, it could reflect the effects of the specific ATM alleles on the overall function of ATM or rely on other, unknown co-mutations. All four of these PARP inhibitors, as well as another PARP inhibitor, Veliparib, continue to be tested in additional Phase 1–3 trials in combination therapies with ARSIs, Temozolomide, and corticosteroids in patients with HRD [99]. As additional clinical data emerges, the ability to both select responsive patient populations, as well as effective drug combinations, will no doubt improve the clinical outcomes of mCRPC.
While many genes associated with HRD are currently screened for in CRPC patients entering clinical trials, the full repertoire of genes that could benefit PARP inhibitor treatment remains to be identified. Recent work in cell lines and mouse models has demonstrated that the loss of MMS22L and RNASEH2B, which both occur frequently in CRPC (~7% and ~12% deep deletions in mCRPC, respectively), may also improve PARP inhibitor efficacy [83]. The loss of MMS22L, or its binding partner TONSL, which work together to facilitate RAD51 deposition in HR, were shown to sensitize PCa cell lines to PARP inhibitors [117,118,119]. It is notable that the deletion of MMS22L or TONSL failed to sensitize two CRPC-SCL cell lines, DU145 and PC3, to PARP inhibitors, potentially due to their lack of functional TP53 signaling [120]. The investigators further demonstrated that the deletion of CHEK2 (encoding CHK2) suppressed PARP inhibitor sensitivity due to the loss of BRCA2 suppression by CHK2-TP53-E2F7. Functional CHK2-TP53 is not required for the synthetic interaction between BRCA1/2 loss and PARP inhibitors, so it remains to be seen if other genetic suppressors may be relevant in the context of MMS22L deficiency in CRPC tumors [92,121].
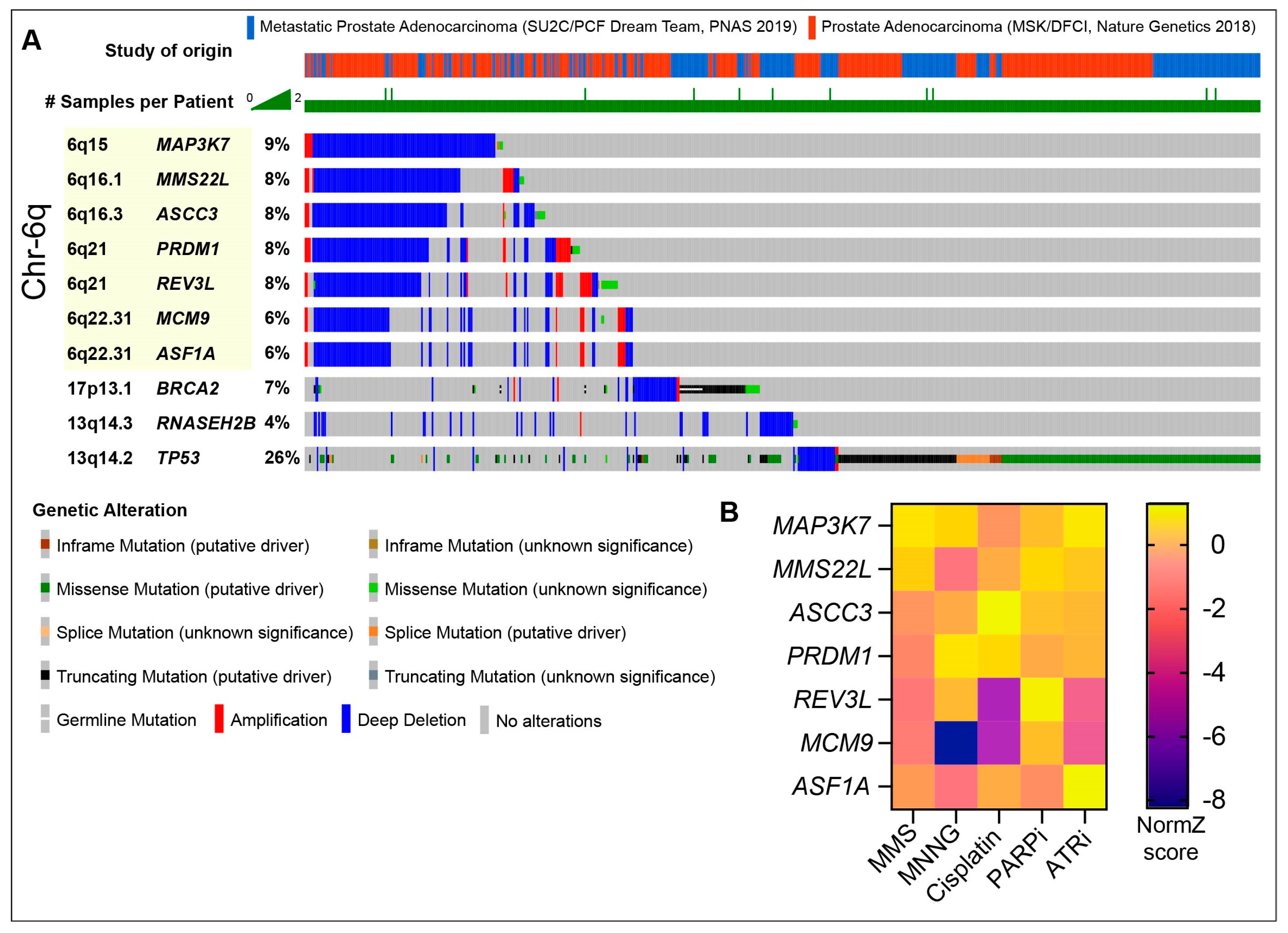
MMS22L deletions appear to occur in PCa tumors primarily in the context of a large recurrent deletion in chromosome 6q, potentially driven by PRDM1 loss (Figure 3A) [122]. This recurrent deletion shows mutual exclusivity with TP53 loss, indicating it may be targetable with PARP inhibitors, and often encompasses additional DDR proteins, including REV3L, ASCC3, ASF1A, MCM9, and MAP3K7 (Figure 3A) [122,123,124,125]. MMS22L derives its name from its identification in yeast screens for sensitivity to the alkylating agent methyl methanesulfonate (MMS) [126]. In CRISPR screens using the human hTERT-RPE1-TP53 null cell line, all of the known DNA repair proteins in this deletion conferred some level of DNA damage sensitivity in vitro, particularly to alkylating agents, including cisplatin, MMS and MNNG (methylnitronitrosoguanidine) (Figure 3B) [127]. ASCC3 is part of the ASCC helicase complex that repairs alkylation damage, including that induced by MMS, in RNA and DNA [128]. REV3L is a subunit of DNA Polymerase Zeta that contributes to replication stress tolerance and lesion bypass, with an important role in ssDNA gap repair in HR-deficient backgrounds [129,130]. MCM9 is a component of a helicase complex, which includes MCM8 and HROB (also referred to as C17orf53 or MCM8IP), which was implicated in HR and interstrand crosslink repair [131,132,133,134,135]. The ASF1A gene is located within MCM9, and ASF1A was implicated in NHEJ [136,137]. The depletion of both ASF1A and its orthologue, ASF1B, also compromised the MMSL22-TONSL-mediated loading of RAD51 in HR [138]. Two papers have recently demonstrated that ASF1A loss conferred resistance to PARP inhibitors in BRCA1-mutant settings, where it was proposed to facilitate heterochromatin formation and inhibit resection, through interactions with RIF1 [139,140]. RIF1, a 53BP1 interactor, is a known suppressor of PARP inhibitor sensitivity in BRCA1 deficiency (Figure 2D). ASF1A loss suppressed PARP inhibitor sensitivity in BRCA1-deficient cells to a similar extent as RIF1 loss, suggesting that ASF1A plays an early role in DSB repair pathway choice and could confound PARP inhibitor sensitivity in the context of the 6q deletion. Finally, the deletion of MAP3K7, also referred to as TAK1, reduced the expression of HR proteins and was shown to sensitize prostate cancer cells to the multi-CDK inhibitor dinaciclib or Olaparib [141]. MAP3K7 was also demonstrated to promote the degradation of AR protein levels in conjunction with a TNFa and NFKB-mediated inflammatory response, indicating that cancers with this Ch6q deletion may have elevated AR and reduced inflammation [142]. As the impact of the loss of this MMS22L-containing 6q locus containing multiple repair genes has not been assessed, it will be interesting to see if it confers clinical responses to PARP inhibitors and other DNA-damaging agents, such as platinum drugs, in future trials in CRPC.
The tripartite Ribonuclease H2 (RNASEH2A/B/C), endonucleolytically removes ribonucleotides from DNA to prevent replication stress, PARP trapping, and DNA damage [144,145]. Recurrent deletions of RNASEH2B, and to a lesser extent RNASEH2A and RNASEH2C (<1% deep deletions), occur in both primary and mCRPC patients, often co-occurring with BRCA2 and RB1 loss in the case of RNASEH2B [120,146]. The deletion of RNASEH2B sensitized PCa cell lines to PARP inhibition, as was observed previously in other cell types [145,146,147]. However, the concurrent loss of RB1, a frequent driver mutation in multiple CRPC subtypes (~10–15%), conferred resistance to PARP inhibition in RNASEH2B-deficient cells by elevating the E2F1-driven expression of BRCA2 [83,147,148,149]. RNASEH2B loss does not impair HR and is synthetically lethal with BRCA1 deficiency, indicating that the distinct mechanisms of PARP inhibitor sensitivity may be differentially affected by the genetic landscape of the particular cancer [145]. This also further points to the complexity of using single genes as biomarkers for DDR inhibitor use and the need for further analysis of diverse CRPC subtypes and their recurrent genomic rearrangements to identify genes that influence sensitivity or resistance to PARP inhibitors and other DDR-targeting agents.
PARP inhibitor efficacy may also be influenced by the TME, with hypoxia proposed to be both a sensitizer of HR-proficient cells to PARP inhibitors, as well as a barrier to PARP inhibitor efficacy in HRD cell lines from various cancers. Chronic hypoxia in cell culture impairs the transcription and translation of DDR genes, including RAD51, thereby reducing HR capacity and causing HR-dependent radiosensitization [150,151,152]. This effect of hypoxia on HR protein translation was demonstrated in multiple cell lines, where it created a contextual lethality that conferred a modest sensitization to PARP inhibitors in cell culture, as well as the killing of hypoxic tumor cells in mouse models in vivo [152]. These data suggest that PARP inhibitors could target chronically hypoxic tumors that are resistant to therapy by inducing a HRD state.
In other experiments under more moderate hypoxia, the PARP inhibitor efficacy decreased in both HR-proficient and HRD cell lines in vitro [153]. Interestingly, this effect was independent of HIF1α and associated with the reduced production of ROS, which cause ssDNA lesions, under hypoxia. Combining PARP inhibitors with the HAP Tirapazamine, which generates ssDNA damage, led to stronger cell killing in vitro and reduced tumor growth in multiple xenograft cancer models. While this approach has not been tested in PCa to our knowledge, it suggests that moderate hypoxia may impede the full potential of PARP inhibitors in PCa, and HAPs that augment the generation of DNA damage could be considered in this context.
5. Targeting DDR Kinases in CRPC Therapy
ATM, ATR, and DNA-PKcs play central roles in the DDR (Figure 2A,D) and the loss of any of these kinases provides numerous opportunities for synthetically lethal interactions [154]. Specific small molecule inhibitors for each kinase were developed and several inhibitors of ATR and DNA-PKcs are currently being tested in many cancer types as monotherapies or in combination therapies with genotoxic agents. In some cases, trials are taking advantage of ATM mutations that occur frequently across cancers and are synthetically lethal with ATR or DNA-PKcs deficiency [154].
Inhibitors of the ATR kinase sensitize cancer cells to replication stress and genotoxic agents have been extensively characterized in cell culture and pre-clinical mouse models, and were analyzed genetically through a number of siRNA and CRISPR screens [146,155,156,157,158,159,160,161,162,163,164]. Genetic sensitizers of ATR inhibitors include some genes that are recurrently mutated in CRPC, such as ATM, BRCA1, BRCA2, RNASESH2B, and ARID1A [122,158,165]. ATR inhibitors also sensitize cells to a number of genotoxic agents, including platinum drugs and topoisomerase inhibitors, both of which are used in a subset of CRPC treatments [166,167].
The direct testing of ATM-deficient PCa models have demonstrated sensitivity to ATR inhibitors that was further potentiated by the addition of PARP inhibitor, a combinatorial effect that has been observed in several cancer types [168,169,170,171]. The co-treatment of cells with both PARP and ATR inhibitors has now been explored in further detail and entered clinical trials for a number of cancers, including CRPC [172]. The combination of the ATR inhibitor AZD6738 and Olaparib is being tested in the Phase 2 TRAP trial (NCT03787680) on resistant mCRPC and AZD6738 is also being evaluated with Olaparib or Durvalumab (anti-PD-1 immunotherapy) in solid tumors including mCRPC (NCT03682289). Additional combination therapies between ATR inhibitors and genotoxic agents, including PARP inhibitors; topoisomerase I inhibitors; and gemcitabine, a cytidine nucleoside analog, are ongoing. The Phase 1/2 TRESR study is evaluating the ATR inhibitor RP-3500 alone or combined with Talazaporib or gemcitabine (NCT04497116) and another Phase 1/2 study is examining the ATR inhibitor ART0380 in combination with the topoisomerase 1 inhibitor irinotecan or gemcitabine (NCT04657068). The combination of ATR inhibitors and topoisomerase 1 inhibitors has shown promise in trials for small cell lung cancer that has included a small number of CRPC-NE patients. A recent Phase 1/2 trial on neuroendocrine cancers using ATR inhibitors in conjunction with sacituzumab govitecan (Trodelvy), an antibody–drug conjugate that couples an antibody for Trop-2 to the topoisomerase inhibitor SN-38, showed tumor regression in two CRPC-NE patients (NCT04826341) and this study is now being extended to include PARP inhibitor-resistant HRD tumors [173]. BRCA1-deficient cells that acquired PARP inhibitor resistance have shown increased dependence on ATR activity, suggesting that resistant HRD tumors may benefit from ATR inhibition [174,175].
Mutations in RNASEH2B, which were investigated in the context of PARP inhibitors in CRPC, have also been identified as sensitizers of ATR inhibition, topoisomerase 1/2 inhibitors, and gemcitabine through whole-genome CRISPR screens in other cell types [146,147,155,176]. As the loss of RNASEH2 function does not impair HR, it remains unclear what genomic features of CRPC may influence this strategy. For example, whether RB1 loss, which rescued PARP inhibitor sensitivity in RNASEH2B-deficient cells, would also abrogate ATR inhibitor or genotoxic agent sensitivity in this background remains to be determined [147].
DNA-PKcs (encoded by PRKDC) is frequently upregulated in prostate cancer [177,178]. In addition to its role in DNA repair, DNA-PKcs plays a variety of transcriptional roles in AR-CRPC, where it was identified as a cofactor for the AR, and more recently, AR-Vs, and its high expression correlated with aggressive and metastatic tumors [177,179]. DNA-PKcs activity has been shown to regulate splicing and the expression of AR-Vs through the control of RBMX expression [179]. The KU70 subunit of DNA-PK has been reported to recognize Topoisomerase 1 cleavage complexes to facilitate enhancer-mediated transcription in breast and prostate cancer [180,181]. DNA-PK has been shown to regulate glycolysis in PCa through the phosphorylation of ALDOA and PKM2 and DNA-PK also has a poorly understood role in translation, where it was shown to be activated by several small RNAs and regulate the processing of the 18S rRNA [182,183].
Numerous DNA-PKcs inhibitors have entered clinical trials for a variety of cancer types, often in combination with RT or other DNA-damaging agents (recently reviewed in reference [37]). Some of these agents also target mTOR1 and 2, structurally related kinases that control cell growth in response to nutrient availability [184]. In CRPC, the dual DNA-PKcs/mTOR targeting agent CC-115 was tested with the ARSI Enzalutamide and is moving to Phase II trials following a successful Phase I safety assessment (NCT02833883) [185]. While DNA-PKcs has not yet been proven to be an effective target as a monotherapy in CRPC, it may prove to be valuable in the context of combination therapies in future studies. Pre-clinical data has shown that DNA-PKcs inhibitors could re-sensitize taxane-resistant cells and restore sensitivity to multiple genotoxic agents, including topoisomerase inhibitors and platinum drugs, and ongoing clinical trials are testing DNA-PKcs inhibitors with RT, PARP inhibitors, and CHK1 inhibitors in a variety of cancer types [37,186].
6. Generating Targeted DNA Damage with Radiopharmaceutical Therapy
Aside from targeting DDR signaling, active efforts to target DNA-damaging radionuclides to cancer cells are underway. Radionuclides can be used for either imaging or therapy depending on the type of radiation emissions intrinsic to the radioisotope. Beta particle-, alpha particle-, or Meitner–Auger-emitting radionuclides can cause single- and double-strand DNA breaks (SSBs and DSBs) and/or damage to critical organelles when delivered to cancer cells as radiopharmaceutical therapy (RPT) [187]. How these RPT agents compare to EBRT with respect to the extent of SSBs or DSBs is challenging given their heterogeneity of distribution within targeted tumors and the differences in dose rates, with radionuclides having much lower dose rates compared to EBRT.
The intrinsic properties of some radionuclides allow for tissue specific targeting. For example, the alpha particle-emitting radium-223 is a calcium mimetic that localizes to areas of bone turnover, including bone metastases. Radium-223 (223RaCl2, Xofigo) has been FDA-approved for use in patients with symptomatic bone-limited mCRPC, following the results of the Phase 3 ALSYMPCA (NCT00699751) trial which showed improved overall survival compared with placebo [188].
To target soft tissue lesions, therapeutic radionuclides need to be coupled to a tumor- or TME-selective targeting molecule. Prostate-specific membrane antigen (PSMA), which is highly expressed in prostate adenocarcinoma, was discovered in the 1990s and has been an attractive target for both imaging and therapy [189]. First, antibodies and, later, peptidomimetics that bind to the extracellular domain were developed and tested as imaging or therapeutic agents for this disease. This culminated in the FDA approval of the PSMA-specific, beta particle-emitting peptidomimetic lutetium-177 vipivotide tetraxetan (a.k.a. 177Lu-PSMA-617, Pluvicto) in 2022, following the pivotal Phase 3 VISION study (NCT03511664), which demonstrated an improved overall survival benefit when compared with the protocol-defined standard of care in patients with mCRPC who had progressed after first-line taxane therapy. Unsurprisingly, 177Lu-PSMA-617 is now being tested prospectively in earlier lines of therapy. For example, a Phase 3 study, PSMAfore, is comparing RPT to ADT change in mCRPC (NCT04689828), one Phase 2 study is evaluating 177Lu-PSMA-617 when combined with first line docetaxel in mCRPC (NCT04663997), and in a Phase 1/2 trial preoperatively (NCT04430192).
While these earlier-line treatments continue to be pursued in potentially less heterogeneous and aggressive prostate adenocarcinoma, additional studies are exploring combinations with other therapies, which may synergize with 177Lu-PSMA-617. One example is the randomized Phase 2 cohort trial of stereotactic ablative radiotherapy versus stereotactic ablative radiotherapy combined with 177Lu-PSMA-617 (NCT05560659) in patients with oligometastatic mCRPC. The PROQURE study is evaluating node-positive hormone-sensitive prostate cancer receiving 177Lu-PSMA-617 with definitive external beam radiotherapy and ADT (NCT05162573). Another trial, PSMAddition, is a Phase 3 trial evaluating the standard of care with or without 177Lu-PSMA-617 in metastatic hormone-sensitive prostate cancer (NCT04720157).
Because the predominant mechanism of cytotoxicity of 177Lu-PSMA-617, and RPT generally, is DNA damage, the combination with DDR inhibitors is a natural line of inquiry. For example, the Phase 1 LuPARP trial (NCT03874884) is evaluating the combination of 177Lu-PSMA-617 and the PARP inhibitor Olaparib. Investigators are also prospectively testing the DNA-PKcs inhibitor, peposertib, in a Phase 1/2 trial combined with 223RaCl2 with or without avelumab (PD-L1 immunotherapy) in patients with mCRPC (NCT04071236). As mutations in ATM, BRCA2, and many other DDR proteins that would be predicted radiosensitizers occur in CRPC with some frequency, future efforts may seek to take further advantage of genetic lesions and DDR inhibitors in RPT.
Beyond clinical studies, answers to other open questions are being pursued. For instance, personalized dosimetry, that is, modulating the activity administered to patients based on patient-specific features (e.g., tumor burden and estimated absorbed dose to tumor and organs at risk), has the potential to improve the therapeutic index of these agents by limiting both over- and underdosing. Derivatives of PSMA-targeting agents containing alpha particle-emitting radionuclides such as actinium-225 (225Ac), including 225Ac-PSMA-617, are being tested in Phase 1 studies (NCT04597411). Alpha particles exhibit high linear energy transfer, that is, deposit high energy over a short pathlength, and they cause several-fold more DNA damage than beta particles. In some studies, patients who were refractory to 177Lu-PSMA-617 demonstrated radiographic and biochemical responses to 225Ac-PSMA-617 [190]. Antibody-based PSMA-targeted RPT agents, which have distinct biodistributions and toxicity profiles from peptidomimetic agents, are also being explored (NCT04876651). If effective, these derivatives will surely be tested in combinations with other modalities or drugs, as have 177Lu-PSMA-617 and 223RaCl2. Other PSMA-targeted ligands, including the well-known PSMA-I&T, are also being coupled to imaging and therapeutic radionuclides for testing in prospective clinical trials. Another PSMA-targeted agent, 18F-rh-PSMA-7.3, was recently FDA-approved for imaging, and its therapeutic counterpart, 177Lu-rh-PSMA-10.1, is being clinically evaluated in a Phase 1/2 clinical trial (NCT05413850) [191,192].
7. Crosstalk between the Immune System and the DDR in CRPC
Immune checkpoint blockade (ICB) immunotherapy has garnered extensive interest, as targeting immune checkpoint receptors, such as PD-1, PD-L1, CTLA4, and LAG-3, effectively suppresses immunosuppressive pathways in the TME in many cancer types [193,194]. ICB aims to disrupt signaling between cancer cells and the immune system that inhibits T-cell proliferation and anti-tumor immune responses. The observation that RT and other DNA-damaging agents induce inflammatory responses, which include the induction of several of these receptors, has launched a large number of studies aimed at understanding how to best combine DDR activation and ICB (recently reviewed in reference [65]).
High TMB, MSI-H, or dMMR are the primary clinical indicators used to guide ICB use, presumably due to the need for neoantigens to drive the immune response [193,195]. Tumor heterogeneity and active nonsense-mediated mRNA-decay have also been shown to limit the capacity of immune responses to engage tumors with high TMB [196,197,198]. The low frequency of dMMR and APOBEC signatures in CRPC represents a potential limitation for ICB approaches [71,72]. Recent work has demonstrated that the sequencing of circulating tumor DNA could be used to identify hypermutation signatures and MMR defects in metastatic PCa, indicating that this could be a powerful screening tool for identifying ICB responders [75]. The genomic analysis of a cohort containing African patients revealed a higher overall TMB, including elevated mutational signatures for both HRD and dMMR, indicating potential opportunities for the use of PARP inhibitors and ICB [199].
Recent work has identified SYNCRIP, an RNA-binding protein implicated in splicing, as a suppressor of APOBEC3B activity in CRPC [77]. The deletion of SYNCRIP occurs frequently in CRPC and it was shown that this enhances APOBEC3B mutagenic activity that can drive resistance to AR-targeted therapies and promote lineage plasticity through the mutagenesis of a number of drivers, including FOXA1, in a pre-clinical model. The treatment of patient-derived explants with ARSIs upregulated APOBEC3B at the RNA and protein levels, suggesting that this could be part of a general response to AR-targeted therapies. APOBEC inhibitors, which are currently under development, could potentially be used to restrain lineage plasticity in future therapeutic approaches using ARSIs and other targeted therapies in CRPC [200,201,202,203,204,205]. How actionable APOBEC-mediated mutagenesis will be in CRPC, either to prevent resistance or enhance ICB, remains to be determined.
While inhibitors of CTLA-4 and PD-L1 have been tested in clinical trials for PCa, the only ICB therapy currently approved in mCRPC with high MSI or dMMR is the PD-1 inhibitor pembrolizumab (Keytruda) that blocks the binding of PD-1 on T-cells to its receptor PD-L1, which is upregulated in some cancer cells [193]. In localized prostate cancer, the loss of BRCA2, RB1, or the chromatin remodeler CHD1, is correlated with PD-L1 expression and have been proposed as patient selection markers for ICB use [206]. However, whether this also applies to mCRPC remains to be determined. As a subset of CRPC patients appear to benefit from anti-PD-1 in unselected patient populations, a large number of clinical trials are ongoing with pembrolizumab and other ICB therapies (ongoing clinical trials were recently reviewed in reference [207]). These include testing patients with HR-proficient and HRD tumors, as well as combination therapies with PARP inhibitors, ARSIs, and RPT. One Phase 2 trial is comparing patients with mCRPC randomized to 177Lu-PSMA-617 or 177Lu-PSMA-617 with ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-L1) (NCT05150236), and another Phase 1b study is assessing the safety and efficacy of 177Lu-PSMA-617 combined with pembrolizumab (NCT03805594). Such trials are of interest in light of preclinical work showing that RPT may enhance ICB in otherwise immunologically cold tumors [208].
Combining ICB with RT, ATR, DNA-PKcs, and PARP inhibitors has been shown to improve outcomes in some cancer types, in large part due the activation of innate immune responses driven by the cGAS-STING pathway [209,210,211,212,213,214,215,216,217,218]. The cGAS protein senses extrachromosomal DNA that can arise from DNA damage and activates the endoplasmic reticulum protein STING through the production of 2′3′-cGAMP [219]. STING activates downstream transcriptional activators, including IRF3 and NFKB, to activate antiviral inflammatory responses. Mutations in RNASEH2B, which occur frequently in CRPC, are associated with Aicardi–Goutieres syndrome, a hereditary inflammatory disease, and similar mutations occur in multiple cancers and have been correlated with inflammatory gene signatures, suggesting that these tumors may respond more readily to ICB [146,220,221]. STING agonists have been shown to improve ICB responses in preclinical CRPC models and have entered clinical use for a number of cancer types [222,223,224].
Recently, targeting B7-H3 (PD-L3; CD274), a member of the PD-L1 family, as well as Myeloid Derived Suppressor cells (MDSCs), has been demonstrated to overcome immunosuppression in CRPC, and other cancer types, in preclinical models [225,226]. The expression of B7-H3 was high in many PCa patient samples and cell lines, notably, higher than PD-L1 (B7-H1), and it correlated with adverse disease events [227,228]. The modulation of B7-H3 levels in multiple cell types, including PTEN-TP53-deficient CRPC, impaired tumor growth [229]. As PD-L1 expression is activated by RT and other DNA-damaging agents, it will be of interest to determine if B7-H3 levels can be similarly manipulated for clinical gain, for example by RPT or ATR/PARP inhibitors. Multiple strategies have been proposed to target the MDSC cell population that suppresses T-cell responses, alone or in combination with ICB, including the use of multi-kinase inhibitors, inhibitors of CXCL5-CXCL2 or co-targeting the MNK1/2 and AKT pathways [230,231]. A clinical trial in ARSI-resistant mCRPC using a combination of Enzalutamide and a CXCR2 inhibitor (AZD5069) showed promising results in a subset of patients, supporting this as a therapeutic strategy for further development (NCT03177187) [232]. Again, whether any of these strategies targeting MDSC-mediated immunosuppression will, like ICB, further benefit from DDR inhibitors or RPT remains to be tested in future work.
8. Targeting the Hypoxic TME in CRPC Therapy
Hypoxia contributes to genomic instability in several tumor types and is associated with high rates of TMB. For instance, hypoxic PCa tumors showed the loss of PTEN function, increased levels of chromothripsis (clustered chromosomal rearrangements), and shorter telomeres in localized PCa [233]. As hypoxia is a barrier to RT, chemotherapies, and immunotherapy, multiple strategies are being employed to directly exploit hypoxia in cancers. In PCa, two HAPs, TH-302/evofosfamide and CP-506, are being tested in clinical trials in patients with PCa. Evofosfamide and CP-506 are DNA-alkylating agents under low oxygen (<0.5% O2) conditions and both reduce tumor growth in a variety of preclinical models, including PCa [234,235,236,237]. CP-506 is being tested in combination with carboplatin or a selection of ICB agents (NCT04954599) and Evofosfamide is being tested with ICB (Ipilimumab/anti-CTLA4) (NCT03098160) in a variety of solid tumors, including PCa.
CRPC-NE is a form of lethal prostate cancer that is resistant to many types of therapy and shares some features with CRPC-SCL [22,238]. The homeodomain TF ONECUT2 (OC2) was recently shown to be a master regulator of neuroendocrine cancer, including CRPC-NE, and additional data have shown that the loss of RB1 is sufficient to increase its expression in CRPC models [239,240]. OC2 depletion in PC3 cells impairs hypoxia-mediated transcriptional programs and drove NE differentiation that was augmented by hypoxia [239]. OC2 regulated HIF1α chromatin occupancy by upregulating SMAD3 expression and OC2 depletion impairs tumor growth in xenografts. OC2 expression has been correlated with high hypoxia in CRPC-NE, and multiple patient-derived xenograft (PDX) models were sensitive to treatment with Evofosfamide. Together, these data suggest that OC2 expression in CRPC-NE may serve as a marker for the use of treatments that take advantage of HAPs and other therapeutic approaches to exploit low oxygen, providing strategies to augment RT and DDR inhibitors that are impaired by hypoxia.
9. Emerging Targets in CRPC
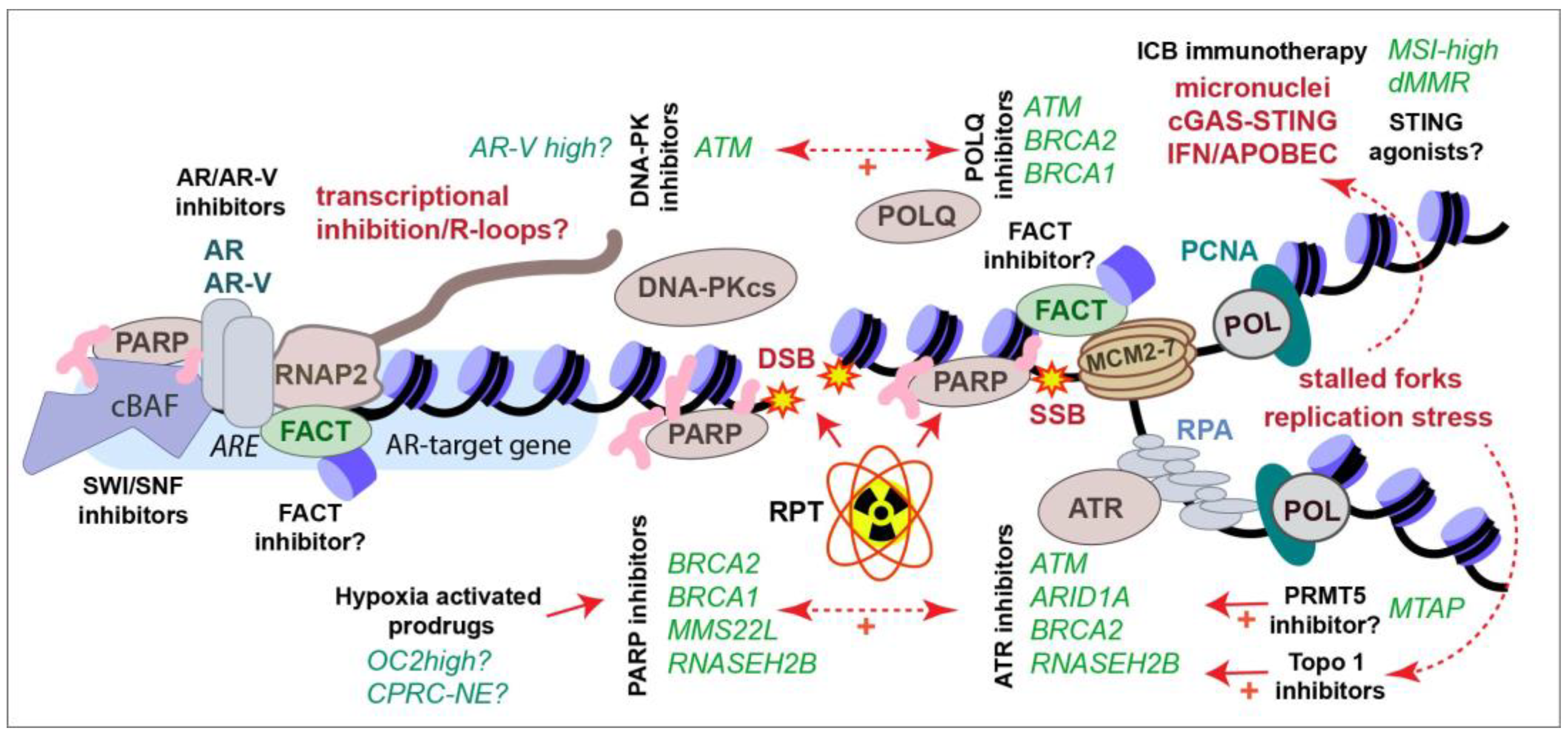
A number of small molecule inhibitors targeting DDR components and chromatin regulators are being used in research and, in some cases, entering clinical trials. Here, we describe in brief a selection of their targets and their potential utility in CRPC treatment. Several are highlighted in Figure 4 in the context of previously described therapies.
9.1. SWI/SNF
Genes that encode members of the SWI/SNF nucleosome remodeling complexes are frequently mutated in many cancer types, including PCa [241]. SWI/SNF complexes have been shown to localize to DNA damage sites and its deficiency sensitizes cells to a number of DNA-damaging agents [242]. SWI/SNF has been implicated in NHEJ, HR, NER, and MMR, although its precise function in these pathways remains unclear [242,243]. Three major subtypes of SWI/SNF complexes have been identified that contain overlapping and unique subunits, and each of these complexes contains one of two ATPase subunits, SMARCA2 (BRM) and SMARCA4 (BRG1) [244]. The depletion of both SMARCA2 and SMARCA4 activity globally reduces chromatin access and leads to increased levels of R-loops, RNA–DNA hybrids, which can cause DNA damage and genomic instability [245,246]. Targeting both of these enzymes, as well as an additional SWI/SNF protein, PBRM1, using the AU-15330 PROTAC in PCa, was demonstrated to have preferential effectiveness in enhancer-addicted AR-dependent PCa [247]. SMARCA4/BRG1 was also identified as a selectively essential gene in PTEN-depleted cells in a CRISPR screen [248]. As the loss of PTEN occurs in up to 50% of CRPCs, and across all subtypes, this may be another vulnerability that can be exploited with agents that target SWI/SNF. The further expanded use of agents targeting SWI/SNF in CRPC may be achievable with combination therapies that exploit R-loop-mediated DNA damage or additional genetic interactions, but this remains to be experimentally determined. Currently, no agents directly targeting SWI/SNF have entered clinical trials, but given the large amount of activity in this space, this will likely change in the near future.
9.2. POLQ
Recently, several inhibitors were identified for POLQ, which is required for TMEJ, that can serve as a backup pathway for DSB repair (Figure 2D) [41,249,250,251,252]. POLQ loss is synthetically lethal with HRD, as well as with the loss or inhibition of ATM or ATR, providing another route to target a subset of CRPCs, including those that become resistant to PARP inhibitors [249,250,253,254,255]. Although not in PCa, several studies have combined POLQ inhibitors with DNA-PKcs inhibitors and observed the increased killing of p53-deficient cells, as well as enhanced radiosensitivity [256,257]. POLQ inhibition also enhances PARP inhibitor effectiveness and activates the cGAS-STING pathway in an HRD pancreatic adenocarcinoma mouse model, suggesting that it may have applications in ICB immunotherapy, in which STING agonists are also under investigation [253]. While POLQ inhibitors have not been explored in PCa to our knowledge, they may be valuable reagents to investigate for future combination therapy approaches involving NHEJ (DNA-PKcs inhibitors), PARP inhibitors, RT, or ICB, as well as mutations in selected DDR genes. Phase I and II trials are ongoing with several POLQ inhibitors in metastatic solid tumors (NCT05898399, NCT04991480, NCT06077877, NCT05687110).
9.3. PRMT1
Protein arginine methyltransferase 1 (PRMT1) has been implicated in cell cycle progression, DNA repair, innate immunity, and RNA processing and proposed as a therapeutic target in several cancer types [258,259,260]. Its precise functions in the DDR remain unclear but it has been shown to methylate key DSBR regulators, BRCA1, MRE11, and 53BP1 and influence DSBR pathway choice [261,262,263,264,265,266]. Moreover, PRMT1 loss in mice caused increased levels of genomic instability and HRD [267]. Multiple small molecule inhibitors of PRMT1 have been developed, the first of which has entered clinical trials for a number of tumor types (NCT03666988) [259]. PRMT1 expression is upregulated in advanced PCa and it has been identified as a regulator of AR-V7 in a whole-genome CRISPR screen [268,269]. The depletion of PRMT1 reduces the occupancy of AR at its target genes, an effect that has also been observed following treatment with the metabolite spermine, which has been shown to inhibit PRMT1 [269,270]. Using a LNCaP-derived cell line with higher AR expression (LNCaP/AR-Enh) it was demonstrated in xenografts that co-targeting AR and PRMT1 was an effective therapeutic approach [269]. Collectively, these data suggest that targeting PRMT1 in combination with ARSIs may be an effective strategy for CRPC-AR and that combining PRMT1 inhibitors with genotoxic agents may be warranted.
9.4. PRMT5
Protein arginine methyltransferase 5 (PRMT5) targets proteins involved in many cellular processes, influences AR signaling and regulates the DDR through direct and indirect mechanisms, leading to radio- and chemosensitivity (recently reviewed in references [271,272]). PRMT5 loss impairs HR and enhances the toxicity of PARP inhibitors in other cancer types [273]. PRMT5 loss is synthetically lethal with MTAP deficiency, which occurs frequently in many cancer types, including PCa, providing an additional rationale for the use of PRMT5 inhibitors in selected patient populations [274]. PRMT5 has also been identified as a suppressor of DNA damage in CRISPR screens and subsequent analysis has demonstrated that PRMT5 inhibitors destabilize ATM in cell cultures, suggesting that it could be used in combination with a variety of DNA-damaging agents, such as ATR or PARP inhibitors, in future therapies [275]. PRMT5 inhibition can also improve ICB outcomes through the augmentation of cGAS-STING responses and PD-L1 expression [276,277]. Two recent reports have demonstrated that PRMT5 inhibition impairs DNA repair, enhances radiosensitivity in multiple CRPC cell lines, and prevents RT-induced neuroendocrine differentiation and tumor growth in CRPC-AR tumor models [278,279]. PRMT5 has also been identified as a suppressor of gemcitabine toxicity in whole-genome CRISPR screens of pancreatic cells [280]. Thus, PRMT5 inhibitors could potentially be useful in treating both castration-sensitive PCa and CRPC and could potentially be enhanced by selection for MTAP loss or combination therapies. Currently, more than 10 Phase 1/2 trials are ongoing with PRMT5 inhibitors in a variety of cancer types, in some cases, utilizing MTAP loss for cohort selection (https://clinicaltrials.gov/ (accessed on 10 December 2023)).
9.5. FACT
The Facilitates Chromatin Transcription (FACT) complex, composed of SSRP1 and SPT16, plays a key role in nucleosome dynamics during transcription, DNA replication, and DNA repair (reviewed in reference [281]). The elevated expression of FACT subunits is often observed in cancer, in some cases correlating with poor clinical outcomes [282]. Both FACT subunits have been identified as common essential genes, indicating that many cancers depend on maintaining FACT function (Depmap.org). The drug CBL0137/Curaxin is a non-mutagenic DNA intercalator that has been shown to trap FACT on chromatin, leading to transcriptional alterations, including the activation of the TP53 response [283]. CBL0137 has been demonstrated to display anti-tumor efficacy as a single agent in glioblastoma mouse models, enhance radiotherapy and histone deacetylase inhibitor responses in CNS tumors, as well as enhance the responses to cisplatin in small-cell lung cancer [284,285,286,287,288,289]. In acute myeloid leukemia cell lines, CBL0137s toxic effects were limited to those with TP53 mutations, suggesting its efficacy may be optimal in TP53-proficient cells [290]. FACT interacts with the TONSL-MMS22L complex that is important for HR and frequently compromised in CRPC, as discussed earlier (Figure 4) [117,118,119,120]. Consistent with this, CBL0137 was shown to impair HR and enhance PARP inhibitor and carboplatin efficacy in serous ovarian carcinoma [291]. The depletion of FACT or treatment with CBL0137 results in the activation of the IFNg response, consistent with previous reports that it is required for endogenous and exogenous viral silencing [292,293,294]. Notably, CBL0137 has been identified in a screen to bypass anti-PD-1 ICB suppression by ADAR1, an RNA-editing enzyme that attenuates antiviral responses [295]. This suggests that it may be interesting to try CBL0137 in combination with ICB, which can be enhanced by innate immune activation, including IFNg [296]. Finally, FACT has been demonstrated to mediate adaptation to hypoxia and CBL0137 impaired tumor growth and enhances the effects of an anti-angiogenic drug, bevacizumab, in a hepatocellular carcinoma model [297]. While the effects of CBL0137 have not been reported in PCa, it has entered clinical trials for a number of other cancer types, and its potential to enhance genotoxic agents, impair transcription, sensitize hypoxic cells, and improve DDR inhibitor and ICB responses may be useful to explore in future work (NCT01905228, NCT02931110, NCT03727789, NCT05498792, NCT04870944).
9.6. The Tousled-Like Kinases (TLKs)
The Tousled Kinase (TSL) was first identified in plants and later the mammalian Tousled like kinases, TLK1 and TLK2, were connected to the DDR when TLK1 was identified as a CHK1 substrate [298,299,300,301]. TLK activity is required for DNA replication and chromatin maintenance, primarily through the regulation of ASF1A and ASF1B [301]. These chaperones facilitate the incorporation of H3/H4 heterodimers during replication, transcription, and DNA repair [301]. TLK1 was shown to regulate additional DDR factors, including RAD9 and RAD54L, which play roles in TMEJ and HR, respectively [302,303,304,305,306]. The depletion of TLK1/2 leads to replication stress and DNA damage that could be enhanced by ATR, CHK1, or PARP inhibitors in several cell lines in vitro [307]. This is accompanied by the activation of cGAS-STING-dependent innate immune responses, including TNFa and IFNg, similar to what has been observed for ASF1A depletion that has been demonstrated to enhance anti-PD-1 ICB immunotherapy via TNFa induction [308,309]. The short hairpin-mediated depletion of TLK2 in breast cancer xenograft models or treatment LNCaP (CRPC-AR) xenografts with TLK1 inhibitors thioridazine or J54-impaired cancer growth, an effect that was enhanced by anti-hormone treatment in both cases [310,311,312]. Additional TLK inhibitor leads based on indirubin E804 have also recently been reported and shown to inhibit ASF1A phosphorylation, impair DNA replication, and induce replication stress and DNA damage [313]. Thioridazine is an antipsychotic medication with a number of potential targets that has been evaluated in a number of clinical trials. However, its effects are unlikely to be restricted to the inhibition of TLK and no clinical trials have employed it for this reason to our knowledge. Existing evidence suggests that the development of more specific and potent inhibitors may be valuable for several cancer types, including CRPC, and that TLK inhibitors could be used in combination with a number of anti-cancer agents including ARSIs, DDR inhibitors, and ICB.
10. Conclusions
There are several opportunities to exploit the DDR in CRPC therapy, taking advantage of recurring genetic lesions or combination therapies (Figure 4). The AR remains a key target in PCa therapy and the continued development of new small molecules to target the AR and AR-Vs may improve future approaches and lend themselves to rational combination therapies, including with DDR-targeting agents. Tumor hypoxia has been associated with worse outcomes at every stage of prostate cancer disease progression and remains a significant barrier to radio- and chemotherapies, including PARP inhibitors. The use of HAPs, and possibly FACT inhibitors, may allow investigators to take advantage of this prominent feature of the PCa TME.
While evidence for HDR and other DDR mutations that facilitate PARP inhibitor use continues to grow, the generally low TMB of CRPC has limited immunotherapy to a subset of patients, with little evidence of clinical efficacy thus far. The identification of new targets, such as B7-H3 and MDSCs, integrated with the manipulation of the DDR, for example via PARP or ATR inhibitors or STING agonists, may determine if more CRPC patients can benefit from immunotherapy approaches in future trials. In addition, the generation of additional syngeneic models of CRPC could aid in the analysis of the interplay between the DDR and immune responses.
The RPT field has undergone a renaissance following recent regulatory approvals based on efficacy in clinically meaningful endpoints; namely quality of life and/or overall survival. Long-known agents are being tested in prospective studies to obtain regulatory approvals. Novel agents and tweaks of existing agents to improve their pharmacokinetic profiles have been synthesized and tested preclinically with the goal of clinical translation. Many opportunities to combine RPT with DDR modulators are waiting to be further explored and the results of open clinical trials will help clarify the best use of these agents.
While agents such as PARP inhibitors appear to have low toxicity based on current clinical information, the variable effects of individual agents or therapeutic combinations on normal tissue toxicity, particularly in diverse genetic backgrounds, will need to be assessed in future studies. Defining the optimal order of treatments to ensure the safest and most efficacious outcomes for patients will also be of high priority in future pre-clinical experiments and clinical trials.
Author Contributions
All authors (T.H.S., O.I.O., F.E.E. and D.E.C.) wrote and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
T.H.S., F.E.E. and D.E.C. are supported by NCI-NIH intramural funding (1ZIABC012012, 1ZIABC011800 and 1ZIABC011552, respectively).
Acknowledgments
We apologize to the many colleagues whose primary work could not be cited due to space constraints. We thank S. Sharma for the critical reading and feedback on the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ADT | Androgen deprivation therapy |
| Alt-NHEJ | Alternative non-homologous end joining |
| APOBEC | Apolipoprotein B mRNA editing enzyme, catalytic polypeptide |
| AR | Androgen receptor |
| AR-V | AR-variants |
| ARE | Androgen response element |
| ARSI | Androgen receptor signaling inhibitor |
| BER | Base excision repair |
| BRCA | Breast cancer associated |
| CE3 | Cryptic exon 3 |
| cNHEJ | Classical non-homologous end-joining |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CRPC | Castration resistant prostate cancer |
| CRPC-AR | CRPC-androgen receptor subtype |
| CRPC-NE | CRPC-neuroendocrine subtype |
| CRPC-SCL | CRPC-stem cell like subtype |
| CRPC-WNT | CRPC-WNT dependent subtype |
| DBD | DNA binding domain |
| DDR | DNA damage response |
| dMMR | Mismatch Repair deficiency |
| DSB | Double-strand break |
| DSBR | DSB repair |
| dsDNA | Double-strand DNA |
| EBRT | External beam radiotherapy |
| HAP | Hypoxia activated prodrug |
| HD | Hinge domain |
| HIF | Hypoxia inducible factor |
| HR | Homologous recombination |
| HRD | Homologous recombination deficiency |
| ICB | Immune checkpoint blockade |
| LBD | Ligand binding domain |
| mCRPC | Metastatic CRPC |
| MMR | Mismatch Repair |
| MMS | Methyl methanesulfonate |
| MNNG | Methylnitronitrosoguanidine |
| MRN | MRE11-RAD50-NBS1 |
| MSI | Microsatellite instability |
| NTD | N-Terminal domain |
| PARP | Poly (ADP ribose) polymerase |
| PCa | Prostate cancer |
| PSA | Prostate specific membrane antigen |
| RNASEH2 | Ribonuclease H2 |
| ROS | Reactive oxygen species |
| RPT | Radiopharmaceutical therapy |
| RT | Radiotherapy |
| SBS | Single base substitution |
| SSB | Single-strand break |
| SSBR | SSB Repair |
| ssDNA | Single-strand DNA |
| TF | Transcription factors |
| TLK | Tousled-like kinase |
| TMB | Tumor mutational burden |
| TME | Tumor microenvironment |
| TMEJ | Polymerase theta-mediated end-joining |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.L.; Antonarakis, E.S.; Armstrong, A.J.; D’Amico, A.V.; Davis, B.J.; Dorff, T.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef]
- Lavaud, P.; Dumont, C.; Thibault, C.; Albiges, L.; Baciarello, G.; Colomba, E.; Flippot, R.; Fuerea, A.; Loriot, Y.; Fizazi, K. Next-Generation Androgen Receptor Inhibitors in Non-Metastatic Castration-Resistant Prostate Cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920978134. [Google Scholar] [CrossRef] [PubMed]
- Estébanez-Perpiñá, E.; Bevan, C.L.; McEwan, I.J. Eighty Years of Targeting Androgen Receptor Activity in Prostate Cancer: The Fight Goes On. Cancers 2021, 13, 509. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Androgen Receptor Splice Variants and Prostate Cancer: From Bench to Bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [PubMed]
- Tietz, K.T.; Dehm, S.M. Androgen Receptor Variants: RNA-Based Mechanisms and Therapeutic Targets. Hum. Mol. Genet. 2020, 29, R19–R26. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Brown, L.C.; Antonarakis, E.S.; Armstrong, A.J.; Luo, J. Androgen Receptor Variant-Driven Prostate Cancer II: Advances in Laboratory Investigations. Prostate Cancer Prostatic Dis. 2020, 23, 381–397. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Graf, R.P.; Hullings, M.; Barnett, E.S.; Carbone, E.; Dittamore, R.; Scher, H.I. Clinical Utility of the Nuclear-Localized AR-V7 Biomarker in Circulating Tumor Cells in Improving Physician Treatment Choice in Castration-Resistant Prostate Cancer. Eur. Urol. 2020, 77, 170–177. [Google Scholar] [CrossRef]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danila, D.C.; Healy, P.; Anand, M.; Rothwell, C.J.; et al. Prospective Multicenter Validation of Androgen Receptor Splice Variant 7 and Hormone Therapy Resistance in High-Risk Castration-Resistant Prostate Cancer: The PROPHECY Study. J. Clin. Oncol. 2019, 37, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; McLaughlin, B.; Lu, D.; Louw, J.; Danila, D.C.; Dugan, L.; Johnson, A.; Heller, G.; et al. Nuclear-Specific AR-V7 Protein Localization Is Necessary to Guide Treatment Selection in Metastatic Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker with Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen Receptor Splice Variant-7 Expression Emerges with Castration Resistance in Prostate Cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Modonutti, D.; Majdalany, S.E.; Corsi, N.; Li, P.; Sood, A.; Dalela, D.; Jamil, M.L.; Hwang, C.; Menon, M.; Rogers, C.G.; et al. A Novel Prognostic Model Predicting Overall Survival in Patients with Metastatic Castration-Resistant Prostate Cancer Receiving Standard Chemotherapy: A Multi-Trial Cohort Analysis. Prostate 2022, 82, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Halabi, S.; Lin, C.-Y.; Kelly, W.K.; Fizazi, K.S.; Moul, J.W.; Kaplan, E.B.; Morris, M.J.; Small, E.J. Updated Prognostic Model for Predicting Overall Survival in First-Line Chemotherapy for Patients with Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2014, 32, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.-K.; Watt, K.; Tam, T.; Yang, Y.C.; Bañuelos, C.A.; et al. Regression of Castrate-Recurrent Prostate Cancer by a Small-Molecule Inhibitor of the Amino-Terminus Domain of the Androgen Receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef]
- Basu, S.; Martinez-Cristobal, P.; Pesarrodona, M.; Frigole-Vivas, M.; Szulc, E.; Lewis, M.; Banuelos, A.; Sanchez-Zarzalejo, C.; Bielskute, S.; Zhu, J.; et al. Androgen Receptor Condensates as Drug Targets. BioRxiv 2022. [Google Scholar] [CrossRef]
- Xie, J.; He, H.; Kong, W.; Li, Z.; Gao, Z.; Xie, D.; Sun, L.; Fan, X.; Jiang, X.; Zheng, Q.; et al. Targeting Androgen Receptor Phase Separation to Overcome Antiandrogen Resistance. Nat. Chem. Biol. 2022, 18, 1341–1350. [Google Scholar] [CrossRef]
- Thiyagarajan, T.; Ponnusamy, S.; Hwang, D.-J.; He, Y.; Asemota, S.; Young, K.L.; Johnson, D.L.; Bocharova, V.; Zhou, W.; Jain, A.K.; et al. Inhibiting Androgen Receptor Splice Variants with Cysteine-Selective Irreversible Covalent Inhibitors to Treat Prostate Cancer. Proc. Natl. Acad. Sci. USA 2023, 120, e2211832120. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Xu, D.; Wang, S.; Wong, C.K.; Martinez-Fundichely, A.; Lee, C.J.; Cohen, S.; Park, J.; Hill, C.E.; Eng, K.; et al. Chromatin Profiles Classify Castration-Resistant Prostate Cancers Suggesting Therapeutic Targets. Science 2022, 376, eabe1505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.D.; Deng, S.; Hoover, E.; Chen, C.-C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 2020, 37, 584–598.e11. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Manzar, N.; Ateeq, B. Dynamics of Cellular Plasticity in Prostate Cancer Progression. Front. Mol. Biosci. 2020, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Abd, G.M.; Laird, M.C.; Ku, J.C.; Li, Y. Hypoxia-Induced Cancer Cell Reprogramming: A Review on How Cancer Stem Cells Arise. Front. Oncol. 2023, 13, 1227884. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.E.; Semenza, G.L. Hypoxia-Inducible Factors: Cancer Progression and Clinical Translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why Is the Partial Oxygen Pressure of Human Tissues a Crucial Parameter? Small Molecules and Hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours-Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Movsas, B.; Chapman, J.D.; Greenberg, R.E.; Hanlon, A.L.; Horwitz, E.M.; Pinover, W.H.; Stobbe, C.; Hanks, G.E. Increasing Levels of Hypoxia in Prostate Carcinoma Correlate Significantly with Increasing Clinical Stage and Patient Age: An Eppendorf pO(2) Study. Cancer 2000, 89, 2018–2024. [Google Scholar] [CrossRef]
- Cameron, S.; Deblois, G.; Hawley, J.R.; Qamra, A.; Zhou, S.; Tonekaboni, S.A.M.; Murison, A.; Van Vliet, R.; Liu, J.; Locasale, J.W.; et al. Chronic Hypoxia Favours Adoption to a Castration-Resistant Cell State in Prostate Cancer. Oncogene 2023, 42, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Xue, C.; Mendonca, J.; Sun, X.-X.; Liu, Q.; Reardon, P.N.; Chen, Y.; Qian, K.; Hua, V.; Chen, A.; et al. Interplay between Hypoxia and Androgen Controls a Metabolic Switch Conferring Resistance to Androgen/AR-Targeted Therapy. Nat. Commun. 2018, 9, 4972. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, K.T.; McCarthy, H.O.; Devlin, A.; Ming, L.; Robson, T.; McKeown, S.R.; Worthington, J. Hypoxia Selects for Androgen Independent LNCaP Cells with a More Malignant Geno- and Phenotype. Int. J. Cancer 2008, 123, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.; Bristow, R. Bad Neighbours: Hypoxia and Genomic Instability in Prostate Cancer. Br. J. Radiol 2020, 93, 20200087. [Google Scholar] [CrossRef] [PubMed]
- Douglass, M.; Hall, E.J.; Amato, J. Giaccia: Radiobiology for the Radiologist. Australas. Phys. Eng. Sci. Med. 2018, 41, 1129–1130. [Google Scholar] [CrossRef]
- Singleton, D.C.; Macann, A.; Wilson, W.R. Therapeutic Targeting of the Hypoxic Tumour Microenvironment. Nat. Rev. Clin. Oncol. 2021, 18, 751–772. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA Damage Response Pathways in Cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human Topoisomerases and Their Roles in Genome Stability and Organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef]
- Semlow, D.R.; Walter, J.C. Mechanisms of Vertebrate DNA Interstrand Cross-Link Repair. Annu. Rev. Biochem. 2021, 90, 107–135. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Caldecott, K.W. DNA Single-Strand Break Repair and Human Genetic Disease. Trends Cell Biol. 2022, 32, 733–745. [Google Scholar] [CrossRef]
- Ramsden, D.A.; Carvajal-Garcia, J.; Gupta, G.P. Mechanism, Cellular Functions and Cancer Roles of Polymerase-Theta-Mediated DNA End Joining. Nat. Rev. Mol. Cell Biol. 2022, 23, 125–140. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Mertz, T.M.; Collins, C.D.; Dennis, M.; Coxon, M.; Roberts, S.A. APOBEC-Induced Mutagenesis in Cancer. Annu. Rev. Genet. 2022, 56, 229–252. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.A.; Lara, P.N.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and Estramustine Compared with Mitoxantrone and Prednisone for Advanced Refractory Prostate Cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Teply, B.A.; Hauke, R.J. Chemotherapy Options in Castration-Resistant Prostate Cancer. Indian J. Urol. 2016, 32, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent Advances in the Management of Metastatic Prostate Cancer. JCO Oncol. Pract. 2022, 18, 45–55. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The Essential Kinase ATR: Ensuring Faithful Duplication of a Challenging Genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- De Marco Zompit, M.; Esteban, M.T.; Mooser, C.; Adam, S.; Rossi, S.E.; Jeanrenaud, A.; Leimbacher, P.-A.; Fink, D.; Shorrocks, A.-M.K.; Blackford, A.N.; et al. The CIP2A-TOPBP1 Complex Safeguards Chromosomal Stability during Mitosis. Nat. Commun. 2022, 13, 4143. [Google Scholar] [CrossRef]
- Trivedi, P.; Steele, C.D.; Au, F.K.C.; Alexandrov, L.B.; Cleveland, D.W. Mitotic Tethering Enables Inheritance of Shattered Micronuclear Chromosomes. Nature 2023, 618, 1049–1056. [Google Scholar] [CrossRef]
- Lin, Y.-F.; Hu, Q.; Mazzagatti, A.; Valle-Inclán, J.E.; Maurais, E.G.; Dahiya, R.; Guyer, A.; Sanders, J.T.; Engel, J.L.; Nguyen, G.; et al. Mitotic Clustering of Pulverized Chromosomes from Micronuclei. Nature 2023, 618, 1041–1048. [Google Scholar] [CrossRef]
- Brambati, A.; Sacco, O.; Porcella, S.; Heyza, J.; Kareh, M.; Schmidt, J.C.; Sfeir, A. RHINO Directs MMEJ to Repair DNA Breaks in Mitosis. Science 2023, 381, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Gelot, C.; Kovacs, M.T.; Miron, S.; Mylne, E.; Haan, A.; Boeffard-Dosierre, L.; Ghouil, R.; Popova, T.; Dingli, F.; Loew, D.; et al. Polθ Is Phosphorylated by PLK1 to Repair Double-Strand Breaks in Mitosis. Nature 2023, 621, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Olave, M.C.; Graham, R.P. Mismatch Repair Deficiency: The What, How and Why It Is Important. Genes Chromosomes Cancer 2022, 61, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.-C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA Damage Response in Immuno-Oncology: Developments and Opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Kanu, N.; Cerone, M.A.; Goh, G.; Zalmas, L.-P.; Bartkova, J.; Dietzen, M.; McGranahan, N.; Rogers, R.; Law, E.K.; Gromova, I.; et al. DNA Replication Stress Mediates APOBEC3 Family Mutagenesis in Breast Cancer. Genome Biol. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.C.; et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019, 176, 1282–1294.e20. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Dananberg, A.; Chu, K.; Bergstrom, E.N.; Striepen, J.; von Morgen, P.; Chen, Y.; Shah, H.; Sale, J.E.; Alexandrov, L.B.; et al. Mechanisms of APOBEC3 Mutagenesis in Human Cancer Cells. Nature 2022, 607, 799–807. [Google Scholar] [CrossRef]
- DeWeerd, R.A.; Németh, E.; Póti, Á.; Petryk, N.; Chen, C.-L.; Hyrien, O.; Szüts, D.; Green, A.M. Prospectively Defined Patterns of APOBEC3A Mutagenesis Are Prevalent in Human Cancers. Cell Rep. 2022, 38, 110555. [Google Scholar] [CrossRef]
- Zou, X.; Koh, G.C.C.; Nanda, A.S.; Degasperi, A.; Urgo, K.; Roumeliotis, T.I.; Agu, C.A.; Badja, C.; Momen, S.; Young, J.; et al. A Systematic CRISPR Screen Defines Mutational Mechanisms Underpinning Signatures Caused by Replication Errors and Endogenous DNA Damage. Nat. Cancer 2021, 2, 643–657. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Wedge, D.C.; Gundem, G.; Mitchell, T.; Woodcock, D.J.; Martincorena, I.; Ghori, M.; Zamora, J.; Butler, A.; Whitaker, H.; Kote-Jarai, Z.; et al. Sequencing of Prostate Cancers Identifies New Cancer Genes, Routes of Progression and Drug Targets. Nat. Genet. 2018, 50, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Ali, S.; Genega, E.; Reed, D.; Sokol, E.; Mathew, P. Aggressive-Variant Microsatellite-Stable POLE Mutant Prostate Cancer with High Mutation Burden and Durable Response to Immune Checkpoint Inhibitor Therapy. JCO Precis. Oncol. 2018, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Wang, H.; He, K.; Peng, M.; Wang, Y.; Li, Y.; Jiang, S.; Li, J.; Yi, L.; Cui, R. Clinical Characterization of Mismatch Repair Gene-Deficient Metastatic Castration-Resistant Prostate Cancer. Front. Oncol. 2020, 10, 533282. [Google Scholar] [CrossRef] [PubMed]
- Ritch, E.; Fu, S.Y.F.; Herberts, C.; Wang, G.; Warner, E.W.; Schönlau, E.; Taavitsainen, S.; Murtha, A.J.; Vandekerkhove, G.; Beja, K.; et al. Identification of Hypermutation and Defective Mismatch Repair in ctDNA from Metastatic Prostate Cancer. Clin. Cancer Res. 2020, 26, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Movasati, A.; Brunner, S.R.; Nguyen, L.; Priestley, P.; Cuppen, E.; Van Hoeck, A. Pan-Cancer Whole-Genome Comparison of Primary and Metastatic Solid Tumours. Nature 2023, 618, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Deng, S.; Zhu, G.; Wang, C.; Johnson, N.A.; Zhang, Z.; Tirado, C.R.; Xu, Y.; Metang, L.A.; et al. Loss of SYNCRIP Unleashes APOBEC-Driven Mutagenesis, Tumor Heterogeneity, and AR-Targeted Therapy Resistance in Prostate Cancer. Cancer Cell 2023, 41, 1427–1449. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef]
- Kohaar, I.; Zhang, X.; Tan, S.-H.; Nousome, D.; Babcock, K.; Ravindranath, L.; Sukumar, G.; Mcgrath-Martinez, E.; Rosenberger, J.; Alba, C.; et al. Germline Mutation Landscape of DNA Damage Repair Genes in African Americans with Prostate Cancer Highlights Potentially Targetable RAD Genes. Nat. Commun. 2022, 13, 1361. [Google Scholar] [CrossRef]
- Zhang, W.; van Gent, D.C.; Incrocci, L.; van Weerden, W.M.; Nonnekens, J. Role of the DNA Damage Response in Prostate Cancer Formation, Progression and Treatment. Prostate Cancer Prostatic Dis. 2020, 23, 24–37. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.-Y.; Gleave, M.E.; Beltran, H. Towards Precision Oncology in Advanced Prostate Cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.T.; Huitema, A.D.R.; Chau, C.H.; Figg, W.D. Resistance to Second-Generation Androgen Receptor Antagonists in Prostate Cancer. Nat. Rev. Urol. 2021, 18, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, R.; Xu, K.; Ding, S.; Li, J.; Baek, G.; Ramanand, S.G.; Ding, S.; Liu, Z.; Gao, Y.; et al. Androgen Receptor Variants Mediate DNA Repair after Prostate Cancer Irradiation. Cancer Res. 2017, 77, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Tolkach, Y.; Kremer, A.; Lotz, G.; Schmid, M.; Mayr, T.; Förster, S.; Garbe, S.; Hosni, S.; Cronauer, M.V.; Kocsmár, I.; et al. Androgen Receptor Splice Variants Contribute to the Upregulation of DNA Repair in Prostate Cancer. Cancers 2022, 14, 4441. [Google Scholar] [CrossRef]
- Basu, S.; Martínez-Cristóbal, P.; Frigolé-Vivas, M.; Pesarrodona, M.; Lewis, M.; Szulc, E.; Bañuelos, C.A.; Sánchez-Zarzalejo, C.; Bielskutė, S.; Zhu, J.; et al. Rational Optimization of a Transcription Factor Activation Domain Inhibitor. Nat. Struct. Mol. Biol. 2023, 30, 1958–1969. [Google Scholar] [CrossRef]
- Kounatidou, E.; Nakjang, S.; McCracken, S.R.C.; Dehm, S.M.; Robson, C.N.; Jones, D.; Gaughan, L. A Novel CRISPR-Engineered Prostate Cancer Cell Line Defines the AR-V Transcriptome and Identifies PARP Inhibitor Sensitivities. Nucleic Acids Res. 2019, 47, 5634–5647. [Google Scholar] [CrossRef]
- Gui, B.; Gui, F.; Takai, T.; Feng, C.; Bai, X.; Fazli, L.; Dong, X.; Liu, S.; Zhang, X.; Zhang, W.; et al. Selective Targeting of PARP-2 Inhibits Androgen Receptor Signaling and Prostate Cancer Growth through Disruption of FOXA1 Function. Proc. Natl. Acad. Sci. USA 2019, 116, 14573–14582. [Google Scholar] [CrossRef]
- Alghoul, E.; Basbous, J.; Constantinou, A. Compartmentalization of the DNA Damage Response: Mechanisms and Functions. DNA Repair 2023, 128, 103524. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers 2022, 14, 1420. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and Overcoming Resistance to PARP Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Ryan, C.J.; Lord, C.J. Exploiting Cancer Synthetic Lethality in Cancer—Lessons Learnt from PARP Inhibitors. In Targeting the DNA Damage Response for Cancer Therapy; Yap, T.A., Shapiro, G.I., Eds.; Cancer Treatment and Research; Springer International Publishing: Cham, Switzerland, 2023; pp. 13–23. ISBN 978-3-031-30065-3. [Google Scholar]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate Cancer and PARP Inhibitors: Progress and Challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Iannantuono, G.M.; Chandran, E.; Floudas, C.S.; Choo-Wosoba, H.; Butera, G.; Roselli, M.; Gulley, J.L.; Karzai, F. Efficacy and Safety of PARP Inhibitors in Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis of Clinical Trials. Cancer Treat. Rev. 2023, 120, 102623. [Google Scholar] [CrossRef]
- Beatson, E.L.; Chau, C.H.; Price, D.K.; Figg, W.D. PARP Inhibitors on the Move in Prostate Cancer: Spotlight on Niraparib & Update on PARP Inhibitor Combination Trials. Am. J. Clin. Exp. Urol. 2022, 10, 252–257. [Google Scholar]
- Mateo, J.; de Bono, J.S.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Agarwal, N.; Olmos, D.; Thiery-Vuillemin, A.; et al. Olaparib for the Treatment of Patients with Metastatic Castration-Resistant Prostate Cancer and Alterations in BRCA1 and/or BRCA2 in the PROfound Trial. JCO 2023. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Bryce, A.H.; Shapiro, J.; Bambury, R.M.; Zhang, J.; Burke, J.M.; Castellano, D.; Font, A.; et al. Rucaparib for the Treatment of Metastatic Castration-Resistant Prostate Cancer Associated with a DNA Damage Repair Gene Alteration: Final Results from the Phase 2 TRITON2 Study. Eur. Urol. 2023, 84, 321–330. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis from the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in Patients with Metastatic Castration-Resistant Prostate Cancer and DNA Repair Gene Defects (GALAHAD): A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib Monotherapy in Metastatic Castration-Resistant Prostate Cancer with DNA Repair Alterations (TALAPRO-1): An Open-Label, Phase 2 Trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Hwang, J.; Shi, X.; Elliott, A.; Arnoff, T.E.; McGrath, J.; Xiu, J.; Walker, P.; Bergom, H.E.; Day, A.; Ahmed, S.; et al. Metastatic Prostate Cancers with BRCA2 versus ATM Mutations Exhibit Divergent Molecular Features and Clinical Outcomes. Clin. Cancer Res. 2023, 29, 2702–2713. [Google Scholar] [CrossRef]
- Su, C.T.; Nizialek, E.; Berchuck, J.E.; Vlachostergios, P.J.; Ashkar, R.; Sokolova, A.; Barata, P.C.; Aggarwal, R.R.; McKay, R.R.; Agarwal, N.; et al. Differential Responses to Taxanes and PARP Inhibitors in ATM-versus BRCA2-Mutated Metastatic Castrate-Resistant Prostate Cancer. Prostate 2023, 83, 227–236. [Google Scholar] [CrossRef]
- Marshall, C.H.; Sokolova, A.O.; McNatty, A.L.; Cheng, H.H.; Eisenberger, M.A.; Bryce, A.H.; Schweizer, M.T.; Antonarakis, E.S. Differential Response to Olaparib Treatment among Men with Metastatic Castration-Resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur. Urol. 2019, 76, 452–458. [Google Scholar] [CrossRef]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM Orchestrates the DNA-Damage Response to Counter Toxic Non-Homologous End-Joining at Broken Replication Forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef]
- Williamson, C.T.; Muzik, H.; Turhan, A.G.; Zamò, A.; O’Connor, M.J.; Bebb, D.G.; Lees-Miller, S.P. ATM Deficiency Sensitizes Mantle Cell Lymphoma Cells to Poly(ADP-Ribose) Polymerase-1 Inhibitors. Mol. Cancer Ther. 2010, 9, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Prodosmo, A.; Stagni, V.; Merli, D.; Monteonofrio, L.; Gatti, V.; Gentileschi, M.P.; Barilà, D.; Soddu, S. ATM-Depletion in Breast Cancer Cells Confers Sensitivity to PARP Inhibition. J. Exp. Clin. Cancer Res. 2013, 32, 95. [Google Scholar] [CrossRef] [PubMed]
- Taza, F.; Holler, A.E.; Fu, W.; Wang, H.; Adra, N.; Albany, C.; Ashkar, R.; Cheng, H.H.; Sokolova, A.O.; Agarwal, N.; et al. Differential Activity of PARP Inhibitors in BRCA1- Versus BRCA2-Altered Metastatic Castration-Resistant Prostate Cancer. JCO Precis. Oncol. 2021, 5, 1200–1220. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Duro, E.; Lundin, C.; Ask, K.; Sanchez-Pulido, L.; MacArtney, T.J.; Toth, R.; Ponting, C.P.; Groth, A.; Helleday, T.; Rouse, J. Identification of the MMS22L-TONSL Complex That Promotes Homologous Recombination. Mol. Cell 2010, 40, 632–644. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.C.; Adamson, B.; Lydeard, J.R.; Sowa, M.E.; Ciccia, A.; Bredemeyer, A.L.; Schlabach, M.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. A Genome-Wide Camptothecin Sensitivity Screen Identifies a Mammalian MMS22L-NFKBIL2 Complex Required for Genomic Stability. Mol. Cell 2010, 40, 645–657. [Google Scholar] [CrossRef]
- O’Donnell, L.; Panier, S.; Wildenhain, J.; Tkach, J.M.; Al-Hakim, A.; Landry, M.-C.; Escribano-Diaz, C.; Szilard, R.K.; Young, J.T.F.; Munro, M.; et al. The MMS22L-TONSL Complex Mediates Recovery from Replication Stress and Homologous Recombination. Mol. Cell 2010, 40, 619–631. [Google Scholar] [CrossRef]
- Tsujino, T.; Takai, T.; Hinohara, K.; Gui, F.; Tsutsumi, T.; Bai, X.; Miao, C.; Feng, C.; Gui, B.; Sztupinszki, Z.; et al. CRISPR Screens Reveal Genetic Determinants of PARP Inhibitor Sensitivity and Resistance in Prostate Cancer. Nat. Commun. 2023, 14, 252. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A Compendium of Mutational Cancer Driver Genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef]
- Bennett, C.B.; Lewis, L.K.; Karthikeyan, G.; Lobachev, K.S.; Jin, Y.H.; Sterling, J.F.; Snipe, J.R.; Resnick, M.A. Genes Required for Ionizing Radiation Resistance in Yeast. Nat. Genet. 2001, 29, 426–434. [Google Scholar] [CrossRef]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef]
- Tsao, N.; Schärer, O.D.; Mosammaparast, N. The Complexity and Regulation of Repair of Alkylation Damage to Nucleic Acids. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 125–136. [Google Scholar] [CrossRef]
- Martin, S.K.; Wood, R.D. DNA Polymerase ζ in DNA Replication and Repair. Nucleic Acids Res. 2019, 47, 8348–8361. [Google Scholar] [CrossRef]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.-W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polζ Maintains the Viability of Homologous Recombination-Deficient Cancer Cells through Mutagenic Repair of PRIMPOL-Dependent ssDNA Gaps. Mol. Cell 2021, 81, 4008–4025.e7. [Google Scholar] [CrossRef]
- Park, J.; Long, D.T.; Lee, K.Y.; Abbas, T.; Shibata, E.; Negishi, M.; Luo, Y.; Schimenti, J.C.; Gambus, A.; Walter, J.C.; et al. The MCM8-MCM9 Complex Promotes RAD51 Recruitment at DNA Damage Sites to Facilitate Homologous Recombination. Mol. Cell Biol. 2013, 33, 1632–1644. [Google Scholar] [CrossRef]
- Lee, K.Y.; Im, J.-S.; Shibata, E.; Park, J.; Handa, N.; Kowalczykowski, S.C.; Dutta, A. MCM8-9 Complex Promotes Resection of Double-Strand Break Ends by MRE11-RAD50-NBS1 Complex. Nat. Commun. 2015, 6, 7744. [Google Scholar] [CrossRef]
- Hustedt, N.; Saito, Y.; Zimmermann, M.; Álvarez-Quilón, A.; Setiaputra, D.; Adam, S.; McEwan, A.; Yuan, J.Y.; Olivieri, M.; Zhao, Y.; et al. Control of Homologous Recombination by the HROB-MCM8-MCM9 Pathway. Genes Dev. 2019, 33, 1397–1415. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Z.; Su, D.; Tang, M.; Nie, L.; Zhang, H.; Feng, X.; Wang, R.; Shen, X.; Srivastava, M.; et al. C17orf53 Is Identified as a Novel Gene Involved in Inter-Strand Crosslink Repair. DNA Repair 2020, 95, 102946. [Google Scholar] [CrossRef]
- Huang, J.-W.; Acharya, A.; Taglialatela, A.; Nambiar, T.S.; Cuella-Martin, R.; Leuzzi, G.; Hayward, S.B.; Joseph, S.A.; Brunette, G.J.; Anand, R.; et al. MCM8IP Activates the MCM8-9 Helicase to Promote DNA Synthesis and Homologous Recombination upon DNA Damage. Nat. Commun. 2020, 11, 2948. [Google Scholar] [CrossRef]
- Lee, K.Y.; Im, J.-S.; Shibata, E.; Dutta, A. ASF1a Promotes Non-Homologous End Joining Repair by Facilitating Phosphorylation of MDC1 by ATM at Double-Strand Breaks. Mol. Cell 2017, 68, 61–75.e5. [Google Scholar] [CrossRef]
- Lee, K.Y.; Dutta, A. Chk1 Promotes Non-Homologous End Joining in G1 through Direct Phosphorylation of ASF1A. Cell Rep. 2021, 34, 108680. [Google Scholar] [CrossRef]
- Huang, T.-H.; Fowler, F.; Chen, C.-C.; Shen, Z.-J.; Sleckman, B.; Tyler, J.K. The Histone Chaperones ASF1 and CAF-1 Promote MMS22L-TONSL-Mediated Rad51 Loading onto ssDNA during Homologous Recombination in Human Cells. Mol. Cell 2018, 69, 879–892.e5. [Google Scholar] [CrossRef]
- Feng, S.; Ma, S.; Li, K.; Gao, S.; Ning, S.; Shang, J.; Guo, R.; Chen, Y.; Blumenfeld, B.; Simon, I.; et al. RIF1-ASF1-Mediated High-Order Chromatin Structure Safeguards Genome Integrity. Nat. Commun. 2022, 13, 957. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Z.; Wang, C.; Feng, X.; Lee, N.; Huang, M.; Zhang, H.; Li, S.; Xiong, Y.; Chen, J. Histone Chaperone ASF1 Acts with RIF1 to Promote DNA End-Joining in BRCA1-Deficient Cells. J. Biol. Chem. 2022, 298, 101979. [Google Scholar] [CrossRef]
- Washino, S.; Rider, L.C.; Romero, L.; Jillson, L.K.; Affandi, T.; Ohm, A.M.; Lam, E.T.; Reyland, M.E.; Costello, J.C.; Cramer, S.D. Loss of MAP3K7 Sensitizes Prostate Cancer Cells to CDK1/2 Inhibition and DNA Damage by Disrupting Homologous Recombination. Mol. Cancer Res. 2019, 17, 1985–1998. [Google Scholar] [CrossRef]
- Huang, Z.; Tang, B.; Yang, Y.; Yang, Z.; Shi, L.; Bai, Y.; Yan, B.; Karnes, R.J.; Zhang, J.; Jimenez, R.; et al. MAP3K7-IKK Inflammatory Signaling Modulates AR Protein Degradation and Prostate Cancer Progression. Cancer Res. 2021, 81, 4471–4484. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The Long Tail of Oncogenic Drivers in Prostate Cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Reijns, M.A.M.; Rabe, B.; Rigby, R.E.; Mill, P.; Astell, K.R.; Lettice, L.A.; Boyle, S.; Leitch, A.; Keighren, M.; Kilanowski, F.; et al. Enzymatic Removal of Ribonucleotides from DNA Is Essential for Mammalian Genome Integrity and Development. Cell 2012, 149, 1008–1022. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR Screens Identify Genomic Ribonucleotides as a Source of PARP-Trapping Lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Wang, C.; Wang, G.; Feng, X.; Shepherd, P.; Zhang, J.; Tang, M.; Chen, Z.; Srivastava, M.; McLaughlin, M.E.; Navone, N.M.; et al. Genome-Wide CRISPR Screens Reveal Synthetic Lethality of RNASEH2 Deficiency and ATR Inhibition. Oncogene 2019, 38, 2451–2463. [Google Scholar] [CrossRef]
- Miao, C.; Tsujino, T.; Takai, T.; Gui, F.; Tsutsumi, T.; Sztupinszki, Z.; Wang, Z.; Azuma, H.; Szallasi, Z.; Mouw, K.W.; et al. RB1 Loss Overrides PARP Inhibitor Sensitivity Driven by RNASEH2B Loss in Prostate Cancer. Sci. Adv. 2022, 8, eabl9794. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 175, 889. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of Lethal Prostate Cancer at Diagnosis and Castration Resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Måseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic Hypoxia Decreases Synthesis of Homologous Recombination Proteins to Offset Chemoresistance and Radioresistance. Cancer Res. 2008, 68, 605–614. [Google Scholar] [CrossRef]
- Chan, N.; Pires, I.M.; Bencokova, Z.; Coackley, C.; Luoto, K.R.; Bhogal, N.; Lakshman, M.; Gottipati, P.; Oliver, F.J.; Helleday, T.; et al. Contextual Synthetic Lethality of Cancer Cell Kill Based on the Tumor Microenvironment. Cancer Res. 2010, 70, 8045–8054. [Google Scholar] [CrossRef]
- Mehibel, M.; Xu, Y.; Li, C.G.; Moon, E.J.; Thakkar, K.N.; Diep, A.N.; Kim, R.K.; Bloomstein, J.D.; Xiao, Y.; Bacal, J.; et al. Eliminating Hypoxic Tumor Cells Improves Response to PARP Inhibitors in Homologous Recombination–Deficient Cancer Models. J. Clin. Investig. 2021, 131, e146256. [Google Scholar] [CrossRef]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef]
- Hustedt, N.; Álvarez-Quilón, A.; McEwan, A.; Yuan, J.Y.; Cho, T.; Koob, L.; Hart, T.; Durocher, D. A Consensus Set of Genetic Vulnerabilities to ATR Inhibition. Open Biol. 2019, 9, 190156. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A Cell-Based Screen Identifies ATR Inhibitors with Synthetic Lethal Properties for Cancer-Associated Mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef]
- Qiu, Z.; Fa, P.; Liu, T.; Prasad, C.B.; Ma, S.; Hong, Z.; Chan, E.R.; Wang, H.; Li, Z.; He, K.; et al. A Genome-Wide Pooled shRNA Screen Identifies PPP2R2A as a Predictive Biomarker for the Response to ATR and CHK1 Inhibitors. Cancer Res. 2020, 80, 3305–3318. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic Stress Sensitizes Murine Cancers to Hypomorphic Suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.D.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting Oncogene-Induced Replicative Stress for the Selective Killing of Myc-Driven Tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR Pathway Inhibition Is Synthetically Lethal in Cancer Cells with ERCC1 Deficiency. Cancer Res. 2014, 74, 2835–2845. [Google Scholar] [CrossRef]
- Mohni, K.N.; Thompson, P.S.; Luzwick, J.W.; Glick, G.G.; Pendleton, C.S.; Lehmann, B.D.; Pietenpol, J.A.; Cortez, D. A Synthetic Lethal Screen Identifies DNA Repair Pathways That Sensitize Cancer Cells to Combined ATR Inhibition and Cisplatin Treatments. PLoS ONE 2015, 10, e0125482. [Google Scholar] [CrossRef]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-Wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef]
- da Costa, A.A.B.A.; Chowdhury, D.; Shapiro, G.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting Replication Stress in Cancer Therapy. Nat. Rev. Drug Discov. 2023, 22, 38–58. [Google Scholar] [CrossRef]
- Roulston, A.; Zimmermann, M.; Papp, R.; Skeldon, A.; Pellerin, C.; Dumas-Bérube, É.; Dumais, V.; Dorich, S.; Fader, L.D.; Fournier, S.; et al. RP-3500: A Novel, Potent, and Selective ATR Inhibitor That Is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Mol. Cancer Ther. 2022, 21, 245–256. [Google Scholar] [CrossRef]
- Barnieh, F.M.; Loadman, P.M.; Falconer, R.A. Progress towards a Clinically-Successful ATR Inhibitor for Cancer Therapy. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100017. [Google Scholar] [CrossRef]
- Jossé, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR Inhibitors VE-821 and VX-970 Sensitize Cancer Cells to Topoisomerase i Inhibitors by Disabling DNA Replication Initiation and Fork Elongation Responses. Cancer Res. 2014, 74, 6968–6979. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR Inhibition Potentiates Genome Instability and Cell Death in ATM-Deficient Cancer Cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Jette, N.R.; Radhamani, S.; Arthur, G.; Ye, R.; Goutam, S.; Bolyos, A.; Petersen, L.F.; Bose, P.; Bebb, D.G.; Lees-Miller, S.P. Combined Poly-ADP Ribose Polymerase and Ataxia-Telangiectasia Mutated/Rad3-Related Inhibition Targets Ataxia-Telangiectasia Mutated-Deficient Lung Cancer Cells. Br. J. Cancer 2019, 121, 600–610. [Google Scholar] [CrossRef]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.-Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition than PARP Inhibition in Prostate Cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Abel, M.L.; Takahashi, N.; Peer, C.; Redon, C.E.; Nichols, S.; Vilimas, R.; Lee, M.-J.; Lee, S.; Shelat, M.; Kattappuram, R.; et al. Targeting Replication Stress and Chemotherapy Resistance with a Combination of Sacituzumab Govitecan and Berzosertib: A Phase I Clinical Trial. Clin. Cancer Res. 2023, 29, 3603–3611. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Bernier, C.; Kaiser, B.; Fournier, S.; Li, L.; Desjardins, J.; Skeldon, A.; Rimkunas, V.; Veloso, A.; Young, J.T.F.; et al. Guiding ATR and PARP Inhibitor Combinationswith Chemogenomic Screens. Cell Rep. 2022, 40, 111081. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Kothari, V.; Drake, J.M.; Zhao, S.; Dylgjeri, E.; Dean, J.L.; Schiewer, M.J.; McNair, C.; Jones, J.K.; Aytes, A.; et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell 2015, 28, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Livingstone, J.; Wrana, J.L.; Finelli, A.; He, H.H.; van der Kwast, T.; Zlotta, A.R.; Bristow, R.G.; Boutros, P.C. Somatic Driver Mutation Prevalence in 1844 Prostate Cancers Identifies ZNRF3 Loss as a Predictor of Metastatic Relapse. Nat. Commun. 2021, 12, 6248. [Google Scholar] [CrossRef]
- Adamson, B.; Brittain, N.; Walker, L.; Duncan, R.; Luzzi, S.; Rescigno, P.; Smith, G.R.; McGill, S.; Burchmore, R.J.; Willmore, E.; et al. The Catalytic Subunit of DNA-PK Regulates Transcription and Splicing of AR in Advanced Prostate Cancer. J. Clin. Investig. 2023, 133, e169200. [Google Scholar] [CrossRef]
- Puc, J.; Kozbial, P.; Li, W.; Tan, Y.; Liu, Z.; Suter, T.; Ohgi, K.A.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Ligand-Dependent Enhancer Activation Regulated by Topoisomerase-I Activity. Cell 2015, 160, 367–380. [Google Scholar] [CrossRef]
- Tan, Y.; Yao, L.; Gamliel, A.; Nair, S.J.; Taylor, H.; Ohgi, K.; Aggarwal, A.K.; Rosenfeld, M.G. Signal-Induced Enhancer Activation Requires Ku70 to Read Topoisomerase1-DNA Covalent Complexes. Nat. Struct. Mol. Biol. 2023, 30, 148–158. [Google Scholar] [CrossRef]
- Shao, Z.; Flynn, R.A.; Crowe, J.L.; Zhu, Y.; Liang, J.; Jiang, W.; Aryan, F.; Aoude, P.; Bertozzi, C.R.; Estes, V.M.; et al. DNA-PKcs Has KU-Dependent Function in rRNA Processing and Haematopoiesis. Nature 2020, 579, 291–296. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Kothari, V.; Shafi, A.A.; Semenova, G.; Gallagher, P.T.; Guan, Y.F.; Pang, A.; Goodwin, J.F.; Irani, S.; McCann, J.J.; et al. A Novel Role for DNA-PK in Metabolism by Regulating Glycolysis in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2022, 28, 1446–1459. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Zhao, J.L.; Antonarakis, E.S.; Cheng, H.H.; George, D.J.; Aggarwal, R.; Riedel, E.; Sumiyoshi, T.; Schonhoft, J.D.; Anderson, A.; Mao, N.; et al. Phase 1b Study of Enzalutamide plus CC-115, a Dual mTORC1/2 and DNA-PK Inhibitor, in Men with Metastatic Castration-Resistant Prostate Cancer (mCRPC). Br. J. Cancer 2023, 1–10. [Google Scholar] [CrossRef]
- Chao, O.S.; Goodman, O.B. DNA-PKc Inhibition Overcomes Taxane Resistance by Promoting Taxane-Induced DNA Damage in Prostate Cancer Cells. Prostate 2021, 81, 1032–1048. [Google Scholar] [CrossRef]
- Salerno, K.E.; Roy, S.; Ribaudo, C.; Fisher, T.; Patel, R.B.; Mena, E.; Escorcia, F.E. A Primer on Radiopharmaceutical Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2023, 115, 48–59. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- Miyahira, A.K.; Soule, H.R. The History of Prostate-Specific Membrane Antigen as a Theranostic Target in Prostate Cancer: The Cornerstone Role of the Prostate Cancer Foundation. J. Nucl. Med. 2022, 63, 331–338. [Google Scholar] [CrossRef]
- Feuerecker, B.; Tauber, R.; Knorr, K.; Heck, M.; Beheshti, A.; Seidl, C.; Bruchertseifer, F.; Pickhard, A.; Gafita, A.; Kratochwil, C.; et al. Activity and Adverse Events of Actinium-225-PSMA-617 in Advanced Metastatic Castration-Resistant Prostate Cancer After Failure of Lutetium-177-PSMA. Eur. Urol. 2021, 79, 343–350. [Google Scholar] [CrossRef]
- Jani, A.B.; Ravizzini, G.C.; Gartrell, B.A.; Siegel, B.A.; Twardowski, P.; Saltzstein, D.; Fleming, M.T.; Chau, A.; Davis, P.; Chapin, B.F.; et al. Diagnostic Performance and Safety of 18F-rhPSMA-7.3 Positron Emission Tomography in Men With Suspected Prostate Cancer Recurrence: Results From a Phase 3, Prospective, Multicenter Study (SPOTLIGHT). J. Urol. 2023, 210, 299–311. [Google Scholar] [CrossRef]
- Bundschuh, R.A.; Pfob, C.H.; Wienand, G.; Dierks, A.; Kircher, M.; Lapa, C. 177 Lu-rhPSMA-10.1 Induces Tumor Response in a Patient with mCRPC After PSMA-Directed Radioligand Therapy with 177 Lu-PSMA-I&T. Clin. Nucl. Med. 2023, 48, 337–338. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Graf, R.P.; Fisher, V.; Weberpals, J.; Gjoerup, O.; Tierno, M.B.; Huang, R.S.P.; Sayegh, N.; Lin, D.I.; Raskina, K.; Schrock, A.B.; et al. Comparative Effectiveness of Immune Checkpoint Inhibitors vs. Chemotherapy by Tumor Mutational Burden in Metastatic Castration-Resistant Prostate Cancer. JAMA Netw. Open 2022, 5, e225394. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The Impact of Nonsense-Mediated mRNA Decay on Genetic Disease, Gene Editing and Cancer Immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Lim, E.L.; Xu, H.; Liu, P.; Al-Bakir, M.; Wong, Y.N.S.; Rowan, A.; Funt, S.A.; Merghoub, T.; et al. Escape from Nonsense-Mediated Decay Associates with Anti-Tumor Immunogenicity. Nat. Commun. 2020, 11, 3800. [Google Scholar] [CrossRef]
- Reading, J.L.; Caswell, D.R.; Swanton, C. Tumor Heterogeneity Impairs Immunogenicity in Mismatch Repair Deficient Tumors. Nat. Genet. 2023, 55, 1610–1612. [Google Scholar] [CrossRef]
- Jaratlerdsiri, W.; Jiang, J.; Gong, T.; Patrick, S.M.; Willet, C.; Chew, T.; Lyons, R.J.; Haynes, A.-M.; Pasqualim, G.; Louw, M.; et al. African-Specific Molecular Taxonomy of Prostate Cancer. Nature 2022, 609, 552–559. [Google Scholar] [CrossRef]
- Serrano, J.C.; von Trentini, D.; Berríos, K.N.; Barka, A.; Dmochowski, I.J.; Kohli, R.M. Structure-Guided Design of a Potent and Specific Inhibitor against the Genomic Mutator APOBEC3A. ACS Chem. Biol. 2022, 17, 3379–3388. [Google Scholar] [CrossRef]
- Petljak, M.; Green, A.M.; Maciejowski, J.; Weitzman, M.D. Addressing the Benefits of Inhibiting APOBEC3-Dependent Mutagenesis in Cancer. Nat. Genet. 2022, 54, 1599–1608. [Google Scholar] [CrossRef]
- Kvach, M.V.; Barzak, F.M.; Harjes, S.; Schares, H.A.M.; Jameson, G.B.; Ayoub, A.M.; Moorthy, R.; Aihara, H.; Harris, R.S.; Filichev, V.V.; et al. Inhibiting APOBEC3 Activity with Single-Stranded DNA Containing 2′-Deoxyzebularine Analogues. Biochemistry 2019, 58, 391–400. [Google Scholar] [CrossRef]
- Barzak, F.M.; Harjes, S.; Kvach, M.V.; Kurup, H.M.; Jameson, G.B.; Filichev, V.V.; Harjes, E. Selective Inhibition of APOBEC3 Enzymes by Single-Stranded DNAs Containing 2′-Deoxyzebularine. Org. Biomol. Chem. 2019, 17, 9435–9441. [Google Scholar] [CrossRef]
- Buisson, R.; Lawrence, M.S.; Benes, C.H.; Zou, L. APOBEC3A and APOBEC3B Activities Render Cancer Cells Susceptible to ATR Inhibition. Cancer Res. 2017, 77, 4567–4578. [Google Scholar] [CrossRef]
- Isozaki, H.; Sakhtemani, R.; Abbasi, A.; Nikpour, N.; Stanzione, M.; Oh, S.; Langenbucher, A.; Monroe, S.; Su, W.; Cabanos, H.F.; et al. Therapy-Induced APOBEC3A Drives Evolution of Persistent Cancer Cells. Nature 2023, 620, 393–401. [Google Scholar] [CrossRef]
- Calagua, C.; Ficial, M.; Jansen, C.S.; Hirz, T.; Del Balzo, L.; Wilkinson, S.; Lake, R.; Ku, A.T.; Voznesensky, O.; Sykes, D.B.; et al. A Subset of Localized Prostate Cancer Displays an Immunogenic Phenotype Associated with Losses of Key Tumor Suppressor Genes. Clin. Cancer Res. 2021, 27, 4836–4847. [Google Scholar] [CrossRef]
- Plas, S.; Pircher, A.; Heidegger, I. Pembrolizumab in mCRPC-Combination Therapies as Breakthrough to Success? Curr. Opin. Urol. 2023, 33, 458–471. [Google Scholar] [CrossRef]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-Dose Targeted Radionuclide Therapy Renders Immunologically Cold Tumors Responsive to Immune Checkpoint Blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef]
- Tang, Z.; Pilié, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-Induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, M.; Bao, X.; Pan, D.; Jiao, M.; Liu, X.; Li, F.; Li, C.-Y. ATM Inhibition Enhances Cancer Immunotherapy by Promoting mtDNA Leakage and cGAS/STING Activation. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 Abrogation Triggers Innate Immune Responses Potentiated by Treatment with PARP Inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef]
- Wang, W.; McMillan, M.T.; Zhao, X.; Wang, Z.; Jiang, L.; Karnak, D.; Lima, F.; Parsels, J.D.; Parsels, L.A.; Lawrence, T.S.; et al. DNA-PK Inhibition and Radiation Promote Antitumoral Immunity through RNA Polymerase III in Pancreatic Cancer. Mol. Cancer Res. 2022, 20, 1137–1150. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib Activates Interferon Signaling and Potentiates Anti-PD-1 Antibody Efficacy in Tumor Models. Sci. Rep. 2019, 9, 1853. [Google Scholar] [CrossRef]
- Carr, M.I.; Chiu, L.-Y.; Guo, Y.; Xu, C.; Lazorchak, A.S.; Yu, H.; Qin, G.; Qi, J.; Marelli, B.; Lan, Y.; et al. DNA-PK Inhibitor Peposertib Amplifies Radiation-Induced Inflammatory Micronucleation and Enhances TGFβ/PD-L1 Targeted Cancer Immunotherapy. Mol. Cancer Res. 2022, 20, 568–582. [Google Scholar] [CrossRef]
- Luo, L.Y.; O’Hara, M.H.; Mitchell, T.C.; Vonderheide, R.H.; Wherry, E.J.; Minn, A.J.; Maity, A. Combining Radiation with Immunotherapy: The University of Pennsylvania Experience. Semin. Radiat. Oncol. 2020, 30, 173–180. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular Mechanisms and Cellular Functions of cGAS-STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Beyer, U.; Brand, F.; Martens, H.; Weder, J.; Christians, A.; Elyan, N.; Hentschel, B.; Westphal, M.; Schackert, G.; Pietsch, T.; et al. Rare ADAR and RNASEH2B Variants and a Type I Interferon Signature in Glioma and Prostate Carcinoma Risk and Tumorigenesis. Acta Neuropathol. 2017, 134, 905–922. [Google Scholar] [CrossRef]
- Liu, A.; Ying, S. Aicardi-Goutières Syndrome: A Monogenic Type I Interferonopathy. Scand. J. Immunol. 2023, 98, e13314. [Google Scholar] [CrossRef]
- Samson, N.; Ablasser, A. The cGAS-STING Pathway and Cancer. Nat. Cancer 2022, 3, 1452–1463. [Google Scholar] [CrossRef]
- Curran, E.; Chen, X.; Corrales, L.; Kline, D.E.; Dubensky, T.W.; Duttagupta, P.; Kortylewski, M.; Kline, J. STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia. Cell Rep. 2016, 15, 2357–2366. [Google Scholar] [CrossRef]
- Le Naour, J.; Zitvogel, L.; Galluzzi, L.; Vacchelli, E.; Kroemer, G. Trial Watch: STING Agonists in Cancer Therapy. Oncoimmunology 2020, 9, 1777624. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Lamb, T.J.; Pawelec, G. Here, There, and Everywhere: Myeloid-Derived Suppressor Cells in Immunology. J. Immunol. 2023, 210, 1183–1197. [Google Scholar] [CrossRef]
- Siemińska, I.; Baran, J. Myeloid-Derived Suppressor Cells as Key Players and Promising Therapy Targets in Prostate Cancer. Front. Oncol. 2022, 12, 862416. [Google Scholar] [CrossRef]
- Roth, T.J.; Sheinin, Y.; Lohse, C.M.; Kuntz, S.M.; Frigola, X.; Inman, B.A.; Krambeck, A.E.; McKenney, M.E.; Karnes, R.J.; Blute, M.L.; et al. B7-H3 Ligand Expression by Prostate Cancer: A Novel Marker of Prognosis and Potential Target for Therapy. Cancer Res. 2007, 67, 7893–7900. [Google Scholar] [CrossRef]
- Guo, C.; Figueiredo, I.; Gurel, B.; Neeb, A.; Seed, G.; Crespo, M.; Carreira, S.; Rekowski, J.; Buroni, L.; Welti, J.; et al. B7-H3 as a Therapeutic Target in Advanced Prostate Cancer. Eur. Urol. 2023, 83, 224–238. [Google Scholar] [CrossRef]
- Shi, W.; Wang, Y.; Zhao, Y.; Kim, J.J.; Li, H.; Meng, C.; Chen, F.; Zhang, J.; Mak, D.H.; Van, V.; et al. Immune Checkpoint B7-H3 Is a Therapeutic Vulnerability in Prostate Cancer Harboring PTEN and TP53 Deficiencies. Sci. Transl. Med. 2023, 15, eadf6724. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective Combinatorial Immunotherapy for Castration-Resistant Prostate Cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef]
- Guo, C.; Sharp, A.; Gurel, B.; Crespo, M.; Figueiredo, I.; Jain, S.; Vogl, U.; Rekowski, J.; Rouhifard, M.; Gallagher, L.; et al. Targeting Myeloid Chemotaxis to Reverse Prostate Cancer Therapy Resistance. Nature 2023, 623, 1053–1061. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular Landmarks of Tumor Hypoxia across Cancer Types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Duan, J.-X.; Jiao, H.; Kaizerman, J.; Stanton, T.; Evans, J.W.; Lan, L.; Lorente, G.; Banica, M.; Jung, D.; Wang, J.; et al. Potent and Highly Selective Hypoxia-Activated Achiral Phosphoramidate Mustards as Anticancer Drugs. J. Med. Chem. 2008, 51, 2412–2420. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, Q.; Zhang, Y.; Liu, Z.; Zheng, Z.; Liu, S.; Meng, L.; Xin, Y.; Jiang, X. Targeting Hypoxia in the Tumor Microenvironment: A Potential Strategy to Improve Cancer Immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 24. [Google Scholar] [CrossRef]
- van der Wiel, A.M.A.; Jackson-Patel, V.; Niemans, R.; Yaromina, A.; Liu, E.; Marcus, D.; Mowday, A.M.; Lieuwes, N.G.; Biemans, R.; Lin, X.; et al. Selectively Targeting Tumor Hypoxia with the Hypoxia-Activated Prodrug CP-506. Mol. Cancer Ther. 2021, 20, 2372–2383. [Google Scholar] [CrossRef]
- Yaromina, A.; Koi, L.; Schuitmaker, L.; van der Wiel, A.M.-M.A.; Dubois, L.J.; Krause, M.; Lambin, P. Overcoming Radioresistance with the Hypoxia-Activated Prodrug CP-506: A Pre-Clinical Study of Local Tumour Control Probability. Radiother. Oncol. 2023, 186, 109738. [Google Scholar] [CrossRef]
- Puca, L.; Vlachostergios, P.J.; Beltran, H. Neuroendocrine Differentiation in Prostate Cancer: Emerging Biology, Models, and Therapies. Cold Spring Harb. Perspect. Med. 2019, 9, a030593. [Google Scholar] [CrossRef]
- Guo, H.; Ci, X.; Ahmed, M.; Hua, J.T.; Soares, F.; Lin, D.; Puca, L.; Vosoughi, A.; Xue, H.; Li, E.; et al. ONECUT2 Is a Driver of Neuroendocrine Prostate Cancer. Nat. Commun. 2019, 10, 278. [Google Scholar] [CrossRef]
- Qian, C.; Yang, Q.; Freedland, S.J.; Di Vizio, D.; Ellis, L.; You, S.; Freeman, M.R. Activation of ONECUT2 by RB1 Loss in Castration-Resistant Prostate Cancer. Am. J. Clin. Exp. Urol. 2022, 10, 397–407. [Google Scholar]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Ribeiro-Silva, C.; Vermeulen, W.; Lans, H. SWI/SNF: Complex Complexes in Genome Stability and Cancer. DNA Repair 2019, 77, 87–95. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A Deficiency Promotes Mutability and Potentiates Therapeutic Antitumor Immunity Unleashed by Immune Checkpoint Blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef]
- Bayona-Feliu, A.; Barroso, S.; Muñoz, S.; Aguilera, A. The SWI/SNF Chromatin Remodeling Complex Helps Resolve R-Loop-Mediated Transcription-Replication Conflicts. Nat. Genet. 2021, 53, 1050–1063. [Google Scholar] [CrossRef]
- Iurlaro, M.; Stadler, M.B.; Masoni, F.; Jagani, Z.; Galli, G.G.; Schübeler, D. Mammalian SWI/SNF Continuously Restores Local Accessibility to Chromatin. Nat. Genet. 2021, 53, 279–287. [Google Scholar] [CrossRef]
- Xiao, L.; Parolia, A.; Qiao, Y.; Bawa, P.; Eyunni, S.; Mannan, R.; Carson, S.E.; Chang, Y.; Wang, X.; Zhang, Y.; et al. Targeting SWI/SNF ATPases in Enhancer-Addicted Prostate Cancer. Nature 2022, 601, 434–439. [Google Scholar] [CrossRef]
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin Remodeling ATPase BRG1 and PTEN Are Synthetic Lethal in Prostate Cancer. J. Clin. Investig. 2019, 129, 759–773. [Google Scholar] [CrossRef]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ Inhibitors Elicit BRCA-Gene Synthetic Lethality and Target PARP Inhibitor Resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Bubenik, M.; Mader, P.; Mochirian, P.; Vallée, F.; Clark, J.; Truchon, J.-F.; Perryman, A.L.; Pau, V.; Kurinov, I.; Zahn, K.E.; et al. Identification of RP-6685, an Orally Bioavailable Compound That Inhibits the DNA Polymerase Activity of Polθ. J. Med. Chem. 2022, 65, 13198–13215. [Google Scholar] [CrossRef]
- Llorens-Agost, M.; Ensminger, M.; Le, H.P.; Gawai, A.; Liu, J.; Cruz-García, A.; Bhetawal, S.; Wood, R.D.; Heyer, W.-D.; Löbrich, M. POLθ-Mediated End Joining Is Restricted by RAD52 and BRCA2 until the Onset of Mitosis. Nat. Cell Biol. 2021, 23, 1095–1104. [Google Scholar] [CrossRef]
- Oh, G.; Wang, A.; Wang, L.; Li, J.; Werba, G.; Weissinger, D.; Zhao, E.; Dhara, S.; Hernandez, R.E.; Ackermann, A.; et al. POLQ Inhibition Elicits an Immune Response in Homologous Recombination–Deficient Pancreatic Adenocarcinoma via cGAS/STING Signaling. J. Clin. Investig. 2023, 133, e165934. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian Polymerase θ Promotes Alternative NHEJ and Suppresses Recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef]
- Wang, Z.; Song, Y.; Li, S.; Kurian, S.; Xiang, R.; Chiba, T.; Wu, X. DNA Polymerase θ (POLQ) Is Important for Repair of DNA Double-Strand Breaks Caused by Fork Collapse. J. Biol. Chem. 2019, 294, 3909–3919. [Google Scholar] [CrossRef]
- Patterson-Fortin, J.; Bose, A.; Tsai, W.-C.; Grochala, C.; Nguyen, H.; Zhou, J.; Parmar, K.; Lazaro, J.-B.; Liu, J.; McQueen, K.; et al. Targeting DNA Repair with Combined Inhibition of NHEJ and MMEJ Induces Synthetic Lethality in TP53-Mutant Cancers. Cancer Res. 2022, 82, 3815–3829. [Google Scholar] [CrossRef]
- Kumar, R.J.; Chao, H.X.; Simpson, D.A.; Feng, W.; Cho, M.-G.; Roberts, V.R.; Sullivan, A.R.; Shah, S.J.; Wozny, A.-S.; Fagan-Solis, K.; et al. Dual Inhibition of DNA-PK and DNA Polymerase Theta Overcomes Radiation Resistance Induced by P53 Deficiency. NAR Cancer 2020, 2, zcaa038. [Google Scholar] [CrossRef]
- Hwang, J.W.; Cho, Y.; Bae, G.-U.; Kim, S.-N.; Kim, Y.K. Protein Arginine Methyltransferases: Promising Targets for Cancer Therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein Arginine Methylation: From Enigmatic Functions to Therapeutic Targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Liu, J.; Bu, X.; Chu, C.; Dai, X.; Asara, J.M.; Sicinski, P.; Freeman, G.J.; Wei, W. PRMT1 Mediated Methylation of cGAS Suppresses Anti-Tumor Immunity. Nat. Commun. 2023, 14, 2806. [Google Scholar] [CrossRef]
- Boisvert, F.-M.; Rhie, A.; Richard, S.; Doherty, A.J. The GAR Motif of 53BP1 Is Arginine Methylated by PRMT1 and Is Necessary for 53BP1 DNA Binding Activity. Cell Cycle 2005, 4, 1834–1841. [Google Scholar] [CrossRef]
- He, L.; Hu, Z.; Sun, Y.; Zhang, M.; Zhu, H.; Jiang, L.; Zhang, Q.; Mu, D.; Zhang, J.; Gu, L.; et al. PRMT1 Is Critical to FEN1 Expression and Drug Resistance in Lung Cancer Cells. DNA Repair 2020, 95, 102953. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, M.F.; González-Guerrero, R.; Sánchez-Del-Campo, L.; Piñero-Madrona, A.; Cabezas-Herrera, J.; Rodríguez-López, J.N. PRMT1-Dependent Methylation of BRCA1 Contributes to the Epigenetic Defense of Breast Cancer Cells against Ionizing Radiation. Sci. Rep. 2020, 10, 13275. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Bailon, M.P.; Choi, S.-Y.; Dufficy, E.R.; Sharma, K.; McNee, G.S.; Gunnell, E.; Chiang, K.; Sahay, D.; Maslen, S.; Stewart, G.S.; et al. Arginine Methylation and Ubiquitylation Crosstalk Controls DNA End-Resection and Homologous Recombination Repair. Nat. Commun. 2021, 12, 6313. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Vogel, G.; Coulombe, Y.; Dubeau, D.; Spehalski, E.; Hébert, J.; Ferguson, D.O.; Masson, J.Y.; Richard, S. The MRE11 GAR Motif Regulates DNA Double-Strand Break Processing and ATR Activation. Cell Res. 2012, 22, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Vadnais, C.; Chen, R.; Fraszczak, J.; Yu, Z.; Boulais, J.; Pinder, J.; Frank, D.; Khandanpour, C.; Hébert, J.; Dellaire, G.; et al. GFI1 Facilitates Efficient DNA Repair by Regulating PRMT1 Dependent Methylation of MRE11 and 53BP1. Nat. Commun. 2018, 9, 1418. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, T.; Hébert, J.; Li, E.; Richard, S. A Mouse PRMT1 Null Allele Defines an Essential Role for Arginine Methylation in Genome Maintenance and Cell Proliferation. Mol. Cell Biol. 2009, 29, 2982–2996. [Google Scholar] [CrossRef] [PubMed]
- Grypari, I.M.; Logotheti, S.; Zolota, V.; Troncoso, P.; Efstathiou, E.; Bravou, V.; Melachrinou, M.; Logothetis, C.; Tzelepi, V. The Protein Arginine Methyltransferases (PRMTs) PRMT1 and CARM1 as Candidate Epigenetic Drivers in Prostate Cancer Progression. Medicine 2021, 100, e27094. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Sethunath, V.; Metaferia, N.Y.; Nogueira, M.F.; Gallant, D.S.; Garner, E.R.; Lairson, L.A.; Penney, C.M.; Li, J.; Gelbard, M.K.; et al. A Genome-Scale CRISPR Screen Reveals PRMT1 as a Critical Regulator of Androgen Receptor Signaling in Prostate Cancer. Cell Rep. 2022, 38, 110417. [Google Scholar] [CrossRef]
- Li, X.; Li, F.; Ye, F.; Guo, H.; Chen, W.; Jin, J.; Wang, Y.; Dai, P.; Shi, H.; Tao, H.; et al. Spermine Is a Natural Suppressor of AR Signaling in Castration-Resistant Prostate Cancer. Cell Rep. 2023, 42, 112798. [Google Scholar] [CrossRef]
- Beketova, E.; Owens, J.L.; Asberry, A.M.; Hu, C.-D. PRMT5: A Putative Oncogene and Therapeutic Target in Prostate Cancer. Cancer Gene Ther. 2021, 29, 264–276. [Google Scholar] [CrossRef]
- Kim, H.; Ronai, Z.A. PRMT5 Function and Targeting in Cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916.e7. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP Deletion Confers Enhanced Dependency on the PRMT5 Arginine Methyltransferase in Cancer Cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Huang, M.; Yao, F.; Nie, L.; Wang, C.; Su, D.; Zhang, H.; Li, S.; Tang, M.; Feng, X.; Yu, B.; et al. FACS-Based Genome-Wide CRISPR Screens Define Key Regulators of DNA Damage Signaling Pathways. Mol. Cell 2023, 83, 2810–2828.e6. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 Control of cGAS/STING and NLRC5 Pathways Defines Melanoma Response to Antitumor Immunity. Sci. Transl. Med. 2020, 12, eaaz5683. [Google Scholar] [CrossRef]
- Hu, R.; Zhou, B.; Chen, Z.; Chen, S.; Chen, N.; Shen, L.; Xiao, H.; Zheng, Y. PRMT5 Inhibition Promotes PD-L1 Expression and Immuno-Resistance in Lung Cancer. Front. Immunol. 2021, 12, 722188. [Google Scholar] [CrossRef]
- Owens, J.L.; Beketova, E.; Liu, S.; Shen, Q.; Pawar, J.S.; Asberry, A.M.; Yang, J.; Deng, X.; Elzey, B.D.; Ratliff, T.L.; et al. Targeting Protein Arginine Methyltransferase 5 Suppresses Radiation-Induced Neuroendocrine Differentiation and Sensitizes Prostate Cancer Cells to Radiation. Mol. Cancer Ther. 2022, 21, 448–459. [Google Scholar] [CrossRef]
- Pawar, J.S.; Al-Amin, M.Y.; Hu, C.-D. JNJ-64619178 Radiosensitizes and Suppresses Fractionated Ionizing Radiation-Induced Neuroendocrine Differentiation (NED) in Prostate Cancer. Front. Oncol. 2023, 13, 1126482. [Google Scholar] [CrossRef]
- Wei, X.; Yang, J.; Adair, S.J.; Ozturk, H.; Kuscu, C.; Lee, K.Y.; Kane, W.J.; O’Hara, P.E.; Liu, D.; Demirlenk, Y.M.; et al. Targeted CRISPR Screening Identifies PRMT5 as Synthetic Lethality Combinatorial Target with Gemcitabine in Pancreatic Cancer Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28068–28079. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Ray, S. The FAcilitates Chromatin Transcription (FACT) Complex: Its Roles in DNA Repair and Implications for Cancer Therapy. DNA Repair 2022, 109, 103246. [Google Scholar] [CrossRef]
- Gurova, K.V.; Garcia, H.; Miecznikowski, J.; Omilian, A.R.; Morrison, C. Level of SSRP1 in Cancer as a Prognostic Marker of Aggressive Disease. Am. J. Clin. Pathol. 2013, 140, A152. [Google Scholar] [CrossRef]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer Compounds That Simultaneously Suppress NF-κB and Activate P53 by Targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef]
- De, S.; Lindner, D.J.; Coleman, C.J.; Wildey, G.; Dowlati, A.; Stark, G.R. The FACT Inhibitor CBL0137 Synergizes with Cisplatin in Small-Cell Lung Cancer by Increasing NOTCH1 Expression and Targeting Tumor-Initiating Cells. Cancer Res. 2018, 78, 2396–2406. [Google Scholar] [CrossRef]
- Song, H.; Xi, S.; Chen, Y.; Pramanik, S.; Zeng, J.; Roychoudhury, S.; Harris, H.; Akbar, A.; Elhag, S.S.; Coulter, D.W.; et al. Histone Chaperone FACT Complex Inhibitor CBL0137 Interferes with DNA Damage Repair and Enhances Sensitivity of Medulloblastoma to Chemotherapy and Radiation. Cancer Lett. 2021, 520, 201–212. [Google Scholar] [CrossRef]
- Tallman, M.M.; Zalenski, A.A.; Deighen, A.M.; Schrock, M.S.; Mortach, S.; Grubb, T.M.; Kastury, P.S.; Huntoon, K.; Summers, M.K.; Venere, M. The Small Molecule Drug CBL0137 Increases the Level of DNA Damage and the Efficacy of Radiotherapy for Glioblastoma. Cancer Lett. 2021, 499, 232–242. [Google Scholar] [CrossRef]
- Somers, K.; Kosciolek, A.; Bongers, A.; El-Ayoubi, A.; Karsa, M.; Mayoh, C.; Wadham, C.; Middlemiss, S.; Neznanov, N.; Kees, U.R.; et al. Potent Antileukemic Activity of Curaxin CBL0137 against MLL-Rearranged Leukemia. Int. J. Cancer 2020, 146, 1902–1916. [Google Scholar] [CrossRef]
- Xiao, L.; Somers, K.; Murray, J.; Pandher, R.; Karsa, M.; Ronca, E.; Bongers, A.; Terry, R.; Ehteda, A.; Gamble, L.D.; et al. Dual Targeting of Chromatin Stability by the Curaxin CBL0137 and Histone Deacetylase Inhibitor Panobinostat Shows Significant Preclinical Efficacy in Neuroblastoma. Clin. Cancer Res. 2021, 27, 4338–4352. [Google Scholar] [CrossRef]
- Ehteda, A.; Simon, S.; Franshaw, L.; Giorgi, F.M.; Liu, J.; Joshi, S.; Rouaen, J.R.C.; Pang, C.N.I.; Pandher, R.; Mayoh, C.; et al. Dual Targeting of the Epigenome via FACT Complex and Histone Deacetylase Is a Potent Treatment Strategy for DIPG. Cell Rep. 2021, 35, 108994. [Google Scholar] [CrossRef]
- Forgione, M.O.; McClure, B.J.; Page, E.C.; Yeung, D.T.; Eadie, L.N.; White, D.L. TP53 Loss-of-function Mutations Reduce Sensitivity of Acute Leukaemia to the Curaxin CBL0137. Oncol. Rep. 2022, 47, 99. [Google Scholar] [CrossRef]
- Lu, X.; He, Y.; Johnston, R.L.; Nanayakarra, D.; Sankarasubramanian, S.; Lopez, J.A.; Friedlander, M.; Kalimutho, M.; Hooper, J.D.; Raninga, P.V.; et al. CBL0137 Impairs Homologous Recombination Repair and Sensitizes High-Grade Serous Ovarian Carcinoma to PARP Inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 355. [Google Scholar] [CrossRef]
- Zhou, D.; Wu, Z.; Park, J.-G.; Fiches, G.N.; Li, T.-W.; Ma, Q.; Huang, H.; Biswas, A.; Martinez-Sobrido, L.; Santoso, N.G.; et al. FACT Subunit SUPT16H Associates with BRD4 and Contributes to Silencing of Interferon Signaling. Nucleic Acids Res. 2022, 50, 8700–8718. [Google Scholar] [CrossRef]
- Huang, H.; Santoso, N.; Power, D.; Simpson, S.; Dieringer, M.; Miao, H.; Gurova, K.; Giam, C.-Z.; Elledge, S.J.; Zhu, J. FACT Proteins, SUPT16H and SSRP1, Are Transcriptional Suppressors of HIV-1 and HTLV-1 That Facilitate Viral Latency. J. Biol. Chem. 2015, 290, 27297–27310. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, W.; Xie, D.; Gao, T.; Dong, Z.; Lu, X. Histone Chaperone FACT Represses Retrotransposon MERVL and MERVL-Derived Cryptic Promoters. Nucleic Acids Res. 2020, 48, 10211–10225. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Fedorov, A.; Qiao, L.; Bao, H.; Beknazarov, N.; Wang, S.; Gautam, A.; Williams, R.M.; Crawford, J.C.; et al. ADAR1 Masks the Cancer Immunotherapeutic Promise of ZBP1-Driven Necroptosis. Nature 2022, 606, 594–602. [Google Scholar] [CrossRef]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or Opponent in the Tumour Microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef]
- Shen, J.; Yang, C.; Zhang, M.S.; Chin, D.W.-C.; Chan, F.-F.; Law, C.-T.; Wang, G.; Cheng, C.L.-H.; Chen, M.; Wan, R.T.-C.; et al. Histone Chaperone FACT Complex Coordinates with HIF to Mediate an Expeditious Transcription Program to Adapt to Poorly Oxygenated Cancers. Cell Rep. 2022, 38, 110304. [Google Scholar] [CrossRef]
- Krause, D.R.; Jonnalagadda, J.C.; Gatei, M.H.; Sillje, H.H.W.; Zhou, B.-B.; Nigg, E.A.; Khanna, K. Suppression of Tousled-like Kinase Activity after DNA Damage or Replication Block Requires ATM, NBS1 and Chk1. Oncogene 2003, 22, 5927–5937. [Google Scholar] [CrossRef]
- Groth, A.; Lukas, J.; Nigg, E.A.; Silljé, H.H.W.; Wernstedt, C.; Bartek, J.; Hansen, K. Human Tousled like Kinases Are Targeted by an ATM- and Chk1-Dependent DNA Damage Checkpoint. EMBO J. 2003, 22, 1676–1687. [Google Scholar] [CrossRef]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A Phospho-Proteomic Screen Identifies Substrates of the Checkpoint Kinase Chk1. Genome Biol. 2011, 12, R78. [Google Scholar] [CrossRef]
- Segura-Bayona, S.; Stracker, T.H. The Tousled-like Kinases Regulate Genome and Epigenome Stability: Implications in Development and Disease. Cell. Mol. Life Sci. 2019, 76, 3827–3841. [Google Scholar] [CrossRef]
- Ghosh, I.; De Benedetti, A. Untousling the Role of Tousled-like Kinase 1 in DNA Damage Repair. Int. J. Mol. Sci. 2023, 24, 13369. [Google Scholar] [CrossRef] [PubMed]
- Canfield, C.; Rains, J.; De Benedetti, A. TLK1B Promotes Repair of DSBs via Its Interaction with Rad9 and Asf1. BMC Mol. Biol. 2009, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Sunavala-Dossabhoy, G.; De Benedetti, A. Tousled Homolog, TLK1, Binds and Phosphorylates Rad9; TLK1 Acts as a Molecular Chaperone in DNA Repair. DNA Repair 2009, 8, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.; Davey, S.K. Tousled-like Kinase-Dependent Phosphorylation of Rad9 Plays a Role in Cell Cycle Progression and G2/M Checkpoint Exit. PLoS ONE 2013, 8, e85859. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Kwon, Y.; Shabestari, A.B.; Chikhale, R.; Chen, J.; Wiese, C.; Sung, P.; De Benedetti, A. TLK1-Mediated RAD54 Phosphorylation Spatio-Temporally Regulates Homologous Recombination Repair. Nucleic Acids Res. 2023, 51, 8643–8662. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-B.; Segura-Bayona, S.; Villamor-Payà, M.; Saredi, G.; Todd, M.A.M.; Attolini, C.S.-O.; Chang, T.-Y.; Stracker, T.H.; Groth, A. Tousled-like Kinases Stabilize Replication Forks and Show Synthetic Lethality with Checkpoint and PARP Inhibitors. Sci. Adv. 2018, 4, eaat4985. [Google Scholar] [CrossRef]
- Segura-Bayona, S.; Villamor-Payà, M.; Attolini, C.S.-O.; Koenig, L.M.; Sanchiz-Calvo, M.; Boulton, S.J.; Stracker, T.H. Tousled-Like Kinases Suppress Innate Immune Signaling Triggered by Alternative Lengthening of Telomeres. Cell Rep. 2020, 32, 107983. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Luster, T.A.; Hu, H.; Zhang, H.; Ng, W.-L.; Khodadadi-Jamayran, A.; Wang, W.; Chen, T.; Deng, J.; et al. In Vivo Epigenetic CRISPR Screen Identifies Asf1a as an Immunotherapeutic Target in Kras-Mutant Lung Adenocarcinoma. Cancer Discov. 2020, 10, 270–287. [Google Scholar] [CrossRef]
- Kim, J.-A.; Tan, Y.; Wang, X.; Cao, X.; Veeraraghavan, J.; Liang, Y.; Edwards, D.P.; Huang, S.; Pan, X.; Li, K.; et al. Comprehensive Functional Analysis of the Tousled-like Kinase 2 Frequently Amplified in Aggressive Luminal Breast Cancers. Nat. Commun. 2016, 7, 12991. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR Axis with Thioridazine Suppresses Outgrowth of Androgen Independent Prostate Tumors. Int. J. Cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef]
- Singh, V.; Bhoir, S.; Chikhale, R.V.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of Phenothiazine with Potent Anti-TLK1 Activity for Prostate Cancer Therapy. iScience 2020, 23, 101474. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-B.; Chang, T.-Y.; Lee, N.-Z.; Yu, Z.-Y.; Liu, C.-Y.; Lee, H.-Y. Design, Synthesis and Biological Evaluation of Bisindole Derivatives as Anticancer Agents against Tousled-like Kinases. Eur. J. Med. Chem. 2022, 227, 113904. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
(A) Schematic of the DNA damage response. DNA lesions are sensed, activating signal transduction networks that trigger cell cycle checkpoints and DNA repair, as well as cell fate decisions like apoptosis and senescence. (B) Example of synthetic lethality. Targeting GENE2 is tolerated by normal cells but is toxic to cancer cells due to a dependency on the function of GENE1 that is mutated (indicated by star) in the cancer cell. (C) Schematic of the major sources of DNA damage (indicated by star) in PCa. Endogenous DSBs arise due to topoisomerase activity during transcription and through replication stress: the slowing or stalling of replication forks and accumulation of ssDNA. Genotoxic treatments used in therapy, including RT, topoisomerase 2 inhibitors (etoposide, mitoxantrone) and platinum agents, such as cisplatin and carboplatin, cause base lesions, DNA interstrand strand crosslinks (ICL), SSBs, and DSBs through distinct mechanisms. (D) Summaries of selected DNA lesions (orange) and repair pathways (blue). Major factors (black) and steps (red) of double-strand break repair (DSBR), single-strand break repair (SSBR), base excision repair (BER) and mismatch repair (MMR) are depicted. Connections between DSBR pathways, classical non-homologous end joining (cNHEJ), homologous recombination (HR), and polymerase theta mediated end joining (TMEJ) are shown. Nucleotide excision repair, ICL repair, and DNA–protein crosslink repair details are omitted for brevity. See text and references for additional information and perspectives [41,42,43,44,45,46,47,48,49,50,51].
Figure 2.
(A) Schematic of the DNA damage response. DNA lesions are sensed, activating signal transduction networks that trigger cell cycle checkpoints and DNA repair, as well as cell fate decisions like apoptosis and senescence. (B) Example of synthetic lethality. Targeting GENE2 is tolerated by normal cells but is toxic to cancer cells due to a dependency on the function of GENE1 that is mutated (indicated by star) in the cancer cell. (C) Schematic of the major sources of DNA damage (indicated by star) in PCa. Endogenous DSBs arise due to topoisomerase activity during transcription and through replication stress: the slowing or stalling of replication forks and accumulation of ssDNA. Genotoxic treatments used in therapy, including RT, topoisomerase 2 inhibitors (etoposide, mitoxantrone) and platinum agents, such as cisplatin and carboplatin, cause base lesions, DNA interstrand strand crosslinks (ICL), SSBs, and DSBs through distinct mechanisms. (D) Summaries of selected DNA lesions (orange) and repair pathways (blue). Major factors (black) and steps (red) of double-strand break repair (DSBR), single-strand break repair (SSBR), base excision repair (BER) and mismatch repair (MMR) are depicted. Connections between DSBR pathways, classical non-homologous end joining (cNHEJ), homologous recombination (HR), and polymerase theta mediated end joining (TMEJ) are shown. Nucleotide excision repair, ICL repair, and DNA–protein crosslink repair details are omitted for brevity. See text and references for additional information and perspectives [41,42,43,44,45,46,47,48,49,50,51].

Figure 3.
(A) Oncoprint of selected DDR genes in the recurrent chromosome 6q deletion in samples from two PCa cohorts; SU2C/PCF Dream Team (mCRPC) and MSK/DFCI (Prostate Adenocarcinoma) [83,123,124,125,143]. Selected DDR genes that are typically lost in the 6q deletion and their relative locations are highlighted (see text for details). PARP sensitizers BRCA2 and RNASEH2B are shown for comparison, along with common driver gene TP53. Note that the 6q locus deletions show mutual exclusivity with TP53 (cbioportal.org). Patient samples without alterations in the selected genes are omitted for brevity. (B) Sensitivity (NormZ-score) to selected agents in RPE1-hTERT-Cas9-TP53-/- cells in CRISPR screens [127]. PARPi = Olaparib and ATRi = AZD6738.
Figure 3.
(A) Oncoprint of selected DDR genes in the recurrent chromosome 6q deletion in samples from two PCa cohorts; SU2C/PCF Dream Team (mCRPC) and MSK/DFCI (Prostate Adenocarcinoma) [83,123,124,125,143]. Selected DDR genes that are typically lost in the 6q deletion and their relative locations are highlighted (see text for details). PARP sensitizers BRCA2 and RNASEH2B are shown for comparison, along with common driver gene TP53. Note that the 6q locus deletions show mutual exclusivity with TP53 (cbioportal.org). Patient samples without alterations in the selected genes are omitted for brevity. (B) Sensitivity (NormZ-score) to selected agents in RPE1-hTERT-Cas9-TP53-/- cells in CRISPR screens [127]. PARPi = Olaparib and ATRi = AZD6738.

Figure 4.
Summary of current and emerging therapies to treat CRPC. Selected genetic alterations (green italics) that sensitize to particular therapies (black bold) are shown. See text for full details.
Figure 4.
Summary of current and emerging therapies to treat CRPC. Selected genetic alterations (green italics) that sensitize to particular therapies (black bold) are shown. See text for full details.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stracker, T.H.; Osagie, O.I.; Escorcia, F.E.; Citrin, D.E. Exploiting the DNA Damage Response for Prostate Cancer Therapy. Cancers 2024, 16, 83. https://doi.org/10.3390/cancers16010083
AMA Style
Stracker TH, Osagie OI, Escorcia FE, Citrin DE. Exploiting the DNA Damage Response for Prostate Cancer Therapy. Cancers. 2024; 16(1):83. https://doi.org/10.3390/cancers16010083
Chicago/Turabian StyleStracker, Travis H., Oloruntoba I. Osagie, Freddy E. Escorcia, and Deborah E. Citrin. 2024. "Exploiting the DNA Damage Response for Prostate Cancer Therapy" Cancers 16, no. 1: 83. https://doi.org/10.3390/cancers16010083
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.