
Synthesis and Validation of a Bioinspired Catechol-Functionalized Pt(IV) Prodrug for Preclinical Intranasal Glioblastoma Treatment


 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Instrumentation
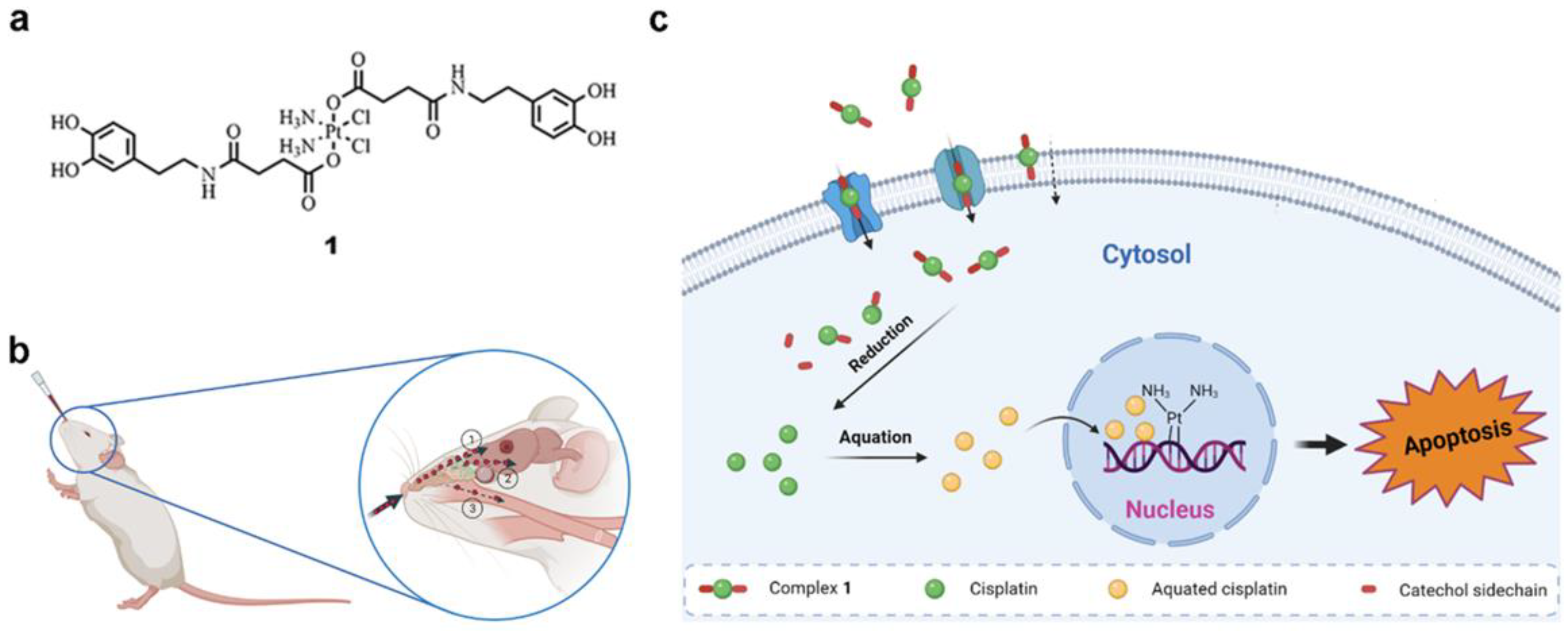
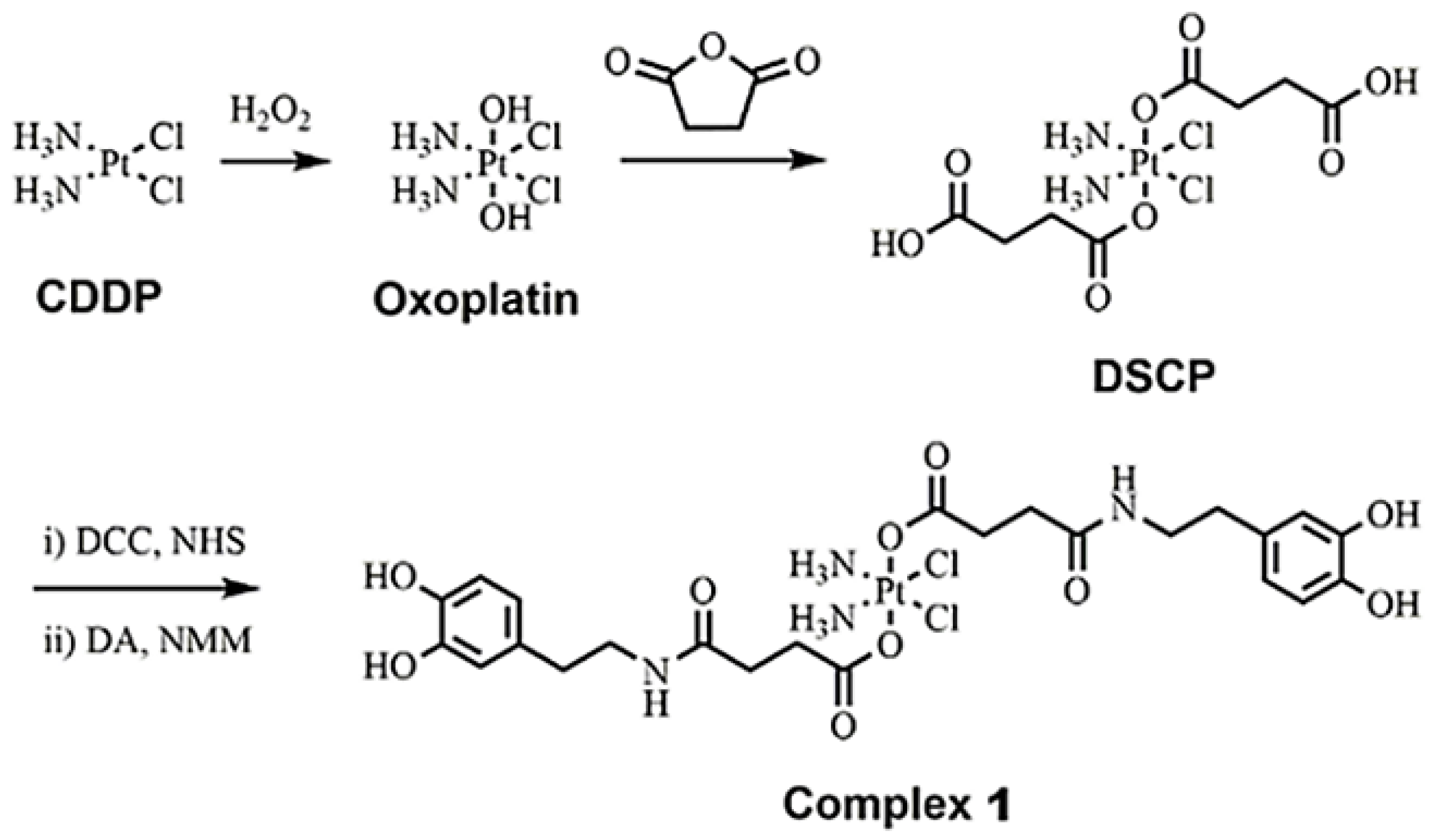
2.2. Synthesis of Complex 1
2.3. In Vitro Studies
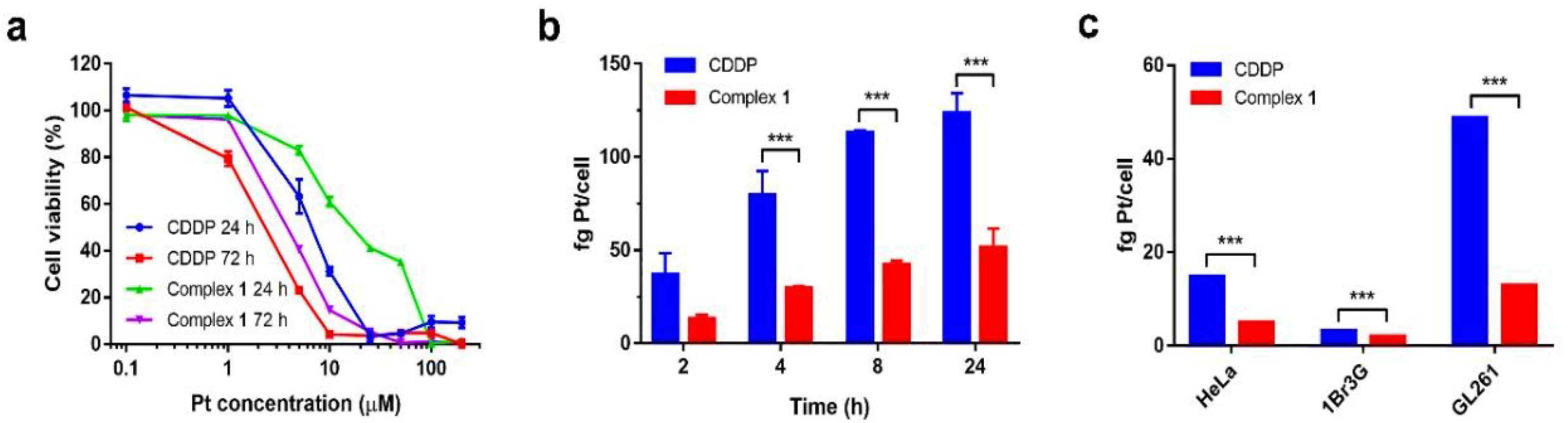
2.3.1. Cell Viability
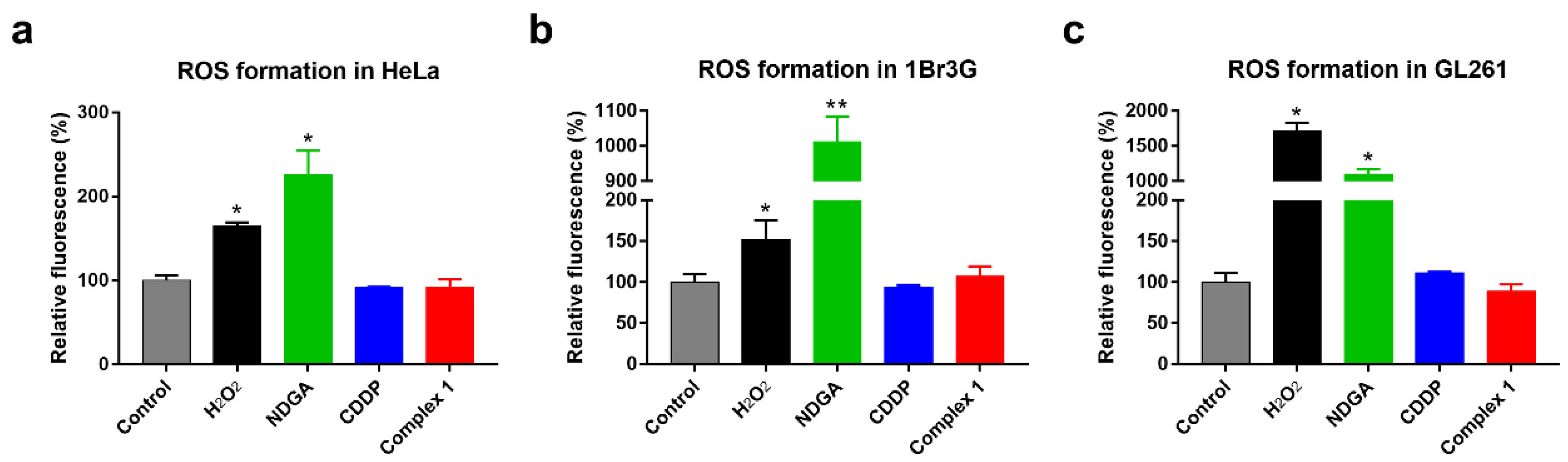
2.3.2. Estimation of ROS Formation
2.3.3. Cellular Internalization
2.3.4. DNA-Bound Pt
2.4. In Vivo Studies
2.4.1. Animal Studies
2.4.2. Tolerability Assessment
2.4.3. Orthotopic Intracranial GB Model by Stereotactic Injection of GL261 Cells
2.4.4. In Vivo MRI Assessment of Tumor Evolution
2.4.5. Antitumor Efficacy In Vivo
2.5. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterization
3.2. In Vitro Studies
3.2.1. Cytotoxicity and Cell Internalization
3.2.2. Pt Bound to DNA
3.2.3. Oxidative Stress Assay
3.3. In Vivo Studies
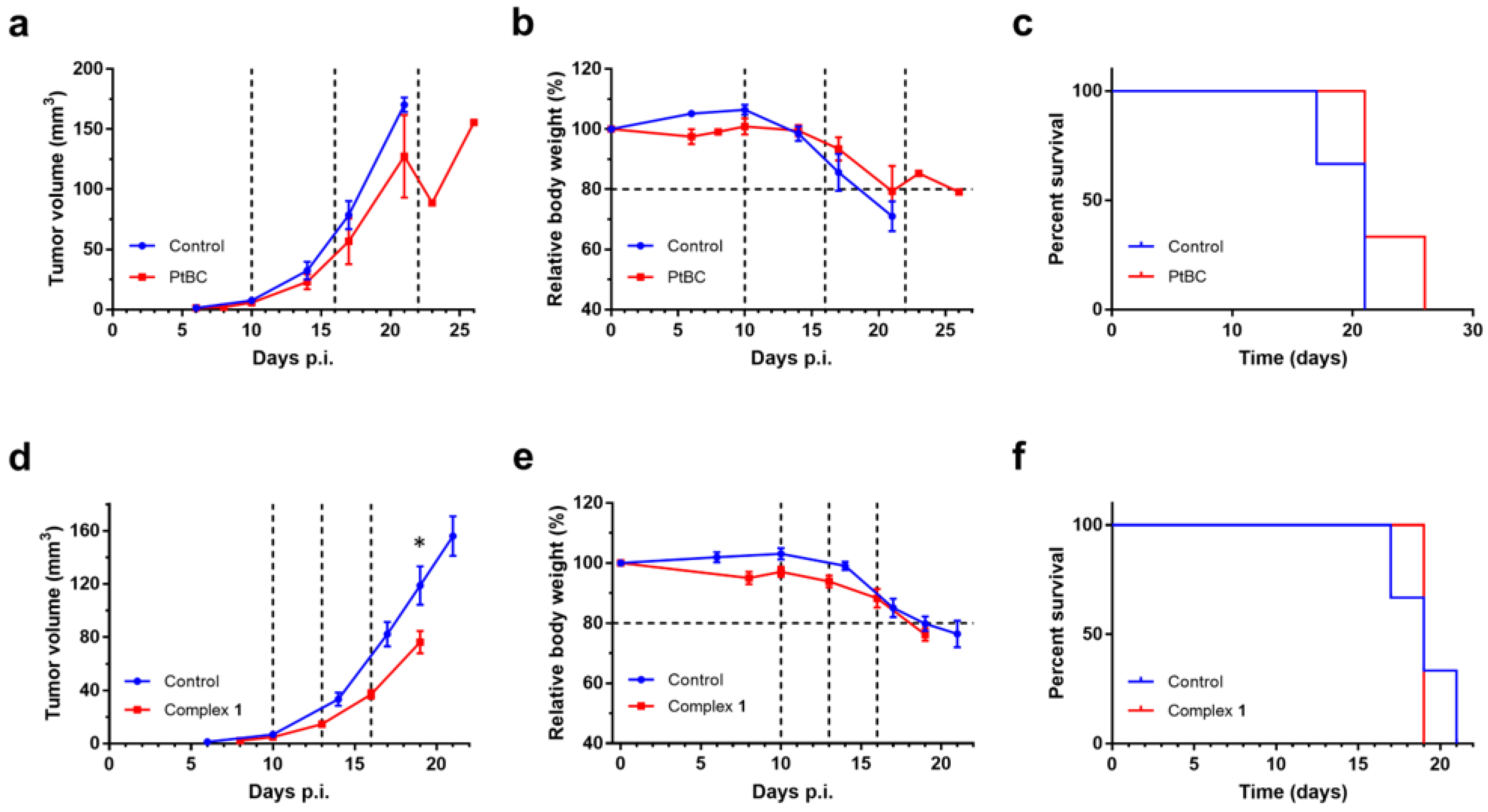
3.3.1. Tolerability Assessment in Mice
3.3.2. Efficacy Evaluation In Vivo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Messali, A.; Villacorta, R.; Hay, J.W. A review of the economic burden of glioblastoma and the cost effectiveness of pharmacologic treatments. Pharmacoeconomics 2014, 32, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Tabatabai, G.; Wakimoto, H. Glioblastoma: State of the art and future perspectives. Cancers 2019, 11, 1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised Phase III study: 5-year analysis of the eortc-ncic trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306. [Google Scholar] [CrossRef] [Green Version]
- Trinh, V.A.; Patel, S.P.; Hwu, W.-J. The safety of temozolomide in the treatment of malignancies. Expert Opin. Drug Saf. 2009, 8, 493–499. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, S. Temozolomide/plga microparticles and antitumor activity against glioma c6 cancer cells in vitro. Int. J. Pharm. 2007, 329, 122. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Del. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.-A.; Merkel, O.; Popp, A. Intranasal drug delivery: Opportunities and toxicologic challenges during drug development. Drug Deliv. Transl. Res. 2021, 1–23. [Google Scholar] [CrossRef]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, A.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef]
- Bruinsmann, F.A.; Richter Vaz, G.; De Cristo Soares Alves, A.; Aguirre, T.; Raffin Pohlmann, A.; Staniscuaski Guterres, S.; Sonvico, F. Nasal drug delivery of anticancer drugs for the treatment of glioblastoma: Preclinical and clinical trials. Molecules 2019, 24, 4312. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Pei, X.; Duan, L.; Zhu, Z.; Liu, Y.; Chen, J.; Chen, T.; Ji, P.; Wan, Q.; Wang, J. A Mussel-inspired film for adhesion to wet buccal tissue and efficient buccal drug delivery. Nat. Commun. 2021, 12, 1689. [Google Scholar] [CrossRef]
- Kim, K.; Kim, K.; Ryu, J.H.; Lee, H. Chitosan-catechol: A polymer with long-lasting mucoadhesive properties. Biomaterials 2015, 52, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Poinard, B.; Lam, S.A.E.; Neoh, K.G.; Kah, J.C.Y. Mucopenetration and biocompatibility of polydopamine surfaces for delivery in an ex vivo porcine bladder. J. Control. Release 2019, 300, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Poinard, B.; Kamaluddin, S.; Tan, A.Q.Q.; Neoh, K.G.; Kah, J.C.Y. Polydopamine coating enhances mucopenetration and cell uptake of nanoparticles. ACS Appl. Mater. Interfaces 2019, 11, 4777–4789. [Google Scholar] [CrossRef]
- Wang, D.; Gao, Y.; Yun, L. Study on brain targeting of raltitrexed following intranasal administration in rats. Cancer Chemother. Pharmacol. 2006, 57, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jiang, X.; Lu, W. Profiles of methotrexate in blood and Csf following intranasal and intravenous administration to rats. Int. J. Pharm. 2003, 263, 1–7. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, H.; Dong, W.; Zhang, M.; Liu, Q.; Wang, X.; Guan, J.; Wu, H.; Mao, S. Design and intestinal mucus penetration mechanism of core-shell nanocomplex. J. Control. Release 2018, 272, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Wang, Q.; Xu, Y.; Cong, L.; Gou, J.; Tao, X.; Zhang, Y.; He, H.; Yin, T.; Zhang, H.; et al. Enhanced oral absorption and anticancer efficacy of cabazitaxel by overcoming intestinal mucus and epithelium barriers using surface polyethylene oxide (peo) decorated positively charged polymer-lipid hybrid nanoparticles. J. Control. Release 2018, 269, 423–438. [Google Scholar] [CrossRef]
- Doz, F.; Berens, M.E.; Dougherty, D.V.; Rosenblum, M.L. Comparison of the cytotoxic activities of cisplatin and carboplatin against glioma cell lines at pharmacologically relevant drug exposures. J. Neurooncol. 1991, 11, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Newton, H.B.; Page, M.A.; Junck, L.; Greenberg, H.S. Intra-arterial cisplatin for the treatment of malignant gliomas. J. Neurooncol. 1989, 7, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Feun, L.G.; Stewart, D.J.; Maor, M.; Leavens, M.; Savaraj, N.; Burgess, M.A.; Yung, W.-K.A.; Benjamin, R.S. A pilot study of cis-diamminedichloroplatinum and radiation therapy in patients with high grade astrocytomas. J. Neurooncol. 1983, 1, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Silvani, A.; Gaviani, P.; Lamperti, E.A.; Eoli, M.; Falcone, C.; Dimeco, F.; Milanesi, I.M.; Erbetta, A.; Boiardi, A.; Fariselli, L.; et al. Cisplatinum and BCNU chemotherapy in primary glioblastoma patients. J. Neurooncol. 2009, 94, 57–62. [Google Scholar] [CrossRef]
- Balaña, C.; López-Pousa, A.; Berrocal, A.; Yaya-Tur, R.; Herrero, A.; García, J.L.; Martín-Broto, J.; Benavides, M.; Cerdá-Nicolás, M.; Ballester, R.; et al. Phase ii study of temozolomide and cisplatin as primary treatment prior to radiotherapy in newly diagnosed glioblastoma multiforme patients with measurable disease. A study of the spanish medical neuro-oncology group (Genom). J. Neurooncol. 2004, 70, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Yap, S.Q.; Chin, C.F.; Hong Thng, A.H.; Pang, Y.Y.; Ho, H.K.; Ang, W.H. Finely tuned asymmetric platinum (IV) anticancer complexes: Structure–Activity relationship and application as orally available prodrugs. Chem. Med. Chem. 2017, 12, 300–311. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, S.-A.; Kerwood, D.J.; Goodisman, J.; Dabrowiak, J.C. Pt (IV) complexes as prodrugs for cisplatin. J. Inorg. Biochem. 2012, 107, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemirovski, A.; Vinograd, I.; Takrouri, K.; Mijovilovich, A.; Rompel, A.; Gibson, D. New reduction pathways for Ctc-[PtCl2(CH3CO2)2(NH3)(Am)] anticancer prodrugs. Chem. Commun. 2010, 46, 1842–1844. [Google Scholar] [CrossRef]
- Lasorsa, A.; Stuchlíková, O.; Brabec, V.; Natile, G.; Arnesano, F. Activation of Platinum (IV) prodrugs by cytochrome C and characterization of the protein binding sites. Mol. Pharm. 2016, 13, 3216–3223. [Google Scholar] [CrossRef]
- Ling, X.; Tu, J.; Wang, J.; Shajii, A.; Kong, N.; Feng, C.; Zhang, Y.; Yu, M.; Xie, T.; Bharwani, Z.; et al. Glutathione-responsive prodrug nanoparticles for effective drug delivery and cancer therapy. ACS Nano 2019, 13, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, B.I.; Jeon, J.H.; Kim, D.K.; Kang, S.G.; Shim, J.K.; Kim, S.Y.; Kang, S.W.; Jang, H. Gossypol suppresses growth of temozolomide-resistant glioblastoma tumor spheres. Biomolecules 2019, 9, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarzabek, M.A.; Amberger-Murphy, V.; Callanan, J.J.; Gao, C.; Zagozdzon, A.M.; Shiels, L.; Wang, J.; Ligon, K.L.; Rich, B.E.; Dicker, P.; et al. Interrogation of gossypol therapy in glioblastoma implementing cell line and patient-derived tumour models. Br. J. Cancer 2014, 111, 2275–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, S.A.; Ye, X.; Peereboom, D.; Rosenfeld, M.R.; Mikkelsen, T.; Supko, J.G.; Desideri, S. Phase I study of terameprocol in patients with recurrent high-grade glioma. Neuro Oncol. 2012, 14, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adarsh, N.N.; Frias, C.; Ponnoth Lohidakshan, T.M.; Lorenzo, J.; Novio, F.; Garcia-Pardo, J.; Ruiz-Molina, D. Pt (IV)-based nanoscale coordination polymers: Antitumor activity, cellular uptake and interactions with nuclear DNA. Chem. Eng. J. 2018, 340, 94–102. [Google Scholar] [CrossRef]
- Saldaña-Ruíz, S.; Soler-Martín, C.; Llorens, J. Role of Cyp2e1-mediated metabolism in the acute and vestibular toxicities of nineteen nitriles in the mouse. Toxicol. Lett. 2012, 208, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Robinson, S.; Chapman, K.; Hudson, S.; Sparrow, S.; Spencer-Briggs, D.; Danks, A.; Hill, R.; Everett, D.; Mulier, B.; Old, S. Guidance on Dose Level Selection for Regulatory General Toxicology Studies for Pharmaceuticals; NC3Rs/LASA: London, UK, 2009. [Google Scholar]
- Zhang, X.; Zhao, S.; Song, X.; Jia, J.; Zhang, Z.; Zhou, H.; Fu, H.; Cui, H.; Hu, S.; Fang, M.; et al. Inhibition effect of glycyrrhiza polysaccharide (gcp) on tumor growth through regulation of the gut microbiota composition. J. Pharmacol. Sci. 2018, 137, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Lippard, S.J. Reinterpretation of the vibrational spectroscopy of the medicinal bioinorganic synthon c,c,t-[Pt(NH3)2Cl2(OH)2]. J. Biol. Inorg. Chem. 2014, 19, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [Green Version]
- Rieter, W.J.; Pott, K.M.; Taylor, K.M.L.; Lin, W. Nanoscale coordination polymers for platinum-based anticancer drug delivery. J. Am. Chem. Soc. 2008, 130, 11584–11585. [Google Scholar] [CrossRef] [PubMed]
- Forooshani, P.K.; Meng, H.; Lee, B.P. Advances in bioinspired and biomedical materials volume 1. Am. Chem. Soc. 2017, 1252, 179. [Google Scholar]
- Yuan, S.; Zhu, Y.; Dai, Y.; Wang, Y.; Jin, D.; Liu, M.; Tang, L.; Arnesano, F.; Natile, G.; Liu, Y. 19F NMR allows the investigation of the fate of Platinum (IV) prodrugs in physiological conditions. Angew. Chem. Int. Ed. 2021, 60, 2–8. [Google Scholar]
- Carr, J.L.; Tingle, M.D.; McKeage, M.J. Rapid biotransformation of satraplatin by human red blood cells in vitro. Cancer Chemother. Pharmacol. 2002, 50, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Parkkila, P.; Viitala, T. Partitioning of catechol derivatives in lipid membranes: Implications for substrate specificity to catechol-o-methyltransferase. ACS Chem. Neurosci. 2020, 11, 969–978. [Google Scholar] [CrossRef] [PubMed]
- García-Pardo, J.; Novio, F.; Nador, F.; Cavaliere, I.; Suárez-García, S.; Lope-Piedrafita, S.; Candiota, A.P.; Romero-Gimenez, J.; Rodríguez-Galván, B.; Bové, J.; et al. Bioinspired theranostic coordination polymer nanoparticles for intranasal dopamine replacement in parkinson’s disease. ACS Nano 2021, 15, 8592–8609. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Cervantesa, G.I.; Villaseñor-Aguayoa, K.; Hernández-Damiána, J.; Aparicio-Trejoa, O.E.; Medina-Camposa, O.N.; López-Marureb, R.; Pedraza-Chaverria, J. Antitumor effects of nordihydroguaiaretic acid (Ndga) in bladder T24 cancer cells are related to increase in ROS production and mitochondrial leak respiration. Nat. Prod. Commun. 2018, 13, 1934578X1801301128. [Google Scholar] [CrossRef]
- Karman, J.; Ling, C.; Sandor, M.; Fabry, Z. Initiation of immune responses in brain is promoted by local dendritic cells. J. Immunol. 2004, 173, 2353–2361. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Calero-Pérez, P.; Villamañan, L.; Arias-Ramos, N.; Pumarola, M.; Ortega-Martorell, S.; Julià-Sapé, M.; Arús, C.; Candiota, A.P. Anti-tumour immune response in Gl261 glioblastoma generated by temozolomide immune-enhancing metronomic schedule monitored with MRSI-based nosological images. NMR Biomed. 2020, 33, e4229. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Wu, X. Ganoderma lucidum polysaccharide (Glp) enhances antitumor immune response by regulating differentiation and inhibition of Mdscs via a Card9-Nf-Κb-Ido pathway. Biosci. Rep. 2020, 40, BSR20201170. [Google Scholar] [CrossRef]
- Qi, X.; Xu, W.; Xie, J.; Wang, Y.; Han, S.; Wei, Z.; Ni, Y.; Dong, Y.; Han, W. Metformin sensitizes the response of oral squamous cell carcinoma to cisplatin treatment through inhibition of Nf-Κb/Hif-1α signal axis. Sci. Rep. 2016, 6, 35788. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Takenaka, T.; Toh, K.; Wu, S.; Nishihara, H.; Kano, M.R.; Ino, Y.; Nomoto, T.; Matsumoto, Y.; Koyama, H.; et al. Cyclic Rgd-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano 2013, 7, 8583–8592. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Telma, K.A.; Chang, K.-E.; Lee, T.D.; Madigan, J.P.; Lloyd, J.R.; Goldlust, I.S.; Hoeschele, J.D.; Gottesman, M.M. Say no to dmso: Dimethylsulfoxide inactivates cisplatin, carboplatin, and other platinum complexes. Cancer Res. 2014, 74, 3913–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Yadavalli, S.; Yenugonda, V.M.; Kesari, S. Repurposed drugs in treating glioblastoma multiforme: Clinical trials update. Cancer J. 2019, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Lee, S.; Kim, H.; Kang, H.; Youn, H.; Jo, S.; Youn, B.; Kim, H.Y. Revisiting platinum-based anticancer drugs to overcome gliomas. Int. J. Mol. Sci. 2021, 22, 5111. [Google Scholar] [CrossRef]
- Alex, A.T.; Joseph, A.; Shavi, G.; Rao, J.V.; Udupa, N. Development and evaluation of carboplatin-loaded pcl nanoparticles for intranasal delivery. Drug Deliv. 2016, 23, 2144–2153. [Google Scholar] [CrossRef]
- Pandey, A.; Singh, K.; Patel, S.; Singh, R.; Patel, K.; Sawant, K. Hyaluronic acid tethered ph-responsive alloy-drug nanoconjugates for multimodal therapy of glioblastoma: An intranasal route approach. Mater. Sci. Eng. C 2019, 98, 419–436. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | ||||
|---|---|---|---|---|
| Time | Cell Lines | |||
| Compounds | 1Br3G | HeLa | GL261 | |
| 24 h | complex 1 | 56.09 ± 1.18 | 29.94 ± 1.04 | 17.40 ± 1.08 |
| CDDP | 45.07 ± 4.60 | 15.98 ± 1.04 | 5.61 ± 0.28 | |
| DSCP | NT b | NT b | NT b | |
| NDGA | NT b | 164.15 ± 0.02 | 109.45 ± 1.95 | |
| 72 h | complex 1 | 10.80 ± 0.60 | 1.85 ± 0.36 | 4.17 ± 0.12 |
| CDDP | 4.63 ± 0.42 | 2.34 ± 0.30 | 2.16 ± 0.26 | |
| DSCP | NT b | NT b | NT b | |
| NDGA | 119.00 ± 14.20 | 55.65 ± 6.21 | 81.73 ± 2.40 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, X.; Wu, S.; Calero-Pérez, P.; Candiota, A.P.; Alfonso, P.; Bruna, J.; Yuste, V.J.; Lorenzo, J.; Novio, F.; Ruiz-Molina, D. Synthesis and Validation of a Bioinspired Catechol-Functionalized Pt(IV) Prodrug for Preclinical Intranasal Glioblastoma Treatment. Cancers 2022, 14, 410. https://doi.org/10.3390/cancers14020410
Mao X, Wu S, Calero-Pérez P, Candiota AP, Alfonso P, Bruna J, Yuste VJ, Lorenzo J, Novio F, Ruiz-Molina D. Synthesis and Validation of a Bioinspired Catechol-Functionalized Pt(IV) Prodrug for Preclinical Intranasal Glioblastoma Treatment. Cancers. 2022; 14(2):410. https://doi.org/10.3390/cancers14020410
Chicago/Turabian StyleMao, Xiaoman, Shuang Wu, Pilar Calero-Pérez, Ana P. Candiota, Paula Alfonso, Jordi Bruna, Victor J. Yuste, Julia Lorenzo, Fernando Novio, and Daniel Ruiz-Molina. 2022. "Synthesis and Validation of a Bioinspired Catechol-Functionalized Pt(IV) Prodrug for Preclinical Intranasal Glioblastoma Treatment" Cancers 14, no. 2: 410. https://doi.org/10.3390/cancers14020410