
High Hemin Concentration Induces Escape from Senescence of Normoxic and Hypoxic Colon Cancer Cells
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
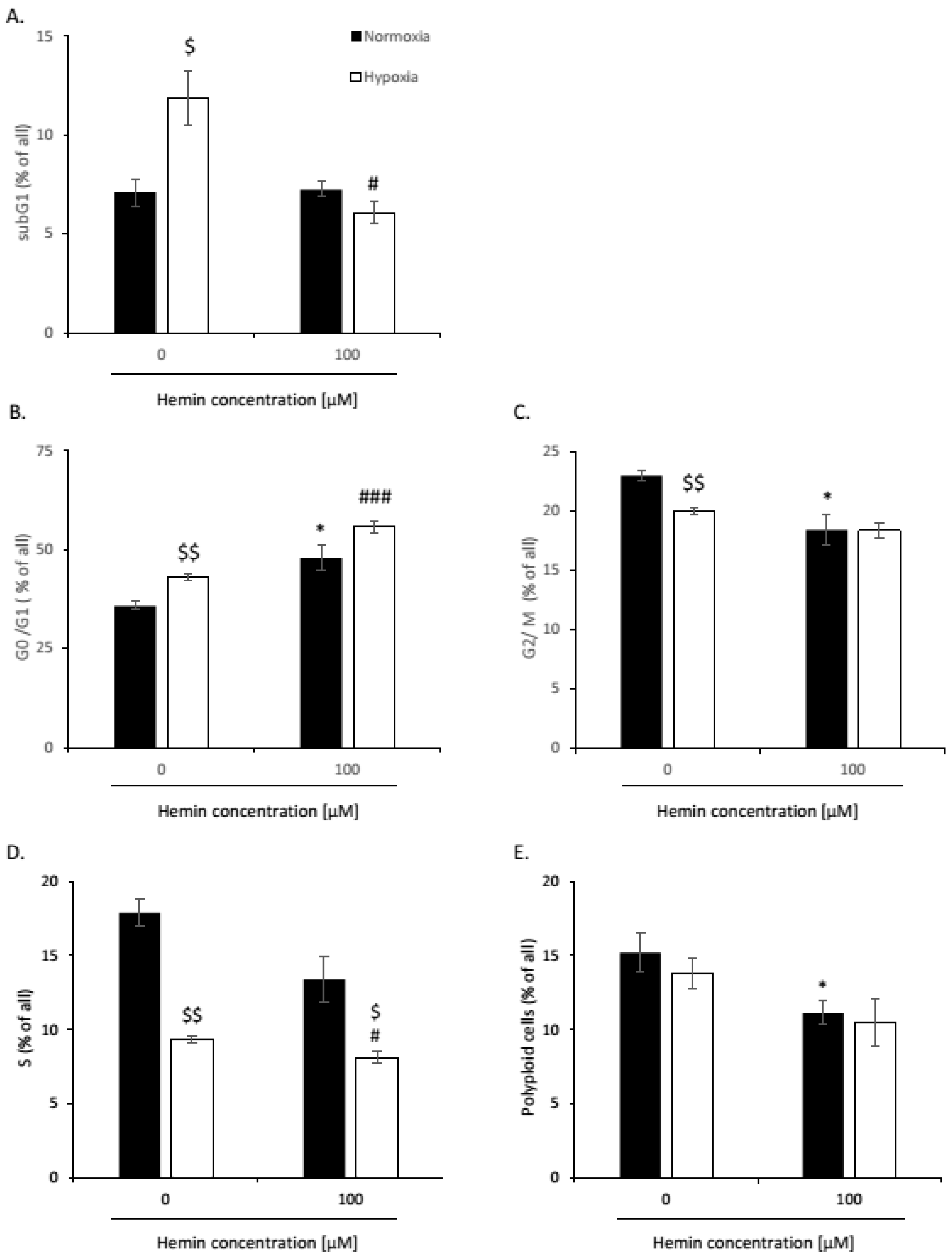
2. Results
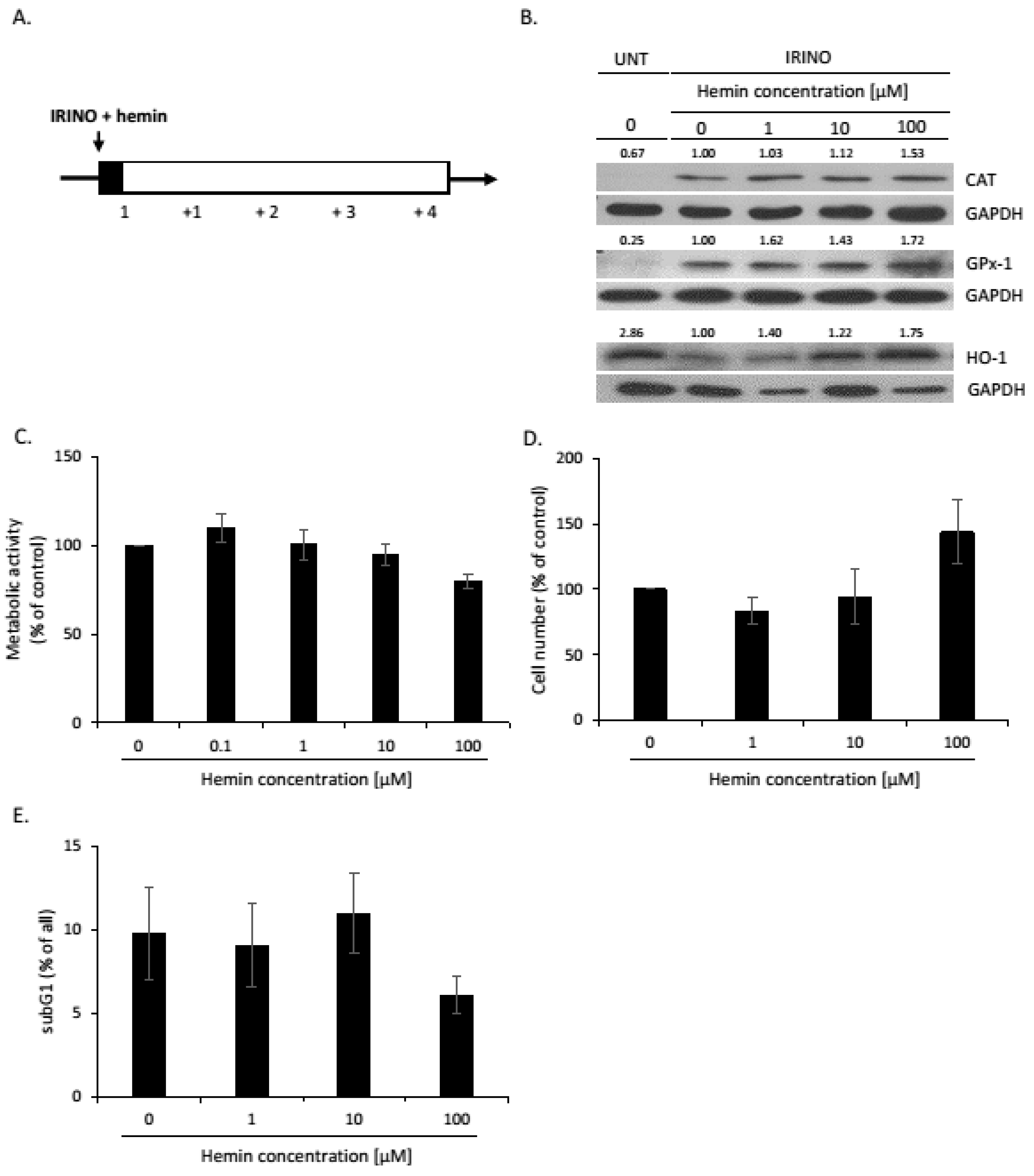
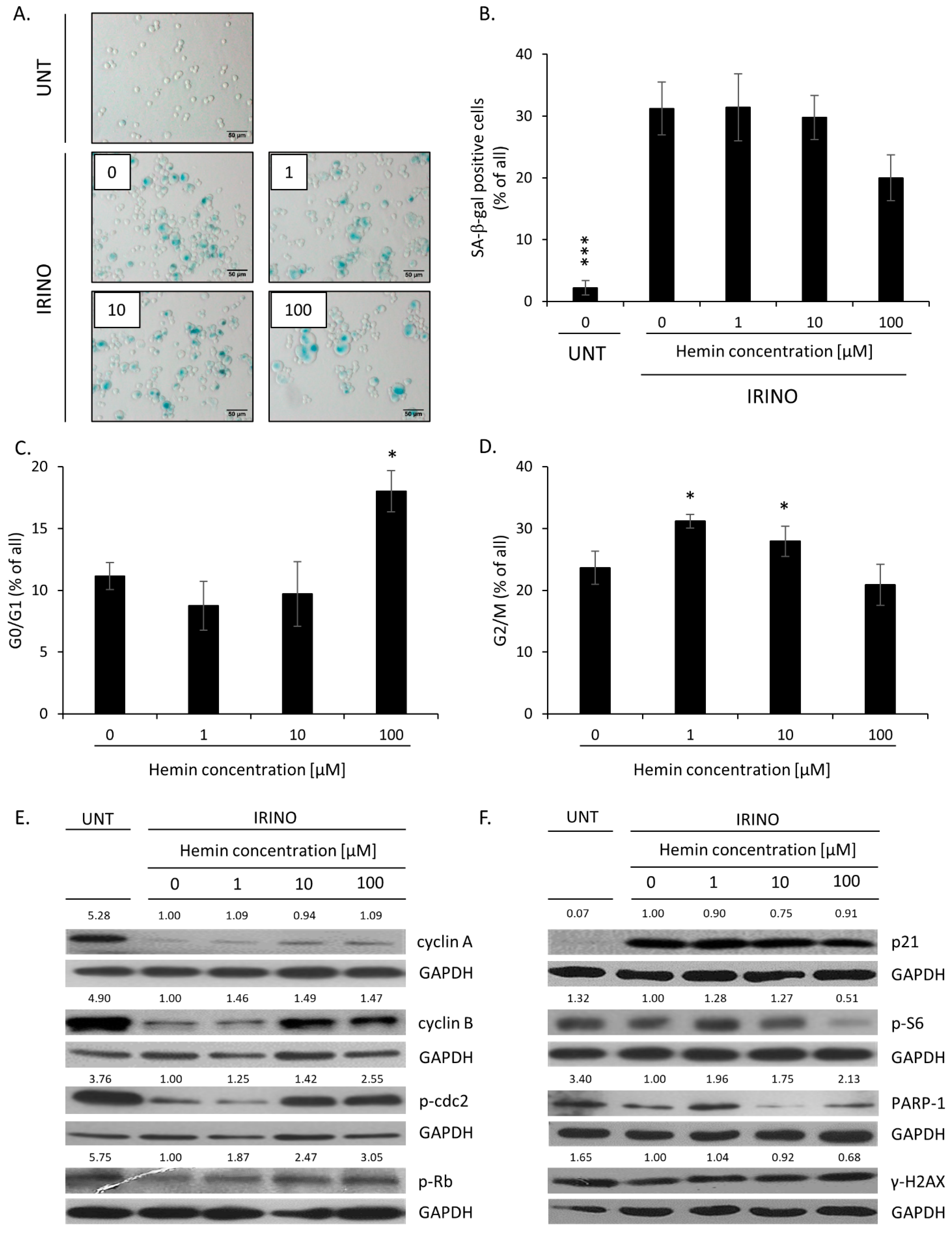
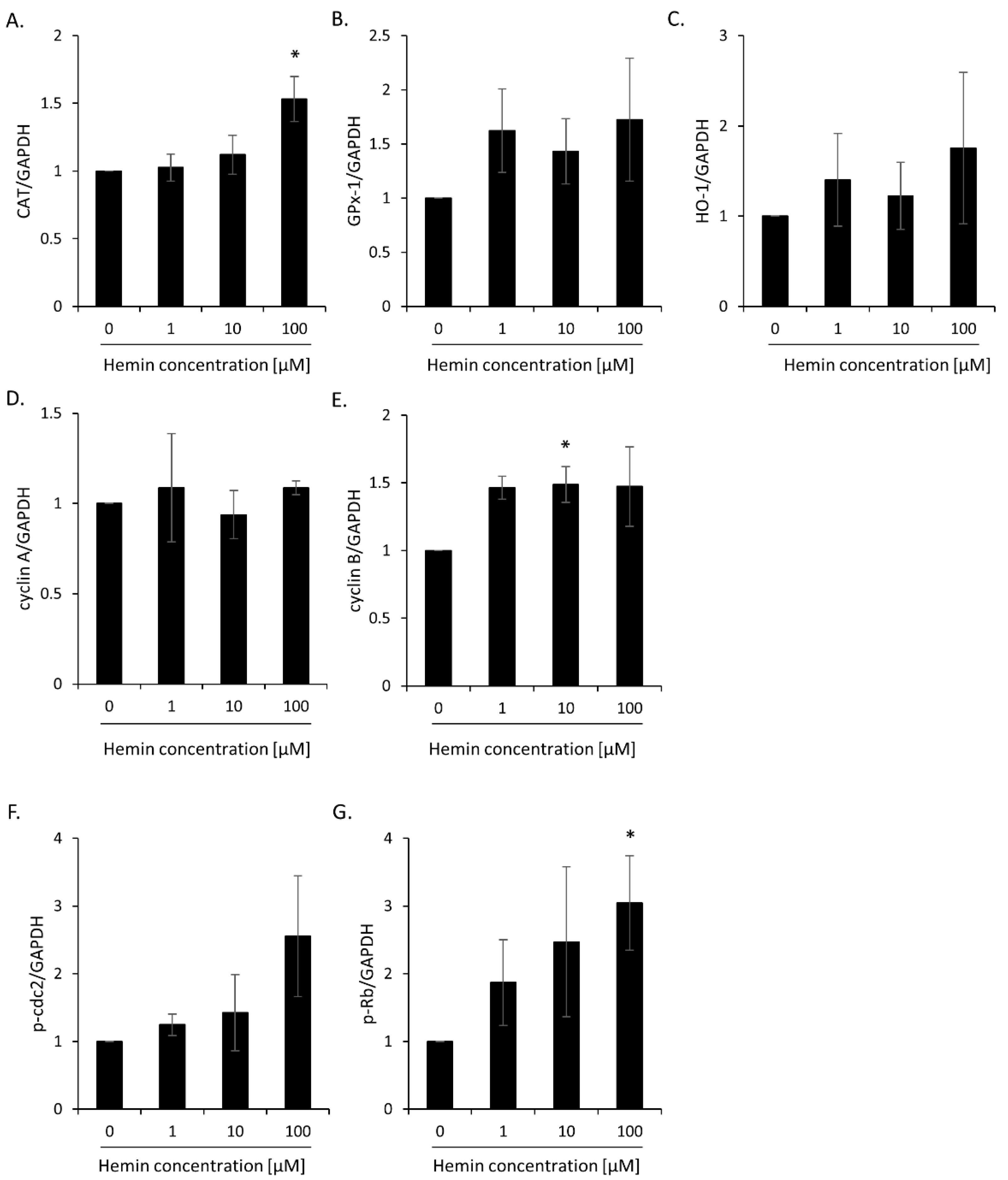
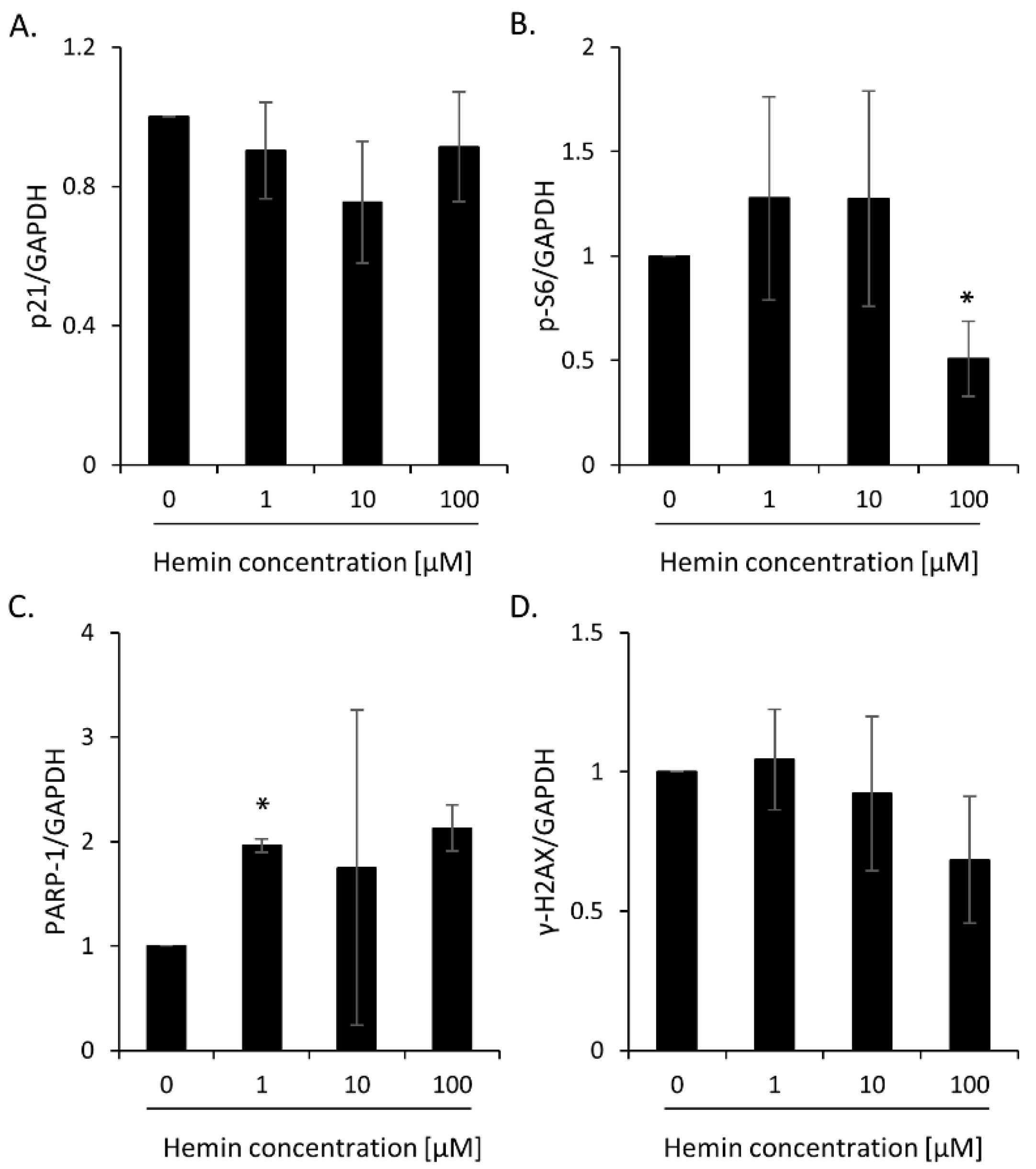
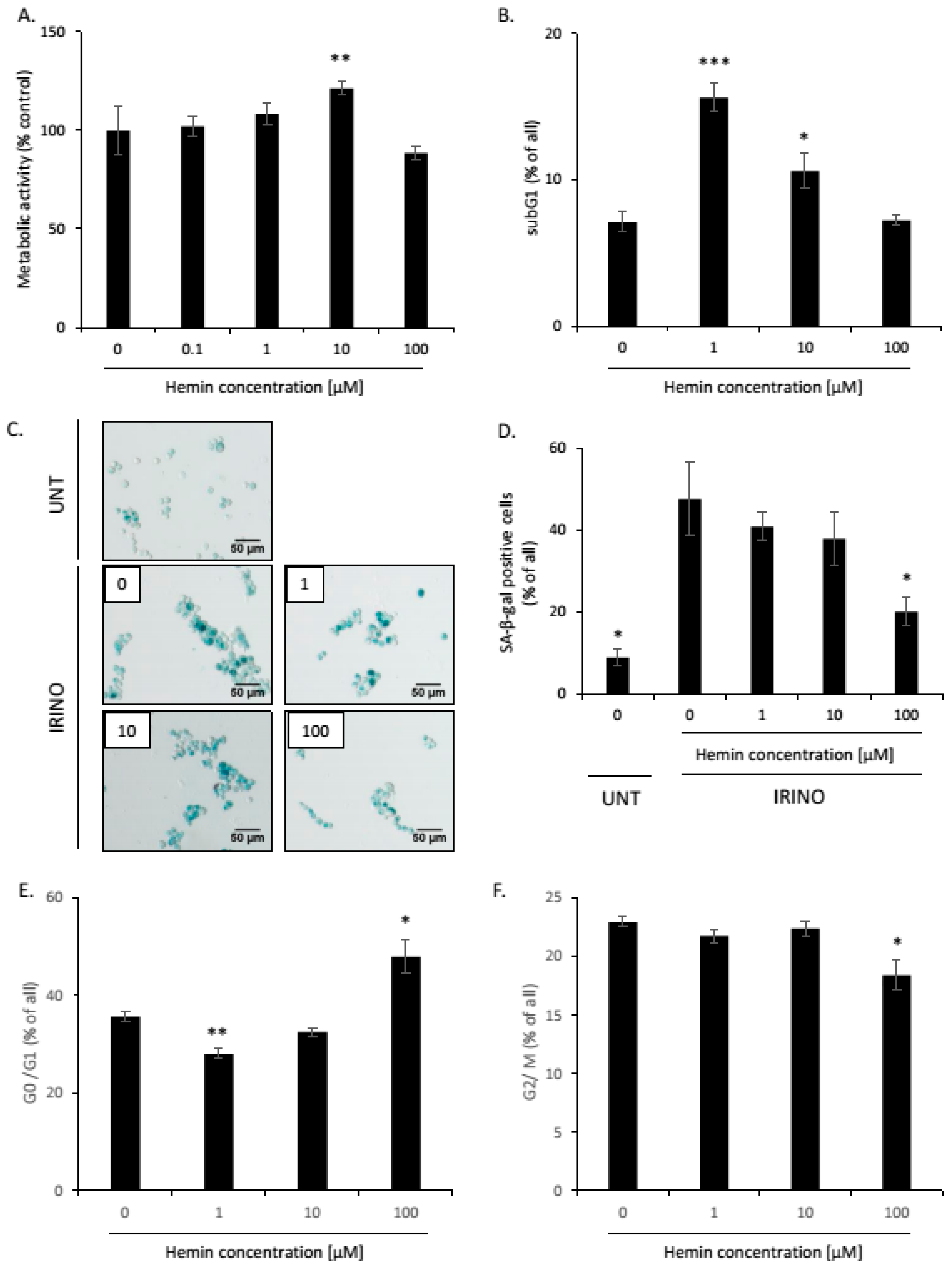
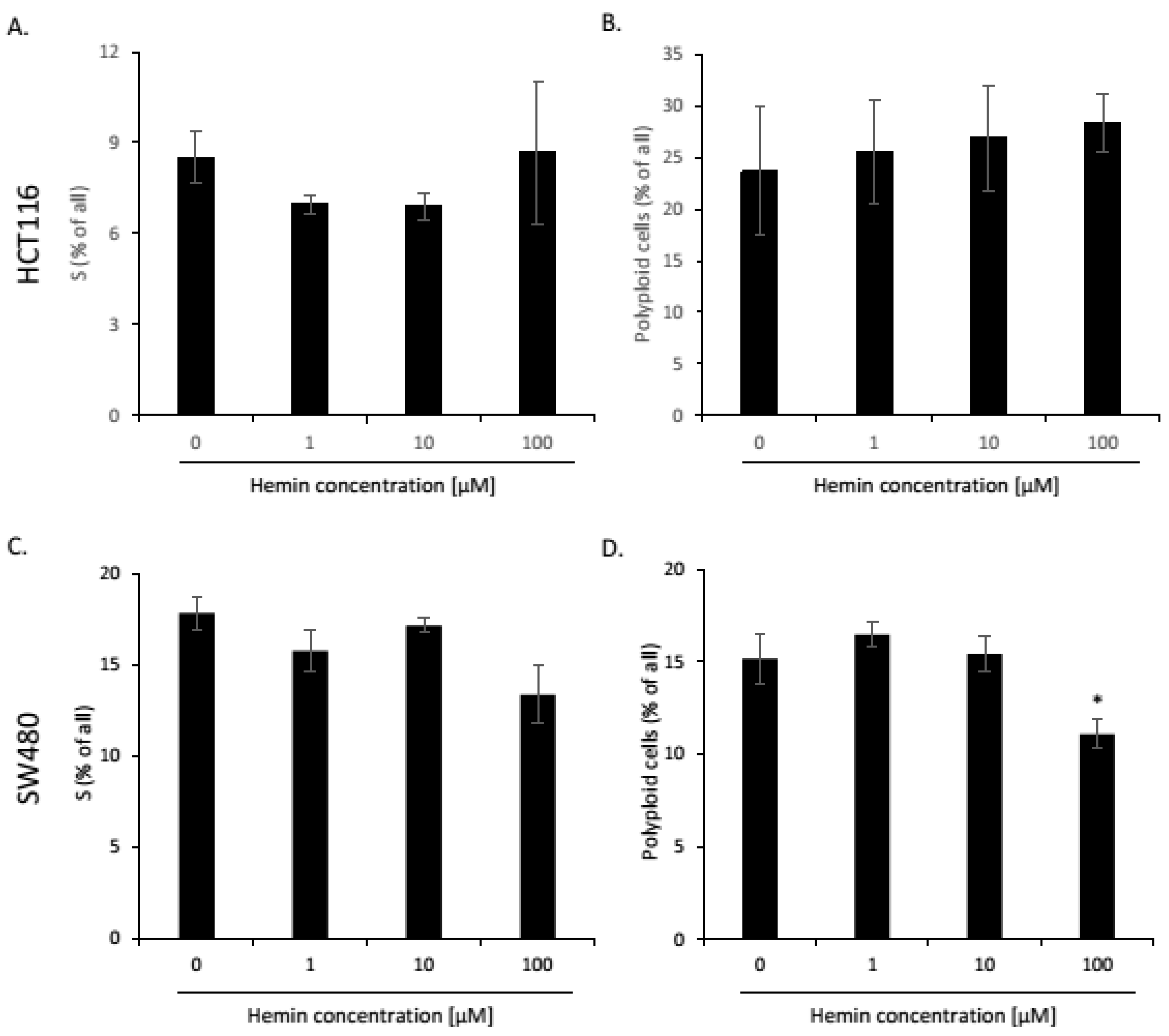
2.1. High Hemin Concentration Causes Escape from Senescence
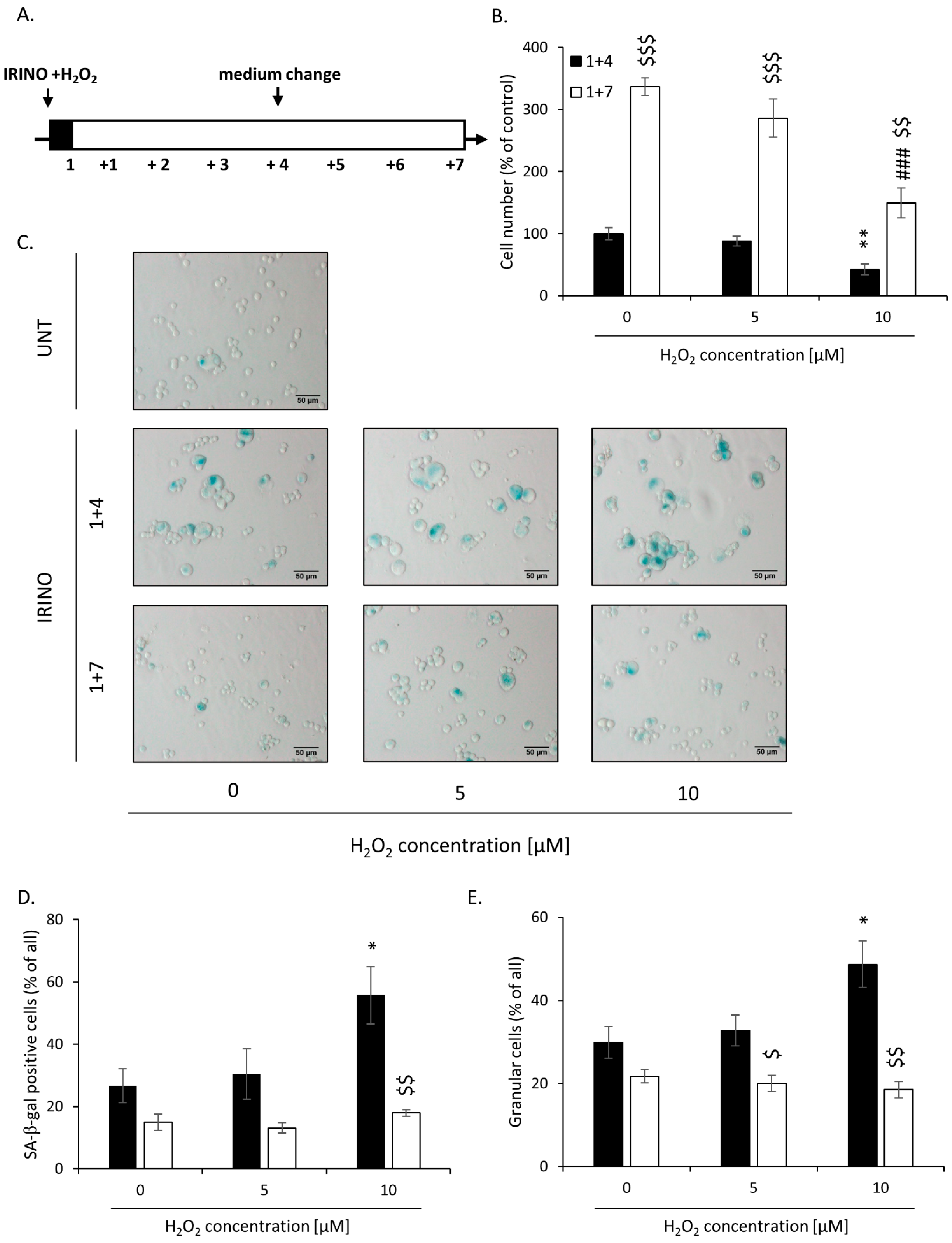
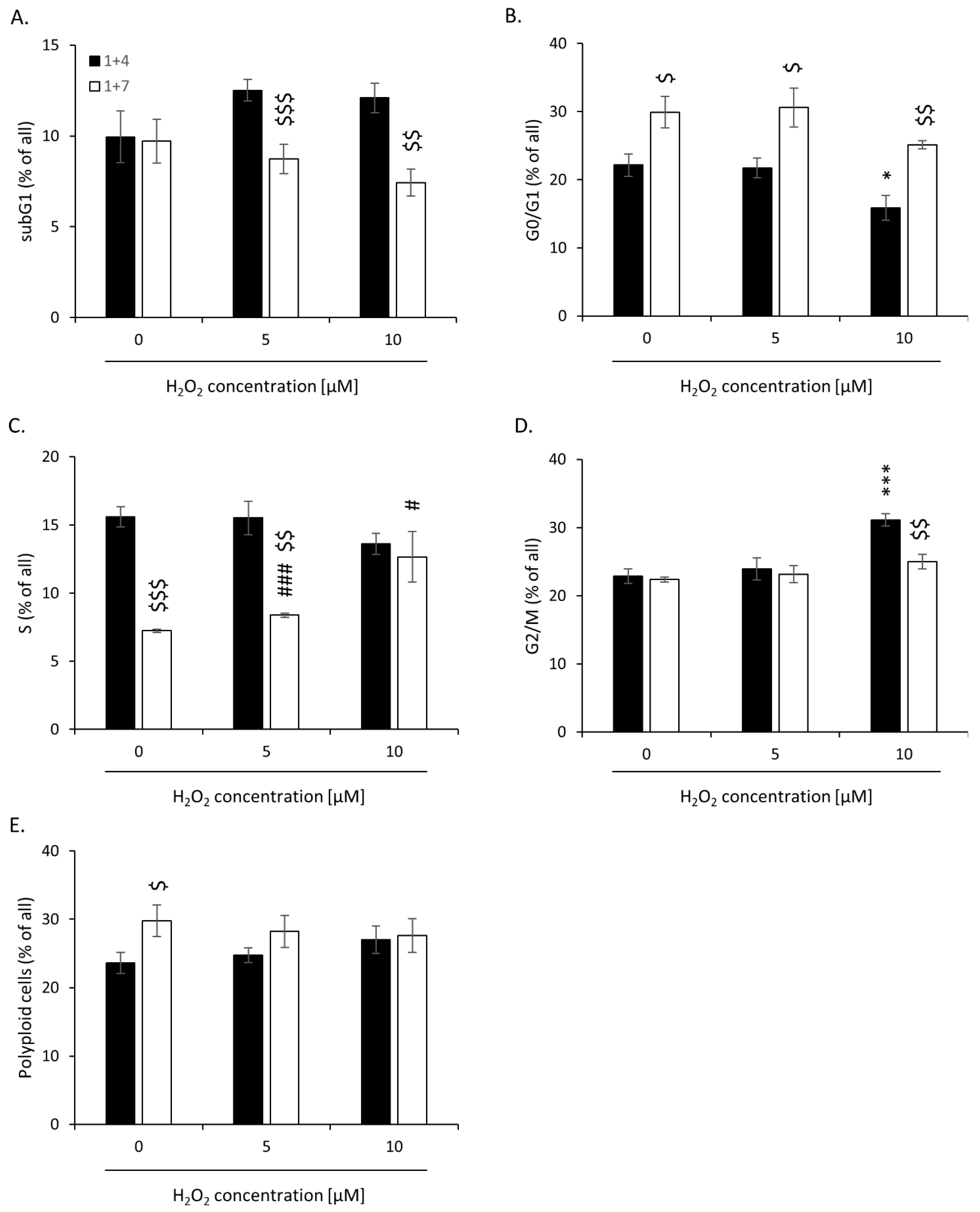
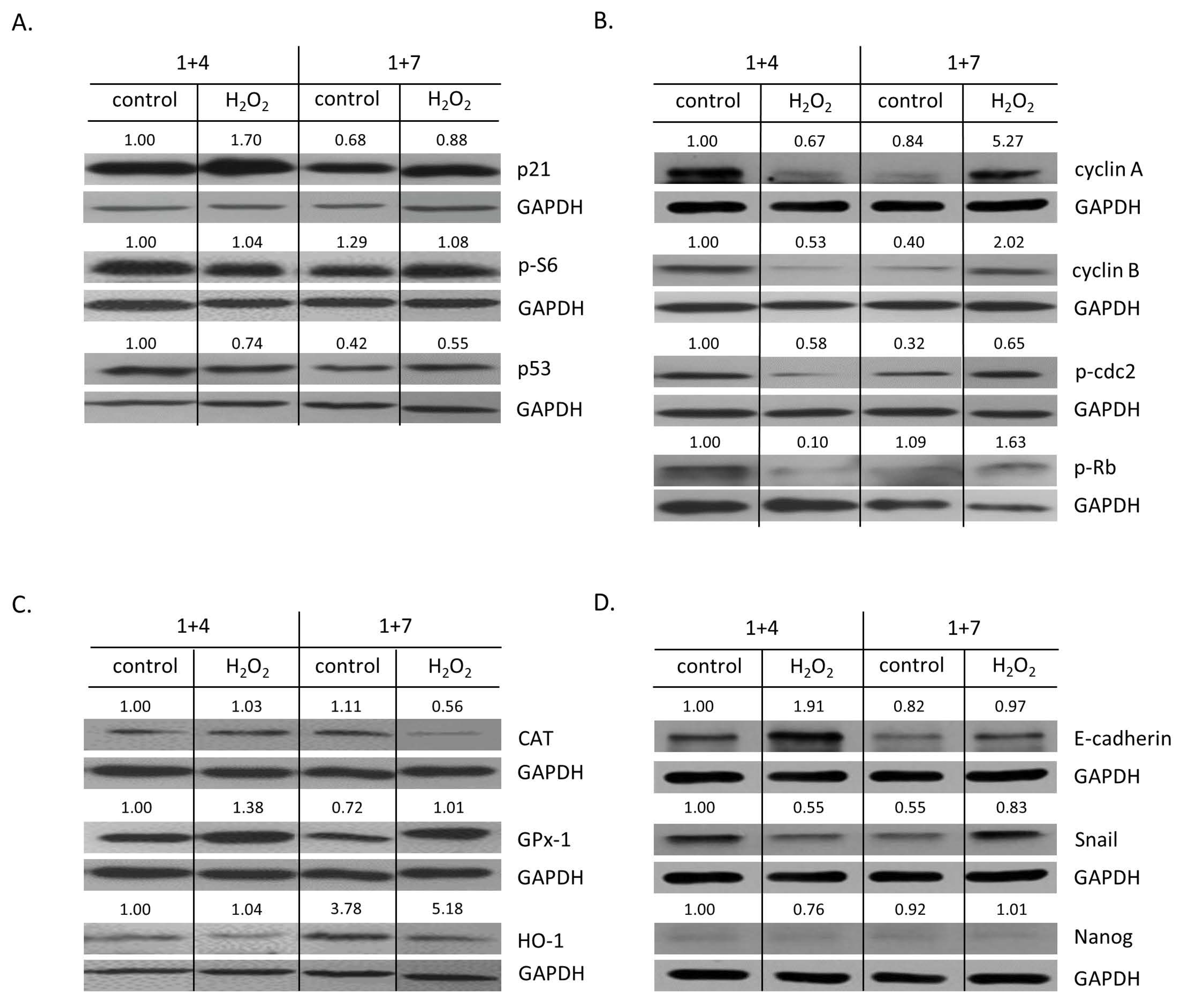
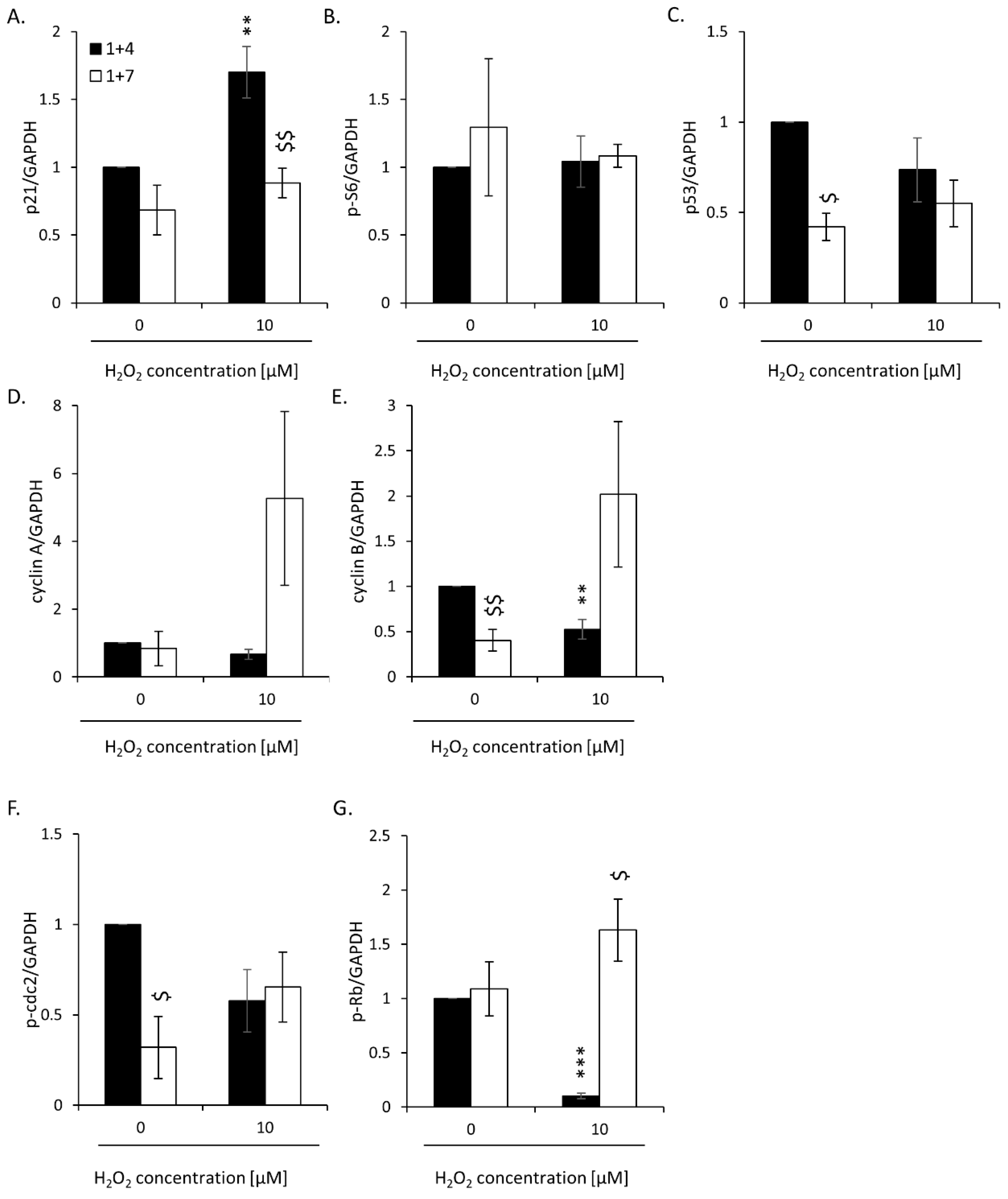
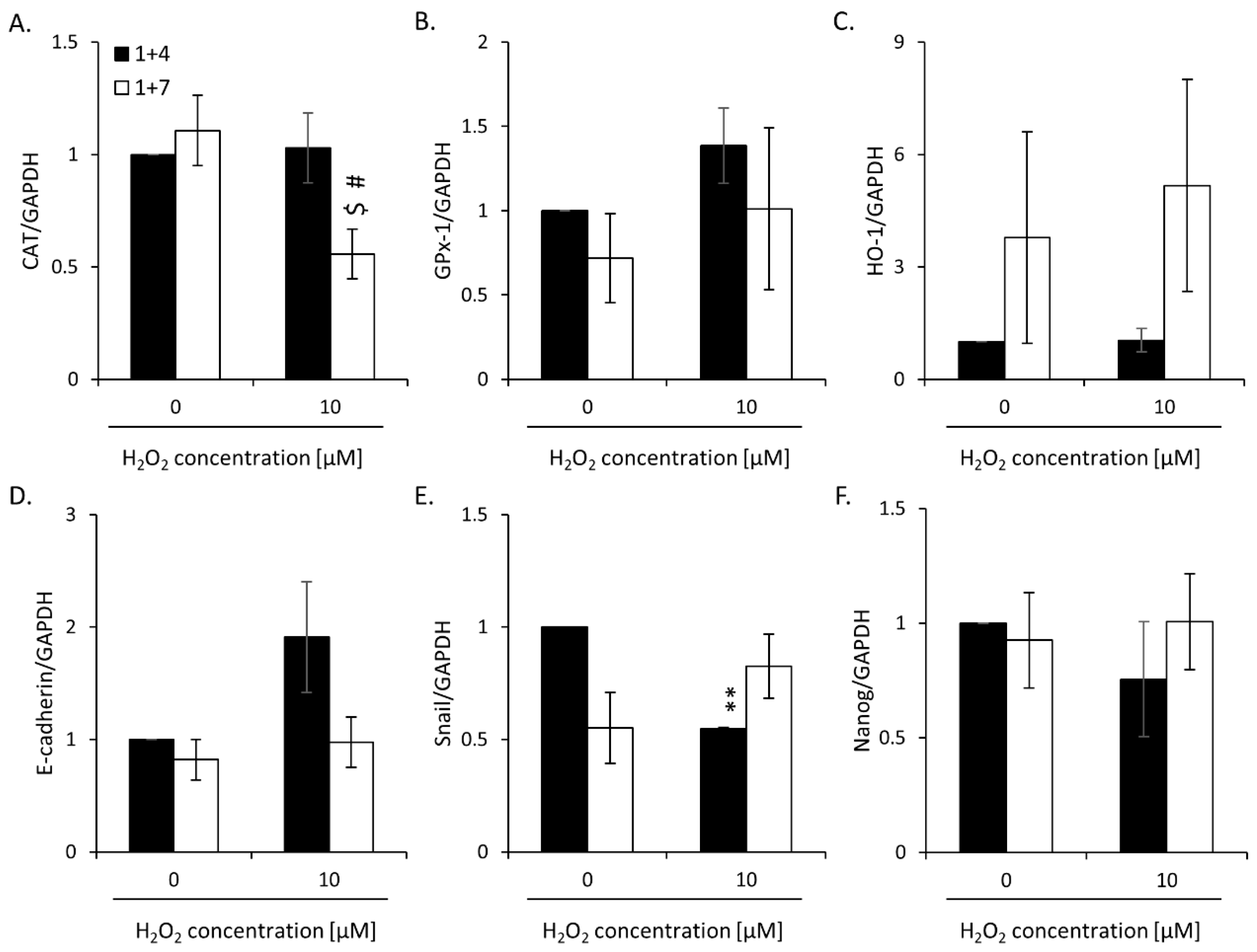
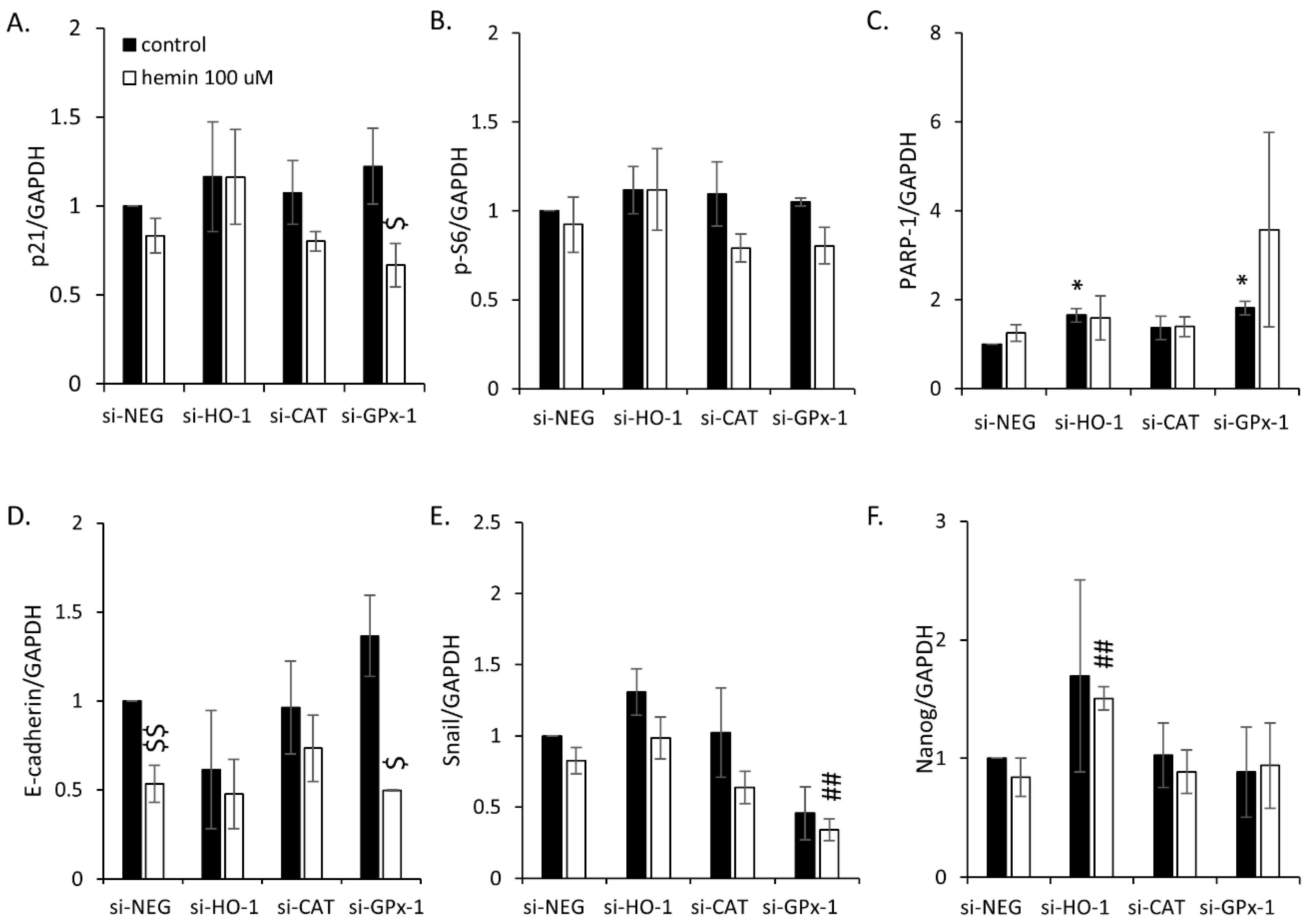
2.2. High H2O2 Level Leads to Escape from Senescence and Induces EMT
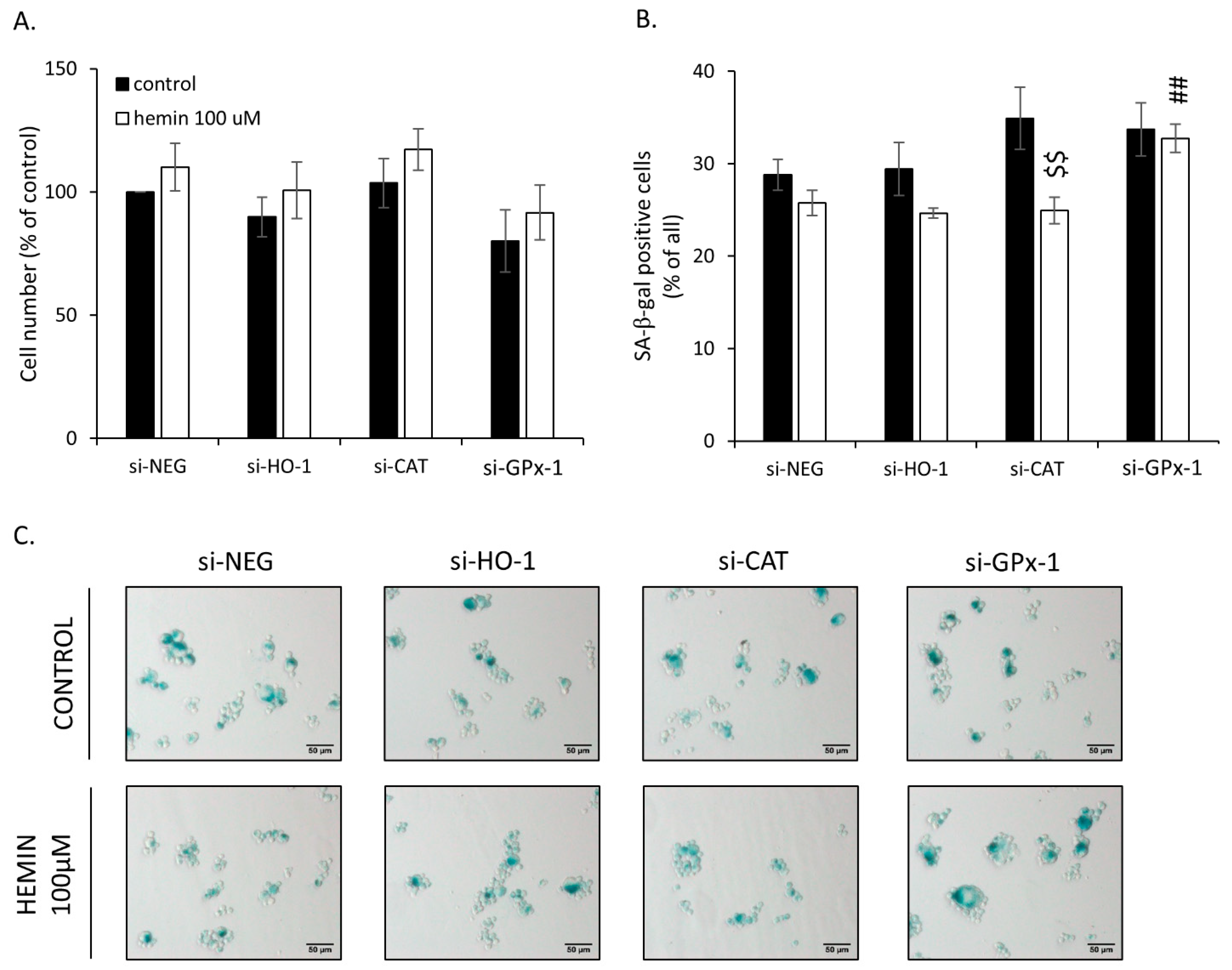
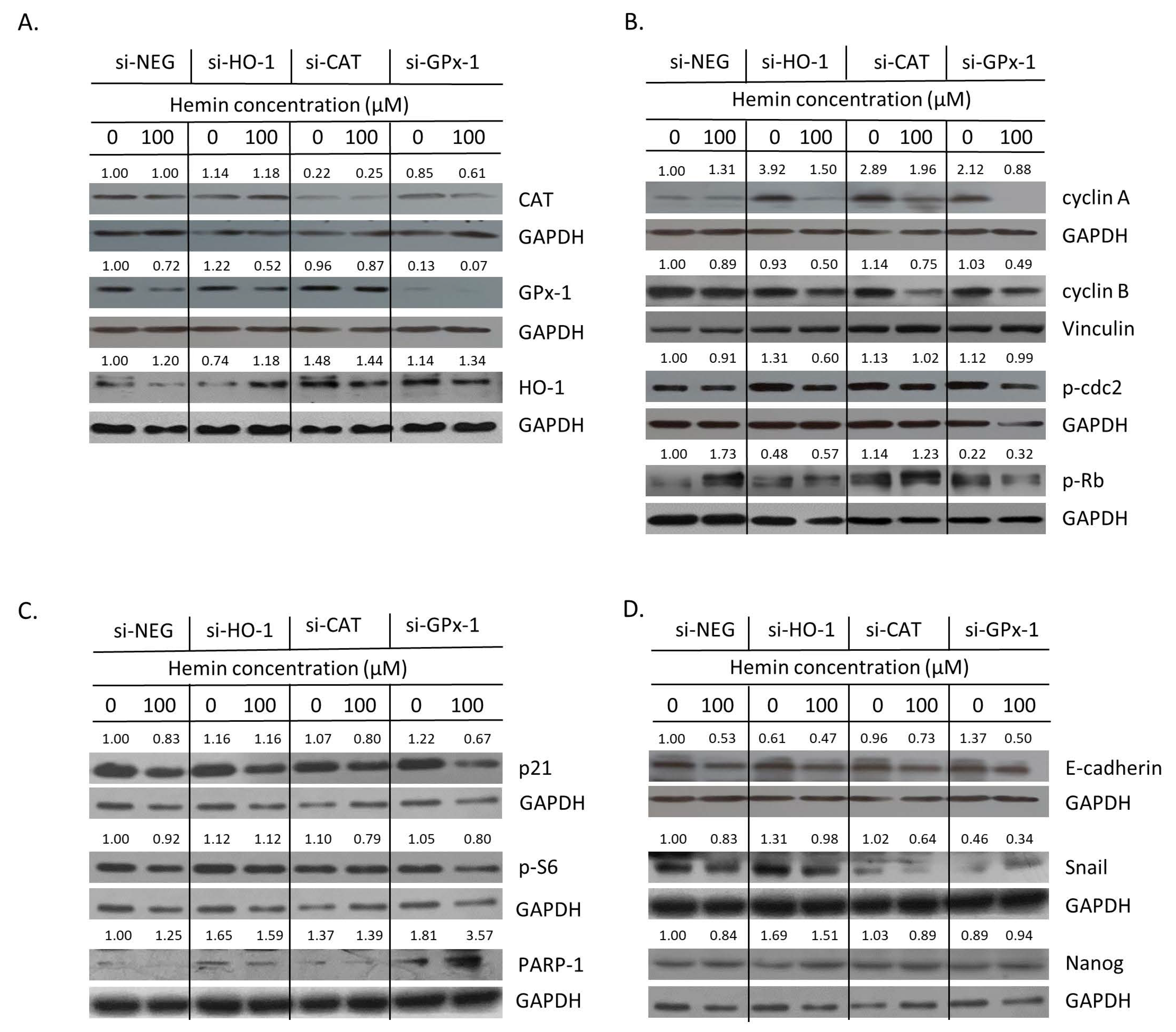
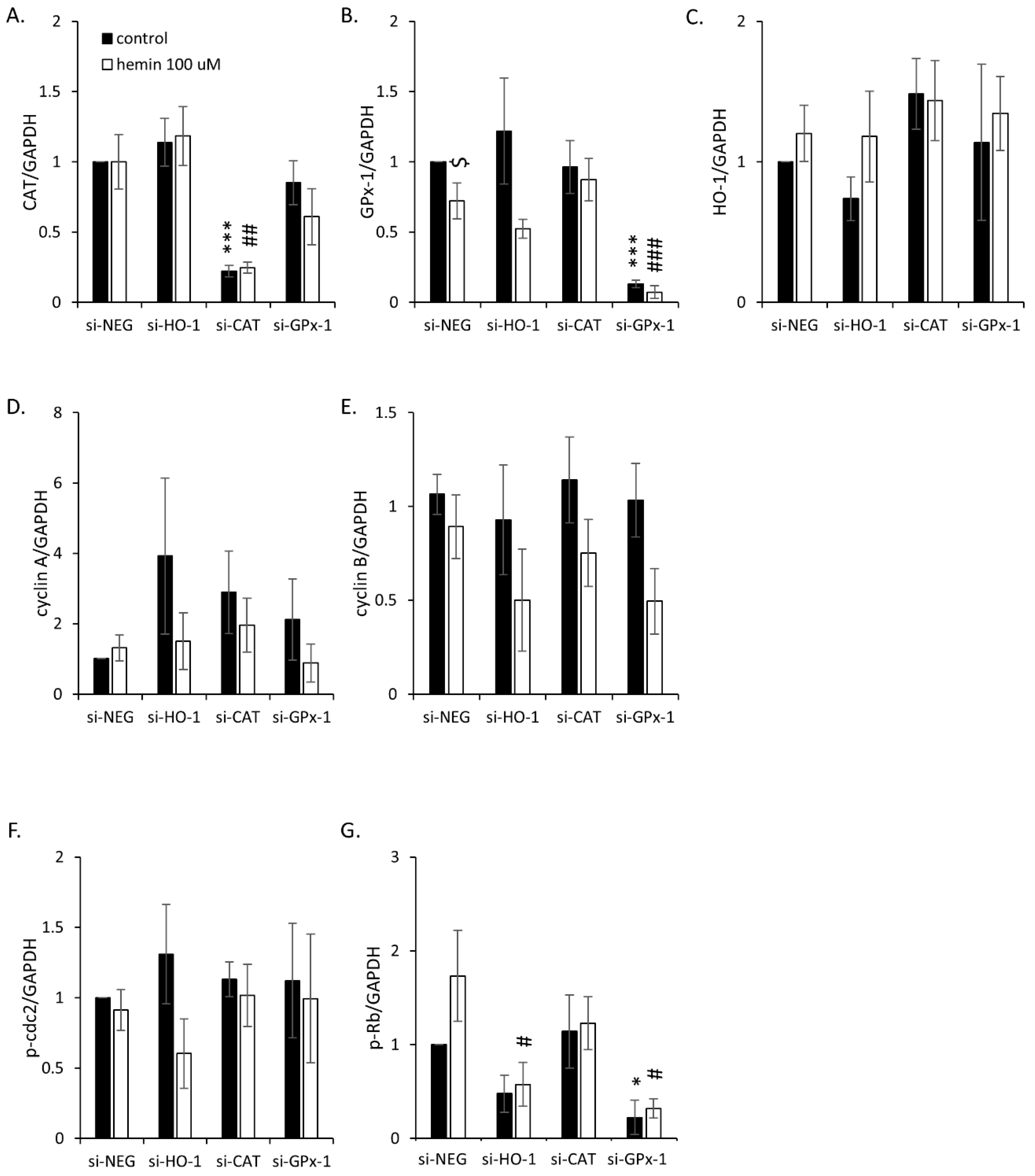
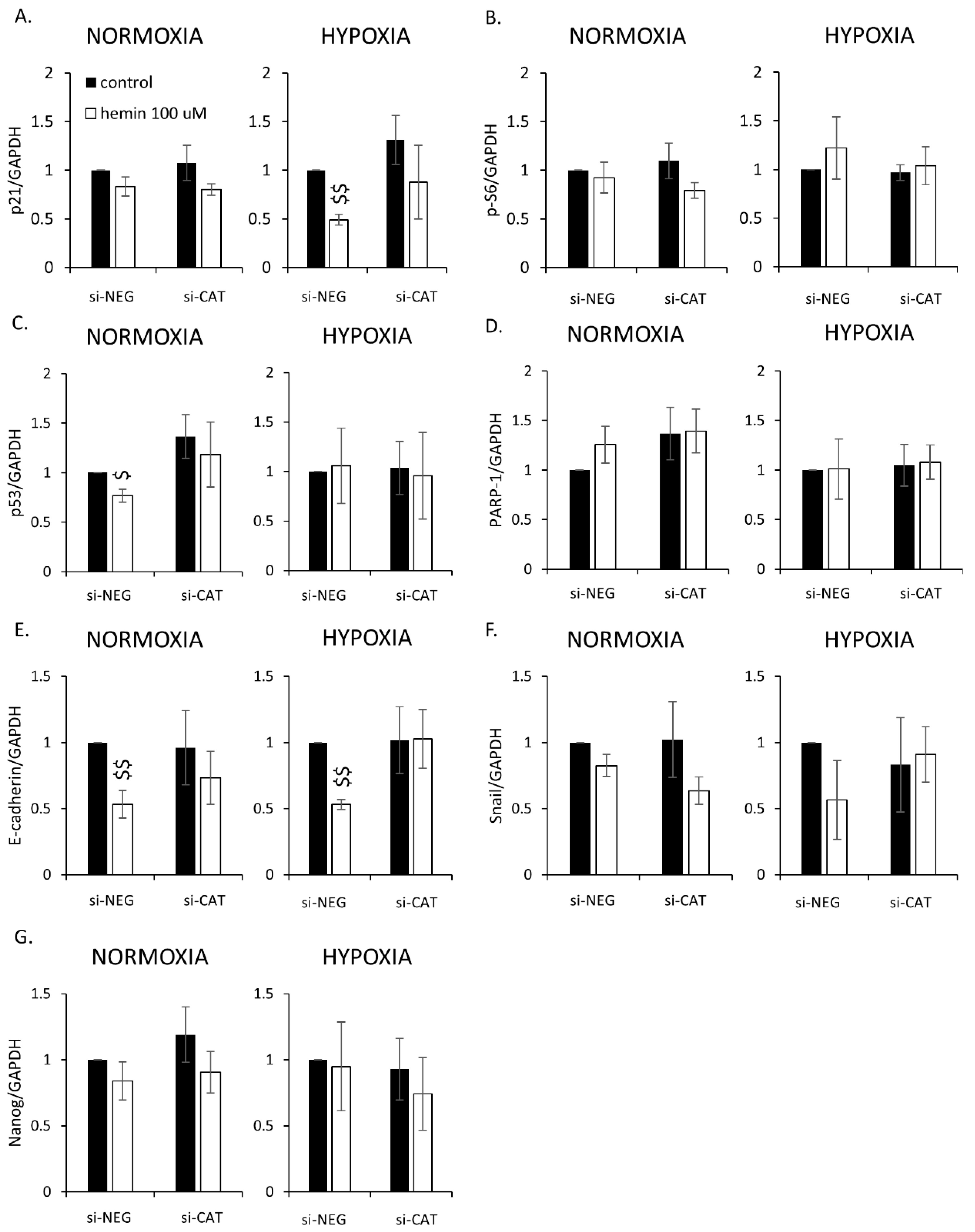
2.3. Silencing of Antioxidative Enzymes Maintains Colon Cancer Cells in a Senescent State
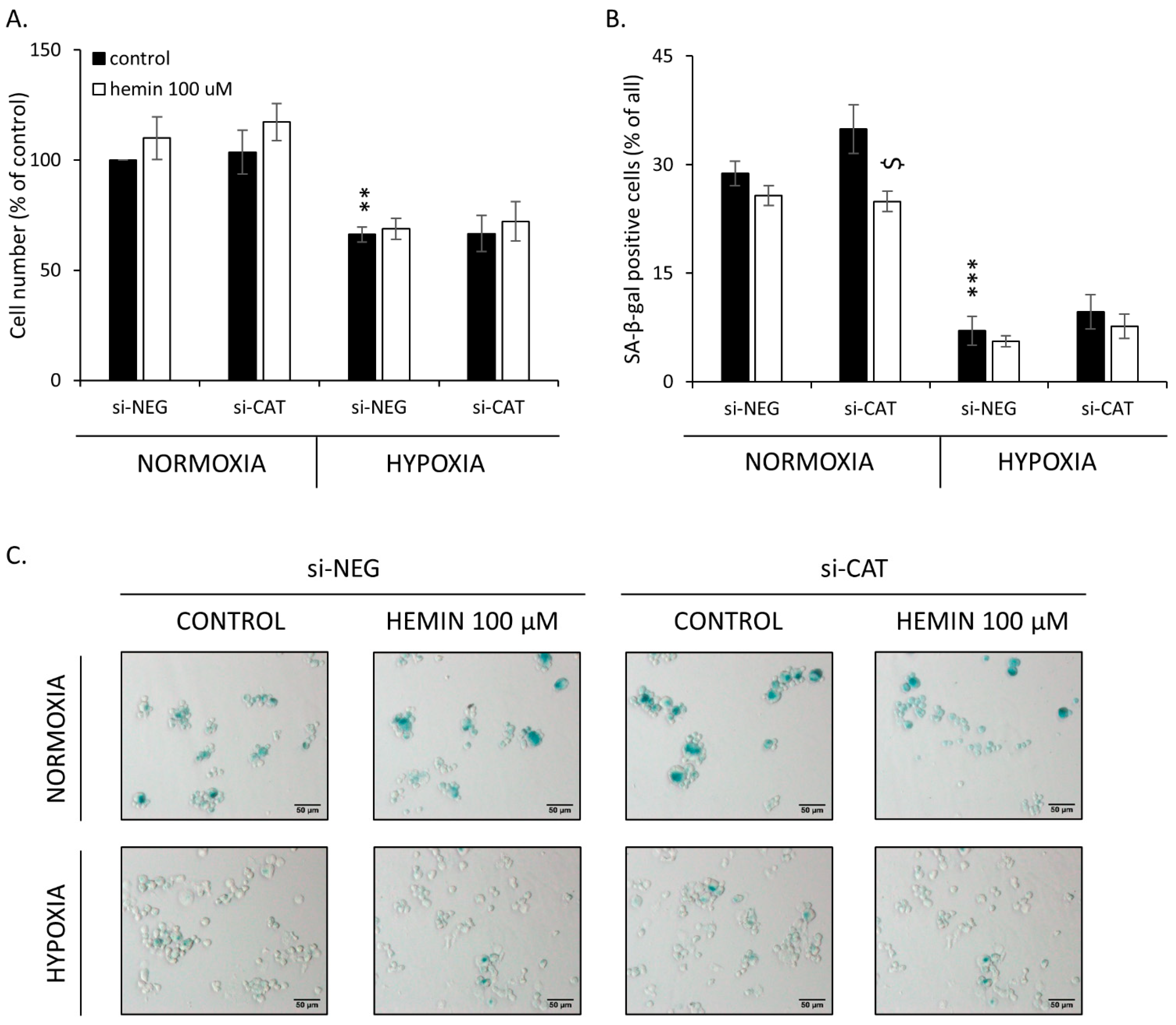
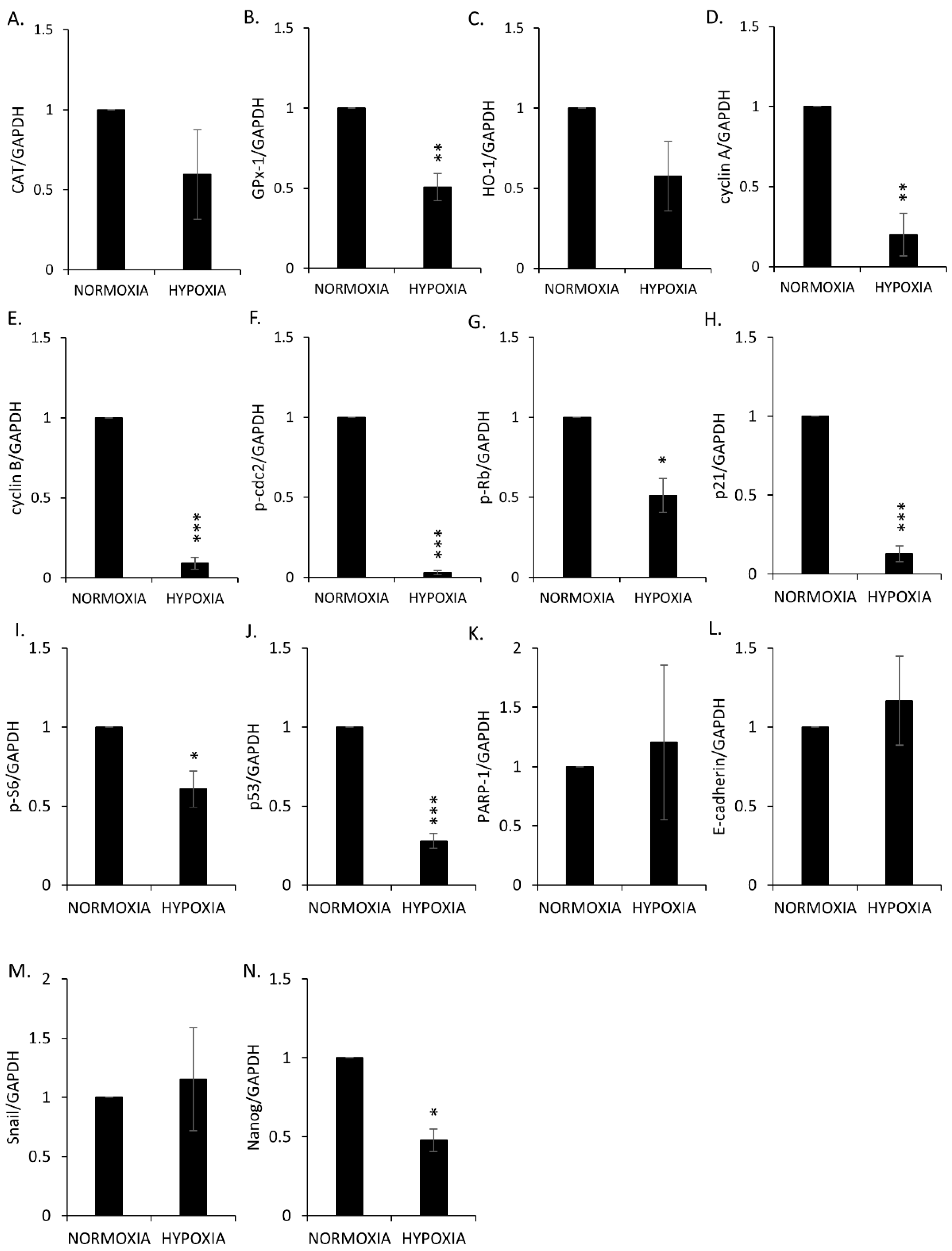
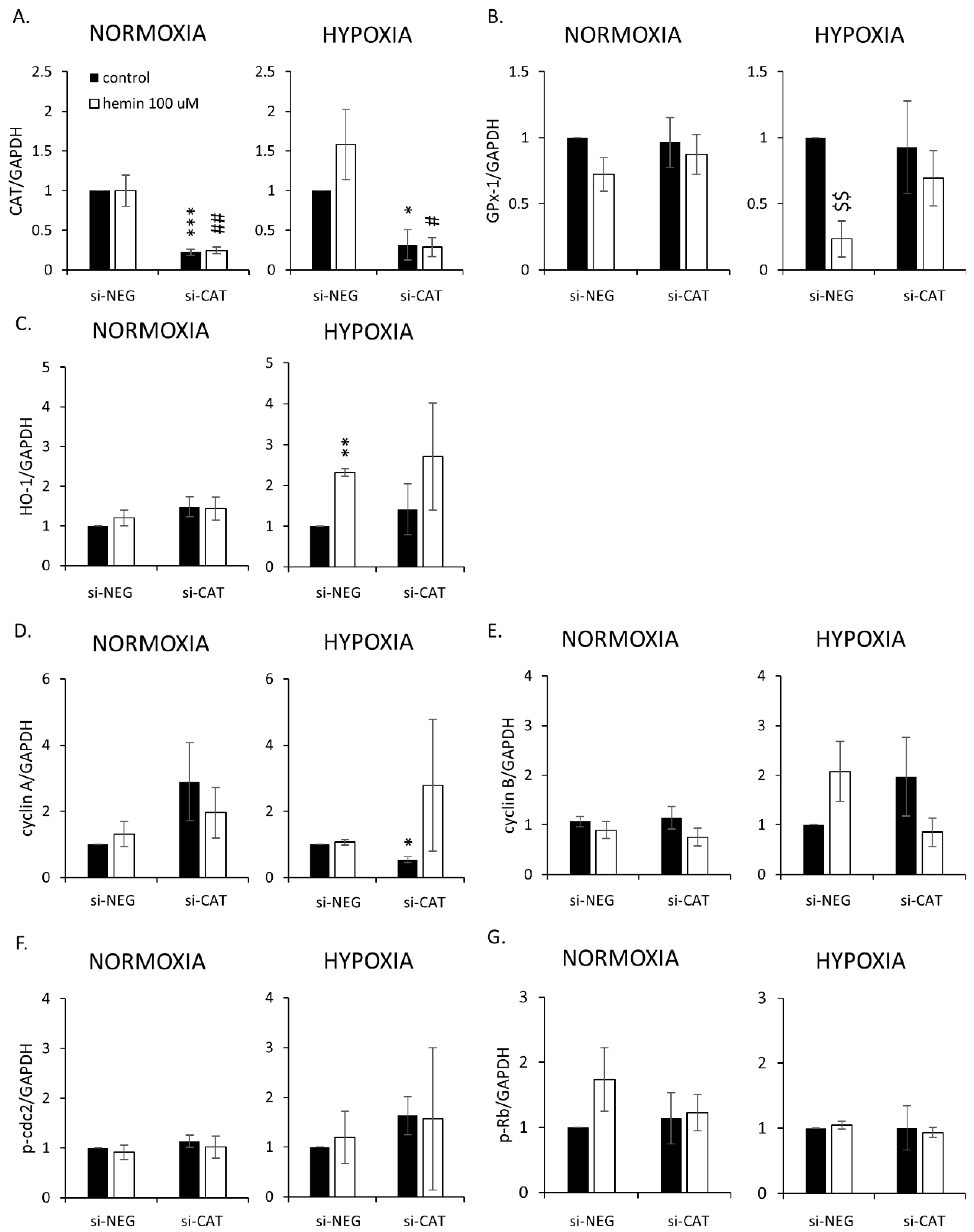
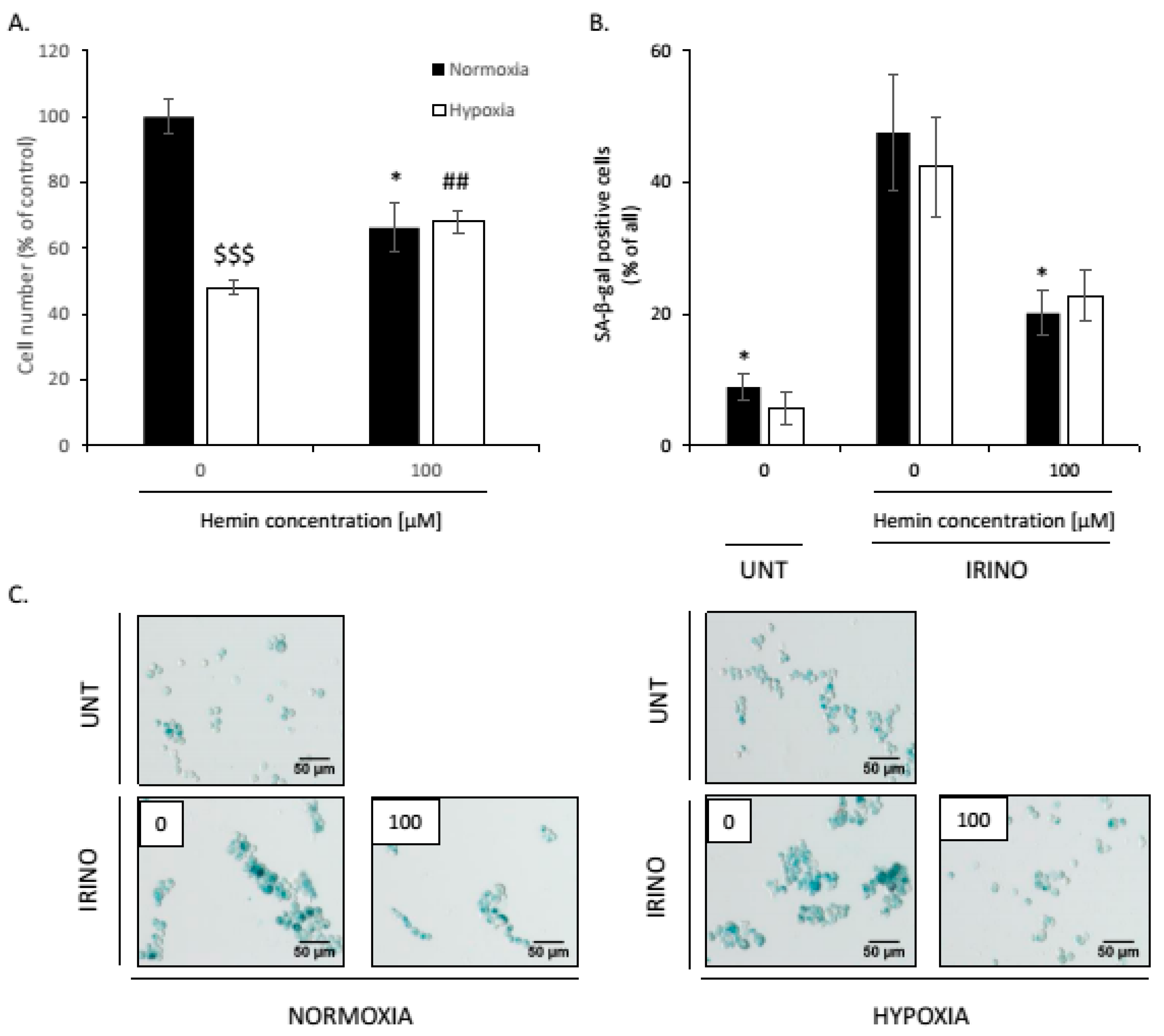
2.4. Hemin Enhances Escape from Senescence in Hypoxic Cells in the Presence of CAT
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cells and Treatment
4.3. Experiments in Hypoxia
4.4. Gene Silencing
4.5. Cell Viability Assay by MTT Metabolism
4.6. Western Blotting
4.7. Detection of Senescence-Associated β-Galactosidase (SA-β-Gal)
4.8. DNA Content Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A













References
- Favoriti, P.; Carbone, G.; Greco, M.; Pirozzi, F.; Pirozzi, R.E.; Corcione, F. Worldwide burden of colorectal cancer: A review. Updates Surg. 2016, 68, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- Montonen, J.; Boeing, H.; Fritsche, A.; Schleicher, E.; Joost, H.G.; Schulze, M.B.; Steffen, A.; Pischon, T. Consumption of red meat and whole-grain bread in relation to biomarkers of obesity, inflammation, glucose metabolism and oxidative stress. Eur. J. Nutr. 2013, 52, 337–345. [Google Scholar] [CrossRef]
- Angeli, J.P.; Garcia, C.C.; Sena, F.; Freitas, F.P.; Miyamoto, S.; Medeiros, M.H.; Di Mascio, P. Lipid hydroperoxide-induced and hemoglobin-enhanced oxidative damage to colon cancer cells. Free Radic. Biol. Med. 2011, 51, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Bastide, N.M.; Pierre, F.H.; Corpet, D.E. Heme iron from meat and risk of colorectal cancer: A meta-analysis and a review of the mechanisms involved. Cancer Prev. Res. (Phila) 2011, 4, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Erdelyi, I.; Levenkova, N.; Lin, E.Y.; Pinto, J.T.; Lipkin, M.; Quimby, F.W.; Holt, P.R. Western-style diets induce oxidative stress and dysregulate immune responses in the colon in a mouse model of sporadic colon cancer. J. Nutr. 2009, 139, 2072–2078. [Google Scholar] [CrossRef]
- Gagniere, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef]
- Estevez, M.; Li, Z.; Soladoye, O.P.; Van-Hecke, T. Health Risks of Food Oxidation. Adv. Food Nutr. Res. 2017, 82, 45–81. [Google Scholar]
- Chandrasekaran, A.; Idelchik, M.; Melendez, J.A.R. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef]
- Mandal, P. Potential biomarkers associated with oxidative stress for risk assessment of colorectal cancer. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 557–565. [Google Scholar] [CrossRef]
- Wu, P.C.; Wang, Q.; Grobman, L.; Chu, E.; Wu, D.Y. Accelerated cellular senescence in solid tumor therapy. Exp. Oncol. 2012, 34, 298–305. [Google Scholar] [PubMed]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.C.; Wang, Q.; Dong, Z.M.; Chu, E.; Roberson, R.S.; Ivanova, I.C.; Wu, D.Y. Expression of coxsackie and adenovirus receptor distinguishes transitional cancer states in therapy-induced cellular senescence. Cell Death Dis. 2010, 1, e70. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Sikora, E.; Czarnecka-Herok, J.; Bojko, A.; Sunderland, P. Therapy-induced polyploidization and senescence: Coincidence or interconnection? Semin Cancer Biol. 2022, 81, 83–95. [Google Scholar] [CrossRef]
- Was, H.; Borkowska, A.; Olszewska, A.; Klemba, A.; Marciniak, M.; Synowiec, A.; Kieda, C. Polyploidy formation in cancer cells: How a Trojan horse is born. Semin Cancer Biol. 2022, 81, 24–36. [Google Scholar] [CrossRef]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A novel type of cell division in cancer. Cancer Biol. 2004, 3, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int. 2006, 6, 25. [Google Scholar] [CrossRef]
- Rajaraman, R.; Rajaraman, M.M.; Rajaraman, S.R.; Guernsey, D.L. Neosis--a paradigm of self-renewal in cancer. Cell Biol. Int. 2005, 29, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Barszcz, K.; Czarnecka, J.; Kowalczyk, A.; Bernas, T.; Uzarowska, E.; Koza, P.; Klejman, A.; Piwocka, K.; Kaminska, B.; et al. Bafilomycin A1 triggers proliferative potential of senescent cancer cells in vitro and in NOD/SCID mice. Oncotarget 2017, 8, 9303–9322. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Czarnecka, J.; Kominek, A.; Barszcz, K.; Bernas, T.; Piwocka, K.; Kaminska, B. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol. 2018, 19, 63–75. [Google Scholar] [CrossRef]
- Chitikova, Z.V.; Gordeev, S.A.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Pospelova, T.V. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle 2014, 13, 1424–1439. [Google Scholar] [CrossRef]
- Mansilla, S.; Bataller, M.; Portugal, J. A nuclear budding mechanism in transiently arrested cells generates drug-sensitive and drug-resistant cells. Biochem Pharm. 2009, 78, 123–132. [Google Scholar] [CrossRef]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Sabisz, M.; Skladanowski, A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: Implications for drug resistance and in vitro drug screening models. Cell Cycle 2009, 8, 3208–3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.T.; Duronio, R.J. Endoreplication and polyploidy: Insights into development and disease. Development 2013, 140, 3–12. [Google Scholar] [CrossRef]
- Coward, J.; Harding, A. Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef]
- Phumala, N.; Porasuphatana, S.; Unchern, S.; Pootrakul, P.; Fucharoen, S.; Chantharaksri, U. Hemin: A possible cause of oxidative stress in blood circulation of beta-thalassemia/hemoglobin E disease. Free Radic. Res. 2003, 37, 129–135. [Google Scholar] [CrossRef]
- Goldstein, L.; Teng, Z.P.; Zeserson, E.; Patel, M.; Regan, R.F. Hemin induces an iron-dependent, oxidative injury to human neuron-like cells. J. NeuroSci. Res. 2003, 73, 113–121. [Google Scholar] [CrossRef]
- Olszewska, A.; Borkowska, A.; Granica, M.; Karolczak, J.; Zglinicki, B.; Kieda, C.; Was, H. Escape From Cisplatin-Induced Senescence of Hypoxic Lung Cancer Cells Can Be Overcome by Hydroxychloroquine. Front. Oncol. 2021, 11, 738385. [Google Scholar] [CrossRef]
- Kong, Y.; Cui, H.; Ramkumar, C.; Zhang, H. Regulation of senescence in cancer and aging. J. Aging Res. 2011, 2011, 963172. [Google Scholar] [CrossRef]
- Sun, H.; Wang, H.; Wang, X.; Aoki, Y.; Wang, X.; Yang, Y.; Cheng, X.; Wang, Z.; Wang, X. Aurora-A/SOX8/FOXK1 signaling axis promotes chemoresistance via suppression of cell senescence and induction of glucose metabolism in ovarian cancer organoids and cells. Theranostics 2020, 10, 6928–6945. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Xu, X.; Liu, C.; Wu, Q.; Wei, J.; Meng, F.; Zhou, L.; Wang, Z.; Lei, L.; Liu, P. Negative regulation of transcription factor FoxM1 by p53 enhances oxaliplatin-induced senescence in hepatocellular carcinoma. Cancer Lett. 2013, 331, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Vassel, F.M.; Bian, K.; Walker, G.C.; Hemann, M.T. Rev7 loss alters cisplatin response and increases drug efficacy in chemotherapy-resistant lung cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 28922–28924. [Google Scholar] [CrossRef]
- Almeida, L.O.; Guimaraes, D.M.; Martins, M.D.; Martins, M.A.T.; Warner, K.A.; Nor, J.E.; Castilho, R.M.; Squarize, C.H. Unlocking the chromatin of adenoid cystic carcinomas using HDAC inhibitors sensitize cancer stem cells to cisplatin and induces tumor senescence. Stem Cell Res. 2017, 21, 94–105. [Google Scholar] [CrossRef]
- Hao, X.; Zhao, B.; Zhou, W.; Liu, H.; Fukumoto, T.; Gabrilovich, D.; Zhang, R. Sensitization of ovarian tumor to immune checkpoint blockade by boosting senescence-associated secretory phenotype. iScience 2021, 24, 102016. [Google Scholar] [CrossRef]
- Bojko, A.; Staniak, K.; Czarnecka-Herok, J.; Sunderland, P.; Dudkowska, M.; Sliwinska, M.A.; Salmina, K.; Sikora, E. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 6084. [Google Scholar] [CrossRef]
- Gerashchenko, B.I.; Salmina, K.; Eglitis, J.; Huna, A.; Grjunberga, V.; Erenpreisa, J. Disentangling the aneuploidy and senescence paradoxes: A study of triploid breast cancers non-responsive to neoadjuvant therapy. Histochem Cell Biol. 2016, 145, 497–508. [Google Scholar] [CrossRef]
- Huna, A.; Salmina, K.; Erenpreisa, J.; Vazquez-Martin, A.; Krigerts, J.; Inashkina, I.; Gerashchenko, B.I.; Townsend, P.A.; Cragg, M.S.; Jackson, T.R. Role of stress-activated OCT4A in the cell fate decisions of embryonal carcinoma cells treated with etoposide. Cell Cycle 2015, 14, 2969–2984. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef]
- Jackson, T.R.; Salmina, K.; Huna, A.; Inashkina, I.; Jankevics, E.; Riekstina, U.; Kalnina, Z.; Ivanov, A.; Townsend, P.A.; Cragg, M.S.; et al. DNA damage causes TP53-dependent coupling of self-renewal and senescence pathways in embryonal carcinoma cells. Cell Cycle 2013, 12, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef] [PubMed]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is Senescence Reversible? Curr Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Torisu-Itakura, H.; Furue, M.; Kuwano, M.; Ono, M. Co-expression of thymidine phosphorylase and heme oxygenase-1 in macrophages in human malignant vertical growth melanomas. Jpn. J. Cancer Res. 2000, 91, 906–910. [Google Scholar] [CrossRef]
- Goodman, A.I.; Choudhury, M.; da Silva, J.L.; Schwartzman, M.L.; Abraham, N.G. Overexpression of the heme oxygenase gene in renal cell carcinoma. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1997, 214, 54–61. [Google Scholar] [CrossRef]
- Maines, M.D.; Abrahamsson, P.A. Expression of heme oxygenase-1 (HSP32) in human prostate: Normal, hyperplastic, and tumor tissue distribution. Urology 1996, 47, 727–733. [Google Scholar] [CrossRef]
- Berberat, P.O.; Dambrauskas, Z.; Gulbinas, A.; Giese, T.; Giese, N.; Kunzli, B.; Autschbach, F.; Meuer, S.; Buchler, M.W.; Friess, H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 2005, 11, 3790–3798. [Google Scholar] [CrossRef]
- Nowis, D.; Legat, M.; Grzela, T.; Niderla, J.; Wilczek, E.; Wilczynski, G.M.; Glodkowska, E.; Mrowka, P.; Issat, T.; Dulak, J.; et al. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene 2006, 25, 3365–3374. [Google Scholar] [CrossRef]
- Kocanova, S.; Buytaert, E.; Matroule, J.Y.; Piette, J.; Golab, J.; de Witte, P.; Agostinis, P. Induction of heme-oxygenase 1 requires the p38MAPK and PI3K pathways and suppresses apoptotic cell death following hypericin-mediated photodynamic therapy. Apoptosis 2007, 12, 731–741. [Google Scholar] [CrossRef]
- Grimm, S.; Mvondo, D.; Grune, T.; Breusing, N. The outcome of 5-ALA-mediated photodynamic treatment in melanoma cells is influenced by vitamin C and heme oxygenase-1. Biofactors 2011, 37, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [PubMed]
- Tertil, M.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Was, H.; Collet, G.; Guichard, A.; Gil, T.; Kuzdzal, J.; Jozkowicz, A.; et al. Regulation and novel action of thymidine phosphorylase in non-small cell lung cancer: Crosstalk with Nrf2 and HO-1. PLoS ONE 2014, 9, e97070. [Google Scholar]
- Skrzypek, K.; Tertil, M.; Golda, S.; Ciesla, M.; Weglarczyk, K.; Collet, G.; Guichard, A.; Kozakowska, M.; Boczkowski, J.; Was, H.; et al. Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid. Redox Signal. 2013, 19, 644–660. [Google Scholar] [CrossRef]
- Was, H.; Sokolowska, M.; Sierpniowska, A.; Dominik, P.; Skrzypek, K.; Lackowska, B.; Pratnicki, A.; Grochot-Przeczek, A.; Taha, H.; Kotlinowski, J.; et al. Effects of heme oxygenase-1 on induction and development of chemically induced squamous cell carcinoma in mice. Free Radic. Biol. Med. 2011, 51, 1717–1726. [Google Scholar] [CrossRef]
- Was, H.; Dulak, J.; Jozkowicz, A. Heme oxygenase-1 in tumor biology and therapy. Curr Drug Targets 2010, 11, 1551–1570. [Google Scholar] [CrossRef]
- Nowis, D.; Bugajski, M.; Winiarska, M.; Bil, J.; Szokalska, A.; Salwa, P.; Issat, T.; Was, H.; Jozkowicz, A.; Dulak, J.; et al. Zinc protoporphyrin IX, a heme oxygenase-1 inhibitor, demonstrates potent antitumor effects but is unable to potentiate antitumor effects of chemotherapeutics in mice. BMC Cancer 2008, 8, 197. [Google Scholar] [CrossRef]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid Redox Signal. 2007, 9, 2099–2117. [Google Scholar] [CrossRef]
- Kusienicka, A.; Bukowska-Strakova, K.; Ciesla, M.; Nowak, W.N.; Bronisz-Budzynska, I.; Seretny, A.; Zukowska, M.; Jez, M.; Krutyholowa, R.; Taha, H.; et al. Heme Oxygenase-1 Has a Greater Effect on Melanoma Stem Cell Properties Than the Expression of Melanoma-Initiating Cell Markers. Int. J. Mol. Sci. 2022, 23, 3596. [Google Scholar] [CrossRef]
- Was, H.; Cichon, T.; Smolarczyk, R.; Lackowska, B.; Mazur-Bialy, A.; Mazur, M.; Szade, A.; Dominik, P.; Mazan, M.; Kotlinowski, J.; et al. Effect of Heme Oxygenase-1 on Melanoma Development in Mice-Role of Tumor-Infiltrating Immune Cells. Antioxidants 2020, 9, 1223. [Google Scholar] [CrossRef]
- Hedblom, A.; Hejazi, S.M.; Canesin, G.; Choudhury, R.; Hanafy, K.A.; Csizmadia, E.; Persson, J.L.; Wegiel, B. Heme detoxification by heme oxygenase-1 reinstates proliferative and immune balances upon genotoxic tissue injury. Cell Death Dis. 2019, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Kihara, M.; Yokomise, H.; Yamauchi, A.; Irie, A.; Matsusaka, K.; Miyauchi, A. Spontaneous rupture of a parathyroid adenoma presenting as a massive cervical hemorrhage: Report of a case. Surg. Today 2001, 31, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Cliff, J.F.; Newman, L.; Malone, M.; Brady, G.; Crean, S.J. Facial features of widespread neuroblastoma: A case report. Int. J. Paediatr. Dent. 2001, 11, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.; Saleh, H.A.; Goh, V.; Ryan, R. A new presentation of angiopathy in neurofibromatosis type 1. J. Laryngol. Otol. 2000, 114, 993–994. [Google Scholar] [CrossRef] [PubMed]
- Connolly, C.M.; Soldin, M.; Dawson, A.; Cooper, A.C. Metastatic malignant melanoma presenting with a bruise. Br. J. Plast Surg. 2003, 56, 725. [Google Scholar] [CrossRef]
- Steele, L.; Yeoh, C.C. Sentinel bruising as a presentation of metastatic melanoma. BMJ Case Rep. 2019, 12. [Google Scholar] [CrossRef]
- Kim, D.H.; Yoon, H.J.; Cha, Y.N.; Surh, Y.J. Role of heme oxygenase-1 and its reaction product, carbon monoxide, in manifestation of breast cancer stem cell-like properties: Notch-1 as a putative target. Free Radic. Res. 2018, 52, 1336–1347. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Liu, J. Tumor stroma and differentiated cancer cells can be originated directly from polyploid giant cancer cells induced by paclitaxel. Int. J. Cancer 2014, 134, 508–518. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Sood, A.; Bast, R.C.; Liu, J. Coevolution of neoplastic epithelial cells and multilineage stroma via polyploid giant cells during immortalization and transformation of mullerian epithelial cells. Genes Cancer 2016, 7, 60–72. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Liu, J. Generation of erythroid cells from fibroblasts and cancer cells in vitro and in vivo. Cancer Lett. 2013, 333, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, L.M.; Jimenez, C.; Valverde, A.; Hernandez, V.; Penarando, J.; Martinez, A.; Lopez-Pedrera, C.; Munoz-Castaneda, J.R.; De la Haba-Rodriguez, J.R.; Aranda, E.; et al. CoCl2, a mimic of hypoxia, induces formation of polyploid giant cells with stem characteristics in colon cancer. PLoS ONE 2014, 9, e99143. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zheng, M.; Zhang, H.; Yang, X.; Fan, L.; Fu, F.; Fu, J.; Niu, R.; Yan, M.; Zhang, S. Arsenic Trioxide Promotes Tumor Progression by Inducing the Formation of PGCCs and Embryonic Hemoglobin in Colon Cancer Cells. Front. Oncol. 2021, 11, 720814. [Google Scholar] [CrossRef] [PubMed]
- Even, B.; Fayad-Kobeissi, S.; Gagliolo, J.M.; Motterlini, R.; Boczkowski, J.; Foresti, R.; Dagouassat, M. Heme oxygenase-1 induction attenuates senescence in chronic obstructive pulmonary disease lung fibroblasts by protecting against mitochondria dysfunction. Aging Cell 2018, 17, e12837. [Google Scholar] [CrossRef]
- Shan, H.; Li, T.; Zhang, L.; Yang, R.; Li, Y.; Zhang, M.; Dong, Y.; Zhou, Y.; Xu, C.; Yang, B.; et al. Heme oxygenase-1 prevents heart against myocardial infarction by attenuating ischemic injury-induced cardiomyocytes senescence. eBioMedicine 2019, 39, 59–68. [Google Scholar] [CrossRef]
- Zheng, H.; Liang, X.; Han, Q.; Shao, Z.; Zhang, Y.; Shi, L.; Hong, Y.; Li, W.; Mai, C.; Mo, Q.; et al. Hemin enhances the cardioprotective effects of mesenchymal stem cell-derived exosomes against infarction via amelioration of cardiomyocyte senescence. J. Nanobiotechnology 2021, 19, 332. [Google Scholar] [CrossRef]
- Luo, W.; Li, J.; Li, Z.; Lin, T.; Zhang, L.; Yang, W.; Mai, Y.; Liu, R.; Chen, M.; Dai, C.; et al. HO-1 nuclear accumulation and interaction with NPM1 protect against stress-induced endothelial senescence independent of its enzymatic activity. Cell Death Dis. 2021, 12, 738. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer 2004, 3, 294–300. [Google Scholar] [CrossRef]
- de Sa Junior, P.L.; Camara, D.A.D.; Porcacchia, A.S.; Fonseca, P.M.M.; Jorge, S.D.; Araldi, R.P.; Ferreira, A.K. The Roles of ROS in Cancer Heterogeneity and Therapy. Oxid. Med. Cell Longev. 2017, 2017, 2467940. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, K.; Martien, S.; Pourtier, A.; Vercamer, C.; Ostoich, P.; Morat, L.; Sabatier, L.; Duprez, L.; T’Kint de Roodenbeke, C.; Gilson, E.; et al. Senescence-associated oxidative DNA damage promotes the generation of neoplastic cells. Cancer Res. 2009, 69, 7917–7925. [Google Scholar] [CrossRef] [Green Version]
- Achuthan, S.; Santhoshkumar, T.R.; Prabhakar, J.; Nair, S.A.; Pillai, M.R. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J. Biol. Chem. 2011, 286, 37813–37829. [Google Scholar] [CrossRef] [PubMed]
- Dharmalingam, P.; Talakatta, G.; Mitra, J.; Wang, H.; Derry, P.J.; Nilewski, L.G.; McHugh, E.A.; Fabian, R.H.; Mendoza, K.; Vasquez, V.; et al. Pervasive Genomic Damage in Experimental Intracerebral Hemorrhage: Therapeutic Potential of a Mechanistic-Based Carbon Nanoparticle. ACS Nano 2020, 14, 2827–2846. [Google Scholar] [CrossRef] [PubMed]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Antibody | Clonality | Clone | Phosphorylation Site | Host | Manufacturer | Reference Number |
|---|---|---|---|---|---|---|
| Catalase | Polyclonal | - | - | Rabbit | Abcam | ab16731 |
| Cyclin A | Monoclonal | BF683 | - | Mouse | Santa Cruz Biotechnology | sc-239 |
| Cyclin B | Monoclonal | D5C10 | - | Rabbit | Cell Signalling | 12231 |
| E-cadherin | Monoclonal | 24E10 | - | Rabbit | Cell Signalling | 3195 |
| GAPDH | Monoclonal | 6C5 | - | Mouse | Millipore | MAB374 |
| GPx-1 | Polyclonal | - | - | Rabbit | Abcam | ab22604 |
| HO-1 | Polyclonal | - | Rabbit | Enzo Life Sciences | ADI-SPA-894 | |
| Nanog | Monoclonal | D73G4 | - | Rabbit | Cell Signalling | 4903 |
| p21 | Monoclonal | 12D1 | - | Rabbit | Cell Signalling | 2947 |
| p21 | Monoclonal | CP74 | Mouse | Sigma-Aldrich | P1484 | |
| p53 | Monoclonal | DO-1 | - | Mouse | Santa Cruz Biotechnology | sc-126 |
| PARP-1 | Monoclonal | C2-10 | - | Mouse | BD-Biosciences | 556362 |
| p-cdc2 | Monoclonal | 10A11 | Tyr15 | Rabbit | Cell Signalling | 4539 |
| p-p53 | Monoclonal | 16G8 | Ser15 | Mouse | Cell Signalling | 9286 |
| p-Rb | Polyclonal | - | Ser807/811 | Rabbit | Cell Signalling | 9308 |
| p-S6 | Monoclonal | D57.2.2E | Ser235/236 | Rabbit | Cell Signalling | 4858 |
| Snail | Monoclonal | C15D3 | - | Rabbit | Cell Signalling | 3879 |
| Vinculin | Monoclonal | V284 | - | Mouse | Sanata Cruz Biotechnology | sc-59803 |
| γ-H2AX | Monoclonal | 9F3 | Ser139 | Mouse | Abcam | ab26350 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borkowska, A.; Olszewska, A.; Skarzynska, W.; Marciniak, M.; Skrzeszewski, M.; Kieda, C.; Was, H. High Hemin Concentration Induces Escape from Senescence of Normoxic and Hypoxic Colon Cancer Cells. Cancers 2022, 14, 4793. https://doi.org/10.3390/cancers14194793
Borkowska A, Olszewska A, Skarzynska W, Marciniak M, Skrzeszewski M, Kieda C, Was H. High Hemin Concentration Induces Escape from Senescence of Normoxic and Hypoxic Colon Cancer Cells. Cancers. 2022; 14(19):4793. https://doi.org/10.3390/cancers14194793
Chicago/Turabian StyleBorkowska, Agata, Aleksandra Olszewska, Weronika Skarzynska, Marta Marciniak, Maciej Skrzeszewski, Claudine Kieda, and Halina Was. 2022. "High Hemin Concentration Induces Escape from Senescence of Normoxic and Hypoxic Colon Cancer Cells" Cancers 14, no. 19: 4793. https://doi.org/10.3390/cancers14194793