

Synthesis and Biological Evaluation of Cyclobutane-Based β3 Integrin Antagonists: A Novel Approach to Targeting Integrins for Cancer Therapy
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General
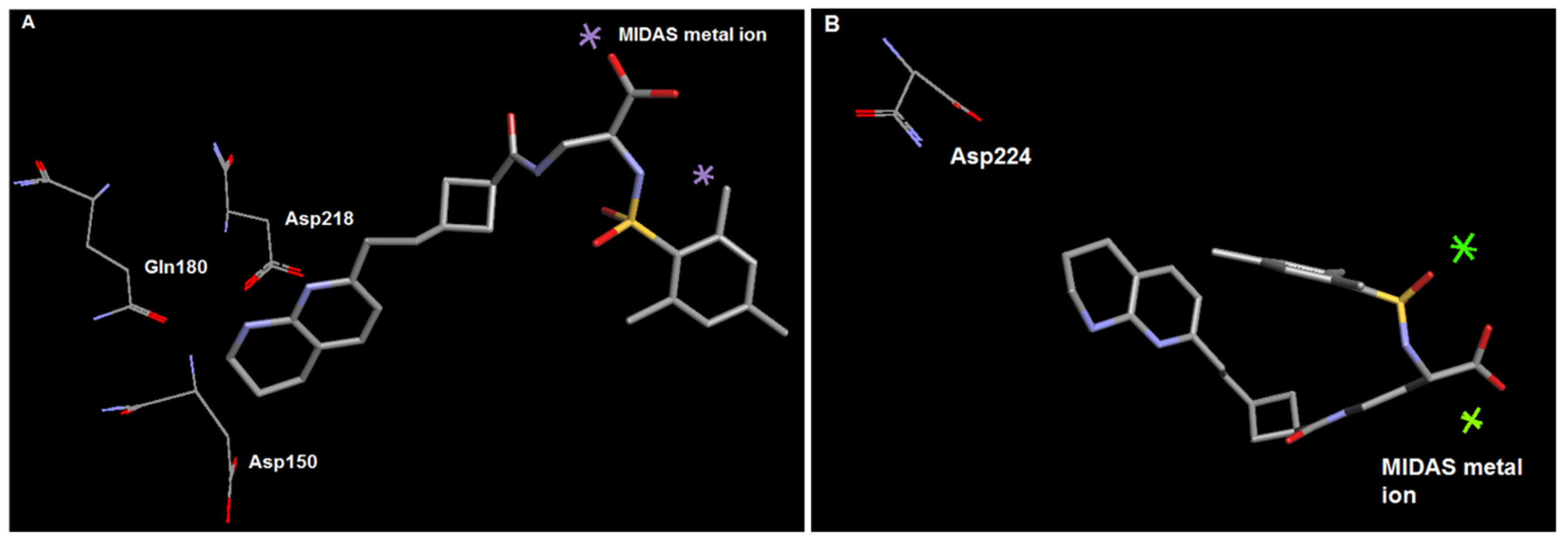
2.2. Molecular Modelling
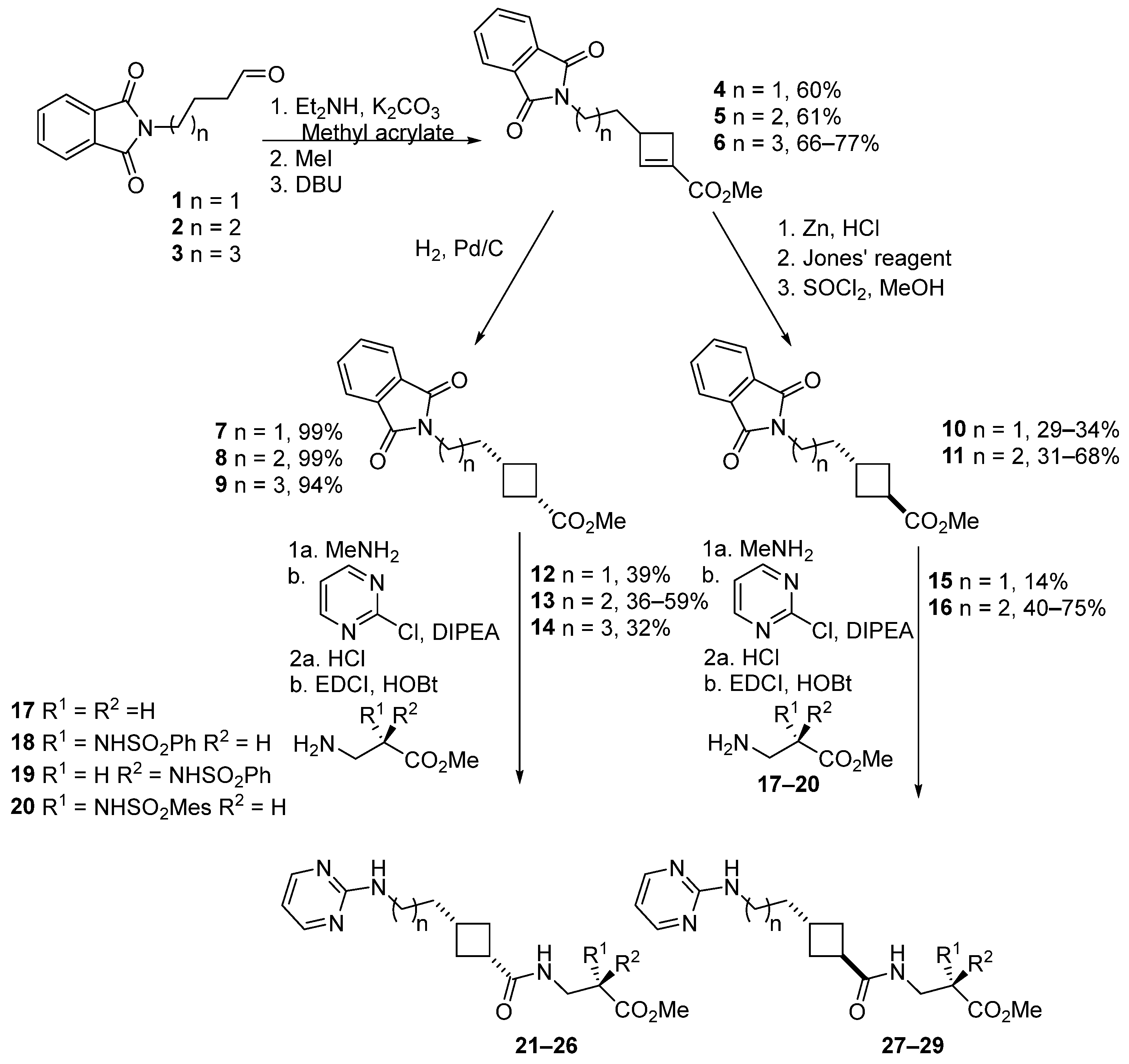
2.3. Chemical Synthesis
2.3.1. 4-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-butyraldehyde 1
2.3.2. 5-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-pentanal 2
2.3.3. General Procedure for Cyclobutene Synthesis
Methyl 3-(2-(1,3-dioxoisoindolin-2-yl)ethyl)cyclobut-1-enecarboxylate 4
2.3.4. General Procedure for Synthesis of Cis-Cyclobutanes
(1s,3s)-methyl 3-(2-(1,3-dioxoisoindolin-2-yl)ethyl)cyclobutanecarboxylate 7
2.3.5. General Procedure for the Synthesis of Trans-Cyclobutanes
3-[3-(1,3-Dioxo-1,3-dihydro-isoindol-2-yl)-propyl]-cyclobutanecarboxylic Acid Methyl Ester 11
(1s,3r)-Methyl 3-(3-oxobutyl)cyclobutanecarboxylate 36
2.3.6. General Procedure for Pyrimidine Incorporation
(1r,3s)-Methyl 3-(3-(pyrimidin-2-ylamino)propyl)cyclobutanecarboxylate 13
2.3.7. General Procedure for Friedlander Synthesis
(1r,3s)-Methyl 3-(2-(1,8-naphthyridin-2-yl)ethyl)cyclobutanecarboxylate 38
2.3.8. Aspartate Mimetic Synthesis
2.3.9. General Procedure for Esterification
3-Amino-2-benzenesulfonylamino-propionic Acid Methyl Ester 18, 19
2.3.10. General Procedure for Coupling Reactions
(S)-Methyl 3-((1s,3r)-3-(2-(pyrimidin-2-ylamino)ethyl)cyclobutanecarboxamido)-2-(2,4,6-trimethylphenylsulfonamido)propanoate 21
2.3.11. General Procedure for Tetrahydronapthyridine Synthesis
(S)-methyl 3-((1s,3r)-3-((5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)methyl)cyclobutanecarboxamido)-2-(2,4,6-trimethylphenylsulfonamido)propanoate 48
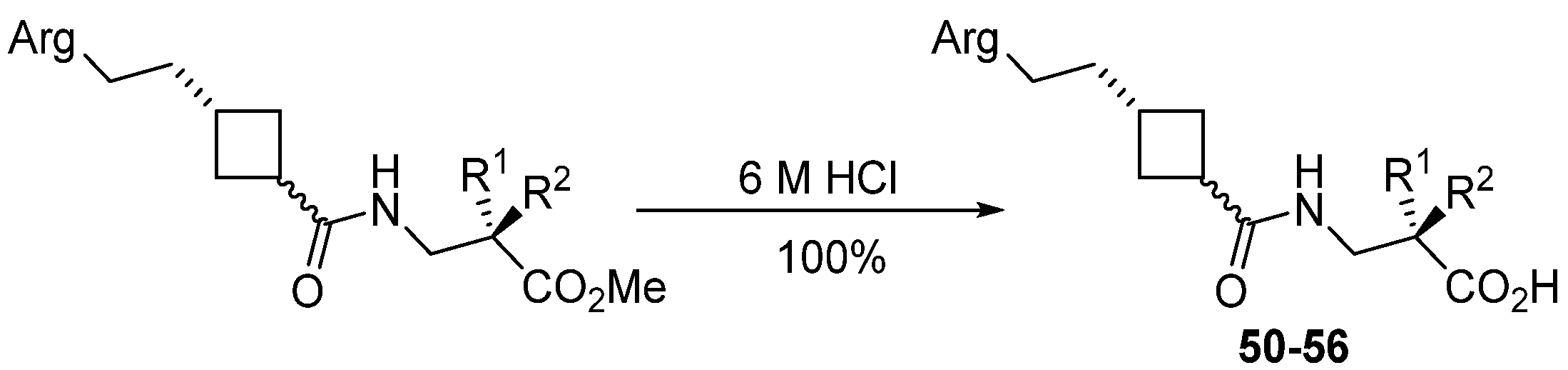
2.3.12. General Procedure for Preparation of Free Acids
2.4. ELISA Assay
2.5. Cell-Binding Assay
2.6. Migration Assay
2.7. Platelet Aggregation Assay
3. Results
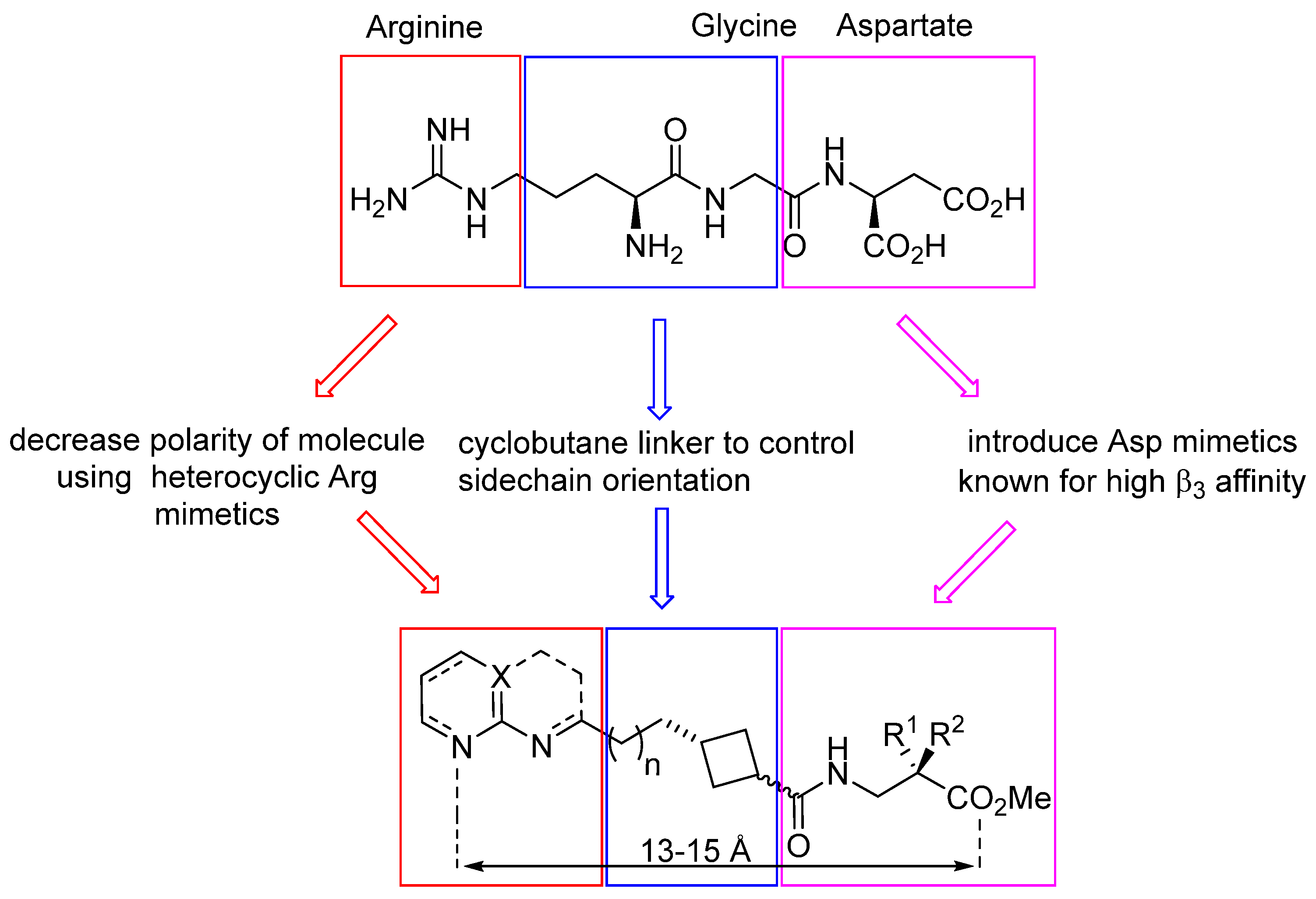
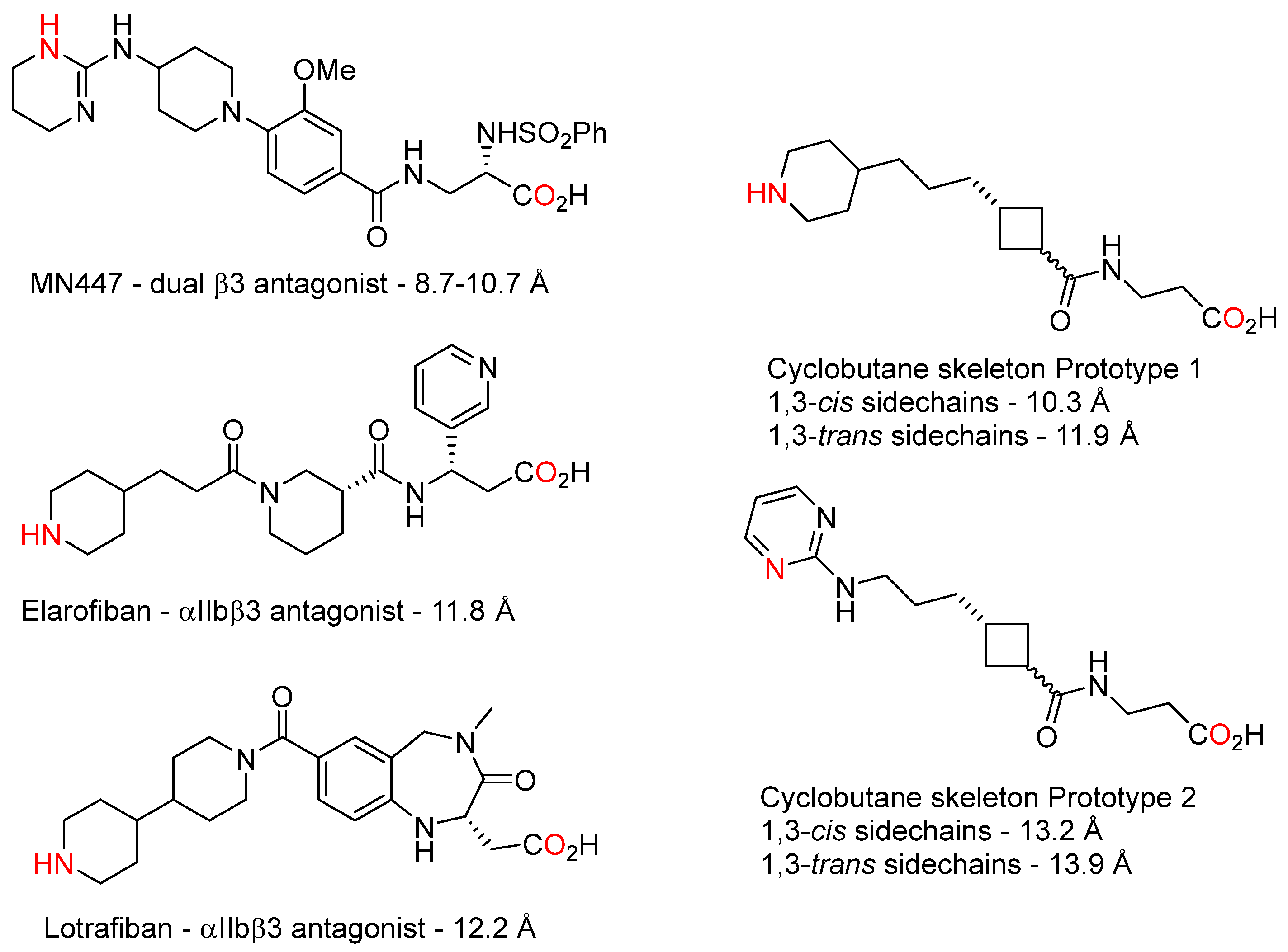
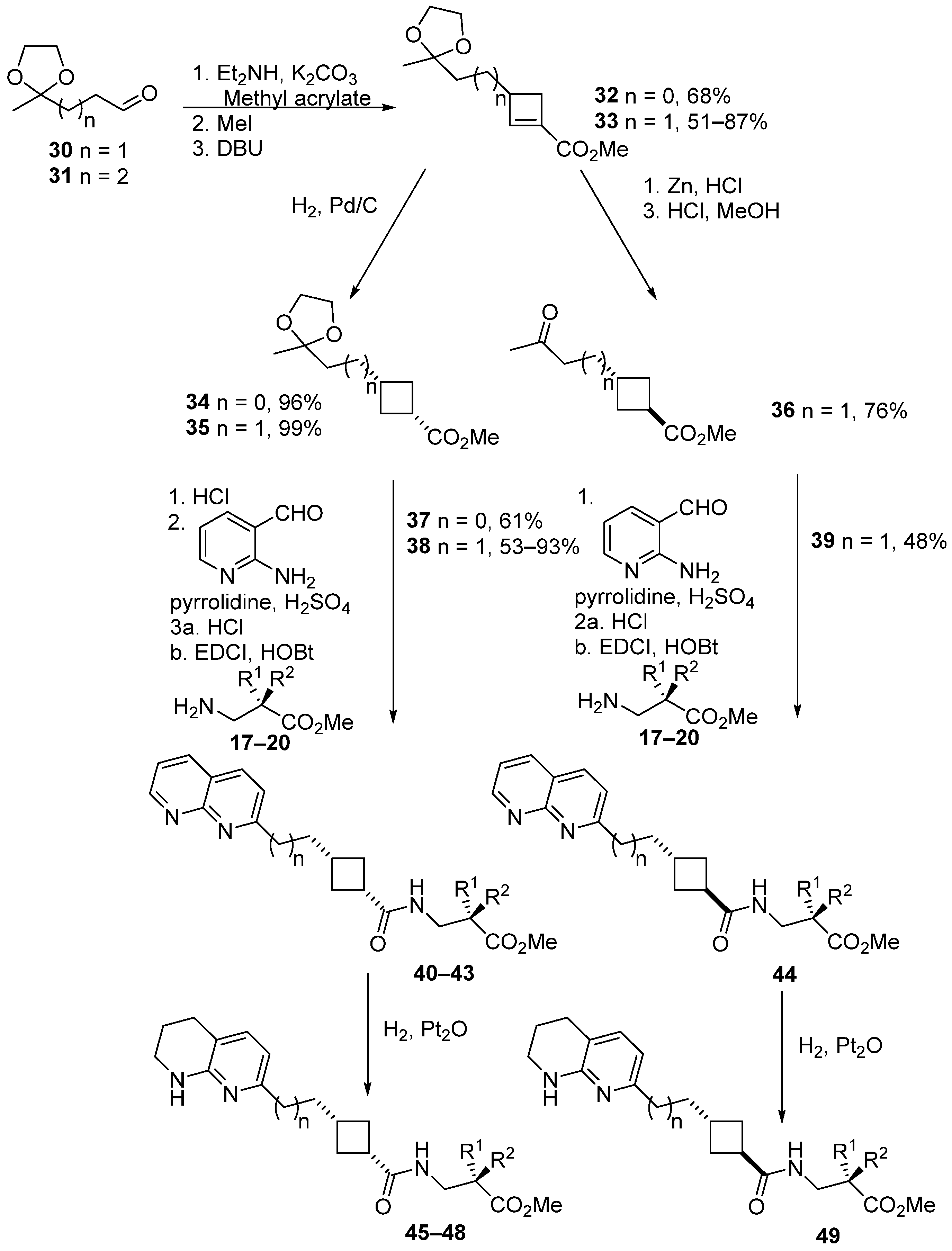
3.1. Design and Synthesis of Cyclobutane-Based RGD-Mimetics
3.2. Investigation of Anti-β3 Integrin Activity
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, M.J. Integrin Structure. Biochem. Soc. Trans. 2000, 28, 311–339. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharm. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2021, 21, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Fullard, J.F. The Role of the Platelet Glycoprotein IIb/IIIa in Thrombosis and Haemostasis. Curr. Pharm. Des. 2004, 10, 1567–1576. [Google Scholar] [CrossRef]
- Bennett, J.S. Structure and Function of the Platelet Integrin αIIbβ3. J. Clin. Investig. 2005, 115, 3363–3369. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.C. Requirement of Vascular Integrin αVβ3 for Angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef]
- Sheldrake, H.M.; Patterson, L.H. Function and antagonism of β3 integrins in the development of cancer therapy. Curr. Cancer Drug Targets 2009, 9, 519–540. [Google Scholar] [CrossRef]
- Mizejewski, G.J. Role of Integrins in Cancer: Survey of Expression Patterns. Proc. Soc. Exp. Biol. Med. 1999, 222, 125–138. [Google Scholar] [CrossRef]
- Hsu, M.-Y.; Shih, D.-T.; Meier, F.E.; Van Belle, P.; Hsu, J.-Y.; Elder, D.E.; Buck, C.A.; Herlyn, M. Adenoviral Gene Transfer of β3 Integrin Subunit Induces Conversion from Radial to Vertical Growth Phase in Primary Human Melanoma. Am. J. Pathol. 1998, 153, 1435–1442. [Google Scholar] [CrossRef]
- Petitclerc, E.; Stromblad, S.; von Schalscha, T.L.; Mitjans, F.; Piulats, J.; Montgomery, A.M.P.; Cheresh, D.A.; Brooks, P.C. Integrin ανβ3 Promotes M21 Melanoma Growth in Human Skin by Regulating Tumor Cell Survival. Cancer Res. 1999, 59, 2724–2730. [Google Scholar]
- Russo, M.A.; Paolillo, M.; Sanchez-Hernandez, Y.; Curti, D.; Ciusanti, E.; Serra, M.; Colombo, L.; Schinelli, S. A small-molecule RGD-integrin antagonist inhibits cell adhesion, cell migration and induces anoikis in glioblastoma cells. Int. J. Oncol. 2013, 42, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felding-Habermann, B.; O’Toole, T.E.; Smith, J.W.; Fransvea, E.; Ruggeri, Z.M.; Ginsberg, M.H.; Hughes, P.E.; Pampori, N.; Shattil, S.J.; Saven, A.; et al. Integrin Activation Controls Metastasis in Human Breast Cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Nasulewicz-Goldeman, A.; Uszczynska, B.; Szczaurska-Nowak, K.; Wietrzyk, J. siRNA-mediated silencing of integrin β3 expression inhibits the metastatic potential of B16 melanoma cells. Oncol. Rep. 2012, 28, 1567–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nip, J.; Shibata, H.; Loskutoff, D.J.; Cheresh, D.A.; Brodt, P. Human Melanoma Cells Derived from Lymphatic Metastases Use Integrin ανβ3 to Adhere to Lymph Node Vitronectin. J. Clin. Inv. 1992, 90, 1406–1413. [Google Scholar] [CrossRef]
- Pecheur, I.; Peyruchaud, O.; Serre, C.-M.; Guglielmi, J.; Voland, C.; Bourre, F.; Margue, C.; Cohen-Solal, M.; Buffet, A.; Clezardin, P. Integrin ανβ3 Expression Confers on Tumor Cells a Greater Propensity to Metastasize to Bone. FASEB J. 2002, 16, 1266–1268. [Google Scholar] [CrossRef]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchand, O.; Baron, R.; Clement-Lacroix, P.; Clezardin, P. Tumor ανβ3 Integrin Is a Therapeutic Target for Breast Cancer Bone Metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef] [Green Version]
- Elgavish, A.; Prince, C.; Chang, P.-L.; Lloyd, K.; Lindsey, R.; Reed, R. Osteopontin Stimulates a Subpopulation of Quiescent Human Prostate Epithelial Cells With High Proliferative Potential to Divide In Vitro. Prostate 1998, 35, 83–94. [Google Scholar] [CrossRef]
- Gordon, J.A.R.; Sodek, J.; Hunter, G.K.; Goldberg, H.A. Bone Sialoprotein Stimulates Focal Adhesion-Related Signaling Pathways: Role in Migration and Survival of Breast and Prostate Cancer Cells. J. Cell. Biochem. 2009, 107, 1118–1128. [Google Scholar] [CrossRef]
- De, S.; Chen, J.; Narizhneva, N.V.; Heston, W.; Brainard, J.; Sage, E.H.; Byzova, T.V. Molecular Pathway for Cancer Metastasis to Bone. J. Biol. Chem. 2003, 278, 39044–39050. [Google Scholar] [CrossRef] [Green Version]
- Barthel, S.R.; Hays, D.L.; Yazawa, E.M.; Opperman, M.; Walley, K.C.; Nimrichter, L.; Burdick, M.M.; Gillard, B.M.; Moser, M.T.; Pantel, K.; et al. Definition of molecular determinants of prostate cancer cell bone extravasation. Cancer Res. 2013, 73, 942–952. [Google Scholar] [CrossRef] [Green Version]
- Bakewell, S.J.; Nestor, P.; Prasad, S.; Tomasson, M.H.; Dowland, N.; Mehrotra, M.; Scarborough, R.M.; Kanter, J.; Abe, K.; Phillips, D.; et al. Platelet and Osteoclast β3 Integrins are Critical for Bone Metastasis. Proc. Natl. Acad. Sci. USA 2003, 100, 14205–14210. [Google Scholar] [CrossRef]
- Leblanc, R.; Lee, S.-C.; David, M.; Bordet, J.-C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clézardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin αvβ3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.R.; Zuka, M.; Lorger, M.; Tschan, M.; Torbett, B.E.; Zijlstra, A.; Quigley, J.P.; Staflin, K.; Eliceiri, B.P.; Krueger, J.S.; et al. Activated tumor cell integrin αvβ3 cooperates with platelets to promote extravasation and metastasis from the blood stream. Thromb. Res. 2016, 140 (Suppl. S1), S27–S36. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Lesperance, J.; Seguin, L.; Gozo, M.; Kato, S.; Franovic, A.; Yebra, M.; Shattil, S.J.; Cheresh, D.A. Integrin αvβ3 Drives Slug Activation and Stemness in the Pregnant and Neoplastic Mammary Gland. Dev. Cell 2014, 30, 295–308. [Google Scholar] [CrossRef] [Green Version]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin β3–KRAS–RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.Y.; Chen, Y.Q.; Fitzgerald, L.A.; Honn, K.V. Analysis of Integrin mRNA in Human and Rodent Tumor Cells. Biochem. Biophys. Res. Comm. 1991, 176, 108–113. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Trikha, M.; Gao, X.; Bazaz, R.; Porter, A.T.; Timar, J.; Honn, K.V. Ectopic Expression of Platelet Integrin αIIbβ3 in Tumor Cells from Various Species and Histological Origin. Int. J. Cancer 1997, 72, 642–648. [Google Scholar] [CrossRef]
- Trikha, M.; Timar, J.; Zacharek, A.; Nemeth, J.A.; Cai, Y.; Dome, B.; Somlai, B.; Raso, E.; Ladanyi, A.; Honn, K.V. Role for β3 Integrins in Human Melanoma Growth and Survival. Int. J. Cancer 2002, 101, 156–167. [Google Scholar] [CrossRef]
- Puerschel, W.C.; Gawaz, M.; Worret, W.-I.; Ring, J. Immunoreactivity of Glycoprotein IIb is Present in Metastasized but not in Non-metastasized Primary Malignant Melanoma. Br. J. Dermatol. 1996, 135, 883–887. [Google Scholar] [CrossRef]
- Trikha, M.; Timar, J.; Lundy, S.K.; Szekeres, K.; Tang, K.; Grignon, D.; Porter, A.T.; Honn, K.V. Human Prostate Carcinoma Cells Express Functional αIIbβ3 Integrin. Cancer Res. 1996, 56, 5071–5078. [Google Scholar]
- Trikha, M.; Raso, E.; Cai, Y.; Fazakas, Z.; Paku, S.; Porter, A.T.; Timar, J.; Honn, K.V. Role of αIIbβ3 Integrin in Prostate Cancer Metastasis. Prostate 1998, 35, 185–192. [Google Scholar] [CrossRef]
- Pontes-Júnior, J.; Reis, S.T.; Neves de Oliveira, L.C.; Sant’Anna, A.C.; Dall’Oglio, M.F.; Antunes, A.A.; Ribeiro-Filho, L.; Carvalho, P.A.; Cury, J.; Srougi, M.; et al. Association Between Integrin Expression and Prognosis in Localized Prostate Cancer. Prostate 2010, 70, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Echtler, K.; Konrad, I.; Lorenz, M.; Schneider, S.; Hofmaier, S.; Plenagl, F.; Stark, K.; Czermak, T.; Tirniceriu, A.; Eichhorn, M.; et al. Platelet GPIIb supports initial pulmonary retention but inhibits subsequent proliferation of melanoma cells during hematogenic metastasis. PLoS ONE 2017, 12, e0172788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felding-Habermann, B.; Fransvea, E.; O’Toole, T.E.; Manzuk, L.; Faha, B.; Hensler, M. Involvement of Tumor Cell Integrin ανβ3 in Hematogenous Metastasis of Human Melanoma Cells. Clin. Exp. Metastasis 2002, 19, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Dardik, R.; Kaufmann, Y.; Savion, N.; Rosenberg, N.; Shenkman, B.; Varon, D. Platelets Mediate Tumor Cell Adhesion to the Subendothelium under Flow Conditions: Involvement of Platelet GPIIb/IIIa and Tumor Cell αν Integrins. Int. J. Cancer 1997, 70, 201–207. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, X.-D.; Liu, Y.-K.; Ren, N.; Chen, J.; Zhao, Y. Platelets Promote the Adhesion of Human Hepatoma Cells with a Highly Metastatic Potential to Extracellular Matrix Protein: Involvement of Platelet P-selectin and GP IIb-IIIa. J. Cancer Res. Clin. Oncol. 2002, 128, 283–287. [Google Scholar] [CrossRef]
- Dashevsky, O.; Varon, D.; Brill, A. Platelet-derived microparticles promote invasiveness of prostate cancer cells via upregulation of MMP-2 production. Int. J. Cancer 2009, 124, 1773–1777. [Google Scholar] [CrossRef]
- Amirkhosravi, A.; Mousa, S.A.; Amaya, M.; Blaydes, S.; Desau, H.; Meyer, T.; Francis, J.L. Inhibition of Tumor Cell-Induced Platelet Aggregation and Lung Metastasis by the Oral GpIIb/IIIa Antagonist XV454. Thromb. Haemost. 2003, 90, 549–554. [Google Scholar] [CrossRef]
- Hardan, I.; Weiss, L.; Hershkoviz, R.; Greenspoon, N.; Alon, R.; Cahalon, L.; Reich, S.; Slavin, S.; Lider, O. Inhibition of Metastatic Cell Colonization in Murine Lungs and Tumor-Induced Morbidity by Non-peptidic Arg-Gly-Asp Mimetics. Int. J. Cancer 1993, 55, 1023–1028. [Google Scholar] [CrossRef]
- Boucharaba, A.; Serre, C.-M.; Gres, S.; Saulnier-Blache, J.S.; Bordet, J.-C.; Guglielmi, J.; Clezardin, P.; Peyruchaud, O. Platelet-derived Lysophosphatidic Acid Supports the Progression of Osteolytic Bone Metastases in Breast Cancer. J. Clin. Investig. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: Results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015, 17, 708–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legler, D.F.; Wiedle, G.; Ross, F.P.; Imhof, B.A. Superactivation of integrin ανβ3 by low antagonist concentrations. J. Cell Sci. 2001, 114, 1545–1553. [Google Scholar] [CrossRef]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nature Med. 2009, 15, 392–400. [Google Scholar] [CrossRef]
- Tucci, M.; Stucci, S.; Silvestris, F. Does Cilengitide deserve another chance? Lancet 2014, 15, e584–e585. [Google Scholar] [CrossRef]
- Cox, D.P. How not to discover a drug integrins. Expert Opin. Drug Discov. 2020, 16, 197–211. [Google Scholar] [CrossRef]
- Chew, D.P.; Bhatt, D.L.; Topol, E.J. Oral Glycoprotein IIb/IIIa Inhibitors. Why Don’t They Work? Amer. J. Cardiovasc. Drugs 2012, 1, 421–428. [Google Scholar] [CrossRef]
- Baba, K.; Aga, Y.; Nakanishi, T.; Motoyama, T.; Ueno, H. UR-3216: A Manageable Oral GPIIb/IIIa Antagonist. Cardiovasc. Drug Rev. 2001, 19, 25–40. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kubota, D.; Yamamoto, M.; Kuroda, C.; Iguchi, M.; Koyanagi, A.; Murakami, S.; Ajito, K. Tricyclic pharmacophore-based molecules as novel integrin ανβ3 antagonists. Part 2: Synthesis of potent ανβ3/αIIbβ3 dual antagonists. Bioorg. Med. Chem. 2006, 14, 2109–2130. [Google Scholar] [CrossRef]
- Meyer dos Santos, S.; Kuczka, K.; Picard-Willems, B.; Nelson, K.; Klinkhardt, U.; Harder, S. The integrin antagonist, cilengitide, is a weak inhibitor of αIIbβ3 mediated platelet activation and inhibits platelet adhesion under flow. Platelets 2015, 26, 59–66. [Google Scholar] [CrossRef]
- Carter, R.Z.; Micocci, K.C.; Natoli, A.; Redvers, R.P.; Paquet-Fifield, S.; Martin, A.C.B.M.; Denoyer, D.; Ling, X.; Kim, S.-H.; Tomasin, R.; et al. Tumour but not stromal expression of β3 integrin is essential, and is required early, for spontaneous dissemination of bone-metastatic breast cancer. J. Pathol. 2015, 235, 760–772. [Google Scholar] [CrossRef]
- Parvani, J.G.; Gujrati, M.D.; Mack, M.A.; Schiemann, W.P.; Lu, Z.-R. Silencing β3 integrin by Targeted ECO/siRNA Nanoparticles Inhibits EMT and Metastasis of Triple Negative Breast Cancer. Cancer Res. 2015, 75, 2316–2325. [Google Scholar] [CrossRef] [Green Version]
- Bosnjak, M.; Dolinsek, T.; Cemazar, M.; Kranjc, S.; Blagus, T.; Markelc, B.; Stimac, M.; Zavrsnik, J.; Kamensek, U.; Heller, L.; et al. Gene electrotransfer of plasmid AMEP, an Integrin targeted therapy, has antitumor and antiangiogenic action in murine B16 melanoma. Gene Ther. 2015, 22, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Trikha, M.; Zhou, Z.; Timar, J.; Raso, E.; Kennel, M.; Emmell, E.; Nakada, M. Multiple Roles for Platelet GPIIb/IIIa and αvβ3 Integrins in Tumor Growth, Angiogenesis, and Metastasis. Cancer Res. 2002, 62, 2824–2833. [Google Scholar]
- Nemeth, J.A.; Cher, M.L.; Zhou, Z.; Mullins, C.; Bhagat, S.; Trikha, M. Inhibition of ανβ3 Integrin Reduces Angiogenesis, Bone Turnover, and Tumor Cell Proliferation in Experimental Prostate Cancer Bone Metastases. Clin. Exp. Metastasis 2003, 20, 413–420. [Google Scholar] [CrossRef]
- Engebraaten, O.; Trikha, M.; Juell, S.; Garman-Vik, S.; Fodstad, O. Inhibition of In Vivo Tumour Growth by the Blocking of Host alpha(v)beta(3) and alpha IIb beta(3) Integrins. Anticancer Res. 2009, 29, 131–137. [Google Scholar] [PubMed]
- Zhang, W.; Dang, S.; Hong, T.; Tang, J.; Fan, J.; Bu, D.; Sun, Y.; Wang, Z.; Wisniewski, T. A humanized single-chain antibody against beta 3 integrin inhibits pulmonary metastasis by preferentially fragmenting activated platelets in the tumor microenvironment. Blood 2012, 120, 2889–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, N.; Vassy, J.; Lebos, C.; Arbeille, B.; Legrand, C.; Fauvel-Lefeve, F. Breast Adenocarcinoma Cell Adhesion to the Vascular Subendothelium in Whole Blood and under Flow Conditions: Effects of αvβ3 and αIIbβ3 Antagonists. Clin. Exp. Metastasis 2004, 21, 553–561. [Google Scholar] [CrossRef]
- Sheldrake, H.M.; Wallace, T.W.; Wilson, C.P. Functionalized Cyclobutenes via Multicomponent Thermal [2+2] Cycloaddition Reactions. Org. Lett. 2005, 7, 4233–4236. [Google Scholar] [CrossRef]
- Throup, A.; Patterson, L.H.; Sheldrake, H.M. Intramolecular thermal stepwise [2+2] cycloadditions: Investigation of a stereoselective synthesis of [n.2.0]-bicyclolactones. Org. Biomol. Chem. 2016, 14, 9554–9559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.R.; Di Fruscia, P.; Lucas, S.C.C.; Michaelides, I.N.; Nelson, J.E.; Ian, S.a.R.; Whitehurst, B.C. Put a ring on it: Application of small aliphatic rings in medicinal chemistry. RSC Med. Chem. 2021, 12, 448–471. [Google Scholar] [CrossRef] [PubMed]
- Kolk, M.R.v.d.; Janssen, M.A.C.H.; Rutjes, F.P.J.T.; Blanco-Ania, D. Cyclobutanes in Small-Molecule Drug Candidates. ChemMedChem 2022, 17, e202200020. [Google Scholar] [CrossRef] [PubMed]
- Bietencourt-Ferreira, G.; Filgueira de Azevedo, W., Jr. Molecular Docking Simulations with ArgusLab. In Docking Screens for Drug Discovery; Filgueira de Azevedo, W., Jr., Ed.; Methods in Molecular Biology; Springer Science + Business Media LLC: Berlin, Germany, 2019; Volume 2053. [Google Scholar] [CrossRef]
- Sall, D.J.; Arfsten, A.E.; Bastian, J.A.; Denney, M.L.; Harms, C.S.; McCowan, J.R.; Morin, J.M.; Rose, J.W.; Scarborough, R.M.; Smyth, M.S.; et al. Use of conformationally restricted benzamidines as arginine surrogates in the design of platelet GPIIb-IIIa receptor antagonists. J. Med. Chem. 1997, 40, 2843–2857. [Google Scholar] [CrossRef]
- Shindo, M.; Matsumoto, K.; Sato, Y.; Shishido, K. The First Tandem [2+2] Cycloaddition-Michael Reaction Using Ynolates: Facile Construction of Substituted Carbocycles. Org. Lett. 2001, 3, 2029–2031. [Google Scholar] [CrossRef] [PubMed]
- Egbertson, M.S.; Cook, J.J.; Bednar, B.; Prugh, J.D.; Bednar, R.A.; Gaul, S.L.; Gould, R.J.; Hartman, G.D.; Homnick, C.F.; Holahan, M.A.; et al. Non-Peptide GPIIb/IIIa Inhibitors. 20. Centrally Constrained Thienothiophene a-Sulfonamides Are Potent, Long Acting in Vivo Inhibitors of Platelet Aggregation. J. Med. Chem. 1999, 42, 2409–2421. [Google Scholar] [CrossRef]
- Pitts, W.J.; Wityak, J.; Smallheer, J.M.; Tobin, A.E.; Jetter, J.W.; Buynitsky, J.S.; Harlow, P.P.; Solomon, K.A.; Corjay, M.H.; Mousa, S.A.; et al. Isoxazolines as Potent Antagonists of the Integrin ανβ3. J. Med. Chem. 2000, 43, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.E.; Sielecki, T.M.; Wityak, J.; Pinto, D.J.; Batt, D.G.; Frietze, W.E.; Liu, J.; Tobin, A.E.; Orwat, M.J.; Di Meo, S.V.; et al. Orally Active Isoxazoline Glycoprotein IIb/IIIa Antagonists with Extended Duration of Action. J. Med. Chem. 1999, 42, 1178–1192. [Google Scholar] [CrossRef]
- Harris, T.D.; Barrett, J.A.; Carpenter, A.P.; Rajopadhye, M. Vitronectin Receptor Antagonist Pharmaceuticals. Patent USP7018611B2, 28 March 2006. [Google Scholar]
- Stragies, R.; Osterkamp, F.; Zischinsky, G.; Vossmeyer, D.; Kalkhof, H.; Reimer, U.; Zahn, G. Design and Synthesis of a New Class of Selective Integrin α5β1 Antagonists. J. Med. Chem. 2007, 50, 3786–3794. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Takagi, J.; Coller, B.S.; Wang, J.-H.; Springer, T.A. Structural Basis for Allostery in Integrins and Binding to Fibrinogen-mimetic Therapeutics. Nature 2004, 432, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.-P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal Structure of the Extracellular Segment of Integrin ανβ3 in Complex with an Arg-Gly-Asp Ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef]
- Pfaff, M.; Tangemann, K.; Müller, M.; Gurrath, M.; Müller, G.; Kessler, H.; Timpl, R.; Engel, J. Selective Recognition of Cyclic RGD Peptides of NMR Defined Conformation by αIIbβ3, ανβ3, and α5β1 Integrins. J. Biol. Chem. 1994, 269, 20233–20238. [Google Scholar] [CrossRef] [PubMed]
- Feuston, B.P.; Culberson, J.C.; Duggan, M.E.; Hartman, G.D.; Leu, C.-T.; Rodan, G.A. Binding Model for Nonpeptide Antagonists of ανβ3 Integrin. J. Med. Chem. 2002, 45, 5640–5648. [Google Scholar] [CrossRef] [PubMed]
- Kubota, D.; Ishikawa, M.; Yamamoto, M.; Murakami, S.; Hachisu, M.; Katano, K.; Ajito, K. Tricyclic pharmacophore-based molecules as novel integrin ανβ3 antagonists. Part 1: Design and synthesis of a lead compound exhibiting ανβ3/αIIbβ3 dual antagonistic activity. Bioorg. Med. Chem. 2006, 14, 2089–2108. [Google Scholar] [CrossRef]
- Thompson, M.A. Arguslab, Version 4.0.1; Planaria Software LLC: Seattle, WA, USA, 2021. [Google Scholar]
- Shannon, K.E.; Keene, J.L.; Settle, S.L.; Duffin, T.D.; Nickols, M.A.; Westlin, M.; Schroeter, S.; Ruminski, P.G.; Griggs, D.W. Anti-Metastatic Properties of RGD-Peptidomimetic Agents S137 and S247. Clin. Exp. Metastasis 2004, 21, 129–138. [Google Scholar] [CrossRef]
- Batt, D.G.; Petraitis, J.J.; Houghton, G.C.; Modi, D.P.; Cain, G.A.; Corjay, M.H.; Mousa, S.A.; Bouchard, P.J.; Forsythe, M.S.; Harlow, P.P.; et al. Disubstituted Indazoles as Potent Antagonists of the Integrin ανβ3. J. Med. Chem. 2000, 43, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Dehmlow, E.V.; Schmidt, S. Synthese von stereoisomeren 3-substituierten Cyclobutancarbonsaure-Derivaten. Liebigs Ann. Chem. 1990, 1990, 411–414. [Google Scholar] [CrossRef]
- Heidler, P.; Zohrabi-Kalantari, V.; Kaiser, M.; Brun, R.; Emmrich, T.; Link, A. Inhibitors of adenosine consuming parasites through polymer-assisted solution phase synthesis of lipophilic 5’-amido-5’-deoxyadenosine derivatives. Bioorg. Med. Chem. 2009, 17, 1428–1436. [Google Scholar] [CrossRef]
- Duggan, M.E.; Duong, L.T.; Fisher, J.E.; Hamill, T.G.; Hoffman, W.F.; Huff, J.R.; Ihle, N.C.; Leu, C.-T.; Nagy, R.M.; Perkins, J.J.; et al. Nonpeptide ανβ3 Antagonists. 1. Transformation of a Potent, Integrin-Selective αIIbβ3 Antagonist into a Potent ανβ3 Antagonist. J. Med. Chem. 2000, 43, 3736–3745. [Google Scholar] [CrossRef]
- Tilley, J.W.; Sidduri, A.; Lou, J.; Kaplan, G.; Tare, N.; Cavallo, G.; Frank, K.; Pamidimukkala, A.; Choi, D.S.; Gerber, L.; et al. Identification of N-acyl 4-(3-pyridonyl)phenylalanine derivatives and their orally active prodrug esters as dual acting α4β1 and α4β7 receptor antagonists. Bioorg. Med. Chem. Lett. 2013, 23, 1036–1040. [Google Scholar] [CrossRef]
- Mousa, S.A.; Bozarth, J.M.; Naik, U.P.; Slee, A. Platelet GPIIb/IIIa binding characteristics of small molecule RGD mimetic: Distinct binding profile for Roxifiban. Brit. J. Pharmacol. 2001, 133, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, N.S.; Panzer-Knodle, S.G.; Salyers, A.K.; Taite, B.B.; Szalony, J.A.; Haas, N.F.; King, L.W.; Zablocki, J.A.; Keller, B.T.; Broschat, K.; et al. SC-54684A: An Orally Active Inhibitor of Platelet Aggregation. Circulation 1995, 91, 403–410. [Google Scholar] [CrossRef]
- Janji, B.; Melchior, C.; Gouon, V.; Vallar, L.; Kieffer, N. Autocrine TGF-b-regulated expression of adhesion receptors and integrin-linked kinase in HT-144 melanoma cells correlates with their metastatic phenotype. Int. J. Cancer 1999, 83, 255–262. [Google Scholar] [CrossRef]
- Putnam, A.J.; Schulz, V.V.; Freiter, E.M.; Bill, H.M.; Miranti, C.K. Src, PKCalpha, and PKCdelta are required for alphavbeta3 integrin-mediated metastatic melanoma invasion. Cell Commun. Signal. 2009, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, A.; Paoletti, I.; Silvestri, I.; Buommino, E.; Carreiro, M.V. Early vitronectin receptor downregulation in a melanoma cell line during all-trans retinoic acid-induced apoptosis. Br. J. Dermatol. 2003, 148, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.-J.; Mas-Moruno, C.; et al. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-binding Integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [Green Version]
- Hieken, T.J.; Farolan, M.; Ronan, S.G.; Shilkaitis, A.; Wild, L.; Das Gupta, T.K. β3 Integrin Expression in Melanoma Predicts Subsequent Metastasis. J. Surg. Res. 1996, 63, 169–173. [Google Scholar] [CrossRef]
- Hieken, T.J.; Ronan, S.G.; Farolan, M.; Shilkaitis, A.L.; Das Gupta, T.K. Molecular Prognostic Markers in Intermediate Thickness Cutaneous Malignant Melanoma. Cancer 1999, 85, 375–382. [Google Scholar] [CrossRef]
- Watson-Hurst, K.; Becker, D. The Role of N-Cadherin, MCAM and β3 integrin in Melanoma Progression, Proliferation, Migration and Invasion. Cancer Biol. Ther. 2006, 5, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Nurzat, Y.; Su, W.; Min, P.; Li, K.; Xu, H.; Zhang, Y. Identification of Therapeutic Targets and Prognostic Biomarkers Among Integrin Subunits in the Skin Cutaneous Melanoma Microenvironment. Front. Oncol. 2021, 11, 751875. [Google Scholar] [CrossRef] [PubMed]
- Neto, D.S.; Pantaleao, L.; Soares de Sa, B.C.; Landman, G. Alpha-v-beta3 Integrin Expression in Melanocytic Nevi and Cutaneous Melanoma. J. Cutan. Pathol. 2007, 34, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Kopatz, V.; Selzer, E. Quantitative and qualitative analysis of integrin subtype expression in melanocytes and melanoma cells. J. Recept. Signal Transduct. 2020, 40, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Delcommenne, M.; Streuli, C.H. Control of Integrin Expression by Extracellular Matrix. J. Biol. Chem. 1995, 270, 26794–26801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, G.D.; Egbertson, M.S.; Halczenko, W.; Laswell, W.L.; Duggan, M.E.; Smith, R.L.; Naylor, A.M.; Manno, P.D.; Lynch, R.J.; Zhang, G.; et al. Non-Peptide Fibrinogen Receptor Antagonists. 1. Discovery and Design of Exosite Inhibitors. J. Med. Chem. 1992, 35, 4640–4642. [Google Scholar] [CrossRef] [PubMed]
- Egbertson, M.S.; Chang, C.T.-C.; Duggan, M.E.; Gould, R.J.; Halczenko, W.; Hartman, G.D.; Laswell, W.L.; Lynch, J.J.J.; Lynch, R.J.; Manno, P.D.; et al. Non-Peptide Fibrinogen Receptor Antagonists. 2. Optimization of a Tyrosine Template as a Mimic for Arg-Gly-Asp. J. Med. Chem. 1994, 37, 2537–2551. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Ranzani, M.; Alifrangis, C.; Perna, D.; Dutton-Regester, K.; Pritchard, A.; Wong, K.; Rashid, M.; Robles-Espinoza, C.D.; Hayward, N.K.; McDermott, U.; et al. BRAF/NRAS wild-type melanoma, NF1 status and sensitivity to trametinib. Pigment Cell Melanoma Res. 2014, 28, 117–119. [Google Scholar] [CrossRef] [Green Version]
- Sloan, E.K.; Pouliot, N.; Stanley, K.L.; Chia, J.; Moseley, J.M.; Hards, D.K.; Anderson, R.L. Tumor-specific expression of αvβ3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006, 8, R20. [Google Scholar] [CrossRef]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef]
- Altei, W.F.; Pachane, B.C.; dos Santos, P.K.; Ribeiro, L.N.M.; Sung, B.H.; Weaver, A.M.; Selistre-de-Araújo, H.S. Inhibition of αvβ3 integrin impairs adhesion and uptake of tumor-derived small extracellular vesicles. Cell Commun. Signal. 2020, 18, 158. [Google Scholar] [CrossRef]
- Joy, S.; Nair, P.S.; Hariharan, R.; Pillai, M.R. Detailed comparison of the protein-ligand docking efficiencies of GOLD, a commercial package and ArgusLab, a licensable freeware. Silico Biol. 2006, 6, 601–605. [Google Scholar]
- Devasthale, P.; Moore, F.; Zhao, G.; Poieniazek, S.N.; Selvakumar, K.; Dhanusu, S.; Panda, M.; Marcin, L.R. WO2018/089355 A1: Cyclobutane- and Azetidinine Containing Mono and Spirocyclic Compounds as Alpha v Integrin Inhibitors; World Intellectual Property Organization: Geneva, Switzerland, 2018. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ester | Yield/% | Acid | Arg Mimetic | n | R1 | R2 |
|---|---|---|---|---|---|---|
| 21 ICT9019 | 34.5 | 50 ICT9030 | Pyrimidine | 1 | NHSO2Mes | H |
| 22 ICT9003 | 65 | - | Pyrimidine | 2 | H | H |
| 23 ICT9023 | 21 | 51 ICT9028 | Pyrimidine | 2 | NHSO2Ph | H |
| 24 ICT9020 | 18 | - | Pyrimidine | 2 | NHSO2Mes | H |
| 25 ICT9021 | 28 | - | Pyrimidine | 2 | H | NHSO2Ph |
| 26 ICT9018 | 59 | - | Pyrimidine | 3 | NHSO2Mes | H |
| 27 ICT9024 | 38 | 52 ICT9031 | Pyrimidine T | 1 | NHSO2Mes | H |
| 28 ICT9025 | 35 | 53 ICT9090 | Pyrimidine T | 2 | NHSO2Mes | H |
| 29 ICT9026 | 41 | 54 ICT9029 | Pyrimidine T | 2 | NHSO2Ph | H |
| 40 ICT9054 | 38 | - | Naphthyridine | 1 | NHSO2Ph | H |
| 41 ICT9053 | 28 | 55 ICT9063 | Naphthyridine | 1 | NHSO2Mes | H |
| 42 ICT9065 | 55 | - | Naphthyridine | 1 | H | H |
| 43 ICT9079 | 23.5 | - | Naphthyridine | 0 | NHSO2Mes | H |
| 44 ICT9061 | 27 | - | Naphthyridine T | 1 | NHSO2Mes | H |
| 45 ICT9057 | 38 | - | THN | 1 | NHSO2Ph | H |
| 46 ICT9055 | 69 | 56 ICT9064 | THN | 1 | NHSO2Mes | H |
| 47 ICT9066 | 77 | - | THN | 1 | H | H |
| 48 ICT9080 | 87.5 | - | THN | 0 | NHSO2Mes | H |
| 49 ICT9062 | 62 | - | THN T | 1 | NHSO2Mes | H |
| Compound | Arg Mimetic | n | R1 | R2 | Acid/Ester | Adhesion % Inhibition @ 50 μM | Adhesion % Inhibition @ 5 μM | Adhesion IC50/µM | Migration IC50/µM |
|---|---|---|---|---|---|---|---|---|---|
| ICT9019 21 | Pyrimidine | 1 | NHSO2Mes | H | Ester | 48.3 ± 4.1 | 35.2 ± 19.1 | 50 ± 5 | 9.5 ± 0.9 |
| ICT9003 22 | Pyrimidine | 2 | H | H | Ester | a | a | - | 4.8 ± 0.2 |
| ICT9023 23 | Pyrimidine | 2 | NHSO2Ph | H | Ester | 7.4 ± 15.1 | b | - | - |
| ICT9020 24 | Pyrimidine | 2 | NHSO2Mes | H | Ester | - | 19.1 ± 14.2 | - | >10 |
| ICT9021 25 | Pyrimidine | 2 | H | NHSO2Ph | Ester | 22.2 ± 22.3 | b | - | - |
| ICT9018 26 | Pyrimidine | 3 | NHSO2Mes | H | Ester | - | 20.9 ± 27.5 | - | >10 |
| ICT9024 27 | Pyrimidine T | 1 | NHSO2Mes | H | Ester | 94.2 ± 4.1 | 39.4 ± 17.1 | 13 ± 3 | - |
| ICT9025 28 | Pyrimidine T | 2 | NHSO2Mes | H | Ester | 92.5 ± 8.1 | 0 ± 19.6 | 38 ± 11 | - |
| ICT9026 29 | Pyrimidine T | 2 | NHSO2Ph | H | Ester | 92.4 ± 8.0 | 0 ± 17.3 | >50 | - |
| ICT9054 40 | Naphthyridine | 1 | NHSO2Ph | H | Ester | - | 48.7 ± 30.6 | - | - |
| ICT9053 41 | Naphthyridine | 1 | NHSO2Mes | H | Ester | - | 27.5 ± 32.9 | >5 | >10 |
| ICT9065 42 | Naphthyridine | 1 | H | H | Ester | - | - | - | - |
| ICT9079 43 | Naphthyridine | 0 | NHSO2Mes | H | Ester | - | 33.5 ± 26.2 | - | - |
| ICT9061 44 | Naphthyridine T | 1 | NHSO2Mes | H | Ester | - | 32.4 ± 16.1 | - | - |
| ICT9057 45 | THN | 1 | NHSO2Ph | H | Ester | - | 59.5 ± 24.0 | - | 1.0 ± 0.09 |
| ICT9055 46 | THN | 1 | NHSO2Mes | H | Ester | - | 98.4 ± 1.9 | 0.34 ± 0.33 | <0.1 |
| ICT9066 47 | THN | 1 | H | H | Ester | 22.4 ± 29.3 | b | - | - |
| ICT9080 48 | THN | 0 | NHSO2Mes | H | Ester | - | 61.5 ± 11.1 | - | - |
| ICT9062 49 | THNT | 1 | NHSO2Mes | H | Ester | - | 72.3 ± 9.4 | - | 0.15 ± 0.03 |
| ICT9030 50 | Pyrimidine | 1 | NHSO2Mes | H | Acid | 51 ± 9.0 | 4.5 ± 19.6 | 50 ± 7 | - |
| ICT9028 51 | Pyrimidine | 2 | NHSO2Ph | H | Acid | 31.8 ± 10.0 | b | >50 | - |
| ICT9031 52 | Pyrimidine T | 1 | NHSO2Mes | H | Acid | 11.1 ± 2.4 | b | >50 | - |
| ICT9090 53 | Pyrimidine T | 2 | NHSO2Mes | H | Acid | 68.9 ± 10.9 | - | 11.0 ± 11.6 | - |
| ICT9029 54 | Pyrimidine T | 2 | NHSO2Ph | H | Acid | 42.5 ± 14.1 | b | >50 | - |
| ICT9063 55 | Naphthyridine | 1 | NHSO2Mes | H | Acid | - | 20.7 ± 11.9 | >5 | - |
| ICT9064 56 | THN | 1 | NHSO2Mes | H | Acid | - | 76.1 ± 20.4 | 3.7 ± 2.3 | 0.2 ± 0.06 |
| cRGDfV | - | 61.5 ± 15.1 | 2.1 ± 0.8 | 4.0 ± 0.07 | |||||
| RGDS | 45.8 ± 16.6 | 68.5 ± 29.1 | 41 ± 13 | - |
| Compound | Arg Mimetic | n | R1 | R2 | % Inhibition (αIIbβ3/Fg) @ 50 μM | IC50/μM (αIIbβ3/Fg) | % Inhibition (Platelet Aggregation) @ 100 μM |
|---|---|---|---|---|---|---|---|
| ICT9019 21 | Pyrimidine | 1 | NHSO2Mes | H | 15.6 ± 14.5 | - | - |
| ICT9030 50 | 91.2 ± 9.6 | 3.5 ± 1.9 | - | ||||
| ICT9023 23 | Pyrimidine | 2 | NHSO2Ph | H | 10.9 ± 15.4 | - | - |
| ICT9028 51 | 93.6 ± 2.2 | 7.8 ± 2.8 | - | ||||
| ICT9024 27 | Pyrimidine T | 1 | NHSO2Mes | H | 33.5 ± 2.8 | 50 ± 5 | - |
| ICT9031 52 | 96.2 ± 6.3 | 1.7 ± 2.4 | 11.1 | ||||
| ICT9025 28 | Pyrimidine T | 2 | NHSO2Mes | H | 35.0 ± 11.3 | >100 | - |
| ICT9090 53 | - | 0.39 ± 0.19 | 21.2 ± 7.8 | ||||
| ICT9026 29 | Pyrimidine T | 2 | NHSO2Ph | H | 31.6 ± 10.7 | - | - |
| ICT9029 54 | 100 ± 6 | 3.3 ± 3.3 | - | ||||
| ICT9053 41 | Naphthyridine | 1 | NHSO2Mes | H | 29.4 ± 17.6 | >100 | - |
| ICT9063 55 | 84.5 ± 8.6 | 4.1 ± 2.2 | 13.0 | ||||
| ICT9055 46 | THN | 1 | NHSO2Mes | H | 0 ± 9.8 | >50 | 5.6 |
| ICT9064 56 | 88.8 ± 2.6 | 1.17 ± 0.9 | 31.4 | ||||
| GR144053 | - | 23.7 ± 3.1 nM | 100 (IC50 240 nM) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutherland, M.; Gordon, A.; Al-Shammari, F.O.F.O.; Throup, A.; Cilia La Corte, A.; Philippou, H.; Shnyder, S.D.; Patterson, L.H.; Sheldrake, H.M. Synthesis and Biological Evaluation of Cyclobutane-Based β3 Integrin Antagonists: A Novel Approach to Targeting Integrins for Cancer Therapy. Cancers 2023, 15, 4023. https://doi.org/10.3390/cancers15164023
Sutherland M, Gordon A, Al-Shammari FOFO, Throup A, Cilia La Corte A, Philippou H, Shnyder SD, Patterson LH, Sheldrake HM. Synthesis and Biological Evaluation of Cyclobutane-Based β3 Integrin Antagonists: A Novel Approach to Targeting Integrins for Cancer Therapy. Cancers. 2023; 15(16):4023. https://doi.org/10.3390/cancers15164023
Chicago/Turabian StyleSutherland, Mark, Andrew Gordon, Fatemah O. F. O. Al-Shammari, Adam Throup, Amy Cilia La Corte, Helen Philippou, Steven D. Shnyder, Laurence H. Patterson, and Helen M. Sheldrake. 2023. "Synthesis and Biological Evaluation of Cyclobutane-Based β3 Integrin Antagonists: A Novel Approach to Targeting Integrins for Cancer Therapy" Cancers 15, no. 16: 4023. https://doi.org/10.3390/cancers15164023