
Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators

1
Department of Life Sciences and Public Health, Università Cattolica del Sacro Cuore, 00168 Rome, Italy
2
Department of Immunology and Regenerative Biology, Weizmann Institute of Science, Rehovot 76100, Israel
*
Author to whom correspondence should be addressed.
Cancers 2023, 15(20), 5009; https://doi.org/10.3390/cancers15205009
Submission received: 31 July 2023
/
Revised: 3 October 2023
/
Accepted: 11 October 2023
/
Published: 16 October 2023
(This article belongs to the Special Issue Growth Factors and Receptor Tyrosine Kinases in Development, Regeneration, and Tumorigenesis)
Abstract
:Simple Summary
Genome-based cancer medicine is becoming the standard of care: the patient’s tumor DNA is first analyzed to identify driver mutations, and this permits later selection of the most effective drugs. Treatment of lung cancer offers many examples. Activating mutations in the epidermal growth factor receptor (EGFR) gene, as well as in other receptor tyrosine kinases (e.g., ALK), are considered actionable candidates, and the respective drugs, called tyrosine kinase inhibitors, are relatively effective. Unfortunately, despite initial activity, the emergence of new, on-target mutations, along with adaptive processes, preempt the anti-cancer effects and necessitate switching to next-generation drugs. This review highlights recent progress in resolving the mechanisms that underlie acquisition of resistance. Specifically, we focus on the endogenous mutators that initiate emergence of new mutations and the potential clinical benefits that may be derived from this new understanding.
Abstract
Epidermal growth factor receptor (EGFR)-specific tyrosine kinase inhibitors (TKIs) have changed the landscape of lung cancer therapy. For patients who are treated with the new TKIs, the current median survival exceeds 3 years, substantially better than the average 20 month survival rate only a decade ago. Unfortunately, despite initial efficacy, nearly all treated patients evolve drug resistance due to the emergence of either new mutations or rewired signaling pathways that engage other receptor tyrosine kinases (RTKs), such as MET, HER3 and AXL. Apparently, the emergence of mutations is preceded by a phase of epigenetic alterations that finely regulate the cell cycle, bias a mesenchymal phenotype and activate antioxidants. Concomitantly, cells that evade TKI-induced apoptosis (i.e., drug-tolerant persister cells) activate an intrinsic mutagenic program reminiscent of the SOS system deployed when bacteria are exposed to antibiotics. This mammalian system imbalances the purine-to-pyrimidine ratio, inhibits DNA repair and boosts expression of mutation-prone DNA polymerases. Thus, the net outcome of the SOS response is a greater probability to evolve new mutations. Deeper understanding of the persister-to-resister transformation, along with the development of next-generation TKIs, EGFR-specific proteolysis targeting chimeras (PROTACs), as well as bispecific antibodies, will permit delaying the onset of relapses and prolonging survival of patients with EGFR+ lung cancer.
1. Introduction
Along with the well-characterized roles for growth factors and their receptor tyrosine kinases (RTKs) in development, physiology and regeneration, these cell-surface molecules have important roles to play in tumorigenesis. Two major modes underlie the ability of growth factors and RTKs to initiate or promote cancer progression. The first involves genetic aberrations of specific RTKs. For example, 10–30% of patients with lung cancer present activating mutations in the epidermal growth factor receptor (EGFR) gene [1,2,3,4,5]. Importantly, the presence of EGFR mutations predicts response to specific tyrosine kinase inhibitors (TKIs), likely due to addiction of the respective tumor cells to an active EGFR pathway [6]. The most frequent gene aberration of HER2/ERBB2, the closest homologue of EGFR, is gene amplification, which occurs in breast, gastric and other tumors, but rare missense mutations have also been reported [7,8]. Importantly, very high expression levels of HER2 predict response to chemotherapy combined with HER2-targeting monoclonal antibodies (mAbs), such as trastuzumab/Herceptin. Similarly, overexpression of EGFR occurs in approximately 50% of brain tumors of glial origin due to gene amplification [9], and a large fraction of these tumors also present internal deletions within the extracellular domain [10]. However, these observations have not been translated to EGFR-targeted treatments of brain malignancies. The other oncogenic roles of growth factors and RTKs involve autocrine or paracrine loops that engage a growth factor and a specific RTK. For example, high expression of EGFR ligands, especially amphiregulin, supports proliferation of colorectal cancer (CRC) cells and might predict response to a combination of chemotherapy plus an anti-EGFR mAb, such as cetuximab [11]. However, the abundance of amphiregulin in CRC specimens has not been translated to a predictive biomarker of response to anti-EGFR antibodies. Yet another clinically approved combination of chemotherapy and an antibody blocking a ligand of RTKs is the combination of bevacizumab/Avastin and chemotherapy, which is used to treat patients with recurrent ovarian cancer. Similar combinations of bevacizumab were approved for breast cancer, non-small-cell lung cancer, glioblastoma, renal cell carcinoma and cervical cancer [12]. Notably, aside from its mitogenic activities, VEGF plays a major role in controlling blood vessel formation (angiogenesis) as well as modulating tumor-induced immunosuppression.
2. Major Classes of Oncogene-Targeted Drugs and Resistance to the Respective Therapeutic Strategies
Only two major classes of molecular targeted treatments currently dominate the field of RTK-centered cancer treatment. These are antibodies targeting either growth factors or RTKs [13], and small-molecule kinase inhibitors, which block tyrosine-specific and other kinases [14]. A third class of inhibitors, which can selectively sort specific proteins for intracellular degradation, is already on the horizon. The key for this strategy is the development of proteolysis-targeting chimeras (PROTACs). While some of the RTK-targeting drugs have greatly changed the landscape of the respective diseases, for example trastuzumab in breast cancer and osimertinib/Tagrisso in lung cancer, drug resistance is considered a major issue. Indeed, overcoming resistance to the latest anti-cancer drugs has been recognized by the Cancer Moonshot Initiative as one of the top ten priorities of contemporary cancer research [15]. This is because the majority of patients with advanced cancer die either because their cancers are inherently resistant to drugs, or their tumors initially respond but they later develop drug resistance [16]. Resistance universally limits applications of many different drugs, including not only chemotherapeutic agents but also kinase inhibitors [16,17], anti-receptor antibodies [13] and immune checkpoint blockers [18,19]. This review will focus on the emergence of resistance of lung cancer to anti-EGFR kinase inhibitors, highlighting the underlying mechanisms and potential strategies to prevent cancer relapses.
3. A Primer to Lung Cancer
Nearly 2 million deaths are expected annually due to lung cancer, the most common cause of cancer-related deaths. Histologically, lung cancer can be divided into two groups: (i) Small-cell lung cancer (SCLC) constitutes approximately 15% of all cases. This deadly tumor is highly different from other lung cancers [20]. Its aggressive nature is attributable to bi-allelic inactivation of TP53 and RB1, along with aberrantly active Notch signaling. (ii) Non-small cell lung cancer (NSCLC) represents the remaining 85% of lung cancers and includes three major histological subtypes: adenocarcinoma, the most common type, squamous cell carcinoma and large cell carcinoma, the least frequent subtype. Unlike squamous NSCLC, which rarely harbors actionable mutations, several such mutations are highly prevalent in adenocarcinomas and include mutant forms of the following oncogenes: KRAS, EGFR, ALK, RET, ROS1, BRAF, HER2 and MET [21]. Although immunotherapy combinations, for example the combination of antibodies targeting CTLA-4 and PD1, seem to benefit some patients presenting with advanced or metastatic NSCLC, tumors with EGFR mutations in general lack abundant infiltrating lymphocytes and have a relatively low tumor mutational burden [22]. Hence these tumors tend not to respond to immune checkpoint blockers. This contrasts with most KRAS- and BRAF-mutated NSCLCs, which are associated with a higher mutational burden [23].
4. EGFR Mutations in Lung Cancer
EGFR and its family members, primarily HER2, are ubiquitously expressed in epithelia, including the epithelium of the lung, and they have been found to be overexpressed or mutated in several types of cancer [24]. Depending on age, gender and ethnicity, 10–30% of all patients with NSCLC present somatic kinase-activating mutations in the gene encoding EGFR [1,2,3,4,5]. Another factor that might increase incidence of EGFR-mutated lung cancer appears to be air pollution. Hill et al. showed that exposure to particulate matter measuring ≤2.5 μm (PM2.5) promotes lung cancer development by recruiting macrophages into the lungs, which in turn release IL-1β. Consequently, a progenitor-like cell state is induced in lung alveolar type II epithelial cells harboring pre-existing EGFR activating mutations [25].
Various short deletions in exon 19 (Del19), along with L858R, a point mutation in exon 21, represent 85–90% of all known EGFR mutations in lung cancer [26]. Rare mutations account for 10–15% of all EGFR mutations in NSCLC and include G719A or G719S, Del18, E709K, exon 19 insertions, S768I, L861Q and exon 20 insertions (Ins20). Two mutations, T790M and C797S, typically emerge when patients are treated with the first/second or third generation TKIs, respectively [27,28,29,30]. It is imperative to note that the mutant forms of EGFR mimic the canonical mechanisms permitting activation of the kinase domain in response to EGF binding. The mutant forms also engage the same signaling pathways, primarily JAK-to-STAT, the RAS-to-mitogen-activated protein kinases (MAPK) pathway and the linear route that enables activation of mTOR by the upstream kinases PI3K and AKT. Although normally kinase activation targets EGFR to CBL-mediated receptor ubiquitination and subsequent degradation in lysosomes, certain EGFR mutants escape this regulation and also display enhanced heterodimerization with HER2, which further leads to persistent stimulation [31]. In addition, while wild-type EGFRs require dimerization for proper signaling [32,33], all kinase-activating mutations, except for L858R, induce an active conformation of the enzyme that is independent of ligand-induced dimerization [34].
5. First and Second Generations of EGFR-Specific Kinase Inhibitors (See Table 1)
It is interesting to draw analogies between TKIs targeting EGFR and TKIs specific to BCR-ABL1, a fusion tyrosine kinase that acts as the driver of chronic myeloid leukemia (CML). The development of imatinib, a BCR-ABL1 inhibitor, allows patients with CML to experience near-normal life expectancy [35]. Although specific point mutations that decrease drug-binding affinity can produce imatinib resistance, second- and third-generation TKIs can largely mitigate this issue. For patients with advanced NSCLC who are treated with a third-generation EGFR inhibitor, the median survival is greater than 3 years [36], significantly better than the <2 year survival rate just a decade ago. When compared to standard chemotherapy, the reversible ATP-competitive first-generation EGFR inhibitors, erlotinib and gefitinib, prolonged progression-free survival (PFS) and overall survival of patients with NSCLC who harbor mutant forms of EGFR [37,38,39]. In comparison to chemotherapy, EGFR TKIs are relatively safe drugs. Nevertheless, several side effects have been reported. They frequently include skin effects and gastrointestinal tract toxicity, such as skin rash and diarrhea, respectively. More severe adverse effects include intestinal obstruction, hepatotoxicity and interstitial lung disease [40]. Unfortunately, despite initial efficacy, treated patients eventually evolve drug resistance. Several mechanisms of acquired resistance have been described, including the gatekeeper T790M mutation within the EGFR kinase domain [27]. Importantly, T790M occurs in >50% of patients who relapse after treatment with the first-generation inhibitors. This mutation increases the affinity of the mutant receptor for ATP, thus preventing drug binding and reducing the potency of ATP competitive drugs. Less frequent routes of evasion have been identified, including amplification of genes encoding compensatory RTKs, such as MET [41] and HER2 [42], as well as overexpression of the hepatocyte growth factor (HGF) [43,44], or up-regulation of another RTK, AXL [45]. The second-generation EGFR inhibitors, such as afatinib and dacomitinib, both bind with EGFR in an irreversible manner, aimed at providing therapeutic answers to T790M-mediated resistance [46,47]. However, although two second-generation TKIs have been approved for clinical use, these drugs also inhibit HER2 and HER4, and they frequently fail to prevent the emergence of T790M in patients [48].

{kind=link}
{kind=link}
{kind=link}
Table 1.
EGFR-specific agents, including clinically approved drugs, targeting mutant forms of EGFR in NSCLC.
Table 1.
EGFR-specific agents, including clinically approved drugs, targeting mutant forms of EGFR in NSCLC.
| Drug | Inhibitor Type | Drug Target | Status | Relevant Studies |
|---|---|---|---|---|
| Gefitinib | 1st generation TKI Competitive Reversible | Del19/L858R-EGFR | Approved by the FDA/EMA in 2003/2009 | NEJ002 [49,50] IPASS [51,52] WJTOG3405 [37,53] |
| Erlotinib | 1st generation TKI Competitive Reversible | Del19/L858R-EGFR | Approved by the FDA/EMA in 2004/2005 | OPTIMAL [39,54] ENSURE [55] EUTARC [38] |
| Icotinib | 1st generation TKI Competitive Reversible | Del19/L858R-EGFR | Approved in China in 2011 | CONVINCE [56] EVIDENCE [57] |
| Afatinib | 2nd generation TKI Covalent Irreversible | Del19/L858R-EGFR | Approved by the FDA/EMA in 2013 | LUX-Lung3 [58,59] LUX-Lung6 [59,60] |
| Dacomitinib | 2nd generation TKI Covalent Irreversible | Del19/L858R-EGFR | Approved by the FDA/EMA in 2018/2019 | ARCHER1050 [61,62] |
| Osimertinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Approved by the FDA/EMA in 2015/2016 | AURA3 [63] FLAURA [64] |
| Aumolertinib * | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Approved in China in 2020 | AENEAS [36,65] APOLLO [66] |
| Furmonertinib $ | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Approved in China in 2021 | FURLONG [67] |
| Lazertinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Approved in South Korea in 2021 | LASER201 [68,69] LASER301 (NCT04248829) |
| Befotertinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase II/III (active) | NCT03861156 [70] NCT04206072 [71] |
| Abivertinib £ | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase I/II (active) | NCT02274337 [72] AEGIS-1 (NCT02330367) |
| Nazartinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase I/II (active) | NCT02108964 [73] |
| Mavelertinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase I/II (terminated) | NCT02349633 [74] |
| Rociletinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Rejected by the FDA in 2016 | NCT01526928 TIGER-1 (NCT02186301) TIGER-3 (NCT02322281) [75] |
| Olmutinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Terminated ° | NCT01588145 NCT02485652 [76] |
| Naquotinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase III (terminated) | NCT02588261 [77] |
| EAI001 | 4th generation TKI Allosteric Reversible | L858R/T790M/C797S-EGFR | Preclinical | [78,79] |
| EAI045 | 4th generation TKI Allosteric Reversible | L858R/T790M/C797S-EGFR | Preclinical | [78,79] |
| JBJ-04-125-02 | 4th generation TKI Allosteric Reversible | L858R/T790M/C797S-EGFR | Preclinical | [80] |
| JBJ-09-063 | 4th generation TKI Allosteric Reversible | L858R/T790M/C797S-EGFR | Preclinical | [81] |
| CH7233163 | 4th generation TKI Non covalent Competitive | Del19/L858R/T790M/C797S-EGFR | Preclinical | [82] |
| BLU-945 | 4th generation TKI Reversible | Del19/L858R/T790M/C797S-EGFR | Phase I/II (Recruiting) | SYMPHONY (NCT04862780) |
| BBT-176 | 4th generation TKI Reversible | Del19/L858R/T790M/C797S-EGFR | Phase I/II (Recruiting) | NCT04820023 [83] |
| TQB3804 | 4th generation TKI | Del19/L858R/T790M/C797S-EGFR | Phase I (Unknown) | NCT04128085 |
| BPI-361175 | 4th generation TKI | Del19/L858R/T790M/C797S-EGFR | Phase I/II (Recruiting) | NCT05329298 |
| HJM-561 | PROTAC | Del19/L858R/T790M/C797S-EGFR | Preclinical | [84] |
| DDC-01-163 | PROTAC | L858R/T790M/C797S-EGFR | Preclinical | [85] |
| Mobocertinib | TKI | Ins20-EGFR | Approved by the FDA in 2021 | EXCLAIM [86] |
| Amivantamab | Bispecific Antibody | Ins20-EGFR MET | Approved by the FDA/EMA in 2021 | CHRYSALIS [87] PAPILLON (NCT04538664) |
The abbreviations used are: TKI, tyrosine kinase inhibitor; Del19, deletions in exon 19; FDA, Food and Drug Administration of the United States of America; EMA, European Medicines Agency. *, previously known as almonertinib; $, previously known as alflutinib; £, also known as avitinib; °, approved in South Korea in 2016 and subsequently terminated following two cases of toxic epidermal necrolisis (one fatal); PROTAC, proteolysis targeting chimera; Ins20, insertions in exon 20.
6. Third-Generation TKIs (See Table 1)
The third-generation EGFR inhibitors were designed to specifically inhibit EGFR-T790M, while sparing wild-type EGFR [88,89]. Similarly to the second-generation drugs, the newer compounds irreversibly bind with the mutant receptor by covalently engaging cysteine 797. Of the three compounds that initially entered pre-clinical tests, WZ4002, rociletinib and osimertinib, only the latter has been approved, in 2015. Although rociletinib demonstrated clinical activity in patients who had relapsed after treatment with the first and second generation TKIs [90], it was eventually discarded due to adverse effects and low efficacy. Two phase III studies established osimertinib as the current drug of choice for patients who progressed under the first-generation TKIs, as well as the drug of choice in the first line scenario. The first trial [63] compared the efficacy of osimertinib and platinum plus pemetrexed-based therapy in patients who had disease progression after first-line EGFR-TKI therapy. In all patients, the median duration of progression-free survival was significantly longer with osimertinib than in the other arm and, likewise, the objective response rate was significantly better with osimertinib. Additionally, the median duration of progression-free survival was longer among patients who received osimertinib, and the proportion of patients with moderate or severe adverse events was lower with osimertinib. The other trial was similarly successful [64]. It compared osimertinib and standard EGFR-TKIs in patients with previously untreated EGFR mutation-positive advanced NSCLC. The patients received either osimertinib or a first-generation TKI. While both arms showed similar safety profiles, the efficacy of osimertinib was superior to that of the standard (first-generation) EGFR-TKIs in the first-line setting. These observations led to the approval of osimertinib not only for patients who progressed following treatment with first-generation TKIs but also as a first-line drug for patients harboring EGFR-activating mutations.
7. Resistance to Osimertinib and Development of Fourth-Generation EGFR Inhibitors (See Table 1)
Despite impressive therapeutic effects, emergence of resistance to osimertinib is nearly inevitable. The most common mechanism of resistance to second-line osimertinib comprises MET amplification (18%), followed by a tertiary mutation within the ATP-binding pocket of the receptor, namely C797S (14%) [91]. Other resistance mechanisms include amplification of HER2, as well as mutations in downstream molecules (i.e., PIK3CA, KRAS and BRAF) [91]. Additionally, approximately half of the patients who progressed following osimertinib treatment within the AURA 3 trial revealed loss of T790M, and at least a third of these patients displayed concurrent resistant mechanisms (e.g., MET, HER2 or PIK3CA amplification) [91]. Moreover, histological transformation from NSCLC to SCLC has been described as one of the mechanisms responsible for osimertinib resistance [92]. In a similar way, the FLAURA 3 trial that tested osimertinib in a first-line setting detected the C797S mutation but no T790M [93]. In addition, this trial identified several off-target mechanisms that might lead to resistance, including amplification of MET and KRAS, HER2 exon 20 insertion and mutations in MEK1, JAK2, KRAS and PIK3CA [93,94]. Note that C797S-EGFR was identified in circulating free DNA (cfDNA) prior to the availability of resistance biopsy specimens [30]. In comparison to the >50% of patients who evolve resistance to the first-generation TKIs due to T790M, the C797S mutation is typically detected in less than a quarter of patients who progress under second-line osimertinib [95]. Fourth-generation inhibitors have been developed with the goal of overcoming C797S-mediated secondary resistance. EAI045 is an allosteric, non-ATP competitive inhibitor able to inhibit not only C797S but also T790M [78]. This compound targets an allosteric site of EGFR but it requires the co-administration of an anti-EGFR antibody. Interestingly, EAI045 is active on L858R-expressing tumors, but not Del19-expressing tumors [78]. A similar compound, JBJ-04-125-02, can inhibit cell proliferation and the triple mutant L858R/T790M/C797S as a single agent [80]. Similarly, BLU-945, a fourth-generation ATP-competitive inhibitor, showed promising in vitro results against C797S-positive tumors [96]. An initial clinical trial is currently evaluating BLU-945 in the TKI resistance setting, as either monotherapy or combined with osimertinib (NCT04862780). Yet another promising fourth-generation TKI is UPR1444, a new sulfonyl fluoride derivative which potently and irreversibly inhibits the triple EGFR mutant through the formation of a sulfonamide bond with the catalytic residue Lys 745 [97]. In summary, several different fourth-generation inhibitors are already in clinical trials.
8. PROTACs
In parallel to the development of fourth-generation EGFR TKIs, a major current effort is directed towards clinical development of proteolysis-targeting chimeras (PROTACs), representing an alternative strategy to directly target EGFR. PROTACs consist of two covalently linked protein ligands: one binds with the target protein (the protein intended to be degraded), while the other molecular arm recruits an E3 ligase that promotes ubiquitination and degradation of the target protein [98]. Notably, the numbers of PROTACs that are currently being evaluated in vitro, in vivo and in clinical studies is increasing [99]. Several PROTACs targeting EGFR have been developed (reviewed in [100]). By using EGFR TKIs as the target protein binder, it is possible to selectively target mutated forms of the receptor, while sparing the wild-type form of EGFR. In this regard, two PROTACs synthetized by using gefitinib as the EGFR binder were able to reduce proliferation of Del19- and L858R-EGFR expressing NSCLC cells, along with the levels of EGFR and the activation of downstream signaling pathways [101]. In a more recent study, Vartak et al. developed a PROTAC containing cetuximab as the EGFR binder, thereby reducing viability of EGFR-mutated NSCLC cells [102]. Despite the promising results showing in vivo efficacy of EGFR-specific PROTACs [103], additional preclinical data are needed prior to translating these findings to clinical applications. For example, membrane permeability and metabolic stability are crucial factors that need to be considered before attempting clinical development of EGFR-specific PROTACs.
9. Darwinian Mechanisms Underlying Resistance to EGFR-Specific TKIs
In the context of anti-cancer therapies, Darwinian mechanisms of drug resistance refer to a selection process that relies on pre-existing genetic variation that is present in a cancer cell population before starting the treatment. The pressure applied by the drug allows cells expressing a resistance-conferring mutation to survive the treatment (selection), leading over time to the establishment of a drug-resistant cancer cell population. Darwinian selection was observed, for instance, in patients with KRAS wild-type CRC that developed resistance after treatment with the anti-EGFR monoclonal antibody panitumumab [104]. While KRAS mutations were undetectable prior to the treatment, they became detectable 5–6 months after treatment with the antibody.
To address the question how resistant clones evolve during TKI treatment, Hata et al. exposed NSCLC cells to a TKI and monitored the development of several resistant clones. This identified early emergence of clones, which represented pre-existing resistant T790M clones, as well as slowly emerging clones that represented de novo acquisition of the T790M mutation within initially T790-WT cells [105]. In a similar way, Turke et al. applied high-throughput FISH analyses on both cell lines and lung cancer patients, and identified subpopulations of cells with MET amplification prior to drug exposure [44]. It is worth noting that emergence of pre-existing mutations post treatment with a TKI has been well-characterized in imatinib-treated patients with Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL). For example, baseline mutations were identified in 21% of imatinib-naïve patients with newly diagnosed Ph+ ALL [106]. In conclusion, the examples from lung cancer, CRC and leukemia exemplify the role played by pre-existing mutations and intra-tumor heterogeneity in resistance to anti-cancer drugs.
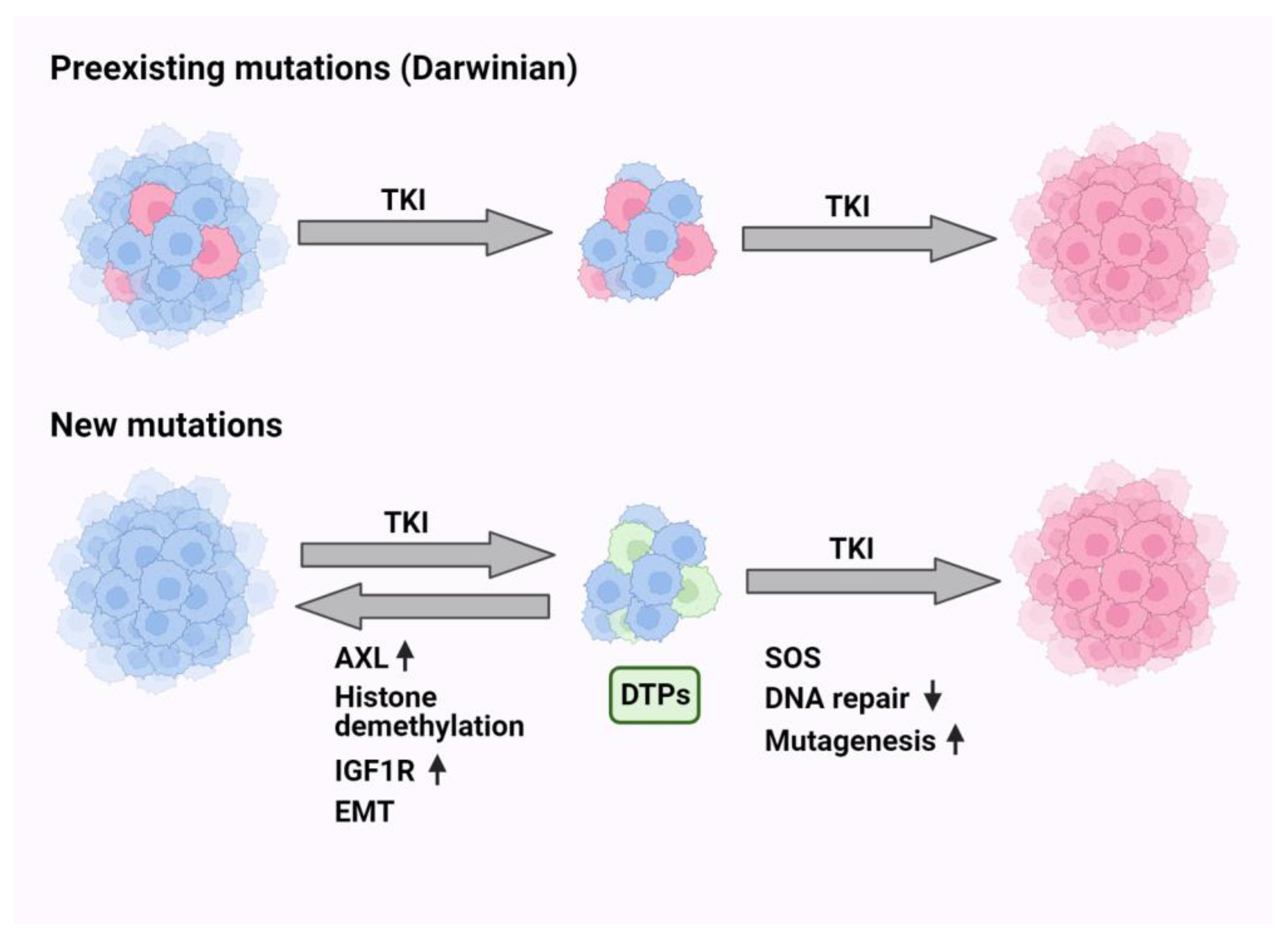
10. Non-Darwinian Mechanisms of Resistance and Drug Tolerance Persistence
Non-Darwinian selection includes all mechanisms that do not involve genomic changes. In this context, epigenetic alterations, factors from the microenvironments, cellular plasticity and phenotypic adaptation are phenomena that might lead to drug resistance. In analogy to antibiotics-tolerant sub-populations of bacteria [107], a small subpopulation of drug-tolerant persister (DTP) cells has been identified in vitro [108]. DTPs are characterized by slow proliferation, reversible phenotypic alterations and altered energy consumption ([109]; see Figure 1). The mechanisms that underlie persistence include diverse epigenetic and transcriptional programs. We recently studied the stepwise transition from DTPs to resisters and detected a host-dependent but non-mutational reversible resister state [110]. In the same vein, single-cell RNA sequencing identified in patient-derived xenografts a rare population of DTP-like cells that converted the major preexisting population of cancer-associated fibroblasts into a state that can promote DTP survival [111]. In analogy, resistance of CRC to cetuximab has been attributed to a transcriptional switch from a cetuximab-sensitive subtype to a fibroblast- and growth factor-rich subtype at progression [112]. These observations propose that DTPs can switch phenotypes or educate their microenvironment. Consistent with this, using a combination of large-scale drug screening and whole-exome sequencing, it was concluded that the DTP state might provide a latent reservoir of cells for the emergence of diverse drug-resistance mechanisms [113]. Although there is no consensus concerning markers of DTP phenotypes, they nevertheless seem to share common features. For example, resistance to various EGFR inhibitors might share aberrant activation of the RAS-to-ERK (extracellular signal-regulated kinase) signaling, and this might be caused by either chromosomal amplification of the MAPK1 gene or by down-regulation of a phosphatase that down-regulates ERK [114].
Another common feature was unraveled by employing barcode labeling, which revealed that antioxidant profiles characterized by increased glutathione metabolism and decreased reactive oxygen species (ROS) are hallmarks of cycling persisters [115]. Similarly, it has been proposed that the epithelial to mesenchymal transition (EMT), a reversible program of trans-differentiation, generates the drug-tolerant cell populations characterized by activation of ABC transporters and AXL, as well as immune evasion and epigenetic reprogramming [116]. Yet another marker emerged from single-cell RNA-seq and single-cell ATAC-seq analyses which confirmed the previously characterized genes AURKA, VIM and AXL, but added CD74, a gene that undergoes up-regulation in the drug-tolerant state [117]. Yet another interesting marker of DTPs is the Wnt/β-catenin signaling pathway, which is activated in response to TKI treatment in a notch3-dependent manner, thereby leading to survival of a subpopulation of stem-like DTP cells [118]. The use of genetically engineered mouse models confirmed that the notch pathway can activate proliferation of TKI-treated cells, such that combined treatments using notch inhibitors plus TKIs could block tumor relapses [119]. In summary, the non-Darwinian mechanisms of drug resistance, especially the emerging common functional features of DTP cells, exemplify the robust and diversified nature of drug tolerance. This offers a spectrum of molecular targets, such as notch and AXL, for combination treatments able to nullify or significantly delay the onset of resistance to drugs, including EGFR inhibitors.
11. Endogenous Mutators Promote the Emergence of New Mutations While under TKI Treatment
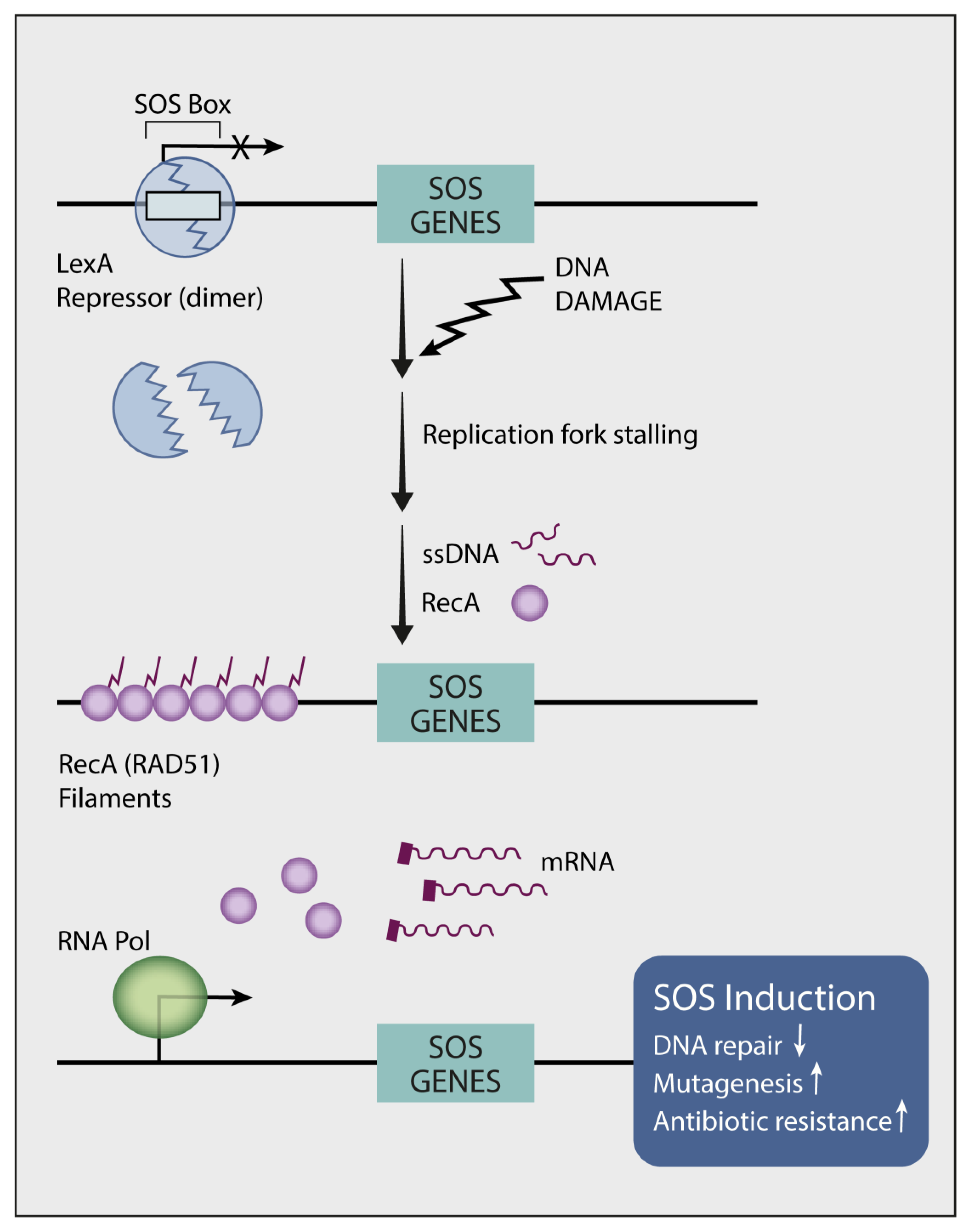
In vitro studies uncovered two routes of resistance to TKIs: (i) a Darwinian route that selects specific clones, which are drug resistant due to a pre-existing secondary mutation, and (ii) a less understood route that promotes de novo emergence of resistance-conferring mutations [105]. Notably, the mechanisms underlying emergence of the secondary (e.g., T790M) and tertiary (e.g., C797S) EGFR mutations are distinct from the mode that generates primary EGFR mutations. The primary mutations likely pre-exist in normal lung tissues but they are later exposed due to the action of tumor promoters, such as air pollutants [25]. What mechanisms could underlie the apparent drug-induced accelerated evolution? In 1975, Radman reported an inducible bacterial DNA mutagenesis system, the SOS response (see Figure 2), which might explain the link between genotoxic stress and adaptive mutagenesis [120]. Following genotoxic stress, bacteria release fragments of single-stranded DNA, which act as sensors that initiate transcriptional programs and mutate the genome [121]. The major endogenous mechanism of mutagenesis (i.e., mutator) in Escherichia coli is DNA polymerase V (polV), which instigates virtually all SOS mutagenesis [122]. PolV belongs to the group called Y family DNA polymerases, which promote translesion synthesis (TLS) of DNA. These polymerases exhibit low fidelity, thereby increasing mutagenesis rates when they are engaged in DNA replication. We previously investigated whether the treatment of lung cancer with TKIs similarly engages hypermutators [123]. Because GAS6 (growth arrest-specific protein 6), AXL’s ligand, is up-regulated in cycling DTP cells and it binds with newly externalized phosphatidylserine of apoptotic bodies, we assumed that the GAS6-AXL module acts as an alarm that stimulates SOS-like reactions in response to TKIs. In line with this prediction and previous reports that associated AXL and GAS6 with intrinsic resistance to TKIs [110,124], we found that AXL overexpression can up-regulate low-fidelity DNA polymerases and down-regulate DNA repair enzymes [123]. Moreover, simultaneously inhibiting AXL and EGFR completely blocked relapses in animal models. Metabolomic analysis uncovered yet another intrinsic mutator that relates to the dependency of DNA replication on balanced pools of deoxyribonucleotides (dNTPs) [125]. By activating MYC and purine synthesis, AXL disbalances the pools of dNTPs, which can influence polymerase proofreading [126] and mutator phenotypes [127,128]. It is worthwhile noting that in similarity to NSCLC cells, human CRC cells exploit adaptive mutability to evade the therapeutic pressure of a combined treatment that used a BRAF kinase inhibitor and an anti-EGFR antibody [129]. This treatment down-regulated mismatch repair (MMR), as well as homologous recombination genes and concomitantly up-regulated error-prone DNA replication. In conclusion, pharmacological stress-induced mutagenesis (SIM) might be shared by eukaryotes and unicellular organisms, arguing against prevailing assumptions that mutations occur purely stochastically [130].
12. A Biomarker Predicting Response of EGFR+ Tumors to Antibody Rather Than TKI Treatment
Although genome-based, personalized cancer medicine is rapidly becoming the standard of medical oncology [131], and with the exception of exon 20 mutation carriers, all patients with EGFR+ NSCLC are treated in the same way. Contrary to this practice, meta-analyses of multiple randomized trials that compared TKIs and chemotherapy confirmed superiority of TKIs and, unexpectedly, revealed that the hazard ratio of progression-free survival for tumors with Del19 was 50% greater than the ratio calculated for tumors with L858R [132]. Importantly, most kinase-activating mutations induce an active conformation of the kinase domain that is independent of ligand-induced EGFR dimerization [133]. For example, exon 19 deletions, exon 20 insertions and the dual L858R/T790M EGFR mutant do not require receptor dimerization. This contrasts with the L858R mutant, which depends on dimerization [34]. These observations raised the possibility that L858R might serve as a biomarker able to predict responses of EGFR+ lung cancer to dimerization-blocking antibodies like cetuximab. As predicted, our recent analyses showed that cetuximab monotherapy completely inhibited relapses of L858R patient-derived xenograft models, but tumors harboring other mutations rapidly relapsed post treatment with either cetuximab or TKIs [134]. Interestingly, unlike TKIs, which elevated reactive oxygen species (ROS) and induced robust cell death, antibody treatments only modestly associated with apoptosis, but they accelerated the rate of EGFR degradation and down-regulated several RTKs that have previously been implicated in drug resistance [134]. Taken together, these observations warrant clinical tests aimed at different, mutation-based immunotherapeutic treatments that would limit the use of TKIs and avoid emergence of secondary EGFR mutations.
13. Individual Bypass Mechanisms and the Respective Combination Therapies (See Figure 3 and Table 2)
13.1. MET Activation
Resistance to the first-, second- and third-generation EGFR TKIs frequently involves activation of compensatory pathways due to amplification and/or overexpression of bypass survival receptors (e.g., HER2, HER3, MET, AXL and IGF1R). As a result, resistant tumors no longer depend on EGFR for survival and proliferation. The MET signaling pathway is the most commonly engaged pathway following treatment with EGFR inhibitors, regardless of TKI type and line of therapy [135]. It has been shown that amplification of the MET gene causes resistance via the HER3/PI3K/AKT pathway [41]. Several MET kinase inhibitors have been developed. Two inhibitors, capmatinib and tepotinib, have been approved for NSCLC carrying the MET exon 14 skipping mutation [136,137]. Additionally, antibodies targeting MET are currently being evaluated in both preclinical and clinical studies. Of note, a bispecific antibody co-targeting MET and EGFR, amivantamab, has been approved for the treatment of NSCLC patients expressing EGFR with exon 20 insertions [138]. Considering the relatively high frequency of MET alterations in lung cancers with EGFR mutations, co-targeting EGFR and MET with either a kinase inhibitor or an antibody appears to be a logical therapeutic strategy that is currently being evaluated. A phase Ib/II trial testing a combination of capmatinib and gefitinib in patients with EGFR-mutated NSCLC that developed resistance to EGFR TKIs showed encouraging results, particularly in tumors with high MET gene copy number [139]. Promising results were also obtained in a phase I trial that combined the MET small-molecule inhibitor savolitinib together with osimertinib and recruited patients with EGFR+ NSCLC harboring MET amplification post treatment with an EGFR inhibitor [140]. Yet another trial combined the EGFR/MET bispecific antibody, amivantamab, and a third-generation EGFR inhibitor, lazertinib [141]. The same drug combination is also being evaluated in a first-line therapy by a phase 3 study, MARIPOSA [142].
Table 2.
Combinatorial approaches under clinical evaluation to overcome resistance to third-generation EGFR-TKIs.
Table 2.
Combinatorial approaches under clinical evaluation to overcome resistance to third-generation EGFR-TKIs.
| Mechanism of Resistance | Strategy to Overcome Resistance | Drugs | Status | Relevant Studies |
|---|---|---|---|---|
| MET alterations | MET TKI | Osimertinib + savolitinib | Phase Ib (active) | NCT02143466 |
| Osimertinib + savolitinib | Phase II (recruiting) | NCT03778229 (SAVANNAH) | ||
| Osimertinib + savolitinib | Phase III (recruiting) | NCT05015608 (SACHI) | ||
| Osimertinib + savolitinib | Phase II (recruiting) | NCT03944772 (ORCHARD) | ||
| Osimertinib + savolitinib | Phase II (not yet recruiting) | NCT05163249 (FLOWERS) | ||
| Osimertinib + savolitinib | Phase III (recruiting) | NCT05261399 (SAFFRON) | ||
| Osimertinib + savolitinib | Phase II (active) | NCT04606771 | ||
| Osimertinib + tepotinib | Phase II (active) | NCT03940703 (INSIGHT) | ||
| Osimertinib + vebreltinib | Phase I/II (recruiting) | NCT04743505 | ||
| Bispecific Antibody (EGFR-MET) | Lazertinib + amivantamab | Phase III (active) | NCT04487080 (MARIPOSA) | |
| Lazertinib ± amivantamab | Phase I (recruiting) | NCT04077463 (CHRYSALIS2) | ||
| Lazertinib + amivantamab | Phase III (recruiting) | NCT05388669 (PALOMA3) | ||
| Lazertinib ± amivantamab ± carboplatin/pemetrexed | Phase I (recruiting) | NCT02609776 (CHRYSALIS) | ||
| Lazertinib + amivantamab + pemetrexed | Phase II (recruiting) | NCT05299125 (AMIGO-1) | ||
| Lazertinib + amivantamab + carboplatin/pemetrexed | Phase III (recruiting) | NCT04988295 (MARIPOSA-2) | ||
| Lazertinib + amivantamab + bevacizumab | Phase II (recruiting) | NCT05601973 (AMAZE-Lung) | ||
| Osimertinib+ EMB-01 | Phase I/II (not yet recruiting) | NCT05498389 | ||
| MET ADC | Osimertinib or erlotinib + telisotuzumab vedotin | Phase I (active) | NCT02099058 | |
| HER2 alterations | HER2 ADC | Trastuzumab deruxtecan | Phase II (active) | NCT03505710 (DESTINY-Lung01) |
| HER2 mAb | Osimertinib + necitumumab + trastuzumab | Phase I/II (recruiting) | NCT04285671 | |
| HER3 alterations | HER3 ADC | Patritumab deruxtecan | Phase II (recruiting) | NCT04619004 (HERTHENA-Lung01) |
| Patritumab deruxtecan | Phase III (recruiting) | NCT05338970 (HERTHENA-Lung02) | ||
| Osimertinib + patritumab deruxtecan | Phase I (recruiting) | NCT04676477 | ||
| Bispecific Antibody (EGFR-HER3) | Osimertinib + izalontamab | Phase II/III (recruiting) | NCT05020769 | |
| EGFR-HER3 ADC (Bispecific Antibody) | Osimertinib + BL-B01D1 | Phase II (not yet recruiting) | NCT05880706 | |
| AXL alterations | AXL TKI | Osimertinib + bemcentinib | Phase I/II (completed) | NCT02424617 |
| Alterations affecting downstream molecules | BRAF inhibitor | Dabrafenib + trametinib | Phase II (recruiting) | NCT04452877 |
| mTOR inhibitor | Osimertinib + sapanisertib | Phase I (recruiting) | NCT02503722 | |
| Osimertinib + sapanisertib | Phase I (not yet recruiting) | NCT04479306 | ||
| JAK inhibitor | Osimertinib + itacitinib | Phase I/II (active) | NCT02917993 | |
| Osimertinib + golidocitinib | Phase I/II (completed) | NCT03450330 (JACKPOT1) | ||
| MEK inhibitor | Osimertinib + selumetinib | Phase I (active) | NCT02143466 (TATTON) | |
| Osimertinib + selumetinib | Phase II (active) | NCT03392246 | ||
| PI3K inhibitor | Osimertinib + TQ-B3525 | Phase I/II (recruiting) | NCT05284994 | |
| RET alterations | RET TKI | Osimertinib + selpercatinib | Phase II (recruiting) | NCT03944772 (ORCHARD) |
| ALK alterations | ALK TKI | Osimertinib + alectinib | Phase II (recruiting) | NCT03944772 (ORCHARD) |
| CDK4/6 amplification | CDK 4/6 inhibitor | Osimertinib + G1738 (lerociclib) | Phase I/II (completed) | NCT03455829 |
| Osimertinib + abemaciclib | Phase II (unknown) | NCT04545710 | ||
| Others | Bcl-2 inhbitor | Osimertinib + navitoclax | Phase I (active) | NCT02520778 |
| Osimertinib + palcitoclax | Phase I (recruiting) | NCT04001777 | ||
| VEGF mAb | Osimertinib + bevacizumab | Phase II (active) | NCT03133546 (BOOSTER) | |
| Erlotinib + bevacizumab | Approved by the EMA in 2016 | BELIEVE JO25567 | ||
| Osimertinib + bevacizumab | Phase I/II (completed) | NCT02803203 | ||
| Osimertinib + bevacizumab | Phase III (recruiting) | NCT04181060 | ||
| Osimertinib + bevacizumab | Phase III (recruiting) | NCT05104281 | ||
| Osimertinib + bevacizumab | Phase II (active) | NCT02971501 | ||
| VEGFR mAb | Osimertinib + ramucirumab | Phase II (recruiting) | NCT03909334 | |
| Osimertinib + ramucirumab or necitumumab | Phase I (completed) | NCT02789345 | ||
| Erlotinib + ramucirumab | Approved by the FDA/EMA in 2020 | NCT02411448 (RELAY) | ||
| VEGFR-PDGFR-FGFR-cKIT TKI | Osimertinib + Anlotinib | Phase I/II (recruiting) | NCT04770688 (AUTOMAN) | |
| Aurora Kinase A inhibitor | Osimertinib + VIC-1911 | Phase I (recruiting) | NCT05489731 | |
| Osimertinib + alisertib | Phase I (not yet recruiting) | NCT04479306 | ||
| Osimertinib + LY3295668 | Phase I/II (active) | NCT05017025 | ||
| MERTK and FLT3 TKI | Osimertinib + MRX-2843 | Phase I (recruiting) | NCT04762199 | |
| ROS17TRK/ALK TKI | Osimertinib + repotrectinib | Phase I (recruiting) | NCT04772235 (TOTEM) |
The abbreviations used are: TKI, tyrosine kinase inhibitor; ADC, antibody-drug conjugate; mAb, monoclonal antibody; FDA, Food and Drug Administration of the United States of America; EMA, European Medicines Agency.
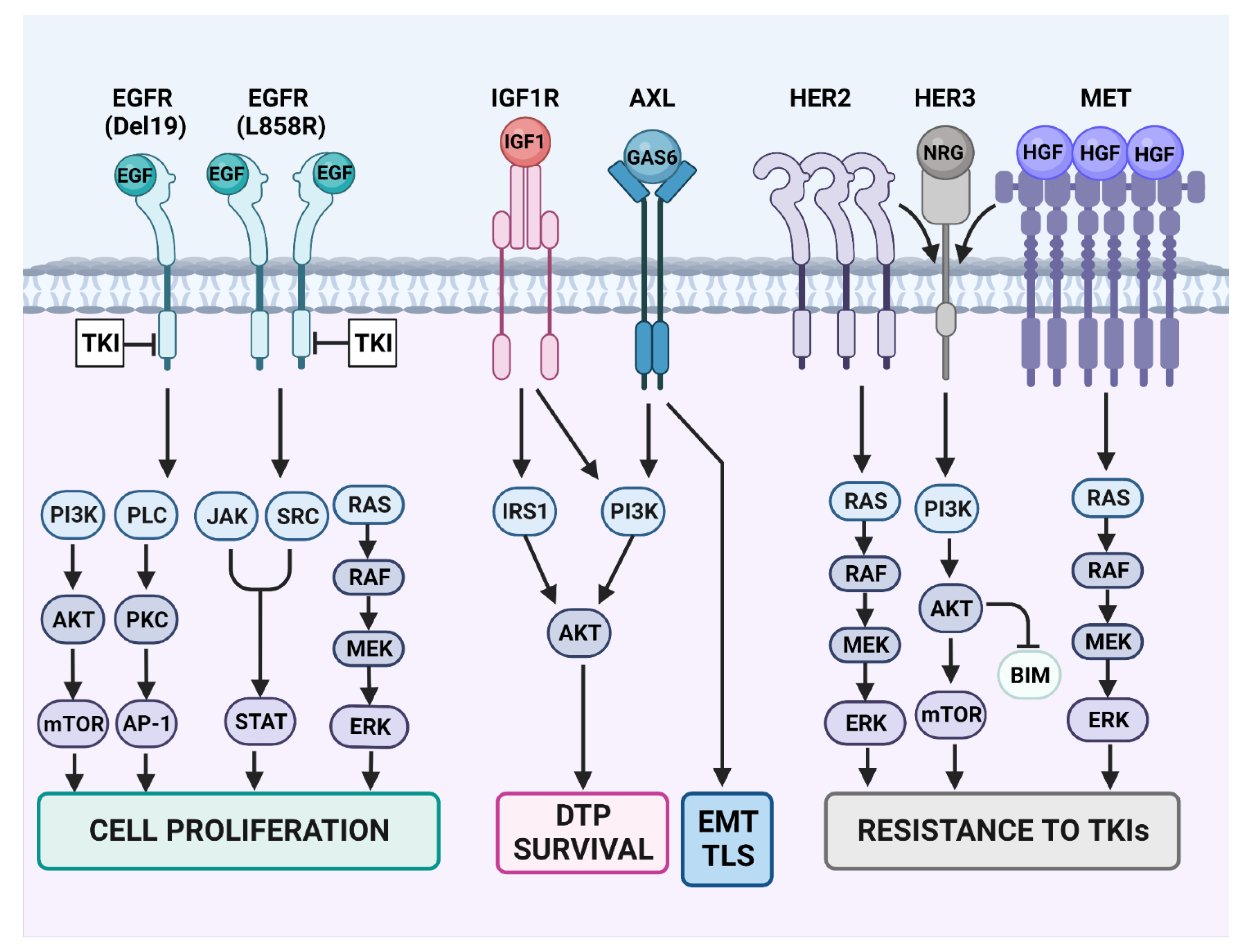
Figure 3.
Resistance-conferring rewiring of RTK signaling in TKI-treated lung cancer. Some of the canonical signaling pathways engaged by EGFR are presented on the left side of the scheme. Note that EGFR dimerization is essential for EGF-induced activation of the wild-type kinase domain. Likewise, EGFR dimerization is crucial for activation of the L858R mutant form of EGFR’s kinase domain. However, all other mutant forms, including the different exon-19 deletion mutants (Del19), need no dimer formation for kinase activation. Because EGFR-specific TKIs can inhibit all EGFR downstream pathways, they usually confer sensitivity to the drugs. However, concomitant with blocking cell proliferation and inducing cell death, the TKIs prompt emergence of drug-tolerant persister (DTP) cells. Survival of the DTP cells is mediated by the insulin-like growth factor 1 (IGF1) pathway and the downstream insulin receptor substrate (IRS). Similarly, adaptive mutagenesis can be promoted by the GAS6-AXL pathway, which stimulates the SOS system (including inhibition of DNA repair and enhancement of trans-lesion synthesis (TLS) of DNA by error prone polymerases). AXL is also involved in another epigenetic program, the epithelial–mesenchymal transition (EMT). TLS and inhibition of DNA repair contribute to drug resistance by means of de novo mutagenesis of EGFR or downstream effectors like RAS and PI3K. Yet another therapy escape route is presented in the right part of the scheme. This route indirectly engages HER3, a kinase-dead member of the EGFR family that strongly activates the PI3K-AKT survival pathway. Constitutive activation of MET due to either MET gene amplification or overexpression of MET’s ligand, the hepatocyte growth factor (HGF), might occur in small populations of cancer cells, prior to treatment with TKIs. Similarly, constitutive activation of HER2 due to either gene amplification, protein overexpression or the HER2 exon 20 mutation may serve as a resistance mechanism, probably due to the tendency of this receptor to form heterodimers with HER3.
Figure 3.
Resistance-conferring rewiring of RTK signaling in TKI-treated lung cancer. Some of the canonical signaling pathways engaged by EGFR are presented on the left side of the scheme. Note that EGFR dimerization is essential for EGF-induced activation of the wild-type kinase domain. Likewise, EGFR dimerization is crucial for activation of the L858R mutant form of EGFR’s kinase domain. However, all other mutant forms, including the different exon-19 deletion mutants (Del19), need no dimer formation for kinase activation. Because EGFR-specific TKIs can inhibit all EGFR downstream pathways, they usually confer sensitivity to the drugs. However, concomitant with blocking cell proliferation and inducing cell death, the TKIs prompt emergence of drug-tolerant persister (DTP) cells. Survival of the DTP cells is mediated by the insulin-like growth factor 1 (IGF1) pathway and the downstream insulin receptor substrate (IRS). Similarly, adaptive mutagenesis can be promoted by the GAS6-AXL pathway, which stimulates the SOS system (including inhibition of DNA repair and enhancement of trans-lesion synthesis (TLS) of DNA by error prone polymerases). AXL is also involved in another epigenetic program, the epithelial–mesenchymal transition (EMT). TLS and inhibition of DNA repair contribute to drug resistance by means of de novo mutagenesis of EGFR or downstream effectors like RAS and PI3K. Yet another therapy escape route is presented in the right part of the scheme. This route indirectly engages HER3, a kinase-dead member of the EGFR family that strongly activates the PI3K-AKT survival pathway. Constitutive activation of MET due to either MET gene amplification or overexpression of MET’s ligand, the hepatocyte growth factor (HGF), might occur in small populations of cancer cells, prior to treatment with TKIs. Similarly, constitutive activation of HER2 due to either gene amplification, protein overexpression or the HER2 exon 20 mutation may serve as a resistance mechanism, probably due to the tendency of this receptor to form heterodimers with HER3.

13.2. HER2 and HER3
EGFR family members, primarily HER2 and HER3, have repeatedly been implicated in resistance to EGFR inhibitors. HER2 amplification is found in higher percentages in resistant tumors treated with the first-generation TKIs, compared to those treated with osimertinib [42,91,93,94]. We previously demonstrated that, when blocking EGFR, not only HER2 but also its kin, HER3, underwent up-regulation that resulted in hyperactivation of the ERK pathway [143]. Accordingly, in animal models, triple combinations targeting EGFR (using both a kinase inhibitor and a monoclonal antibody), as well as HER2 (using a monoclonal antibody), showed efficacy in first- and in second-line treatment scenarios [144,145]. These observations suggested that the combination approach may be a suitable way to overcome resistance to EGFR inhibitors. In line with these findings, co-targeting EGFR and HER2 using antibody-drug conjugates (ADCs) might represent an alternative strategy. Thus, the combination of osimertinib and the HER2-specific ADC trastuzumab emtansine showed efficacy in overcoming resistance to osimertinib in EGFR-mutated lung cancer [146].
In similarity to HER2, HER3 expression is detected in a large fraction of NSCLC [147], and in EGFR-mutated lung cancers the levels are higher when compared to EGFR-wild type lung cancers [148]. In addition, HER3 overexpression has been observed in EGFR-mutated lung cancer models treated with osimertinib [149,150]. Along this line, it has been reported that resistance to EGFR TKIs can arise from HER3 via heterodimerization with other RTKs, such as HER2 and MET [151]. In this context, the anti-HER3 antibody patritumab is able to overcome TKI resistance mediated by neuregulin, the HER3 ligand, in EGFR-mutated lung cancer models [152]. Moreover, the levels of soluble neuregulin in the serum of patients with NSCLC appear to associate with better responses to patritumab [153]. In similarity to therapies targeting HER2 in NSCLC, ADCs directed against HER3 were also developed. Patritumab deruxtecan demonstrated both efficacy and safety in a phase I trial involving patients with EGFR-mutated lung cancer that progressed after TKI treatment [154]. Based on this encouraging result, the phase 3 trial, HERTHENA-Lung02, currently examines patritumab deruxtecan versus platinum-based chemotherapy in patients with EGFR-mutated NSCLC after failure of EGFR TKIs (NCT05338970).
13.3. Activation of AXL
AXL is overexpressed in several types of tumors, including lung cancer, and this feature is correlated with poor prognosis [155]. Additionally, AXL-positive tumors are more frequent among NSCLC tumors harboring mutant EGFR, compared to tumors expressing wild-type EGFR [156]. Overexpression of AXL has been found in EGFR-mutated lung cancer models that acquired resistance to the first-, second- or third-generation TKIs [45,110,124]. Of note, this receptor seems to be involved in the generation of DTPs, following treatment with EGFR TKIs [110,124]. Moreover, AXL is up-regulated in samples obtained from patients with EGFR-mutated NSCLC, after they developed resistance to TKIs [45]. In this respect, combination therapies involving EGFR TKIs and an inhibitor of AXL (either an antibody or a small molecule) were able to prevent resistance to EGFR TKIs in EGFR-mutated NSCLC models [123,157]. Consistent with these observations, several AXL inhibitors are being tested in clinical studies, in combination with EGFR TKIs [158].
13.4. IGF1-Receptor (IGF1R)
EGFR inhibition often leads to compensatory activation of the IGF1R pathway [159,160], with a potential benefit arising from the inhibition of this receptor [160]. In this regard, IGF1R is crucial for the establishment of DTPs following treatment of EGFR-mutated lung cancer cells with an EGFR-specific TKI [108,161]. Importantly, inhibition of IGF1R completely eliminates the ability of EGFR-mutated cancer cells to generate DTPs [108]. Thus, combinations of EGFR and IGF1R inhibitors might prevent the onset of resistance. Indeed, blocking both EGFR and IGF1R was able to inhibit EGFR-mutated lung xenografts [161]. Moreover, the insulin receptor substrate 1 (IRS1) has been shown to be crucial for the generation of DTPs, and combinations of EGFR and IRS1 inhibitors were highly effective in models of EGFR-mutated lung cancer that were examined in animals [162].
13.5. Fibroblast Growth Factor Receptors (FGFR)
Oncogenic fusions involving the fibroblast growth factor receptor (FGFR) have been reported to confer resistance to osimertinib [91,95]. In line with this, whole-genome CRISPR screening identified FGFR1 as the top target promoting survival of mesenchymal EGFR mutant cancers. Subsequently, testing combinations of EGFR and FGFR inhibitors revealed that the combination was able to overcome EMT-dependent resistance to EGFR-specific TKIs in models of EGFR-mutated lung cancer [163].
14. Conclusions
Although the two major oncogenes that drive NSCLC, KRAS and EGFR, activate largely overlapping biochemical pathways, their co-expression in tumors is very rare [164]. This mutual exclusivity is likely due to the toxic effects of their co-expression. In addition, the main carcinogen driving KRAS mutations is tobacco smoke, but the identity of the carcinogen(s) responsible for EGFR mutations is less characterized. Hence, it is predictable that prevention efforts will eventually decrease the absolute incidence of KRAS mutations, thereby increase the relative fraction of patients with EGFR-mutated NSCLC. Moreover, because KRAS+ tumors, better than EGFR+ tumors, respond to immune checkpoint inhibitors [165], the issue of resistance to EGFR inhibitors will likely remain one of the major challenges of medical oncology. Despite this gloomy scenario, the approval of osimertinib, the pioneer third-generation inhibitor, and its later application in first-line settings, promise that improved chemical designs, along with better understanding of the complex cascade that precedes emergence of new mutations, will enhance the success of future attempts to inhibit resistance to the TKIs. Unlike KRAS, which resides in the cytoplasm, the transmembrane localization of EGFR might become a key for effective targeting of the mutant forms using synthetic degraders, especially lysosome-targeting PROTACs [166]. In the same vein, the many lines of evidence linking up-regulation of compensatory RTKs (e.g., HER3 and HER2) and resistance to TKIs offer combinations of antibodies, for example cetuximab (anti-EGFR) plus trastuzumab (anti-HER2), as potential resistance-nullifying approaches. Likewise, the clinical approval of amivantamab, a bispecific antibody co-targeting MET and EGFR [138], is yet another source of hope that overcoming resistance to EGFR-specific TKIs is within reach.
Author Contributions
Both I.M. and Y.Y. outlined the review, performed the literature search, wrote the text and designed the figures. All authors have read and agreed to the published version of the manuscript.
Funding
Y.Y. is the incumbent of the Harold and Zelda Goldenberg Professorial Chair in Molecular Cell Biology. Our studies were supported by the Israel Science Foundation (Grant #1931/20), the European Research Council (ERC, Grant #740469), the Israel Cancer Research Fund (ICRF, Grant #946436) and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (Grant #15A). Some Figures of this manuscript were prepared with the help of Biorender.com.
Acknowledgments
We thank members of our teams and many colleagues for their input.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef]
- Wang, Z.; Longo, P.A.; Tarrant, M.K.; Kim, K.; Head, S.; Leahy, D.J.; Cole, P.A. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat. Struct. Mol. Biol. 2011, 18, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Yuan, J.Q.; Wang, K.F.; Fu, X.H.; Han, X.R.; Threapleton, D.; Yang, Z.Y.; Mao, C.; Tang, J.L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Zabransky, D.J.; Yankaskas, C.L.; Cochran, R.L.; Wong, H.Y.; Croessmann, S.; Chu, D.; Kavuri, S.M.; Brewer, M.R.; Rosen, D.M.; Dalton, W.B.; et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc. Natl. Acad. Sci. USA 2015, 112, E6205–E6214. [Google Scholar] [CrossRef]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef]
- Jeuken, J.; Sijben, A.; Alenda, C.; Rijntjes, J.; Dekkers, M.; Boots-Sprenger, S.; McLendon, R.; Wesseling, P. Robust detection of EGFR copy number changes and EGFR variant III: Technical aspects and relevance for glioma diagnostics. Brain Pathol. 2009, 19, 661–671. [Google Scholar] [CrossRef]
- Stahler, A.; Stintzing, S.; Modest, D.P.; Ricard, I.; Giessen-Jung, C.; Kapaun, C.; Ivanova, B.; Kaiser, F.; Fischer von Weikersthal, L.; Moosmann, N.; et al. Amphiregulin Expression Is a Predictive Biomarker for EGFR Inhibition in Metastatic Colorectal Cancer: Combined Analysis of Three Randomized Trials. Clin. Cancer Res. 2020, 26, 6559–6567. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Levi-Schaffer, F.; Sela, M.; Yarden, Y. Immunotherapy of cancer: From monoclonal to oligoclonal cocktails of anti-cancer antibodies: IUPHAR Review 18. Br. J. Pharmacol. 2016, 173, 1407–1424. [Google Scholar] [CrossRef]
- Pujol, J.L.; Pirker, R.; Lynch, T.J.; Butts, C.A.; Rosell, R.; Shepherd, F.A.; Vansteenkiste, J.; O’Byrne, K.J.; de Blas, B.; Heighway, J.; et al. Meta-analysis of individual patient data from randomized trials of chemotherapy plus cetuximab as first-line treatment for advanced non-small cell lung cancer. Lung Cancer 2014, 83, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, E.M.; Dang, C.V.; Agus, D.B.; Alexander, B.M.; Anderson, K.C.; Ashworth, A.; Barker, A.D.; Bastani, R.; Bhatia, S.; Bluestone, J.A.; et al. Future cancer research priorities in the USA: A Lancet Oncology Commission. Lancet Oncol. 2017, 18, e653–e706. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef]
- Mancini, M.; Yarden, Y. Mutational and network level mechanisms underlying resistance to anti-cancer kinase inhibitors. Semin. Cell Dev. Biol. 2016, 50, 164–176. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 765. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.D.; He, X.; Qin, B.D.; Liu, K.; Wu, Y.; Liu, J.; Hou, T.; Zang, Y.S. The prognostic value of tumor mutation burden in EGFR-mutant advanced lung adenocarcinoma, an analysis based on cBioPortal data base. J. Thorac. Dis. 2019, 11, 4507–4515. [Google Scholar] [CrossRef] [PubMed]
- Negrao, M.V.; Skoulidis, F.; Montesion, M.; Schulze, K.; Bara, I.; Shen, V.; Xu, H.; Hu, S.; Sui, D.; Elamin, Y.Y.; et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J. Immunother. Cancer 2021, 9, e002891. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Hill, W.; Lim, E.L.; Weeden, C.E.; Lee, C.; Augustine, M.; Chen, K.; Kuan, F.C.; Marongiu, F.; Evans, E.J., Jr.; Moore, D.A.; et al. Lung adenocarcinoma promotion by air pollutants. Nature 2023, 616, 159–167. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. S1), S24–S31. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The allelic context of the C797S mutation acquired upon treatment with third generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Shtiegman, K.; Kochupurakkal, B.S.; Zwang, Y.; Pines, G.; Starr, A.; Vexler, A.; Citri, A.; Katz, M.; Lavi, S.; Ben-Basat, Y.; et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene 2007, 26, 6968–6978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Schlessinger, J. Self-phosphorylation of epidermal growth factor receptor: Evidence for a model of intermolecular allosteric activation. Biochemistry 1987, 26, 1434–1442. [Google Scholar] [CrossRef]
- Cho, J.; Chen, L.; Sangji, N.; Okabe, T.; Yonesaka, K.; Francis, J.M.; Flavin, R.J.; Johnson, W.; Kwon, J.; Yu, S.; et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013, 73, 6770–6779. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Huang, J.; Meng, L.; Yang, B.; Sun, S.; Luo, Z.; Chen, H. Safety Profile of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors: A Disproportionality Analysis of FDA Adverse Event Reporting System. Sci. Rep. 2020, 10, 4803. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discovery 2012, 2, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Sequist, L.V.; Besse, B.; Lynch, T.J.; Miller, V.A.; Wong, K.K.; Gitlitz, B.; Eaton, K.; Zacharchuk, C.; Freyman, A.; Powell, C.; et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: Results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3076–3083. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Su, W.C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Yang, Z.; Hackshaw, A.; Feng, Q.; Fu, X.; Zhang, Y.; Mao, C.; Tang, J. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: A meta-analysis. Int. J. Cancer 2017, 140, 2805–2819. [Google Scholar] [CrossRef]
- Miyauchi, E.; Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Saijo, Y.; Yoshizawa, H.; et al. Efficacy of chemotherapy after first-line gefitinib therapy in EGFR mutation-positive advanced non-small cell lung cancer-data from a randomized Phase III study comparing gefitinib with carboplatin plus paclitaxel (NEJ002). Jpn. J. Clin. Oncol. 2015, 45, 670–676. [Google Scholar] [CrossRef]
- Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 54–59. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Wu, Y.L.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.S.; Sriuranpong, V.; Chao, T.Y.; Nakagawa, K.; Chu, D.T.; Saijo, N.; et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, H.; Shimokawa, M.; Seto, T.; Morita, S.; Yatabe, Y.; Okamoto, I.; Tsurutani, J.; Satouchi, M.; Hirashima, T.; Atagi, S.; et al. Final overall survival results of WJTOG3405, a randomized phase III trial comparing gefitinib versus cisplatin with docetaxel as the first-line treatment for patients with stage IIIB/IV or postoperative recurrent EGFR mutation-positive non-small-cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1978–1984. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhou, C.; Liam, C.K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.K.; Wang, L.; Han, B.H.; Li, W.; Yu, P.; Liu, Y.P.; Ding, C.M.; Song, X.; Ma, Z.Y.; Ren, X.L.; et al. First-line icotinib versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for patients with advanced EGFR mutation-positive lung adenocarcinoma (CONVINCE: A phase 3, open-label, randomized study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2443–2450. [Google Scholar] [CrossRef]
- He, J.; Su, C.; Liang, W.; Xu, S.; Wu, L.; Fu, X.; Zhang, X.; Ge, D.; Chen, Q.; Mao, W.; et al. Icotinib versus chemotherapy as adjuvant treatment for stage II-IIIA EGFR-mutant non-small-cell lung cancer (EVIDENCE): A randomised, open-label, phase 3 trial. Lancet Respir. Med. 2021, 9, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2023, 41, 2869–2876. [Google Scholar] [CrossRef]
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet. Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet. Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet. Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Chawla, A.; Rosell, R.; Corral, J.; Migliorino, M.R.; et al. Updated Overall Survival in a Randomized Study Comparing Dacomitinib with Gefitinib as First-Line Treatment in Patients with Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. Drugs 2021, 81, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Dong, X.; Jian, H.; Chen, J.; Chen, G.; Sun, Y.; Ji, Y.; Wang, Z.; Shi, J.; Lu, J.; et al. AENEAS: A Randomized Phase III Trial of Aumolertinib Versus Gefitinib as First-Line Therapy for Locally Advanced or MetastaticNon-Small-Cell Lung Cancer with EGFR Exon 19 Deletion or L858R Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 3162–3171. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Q.; Zhang, G.; Dong, X.; Yang, C.T.; Song, Y.; Chang, G.C.; Lu, Y.; Pan, H.; Chiu, C.H.; et al. Efficacy of Aumolertinib (HS-10296) in Patients with Advanced EGFR T790M+ NSCLC: Updated Post-National Medical Products Administration Approval Results From the APOLLO Registrational Trial. J. Thorac. Oncol. 2022, 17, 411–422. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, G.; Wang, X.; Liu, Y.; Wu, L.; Hao, Y.; Liu, C.; Zhu, S.; Zhang, X.; Li, Y.; et al. Furmonertinib (AST2818) versus gefitinib as first-line therapy for Chinese patients with locally advanced or metastatic EGFR mutation-positive non-small-cell lung cancer (FURLONG): A multicentre, double-blind, randomised phase 3 study. Lancet Respir. Med. 2022, 10, 1019–1028. [Google Scholar] [CrossRef]
- Ahn, M.J.; Han, J.Y.; Lee, K.H.; Kim, S.W.; Kim, D.W.; Lee, Y.G.; Cho, E.K.; Kim, J.H.; Lee, G.W.; Lee, J.S.; et al. Lazertinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Results from the dose escalation and dose expansion parts of a first-in-human, open-label, multicentre, phase 1–2 study. Lancet. Oncol. 2019, 20, 1681–1690. [Google Scholar] [CrossRef]
- Cho, B.C.; Han, J.Y.; Kim, S.W.; Lee, K.H.; Cho, E.K.; Lee, Y.G.; Kim, D.W.; Kim, J.H.; Lee, G.W.; Lee, J.S.; et al. A Phase 1/2 Study of Lazertinib 240 mg in Patients with Advanced EGFR T790M-Positive NSCLC after Previous EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2022, 17, 558–567. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, Y.; Zhang, G.; Zhou, J.; Cang, S.; Cheng, Y.; Wu, G.; Cao, P.; Lv, D.; Jian, H.; et al. Efficacy and Safety of Befotertinib (D-0316) in Patients with EGFR T790M-Mutated NSCLC That Had Progressed after Prior EGFR Tyrosine Kinase Inhibitor Therapy: A Phase 2, Multicenter, Single-Arm, Open-Label Study. J. Thorac. Oncol. 2022, 17, 1192–1204. [Google Scholar] [CrossRef]
- Lu, S.; Zhou, J.; Jian, H.; Wu, L.; Cheng, Y.; Fan, Y.; Fang, J.; Chen, G.; Zhang, Z.; Lv, D.; et al. Befotertinib (D-0316) versus icotinib as first-line therapy for patients with EGFR-mutated locally advanced or metastatic non-small-cell lung cancer: A multicentre, open-label, randomised phase 3 study. Lancet Respir. Med. 2023, 11, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zheng, X.; Zhao, H.; Fang, W.; Zhang, Y.; Ge, J.; Wang, L.; Wang, W.; Jiang, J.; Chuai, S.; et al. First-in-Human Phase I Study of AC0010, a Mutant-Selective EGFR Inhibitor in Non-Small Cell Lung Cancer: Safety, Efficacy, and Potential Mechanism of Resistance. J. Thorac. Oncol. 2018, 13, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.W.; Kim, S.W.; Ponce Aix, S.; Sequist, L.V.; Smit, E.F.; Yang, J.C.H.; Hida, T.; Toyozawa, R.; Felip, E.; Wolf, J.; et al. Nazartinib for treatment-naive EGFR-mutant non-small cell lung cancer: Results of a phase 2, single-arm, open-label study. Eur. J. Cancer 2022, 172, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.; Goldberg, S.B.; Kim, D.W.; Socinski, M.A.; Burns, T.F.; Lwin, Z.; Pathan, N.; Ma, W.D.; Masters, J.C.; Cossons, N.; et al. A phase 1b/2 study of PF-06747775 as monotherapy or in combination with Palbociclib in patients with epidermal growth factor receptor mutant advanced non-small cell lung cancer. Expert. Opin. Investig. Drugs 2022, 31, 747–757. [Google Scholar] [CrossRef]
- Yang, J.C.; Reckamp, K.L.; Kim, Y.C.; Novello, S.; Smit, E.F.; Lee, J.S.; Su, W.C.; Akerley, W.L.; Blakely, C.M.; Groen, H.J.M.; et al. Efficacy and Safety of Rociletinib Versus Chemotherapy in Patients with EGFR-Mutated NSCLC: The Results of TIGER-3, a Phase 3 Randomized Study. JTO Clin. Res. Rep. 2021, 2, 100114. [Google Scholar] [CrossRef]
- Park, K.; Jӓnne, P.A.; Kim, D.W.; Han, J.Y.; Wu, M.F.; Lee, J.S.; Kang, J.H.; Lee, D.H.; Cho, B.C.; Yu, C.J.; et al. Olmutinib in T790M-positive non-small cell lung cancer after failure of first-line epidermal growth factor receptor-tyrosine kinase inhibitor therapy: A global, phase 2 study. Cancer 2021, 127, 1407–1416. [Google Scholar] [CrossRef]
- Kelly, R.J.; Shepherd, F.A.; Krivoshik, A.; Jie, F.; Horn, L. A phase III, randomized, open-label study of ASP8273 versus erlotinib or gefitinib in patients with advanced stage IIIB/IV non-small-cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1127–1133. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Zhao, P.; Yao, M.Y.; Zhu, S.J.; Chen, J.Y.; Yun, C.H. Crystal structure of EGFR T790M/C797S/V948R in complex with EAI045. Biochem. Biophys. Res. Commun. 2018, 502, 332–337. [Google Scholar] [CrossRef]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef]
- To, C.; Beyett, T.S.; Jang, J.; Feng, W.W.; Bahcall, M.; Haikala, H.M.; Shin, B.H.; Heppner, D.E.; Rana, J.K.; Leeper, B.A.; et al. An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nat. Cancer 2022, 3, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Kawauchi, H.; Tanimura, H.; Tachibana, Y.; Chiba, T.; Torizawa, T.; Sakamoto, H. CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Mol. Cancer Ther. 2020, 19, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Fujino, T.; Kim, C.; Lee, G.; Lee, Y.H.; Kim, D.W.; Ahn, J.S.; Mitsudomi, T.; Jin, T.; Lee, S.Y. BBT-176, a Novel Fourth-Generation Tyrosine Kinase Inhibitor for Osimertinib-Resistant Epidermal Growth Factor Receptor Mutations in Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Chen, Y.; Wang, Y.; Chen, J.; Lu, X.; Zhang, L.; Li, Y.; Wang, Z.; Ye, G.; Zhang, G. HJM-561, a Potent, Selective, and Orally Bioavailable EGFR PROTAC that Overcomes Osimertinib-Resistant EGFR Triple Mutations. Mol. Cancer Ther. 2022, 21, 1060–1066. [Google Scholar] [CrossRef]
- Jang, J.; To, C.; De Clercq, D.J.H.; Park, E.; Ponthier, C.M.; Shin, B.H.; Mushajiang, M.; Nowak, R.P.; Fischer, E.S.; Eck, M.J.; et al. Mutant-Selective Allosteric EGFR Degraders are Effective Against a Broad Range of Drug-Resistant Mutations. Angew. Chem. Int. Ed. Engl. 2020, 59, 14481–14489. [Google Scholar] [CrossRef]
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.W.; Yang, J.C.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Garcia Campelo, M.R.; Felip, E.; et al. Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients with EGFR Exon 20 Insertion-Positive Metastatic Non-Small Cell Lung Cancer: A Phase 1/2 Open-label Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, e214761. [Google Scholar] [CrossRef]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.Y.; Kim, S.W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results from the CHRYSALIS Phase I Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Sequist, L.V.; Rolfe, L.; Allen, A.R. Rociletinib in EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 578–579. [Google Scholar] [CrossRef]
- Chmielecki, J.; Mok, T.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.; Shepherd, F.A.; et al. Analysis of acquired resistance mechanisms to osimertinib in patients with EGFR-mutated advanced non-small cell lung cancer from the AURA3 trial. Nat. Commun. 2023, 14, 1071. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Minari, R.; Mazzaschi, G.; Gnetti, L.; La Monica, S.; Alfieri, R.; Campanini, N.; Verzè, M.; Olivani, A.; Ventura, L.; et al. Small Cell Lung Cancer Transformation as a Resistance Mechanism to Osimertinib in Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: Case Report and Literature Review. Front. Oncol. 2021, 11, 642190. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Yang, J.C.; Lee, C.K.; Kurata, T.; Kim, D.W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Gray, J.E.; Cheng, Y.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer. Nat. Commun. 2023, 14, 1070. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef]
- Eno, M.S.; Brubaker, J.D.; Campbell, J.E.; De Savi, C.; Guzi, T.J.; Williams, B.D.; Wilson, D.; Wilson, K.; Brooijmans, N.; Kim, J.; et al. Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 9662–9677. [Google Scholar] [CrossRef]
- Ferlenghi, F.; Scalvini, L.; Vacondio, F.; Castelli, R.; Bozza, N.; Marseglia, G.; Rivara, S.; Lodola, A.; La Monica, S.; Minari, R.; et al. A sulfonyl fluoride derivative inhibits EGFR(L858R/T790M/C797S) by covalent modification of the catalytic lysine. Eur. J. Med. Chem. 2021, 225, 113786. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Qi, S.M.; Dong, J.; Xu, Z.Y.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Chen, W.; Wang, Y.; Xing, D. Epidermal growth factor receptor PROTACs as an effective strategy for cancer therapy: A review. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188927. [Google Scholar] [CrossRef]
- Cheng, M.; Yu, X.; Lu, K.; Xie, L.; Wang, L.; Meng, F.; Han, X.; Chen, X.; Liu, J.; Xiong, Y.; et al. Discovery of Potent and Selective Epidermal Growth Factor Receptor (EGFR) Bifunctional Small-Molecule Degraders. J. Med. Chem. 2020, 63, 1216–1232. [Google Scholar] [CrossRef] [PubMed]
- Vartak, R.; Deore, B.; Sanhueza, C.A.; Patel, K. Cetuximab-based PROteolysis targeting chimera for effectual downregulation of NSCLC with varied EGFR mutations. Int. J. Biol. Macromol. 2023, 252, 126413. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Du, Y.; Huang, L.; Cui, J.; Niu, J.; Xu, Y.; Zhu, Q. Discovery of novel potent covalent inhibitor-based EGFR degrader with excellent in vivo efficacy. Bioorg Chem. 2022, 120, 105605. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-E.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [PubMed]
- Pfeifer, H.; Lange, T.; Wystub, S.; Wassmann, B.; Maier, J.; Binckebanck, A.; Giagounidis, A.; Stelljes, M.; Schmalzing, M.; Duhrsen, U.; et al. Prevalence and dynamics of bcr-abl kinase domain mutations during imatinib treatment differ in patients with newly diagnosed and recurrent bcr-abl positive acute lymphoblastic leukemia. Leukemia 2012, 26, 1475–1481. [Google Scholar] [CrossRef]
- Kaldalu, N.; Hauryliuk, V.; Tenson, T. Persisters—As elusive as ever. Appl. Microbiol. Biotechnol. 2016, 100, 6545–6553. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef]
- Haga, Y.; Marrocco, I.; Noronha, A.; Uribe, M.L.; Nataraj, N.B.; Sekar, A.; Drago-Garcia, D.; Borgoni, S.; Lindzen, M.; Giri, S.; et al. Host-Dependent Phenotypic Resistance to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2021, 81, 3862–3875. [Google Scholar] [CrossRef]
- Moghal, N.; Li, Q.; Stewart, E.L.; Navab, R.; Mikubo, M.; D’Arcangelo, E.; Martins-Filho, S.N.; Raghavan, V.; Pham, N.A.; Li, M.; et al. Single-Cell Analysis Reveals Transcriptomic Features of Drug-Tolerant Persisters and Stromal Adaptation in a Patient-Derived EGFR-Mutated Lung Adenocarcinoma Xenograft Model. J. Thorac. Oncol. 2023, 18, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Woolston, A.; Khan, K.; Spain, G.; Barber, L.J.; Griffiths, B.; Gonzalez-Exposito, R.; Hornsteiner, L.; Punta, M.; Patil, Y.; Newey, A.; et al. Genomic and Transcriptomic Determinants of Therapy Resistance and Immune Landscape Evolution during Anti-EGFR Treatment in Colorectal Cancer. Cancer Cell 2019, 36, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef] [PubMed]
- Ercan, D.; Xu, C.; Yanagita, M.; Monast, C.S.; Pratilas, C.A.; Montero, J.; Butaney, M.; Shimamura, T.; Sholl, L.; Ivanova, E.V.; et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 29–39. [Google Scholar] [CrossRef]
- Kashima, Y.; Shibahara, D.; Suzuki, A.; Muto, K.; Kobayashi, I.S.; Plotnick, D.; Udagawa, H.; Izumi, H.; Shibata, Y.; Tanaka, K.; et al. Single-Cell Analyses Reveal Diverse Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors in Lung Cancer. Cancer Res. 2021, 81, 4835–4848. [Google Scholar] [CrossRef]
- Arasada, R.R.; Shilo, K.; Yamada, T.; Zhang, J.; Yano, S.; Ghanem, R.; Wang, W.; Takeuchi, S.; Fukuda, K.; Katakami, N.; et al. Notch3-dependent beta-catenin signaling mediates EGFR TKI drug persistence in EGFR mutant NSCLC. Nat. Commun. 2018, 9, 3198. [Google Scholar] [CrossRef]
- Bousquet Mur, E.; Bernardo, S.; Papon, L.; Mancini, M.; Fabbrizio, E.; Goussard, M.; Ferrer, I.; Giry, A.; Quantin, X.; Pujol, J.L.; et al. Notch inhibition overcomes resistance to tyrosine kinase inhibitors in EGFR-driven lung adenocarcinoma. J. Clin. Investig. 2020, 130, 612–624. [Google Scholar] [CrossRef]
- Radman, M. SOS repair hypothesis: Phenomenology of an inducible DNA repair which is accompanied by mutagenesis. In Molecular Mechanisms for Repair of DNA: Part A; Basic Life Science Book Series; Springer: Boston, MA, USA, 1975; pp. 355–367. [Google Scholar] [CrossRef]
- wBaharoglu, Z.; Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef]
- Noronha, A.; Belugali Nataraj, N.; Lee, J.S.; Zhitomirsky, B.; Oren, Y.; Oster, S.; Lindzen, M.; Mukherjee, S.; Will, R.; Ghosh, S.; et al. AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer. Cancer Discov. 2022, 12, 2666–2683. [Google Scholar] [CrossRef]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.T.; Reyes, G.; Gries, K.; Ceylan, C.U.; Sharma, S.; Meurer, M.; Knop, M.; Chabes, A.; Hombauer, H. Alterations in cellular metabolism triggered by URA7 or GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis. Proc. Natl. Acad. Sci. USA 2017, 114, E4442–E4451. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Elledge, S.J. Identification of RNR4, encoding a second essential small subunit of ribonucleotide reductase in Saccharomyces cerevisiae. Mol. Cell Biol. 1997, 17, 6105–6113. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Adler, L.; Karathia, H.; Carmel, N.; Rabinovich, S.; Auslander, N.; Keshet, R.; Stettner, N.; Silberman, A.; Agemy, L.; et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 2018, 174, 1559–1570.e1522. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magri, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Hastings, P.J.; Rosenberg, S.M. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef]
- Garraway, L.A.; Verweij, J.; Ballman, K.V. Precision oncology: An overview. J. Clin. Oncol. 2013, 31, 1803–1805. [Google Scholar] [CrossRef]
- Lee, C.K.; Wu, Y.L.; Ding, P.N.; Lord, S.J.; Inoue, A.; Zhou, C.; Mitsudomi, T.; Rosell, R.; Pavlakis, N.; Links, M.; et al. Impact of Specific Epidermal Growth Factor Receptor (EGFR) Mutations and Clinical Characteristics on Outcomes after Treatment with EGFR Tyrosine Kinase Inhibitors Versus Chemotherapy in EGFR-Mutant Lung Cancer: A Meta-Analysis. J. Clin. Oncol. 2015, 33, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Papakyriakou, A.; Vourloumis, D.; Tzortzatou-Stathopoulou, F.; Karpusas, M. Conformational dynamics of the EGFR kinase domain reveals structural features involved in activation. Proteins Struct. Funct. Bioinform. 2009, 76, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Giri, S.; Simoni-Nieves, A.; Gupta, N.; Rudnitsky, A.; Haga, Y.; Romaniello, D.; Sekar, A.; Zerbib, M.; Oren, R.; et al. L858R emerges as a potential biomarker predicting response of lung cancer models to anti-EGFR antibodies: Comparison of osimertinib vs. cetuximab. Cell Rep. Med. 2023, 4, 101142. [Google Scholar] [CrossRef]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Dhillon, S. Capmatinib: First Approval. Drugs 2020, 80, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Tepotinib: First Approval. Drugs 2020, 80, 829–833. [Google Scholar] [CrossRef]
- Syed, Y.Y. Amivantamab: First Approval. Drugs 2021, 81, 1349–1353. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib after Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients with EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet. Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Shu, C.A.; Goto, K.; Ohe, Y.; Besse, B.; Lee, S.-H.; Wang, Y.; Griesinger, F.; Yang, J.C.-H.; Felip, E.; Sanborn, R.E.; et al. Amivantamab and lazertinib in patients with EGFR-mutant non–small cell lung (NSCLC) after progression on osimertinib and platinum-based chemotherapy: Updated results from CHRYSALIS-2. J. Clin. Oncol. 2022, 40, 9006. [Google Scholar] [CrossRef]
- Cho, B.C.; Felip, E.; Hayashi, H.; Thomas, M.; Lu, S.; Besse, B.; Sun, T.; Martinez, M.; Sethi, S.N.; Shreeve, S.M.; et al. MARIPOSA: Phase 3 study of first-line amivantamab + lazertinib versus osimertinib in EGFR-mutant non-small-cell lung cancer. Future Oncol. 2022, 18, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Gaborit, N.; Lindzen, M.; Salame, T.M.; Dall’Ora, M.; Sevilla-Sharon, M.; Abdul-Hai, A.; Downward, J.; Yarden, Y. Combining three antibodies nullifies feedback-mediated resistance to erlotinib in lung cancer. Sci. Signal 2015, 8, ra53. [Google Scholar] [CrossRef]
- Romaniello, D.; Mazzeo, L.; Mancini, M.; Marrocco, I.; Noronha, A.; Kreitman, M.; Srivastava, S.; Ghosh, S.; Lindzen, M.; Salame, T.M.; et al. A Combination of Approved Antibodies Overcomes Resistance of Lung Cancer to Osimertinib by Blocking Bypass Pathways. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5610–5621. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Romaniello, D.; Vaknin, I.; Drago-Garcia, D.; Oren, R.; Uribe, M.L.; Belugali Nataraj, N.; Ghosh, S.; Eilam, R.; Salame, T.M.; et al. Upfront admixing antibodies and EGFR inhibitors preempts sequential treatments in lung cancer models. EMBO Mol. Med. 2021, 13, e13144. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Digiacomo, G.; Flammini, L.; Barocelli, E.; Minari, R.; Naldi, N.; et al. Trastuzumab emtansine delays and overcomes resistance to the third-generation EGFR-TKI osimertinib in NSCLC EGFR mutated cell lines. J. Exp. Clin. Cancer Res. 2017, 36, 174. [Google Scholar] [CrossRef] [PubMed]
- Scharpenseel, H.; Hanssen, A.; Loges, S.; Mohme, M.; Bernreuther, C.; Peine, S.; Lamszus, K.; Goy, Y.; Petersen, C.; Westphal, M.; et al. EGFR and HER3 expression in circulating tumor cells and tumor tissue from non-small cell lung cancer patients. Sci. Rep. 2019, 9, 7406. [Google Scholar] [CrossRef]
- Kawano, O.; Sasaki, H.; Endo, K.; Suzuki, E.; Haneda, H.; Yukiue, H.; Kobayashi, Y.; Yano, M.; Fujii, Y. ErbB3 mRNA expression correlated with specific clinicopathologic features of Japanese lung cancers. J. Surg. Res. 2008, 146, 43–48. [Google Scholar] [CrossRef]
- Mancini, M.; Gal, H.; Gaborit, N.; Mazzeo, L.; Romaniello, D.; Salame, T.M.; Lindzen, M.; Mahlknecht, G.; Enuka, Y.; Burton, D.G.; et al. An oligoclonal antibody durably overcomes resistance of lung cancer to third-generation EGFR inhibitors. EMBO Mol. Med. 2018, 10, 294–308. [Google Scholar] [CrossRef]
- Romaniello, D.; Marrocco, I.; Belugali Nataraj, N.; Ferrer, I.; Drago-Garcia, D.; Vaknin, I.; Oren, R.; Lindzen, M.; Ghosh, S.; Kreitman, M.; et al. Targeting HER3, a Catalytically Defective Receptor Tyrosine Kinase, Prevents Resistance of Lung Cancer to a Third-Generation EGFR Kinase Inhibitor. Cancers 2020, 12, 2394. [Google Scholar] [CrossRef]
- Haikala, H.M.; Jänne, P.A. Thirty Years of HER3: From Basic Biology to Therapeutic Interventions. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 3528–3539. [Google Scholar] [CrossRef]
- Yonesaka, K.; Hirotani, K.; Kawakami, H.; Takeda, M.; Kaneda, H.; Sakai, K.; Okamoto, I.; Nishio, K.; Janne, P.A.; Nakagawa, K. Anti-HER3 monoclonal antibody patritumab sensitizes refractory non-small cell lung cancer to the epidermal growth factor receptor inhibitor erlotinib. Oncogene 2016, 35, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Yonesaka, K.; Hirotani, K.; von Pawel, J.; Dediu, M.; Chen, S.; Copigneaux, C.; Nakagawa, K. Circulating heregulin level is associated with the efficacy of patritumab combined with erlotinib in patients with non-small cell lung cancer. Lung Cancer 2017, 105, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Baik, C.; Su, W.C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discov. 2022, 12, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suda, K.; Shimizu, S.; Sakai, K.; Mizuuchi, H.; Tomizawa, K.; Takemoto, T.; Nishio, K.; Mitsudomi, T. Clinical, Pathological, and Molecular Features of Lung Adenocarcinomas with AXL Expression. PLoS ONE 2016, 11, e0154186. [Google Scholar] [CrossRef]
- Wu, Z.; Bai, F.; Fan, L.; Pang, W.; Han, R.; Wang, J.; Liu, Y.; Yan, X.; Duan, H.; Xing, L. Coexpression of receptor tyrosine kinase AXL and EGFR in human primary lung adenocarcinomas. Hum. Pathol. 2015, 46, 1935–1944. [Google Scholar] [CrossRef]
- Wang, F.; Liu, X.; Bartholdy, B.A.; Cheng, H.; Halmos, B. Blockade of AXL activation overcomes acquired resistance to EGFR tyrosine kinase inhibition in non-small cell lung cancer. Transl. Cancer Res. 2019, 8, 2425–2438. [Google Scholar] [CrossRef]
- Zaman, A.; Bivona, T.G. Targeting AXL in NSCLC. Lung Cancer Targets Ther. 2021, 12, 67–79. [Google Scholar] [CrossRef]
- Lee, Y.; Wang, Y.; James, M.; Jeong, J.H.; You, M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol. Carcinog. 2016, 55, 991–1001. [Google Scholar] [CrossRef]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Murata, Y.; Kusumoto, S.; Ando, K.; Ishida, H.; Ohnishi, T.; Sasaki, Y. Distinct Afatinib Resistance Mechanisms Identified in Lung Adenocarcinoma Harboring an EGFR Mutation. Mol. Cancer Res. 2017, 15, 915–928. [Google Scholar] [CrossRef]
- Wang, R.; Yamada, T.; Kita, K.; Taniguchi, H.; Arai, S.; Fukuda, K.; Terashima, M.; Ishimura, A.; Nishiyama, A.; Tanimoto, A.; et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat. Commun. 2020, 11, 4607. [Google Scholar] [CrossRef]
- Jacob Berger, A.; Gigi, E.; Kupershmidt, L.; Meir, Z.; Gavert, N.; Zwang, Y.; Prior, A.; Gilad, S.; Harush, U.; Haviv, I.; et al. IRS1 phosphorylation underlies the non-stochastic probability of cancer cells to persist during EGFR inhibition therapy. Nat. Cancer 2021, 2, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef] [PubMed]
- Varmus, H.; Unni, A.M.; Lockwood, W.W. How Cancer Genomics Drives Cancer Biology: Does Synthetic Lethality Explain Mutually Exclusive Oncogenic Mutations? Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
Figure 1.
Darwinian and non-Darwinian models of resistance to tyrosine kinase inhibitors (TKIs). According to the Darwinian model (upper part), a few TKI-resistant cells preexist in tumors. Hence, treatment with TKIs kills the sensitive cells and repopulates the whole tumor with drug-resistant cells (shown in red). According to the alternative model, exposure to the drug induces the generation of reactive oxygen species (ROS) and extensive cell death. Concurrently, the drug instigates epigenetic changes, especially epithelial-mesenchymal transition (EMT), in a small population of cells called drug tolerant persister cells (DTPs). Note that this transition is reversible and involves histone demethylation, up-regulation of vimentin, AXL and additional RTKs. Continuous exposure to the drug elevates antioxidants, along with the SOS-like system. This system comprises down-regulation of both DNA repair and high-fidelity DNA polymerases, which are replaced by a group of low-fidelity (error prone) polymerases. Thus, DTPs might irreversibly acquire resistance due to on-target or off-target new mutations.
Figure 1.
Darwinian and non-Darwinian models of resistance to tyrosine kinase inhibitors (TKIs). According to the Darwinian model (upper part), a few TKI-resistant cells preexist in tumors. Hence, treatment with TKIs kills the sensitive cells and repopulates the whole tumor with drug-resistant cells (shown in red). According to the alternative model, exposure to the drug induces the generation of reactive oxygen species (ROS) and extensive cell death. Concurrently, the drug instigates epigenetic changes, especially epithelial-mesenchymal transition (EMT), in a small population of cells called drug tolerant persister cells (DTPs). Note that this transition is reversible and involves histone demethylation, up-regulation of vimentin, AXL and additional RTKs. Continuous exposure to the drug elevates antioxidants, along with the SOS-like system. This system comprises down-regulation of both DNA repair and high-fidelity DNA polymerases, which are replaced by a group of low-fidelity (error prone) polymerases. Thus, DTPs might irreversibly acquire resistance due to on-target or off-target new mutations.

Figure 2.
The bacterial SOS system. The outlined error-prone DNA repair system contributes significantly to DNA changes in bacteria. Normally, the dimeric LexA repressor binds with the SOS boxes of 20 nucleotide pairs found in the operator region of SOS genes, thereby preventing their action. Activation of the SOS genes occurs after genotoxic stress and DNA damage. Upon damage, newly formed single-stranded DNA (ssDNA) fragments that accumulate at stalled replication forks bind with RecA (the orthologue of RAD51 in mammals), an inducer. RecA forms a filament around these ssDNA regions. The ssDNA/RecA complex promotes auto-cleavage of the LexA repressor to permit recruitment of RNA polymerase and facilitate expression of genes under LexA control. Collectively, the newly translated proteins inhibit DNA repair while increasing mutagenesis, which confers resistance to antibiotics.
Figure 2.
The bacterial SOS system. The outlined error-prone DNA repair system contributes significantly to DNA changes in bacteria. Normally, the dimeric LexA repressor binds with the SOS boxes of 20 nucleotide pairs found in the operator region of SOS genes, thereby preventing their action. Activation of the SOS genes occurs after genotoxic stress and DNA damage. Upon damage, newly formed single-stranded DNA (ssDNA) fragments that accumulate at stalled replication forks bind with RecA (the orthologue of RAD51 in mammals), an inducer. RecA forms a filament around these ssDNA regions. The ssDNA/RecA complex promotes auto-cleavage of the LexA repressor to permit recruitment of RNA polymerase and facilitate expression of genes under LexA control. Collectively, the newly translated proteins inhibit DNA repair while increasing mutagenesis, which confers resistance to antibiotics.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marrocco, I.; Yarden, Y. Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators. Cancers 2023, 15, 5009. https://doi.org/10.3390/cancers15205009
AMA Style
Marrocco I, Yarden Y. Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators. Cancers. 2023; 15(20):5009. https://doi.org/10.3390/cancers15205009
Chicago/Turabian StyleMarrocco, Ilaria, and Yosef Yarden. 2023. "Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators" Cancers 15, no. 20: 5009. https://doi.org/10.3390/cancers15205009
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.