
Analysis of NIS Plasma Membrane Interactors Discloses Key Regulation by a SRC/RAC1/PAK1/PIP5K/EZRIN Pathway with Potential Implications for Radioiodine Re-Sensitization Therapy in Thyroid Cancer


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of the HA-NIS Construct
2.2. Cell Line Generation
2.3. Cell Culture, Treatment and Transfection
2.4. Co-Immunoprecipitation of Plasma Membrane-Specific NIS Interactors
2.5. Co-Immunoprecipitation of the Whole Cell NIS Interactors
2.6. Western Blot Analysis
2.7. Mass Spectrometry (MS) and MS Data Analysis
2.8. Biotinylation of the Cell-Surface Proteins
2.9. Immunofluorescence and Confocal Microscopy
2.10. HS-YFP-Based NIS Functional Assay
2.11. Active RAC Pull-Down Assay
2.12. TCGA Data Extraction and General Statistical Analysis
3. Results
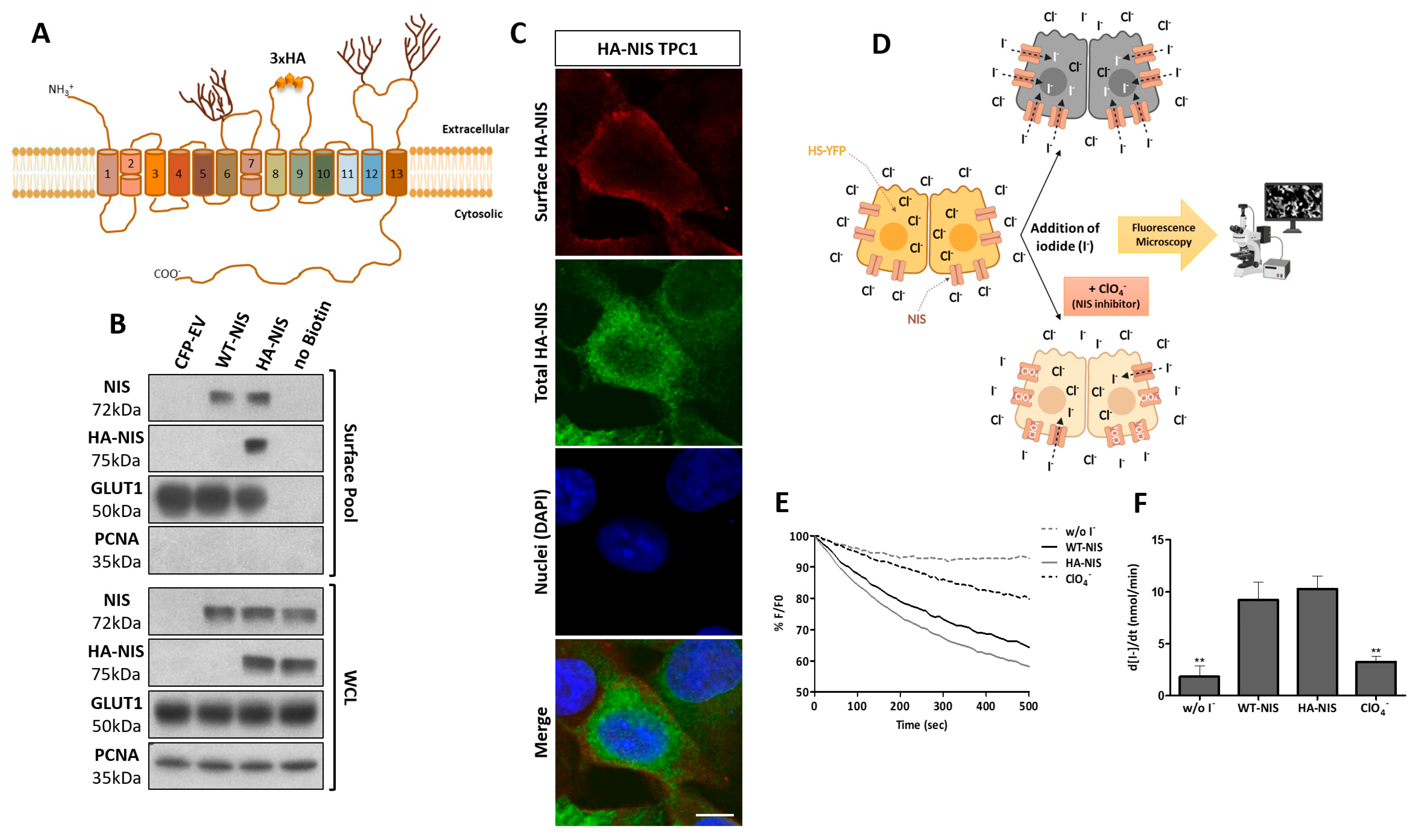
3.1. Production and Functional Validation of the Stable HA-NIS-Expressing TPC1 Cell Line
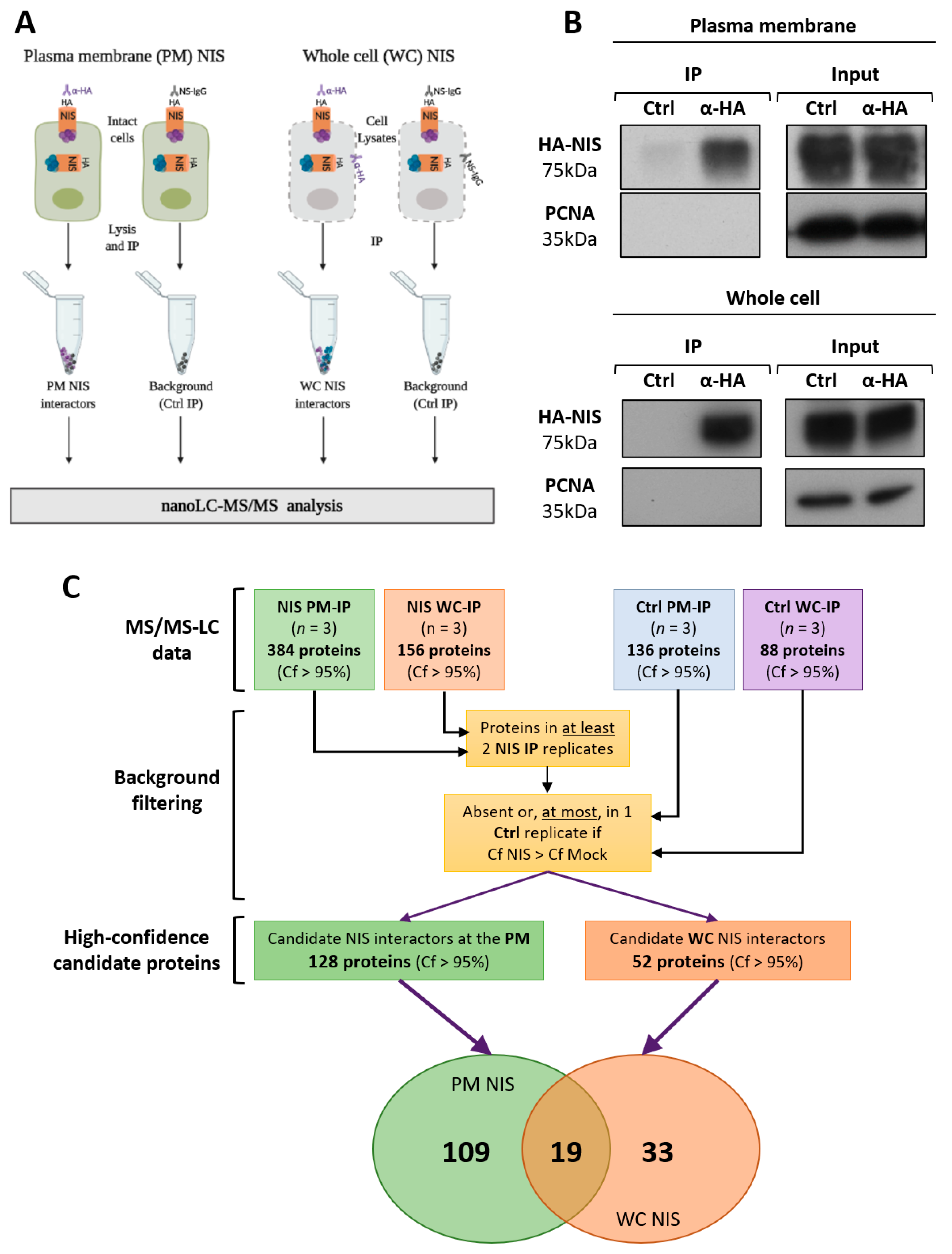
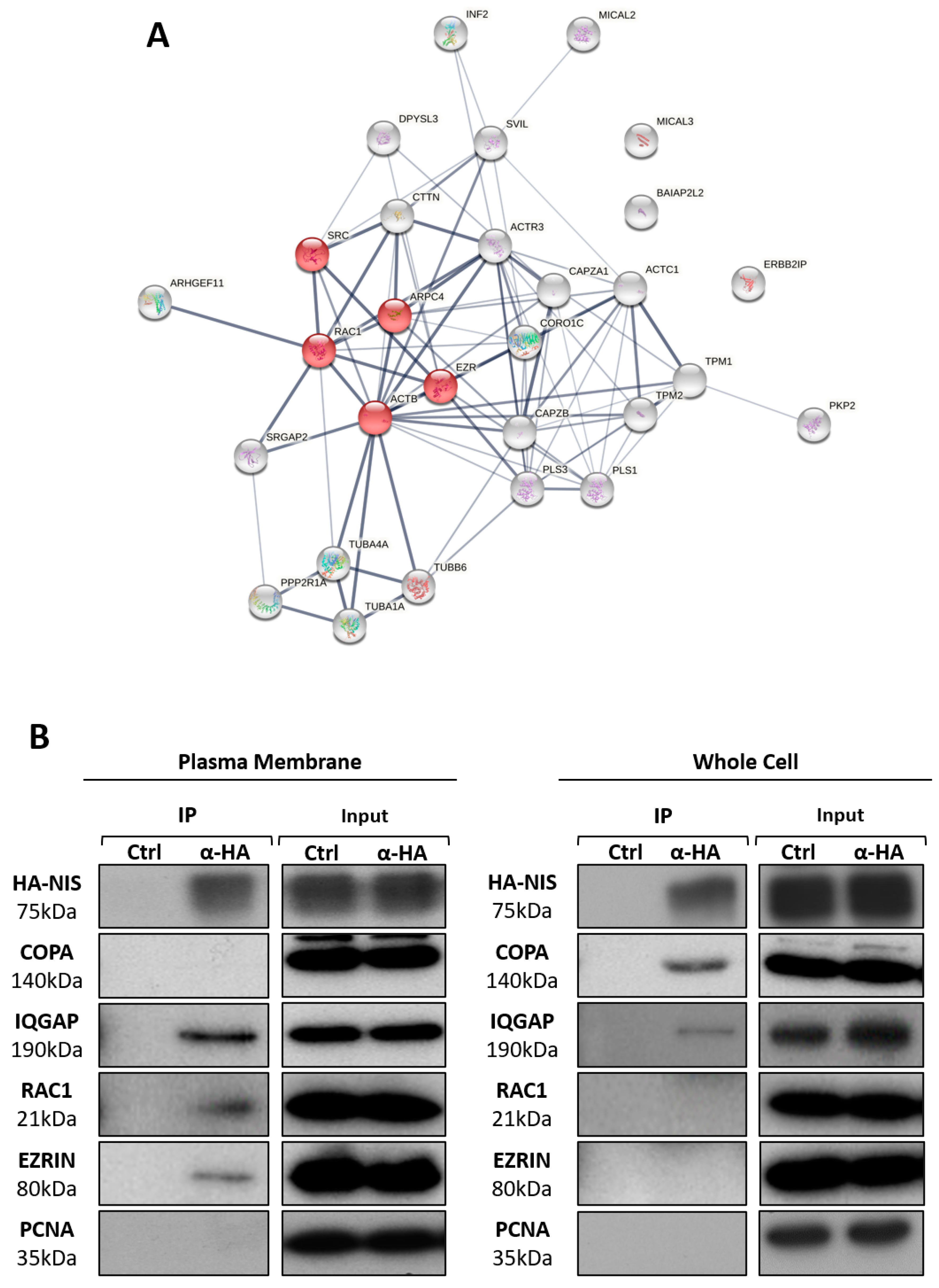
3.2. NIS PM Interactome Is Enriched in Proteins Associated with Actin Cytoskeleton Dynamics
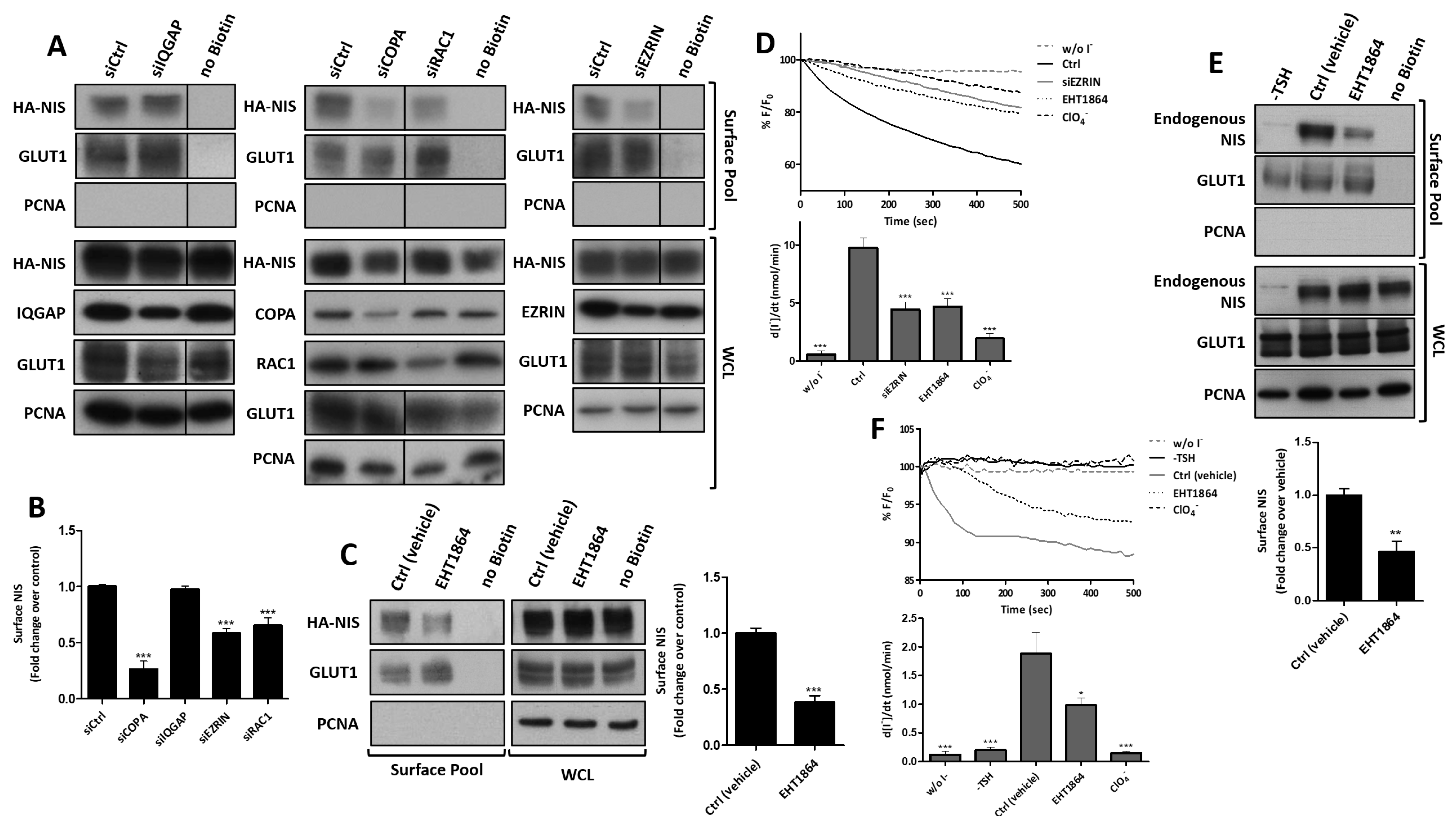
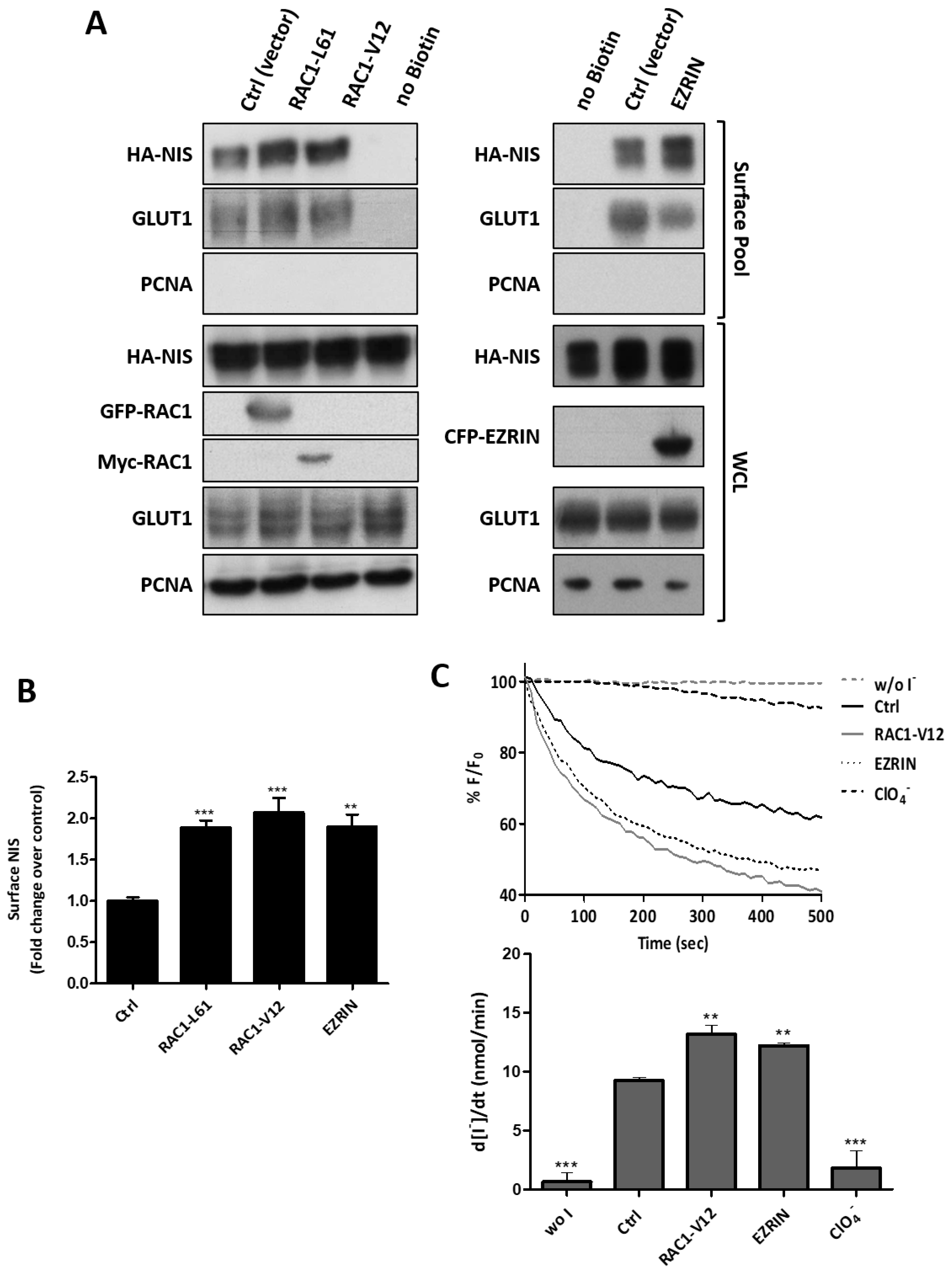
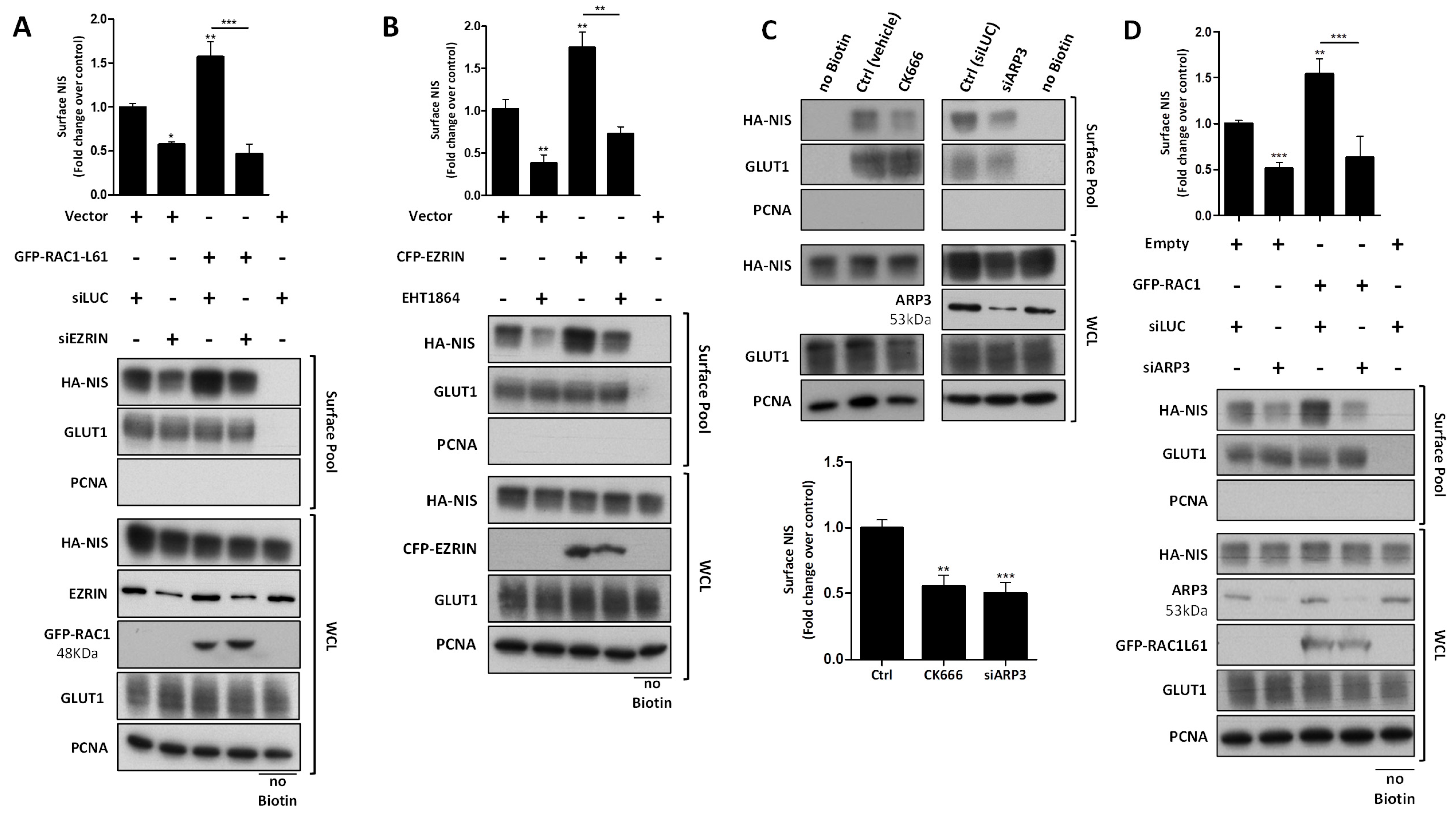
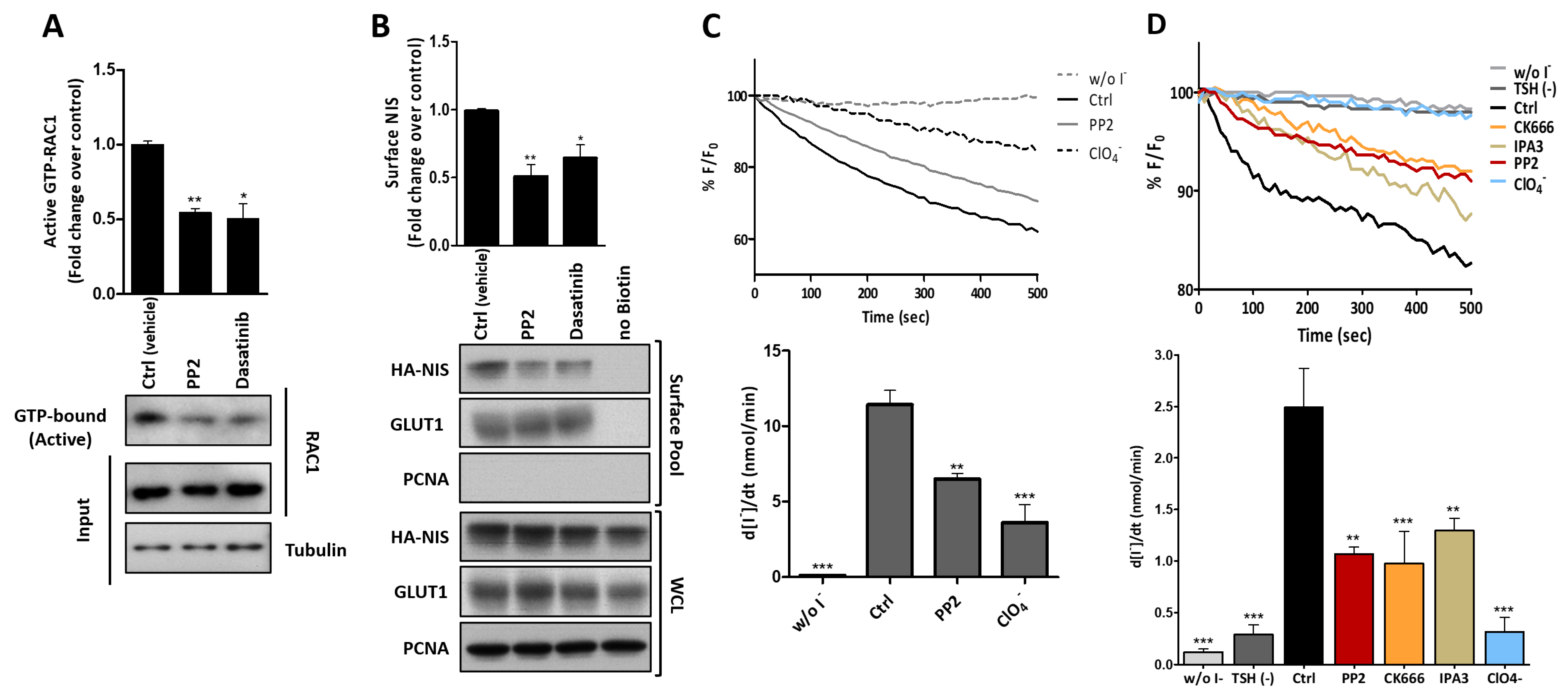
3.3. RAC1 Upregulates NIS Plasma Membrane Levels by Promoting Actin Polymerization and EZRIN Recruitment
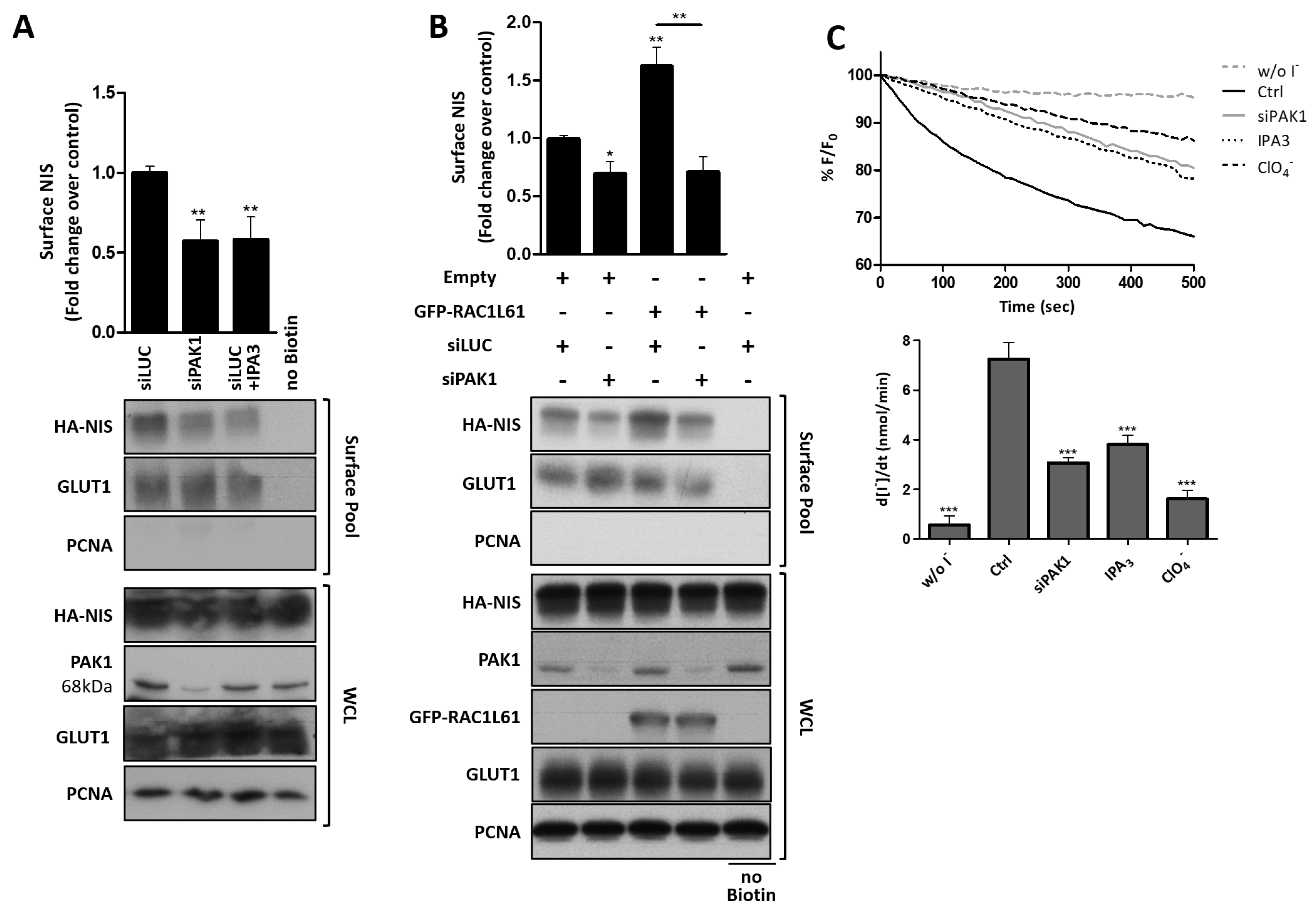
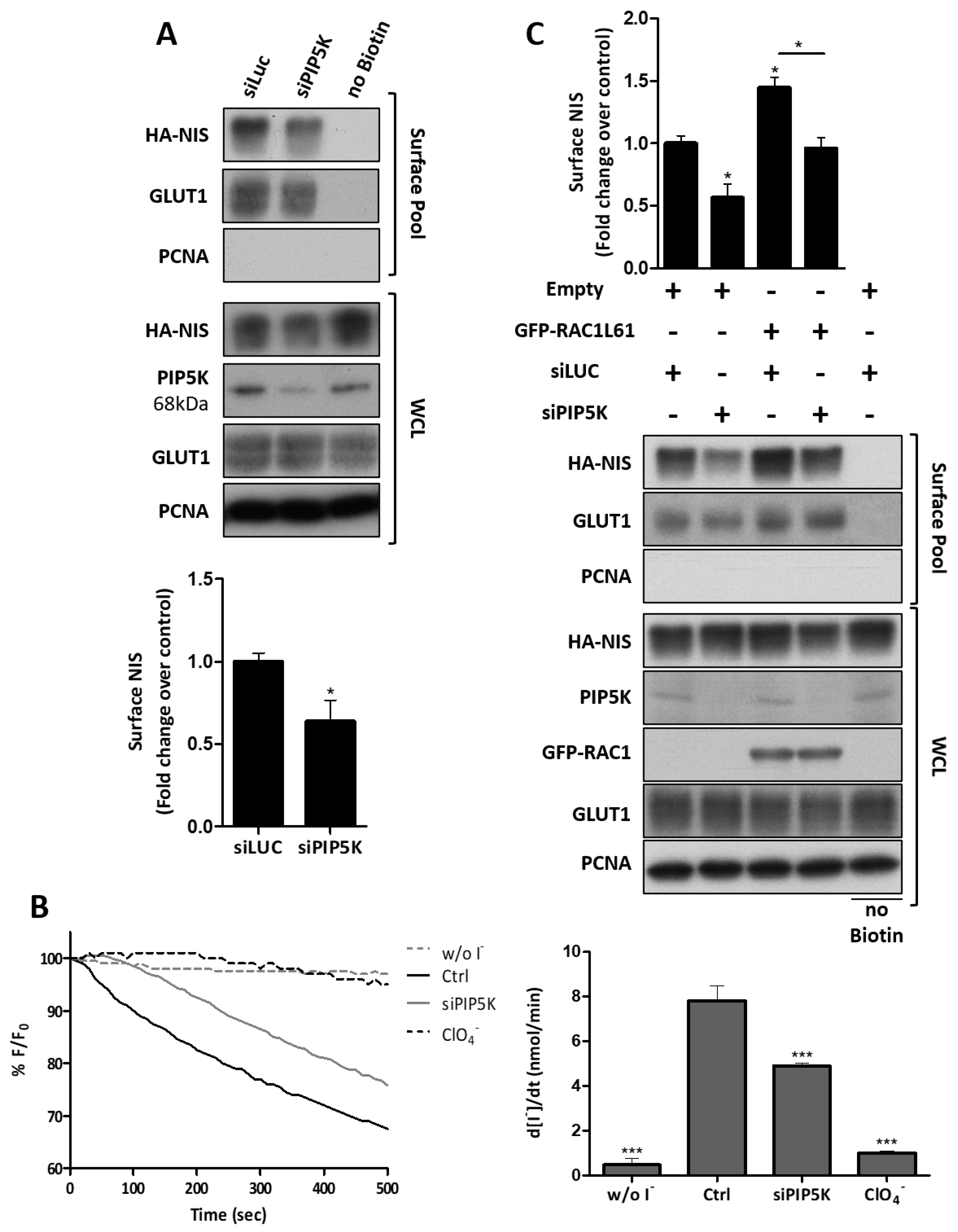
3.4. RAC1 Signals through PAK1 and PIP5K Kinases to Upregulate NIS PM Levels
3.5. SRC Kinase Functions Upstream of RAC1 to Increase the NIS Cell Surface Levels
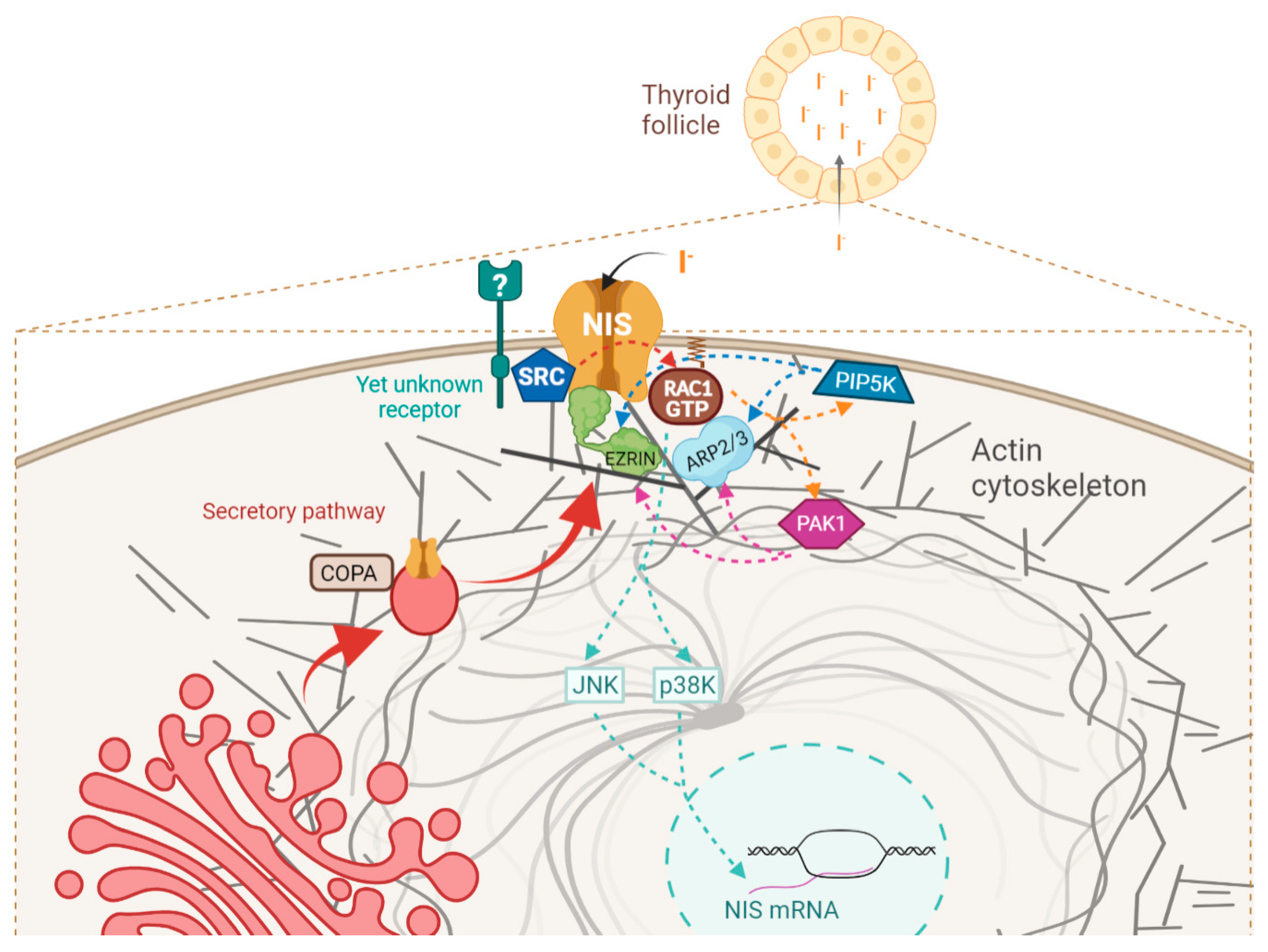
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colin, I.M.; Denef, J.-F.; Lengelé, B.; Many, M.-C.; Gérard, A.-C. Recent Insights into the Cell Biology of Thyroid Angiofollicular Units. Endocr. Rev. 2013, 34, 209–238. [Google Scholar] [CrossRef] [Green Version]
- Eskandari, S.; Loo, D.D.; Dai, G.; Levy, O.; Wright, E.M.; Carrasco, N. Thyroid Na+/I− Symporter. Mechanism, Stoichiometry, and Specificity. J. Biol. Chem. 1997, 272, 27230–27238. [Google Scholar] [CrossRef] [Green Version]
- Nicola, J.P.; Carrasco, N.; Amzel, L.M. Physiological Sodium Concentrations Enhance the Iodide Affinity of the Na+/I− Symporter. Nat. Commun. 2014, 5, 3948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, E.K.; Larsen, P.R. Radioiodine for Thyroid Cancer—Is Less More? N. Engl. J. Med. 2012, 366, 1732–1733. [Google Scholar] [CrossRef] [Green Version]
- Leitha, T.; Schartel, M.; Hurtl, I.; Bednar, R.; König, F.; Mayrhofer, K.; Pietschmann, F. Dosimetry-Guided Radioiodine Therapy of Hyperthyroidism: Long-Term Experience and Implications for Guidelines. Nucl. Med. Commun. 2021, 42, 150–159. [Google Scholar] [CrossRef]
- Martín, M.; Geysels, R.C.; Peyret, V.; Bernal Barquero, C.E.; Masini-Repiso, A.M.; Nicola, J.P. Implications of Na+/I− Symporter Transport to the Plasma Membrane for Thyroid Hormonogenesis and Radioiodide Therapy. J. Endocr. Soc. 2019, 3, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Tavares, C.; Coelho, M.J.; Eloy, C.; Melo, M.; da Rocha, A.G.; Pestana, A.; Batista, R.; Ferreira, L.B.; Rios, E.; Selmi-Ruby, S.; et al. NIS Expression in Thyroid Tumors, Relation with Prognosis Clinicopathological and Molecular Features. Endocr. Connect. 2018, 7, 78–90. [Google Scholar] [CrossRef]
- Kogai, T.; Taki, K.; Brent, G.A. Enhancement of Sodium/Iodide Symporter Expression in Thyroid and Breast Cancer. Endocr. Relat. Cancer 2006, 13, 797–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlumberger, M.; Lacroix, L.; Russo, D.; Filetti, S.; Bidart, J.-M. Defects in Iodide Metabolism in Thyroid Cancer and Implications for the Follow-up and Treatment of Patients. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 260–269. [Google Scholar] [CrossRef]
- Zarnegar, R.; Brunaud, L.; Kanauchi, H.; Wong, M.; Fung, M.; Ginzinger, D.; Duh, Q.-Y.; Clark, O.H. Increasing the Effectiveness of Radioactive Iodine Therapy in the Treatment of Thyroid Cancer Using Trichostatin A, a Histone Deacetylase Inhibitor. Surgery 2002, 132, 984–990. [Google Scholar] [CrossRef]
- Worden, F. Treatment Strategies for Radioactive Iodine-Refractory Differentiated Thyroid Cancer. Ther. Adv. Med. Oncol. 2014, 6, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, M.; Haugen, B.R.; Schlumberger, M. Progress in Molecular-Based Management of Differentiated Thyroid Cancer. Lancet 2013, 381, 1058–1069. [Google Scholar] [CrossRef] [Green Version]
- Nixon, I.J.; Whitcher, M.M.; Palmer, F.L.; Tuttle, R.M.; Shaha, A.R.; Shah, J.P.; Patel, S.G.; Ganly, I. The Impact of Distant Metastases at Presentation on Prognosis in Patients with Differentiated Carcinoma of the Thyroid Gland. Thyroid 2012, 22, 884–889. [Google Scholar] [CrossRef] [Green Version]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-Term Outcome of 444 Patients with Distant Metastases from Papillary and Follicular Thyroid Carcinoma: Benefits and Limits of Radioiodine Therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, M.; Spitzweg, C.; Vassaux, G.; Newbold, K.; Melcher, A.; Pandha, H.; Vile, R.; Harrington, K. The Biology of the Sodium Iodide Symporter and Its Potential for Targeted Gene Delivery. Curr. Cancer Drug Targets 2010, 10, 242–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogai, T.; Brent, G.A. The Sodium Iodide Symporter (NIS): Regulation and Approaches to Targeting for Cancer Therapeutics. Pharmacol. Ther. 2012, 135, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Dohán, O.; Baloch, Z.; Bánrévi, Z.; Livolsi, V.; Carrasco, N. Rapid Communication: Predominant Intracellular Overexpression of the Na(+)/I(−) Symporter (NIS) in a Large Sampling of Thyroid Cancer Cases. J. Clin. Endocrinol. Metab. 2001, 86, 2697–2700. [Google Scholar] [CrossRef]
- Wapnir, I.L.; van de Rijn, M.; Nowels, K.; Amenta, P.S.; Walton, K.; Montgomery, K.; Greco, R.S.; Dohán, O.; Carrasco, N. Immunohistochemical Profile of the Sodium/Iodide Symporter in Thyroid, Breast, and Other Carcinomas Using High Density Tissue Microarrays and Conventional Sections. J. Clin. Endocrinol. Metab. 2003, 88, 1880–1888. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.E.; Read, M.L.; Turnell, A.S.; Watkins, R.J.; Watkinson, J.C.; Lewy, G.D.; Fong, J.C.W.; James, S.R.; Eggo, M.C.; Boelaert, K.; et al. A Novel Mechanism of Sodium Iodide Symporter Repression in Differentiated Thyroid Cancer. J. Cell Sci. 2009, 122, 3393–3402. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.E.; Franklyn, J.A.; McCabe, C.J. Expression and Function of the Novel Proto-Oncogene PBF in Thyroid Cancer: A New Target for Augmenting Radioiodine Uptake. J. Endocrinol. 2011, 210, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.E.; Sharma, N.; Watkins, R.J.; Read, M.L.; Ryan, G.A.; Kwan, P.P.; Martin, A.; Watkinson, J.C.; Boelaert, K.; Franklyn, J.A.; et al. Manipulation of PBF/PTTG1IP Phosphorylation Status; a Potential New Therapeutic Strategy for Improving Radioiodine Uptake in Thyroid and Other Tumors. J. Clin. Endocrinol. Metab. 2013, 98, 2876–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, A.; Read, M.L.; Thornton, C.E.M.; Larner, D.P.; Poole, V.L.; Brookes, K.; Nieto, H.R.; Alshahrani, M.; Thompson, R.J.; Lavery, G.G.; et al. Targeting Novel Sodium Iodide Symporter Interactors ADP-Ribosylation Factor 4 and Valosin-Containing Protein Enhances Radioiodine Uptake. Cancer Res. 2020, 80, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Faria, M.; Félix, D.; Domingues, R.; Bugalho, M.J.; Matos, P.; Silva, A.L. Antagonistic Effects of RAC1 and Tumor-Related RAC1b on NIS Expression in Thyroid. J. Mol. Endocrinol. 2019, 63, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.; Collard, J.G.; Jordan, P. Tumor-Related Alternatively Spliced Rac1b Is Not Regulated by Rho-GDP Dissociation Inhibitors and Exhibits Selective Downstream Signaling. J. Biol. Chem. 2003, 278, 50442–50448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, P.; Jordan, P.; Matos, P. Rac1 Signaling Modulates BCL-6-Mediated Repression of Gene Transcription. Mol. Cell Biol. 2009, 29, 4156–4166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, C.A.; Matos, A.M.; Dias-Alves, Â.; Pereira, J.F.; Uliyakina, I.; Barros, P.; Amaral, M.D.; Matos, P. A Molecular Switch in the Scaffold NHERF1 Enables Misfolded CFTR to Evade the Peripheral Quality Control Checkpoint. Sci. Signal. 2015, 8, ra48. [Google Scholar] [CrossRef]
- Moniz, S.; Sousa, M.; Moraes, B.J.; Mendes, A.I.; Palma, M.; Barreto, C.; Fragata, J.I.; Amaral, M.D.; Matos, P. HGF Stimulation of Rac1 Signaling Enhances Pharmacological Correction of the Most Prevalent Cystic Fibrosis Mutant F508del-CFTR. ACS Chem. Biol. 2013, 8, 432–442. [Google Scholar] [CrossRef]
- Galietta, L.J.V.; Haggie, P.M.; Verkman, A.S. Green Fluorescent Protein-Based Halide Indicators with Improved Chloride and Iodide Affinities. FEBS Lett. 2001, 499, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Matos, P.; Jordan, P. Rac1, but Not Rac1B, Stimulates RelB-Mediated Gene Transcription in Colorectal Cancer Cells. J. Biol. Chem. 2006, 281, 13724–13732. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Ishizaka, Y.; Itoh, F.; Tahira, T.; Ikeda, I.; Ogura, T.; Sugimura, T.; Nagao, M. Presence of Aberrant Transcripts of Ret Proto-Oncogene in a Human Papillary Thyroid Carcinoma Cell Line. Jpn. J. Cancer Res. 1989, 80, 1149–1152. [Google Scholar] [CrossRef]
- Landa, I.; Pozdeyev, N.; Korch, C.; Marlow, L.A.; Smallridge, R.C.; Copland, J.A.; Henderson, Y.C.; Lai, S.Y.; Clayman, G.L.; Onoda, N.; et al. Comprehensive Genetic Characterization of Human Thyroid Cancer Cell Lines: A Validated Panel for Preclinical Studies. Clin. Cancer Res. 2019, 25, 3141–3151. [Google Scholar] [CrossRef]
- Wächter, S.; Damanakis, A.I.; Elxnat, M.; Roth, S.; Wunderlich, A.; Verburg, F.A.; Fellinger, S.A.; Bartsch, D.K.; Di Fazio, P. Epigenetic Modifications in Thyroid Cancer Cells Restore NIS and Radio-Iodine Uptake and Promote Cell Death. J. Clin. Med. 2018, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Rhoden, K.J.; Cianchetta, S.; Stivani, V.; Portulano, C.; Galietta, L.J.V.; Romeo, G. Cell-Based Imaging of Sodium Iodide Symporter Activity with the Yellow Fluorescent Protein Variant YFP-H148Q/I152L. Am. J. Physiol. Cell Physiol. 2007, 292, C814–C823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianchetta, S.; di Bernardo, J.; Romeo, G.; Rhoden, K.J. Perchlorate Transport and Inhibition of the Sodium Iodide Symporter Measured with the Yellow Fluorescent Protein Variant YFP-H148Q/I152L. Toxicol. Appl. Pharmacol. 2010, 243, 372–380. [Google Scholar] [CrossRef]
- Beck, R.; Ravet, M.; Wieland, F.T.; Cassel, D. The COPI System: Molecular Mechanisms and Function. FEBS Lett. 2009, 583, 2701–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, M.W.; Sacks, D.B. IQGAP Proteins Are Integral Components of Cytoskeletal Regulation. EMBO Rep. 2003, 4, 571–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orci, L.; Stamnes, M.; Ravazzola, M.; Amherdt, M.; Perrelet, A.; Söllner, T.H.; Rothman, J.E. Bidirectional Transport by Distinct Populations of COPI-Coated Vesicles. Cell 1997, 90, 335–349. [Google Scholar] [CrossRef] [Green Version]
- Barlowe, C. Traffic COPs of the Early Secretory Pathway. Traffic 2000, 1, 371–377. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Transport from the ER through the Golgi Apparatus. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Jung, H.; Yoon, S.R.; Lim, J.; Cho, H.J.; Lee, H.G. Dysregulation of Rho GTPases in Human Cancers. Cancers 2020, 12, 1179. [Google Scholar] [CrossRef]
- Tang, S.; Jiang, J.; Zhang, N.; Sun, J.; Sun, G. Tumor Necrosis Factor-α Requires Ezrin to Regulate the Cytoskeleton and Cause Pulmonary Microvascular Endothelial Barrier Damage. Microvasc. Res. 2021, 133, 104093. [Google Scholar] [CrossRef]
- Michie, K.A.; Bermeister, A.; Robertson, N.O.; Goodchild, S.C.; Curmi, P.M.G. Two Sides of the Coin: Ezrin/Radixin/Moesin and Merlin Control Membrane Structure and Contact Inhibition. Int. J. Mol. Sci. 2019, 20, 1996. [Google Scholar] [CrossRef] [Green Version]
- Ogihara, T.; Mizoi, K.; Kamioka, H.; Yano, K. Physiological Roles of ERM Proteins and Transcriptional Regulators in Supporting Membrane Expression of Efflux Transporters as Factors of Drug Resistance in Cancer. Cancers 2020, 12, 3352. [Google Scholar] [CrossRef]
- Song, Y.; Ma, X.; Zhang, M.; Wang, M.; Wang, G.; Ye, Y.; Xia, W. Ezrin Mediates Invasion and Metastasis in Tumorigenesis: A Review. Front. Cell Dev. Biol. 2020, 8, 588801. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The Small GTP-Binding Protein Rac Regulates Growth Factor-Induced Membrane Ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Sit, S.-T.; Manser, E. Rho GTPases and Their Role in Organizing the Actin Cytoskeleton. J. Cell Sci. 2011, 124, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, M.; Nakagawa, M.; Kaibuchi, K. Roles of Rho-Family GTPases in Cell Polarisation and Directional Migration. Curr. Opin. Cell Biol. 2003, 15, 590–597. [Google Scholar] [CrossRef]
- Dominguez, R. Structural Insights into de Novo Actin Polymerization. Curr. Opin. Struct. Biol. 2010, 20, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Chou, H.-T.; Brautigam, C.A.; Xing, W.; Yang, S.; Henry, L.; Doolittle, L.K.; Walz, T.; Rosen, M.K. Rac1 GTPase Activates the WAVE Regulatory Complex through Two Distinct Binding Sites. eLife 2017, 6, e29795. [Google Scholar] [CrossRef] [PubMed]
- Goley, E.D.; Welch, M.D. The ARP2/3 Complex: An Actin Nucleator Comes of Age. Nat. Rev. Mol. Cell Biol. 2006, 7, 713–726. [Google Scholar] [CrossRef]
- Goley, E.D.; Rammohan, A.; Znameroski, E.A.; Firat-Karalar, E.N.; Sept, D.; Welch, M.D. An Actin-Filament-Binding Interface on the Arp2/3 Complex Is Critical for Nucleation and Branch Stability. Proc. Natl. Acad. Sci. USA 2010, 107, 8159–8164. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho GTPases and the Actin Cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J. Rho GTPases and Actin Dynamics in Membrane Protrusions and Vesicle Trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef]
- Menke, A.; Giehl, K. Regulation of Adherens Junctions by Rho GTPases and P120-Catenin. Arch. Biochem. Biophys. 2012, 524, 48–55. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New Insights into Their Functions from in Vivo Studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Kogai, T.; Ohashi, E.; Jacobs, M.S.; Sajid-Crockett, S.; Fisher, M.L.; Kanamoto, Y.; Brent, G.A. Retinoic Acid Stimulation of the Sodium/Iodide Symporter in MCF-7 Breast Cancer Cells Is Mediated by the Insulin Growth Factor-I/Phosphatidylinositol 3-Kinase and P38 Mitogen-Activated Protein Kinase Signaling Pathways. J. Clin. Endocrinol. Metab. 2008, 93, 1884–1892. [Google Scholar] [CrossRef] [Green Version]
- Kogai, T.; Liu, Y.-Y.; Mody, K.; Shamsian, D.V.; Brent, G.A. Regulation of Sodium Iodide Symporter Gene Expression by Rac1/P38β Mitogen-Activated Protein Kinase Signaling Pathway in MCF-7 Breast Cancer Cells. J. Biol. Chem. 2012, 287, 3292–3300. [Google Scholar] [CrossRef] [Green Version]
- Fenton, M.S.; Marion, K.M.; Salem, A.K.; Hogen, R.; Naeim, F.; Hershman, J.M. Sunitinib Inhibits MEK/ERK and SAPK/JNK Pathways and Increases Sodium/Iodide Symporter Expression in Papillary Thyroid Cancer. Thyroid 2010, 20, 965–974. [Google Scholar] [CrossRef]
- Manser, E.; Huang, H.Y.; Loo, T.H.; Chen, X.Q.; Dong, J.M.; Leung, T.; Lim, L. Expression of Constitutively Active Alpha-PAK Reveals Effects of the Kinase on Actin and Focal Complexes. Mol. Cell. Biol. 1997, 17, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, D.C.; Sanders, L.C.; Bokoch, G.M.; Gill, G.N. Activation of LIM-Kinase by Pak1 Couples Rac/Cdc42 GTPase Signalling to Actin Cytoskeletal Dynamics. Nat. Cell Biol. 1999, 1, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, J. Involvement of Rac/Cdc42/PAK Pathway in Cytoskeletal Rearrangements. Acta Biochim. Pol. 2009, 56, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Staser, K.; Shew, M.A.; Michels, E.G.; Mwanthi, M.M.; Yang, F.-C.; Clapp, D.W.; Park, S.-J. A Pak1-PP2A-ERM Signaling Axis Mediates F-Actin Rearrangement and Degranulation in Mast Cells. Exp. Hematol 2013, 41, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Tavasoli, M.; Li, L.; Al-Momany, A.; Zhu, L.-F.; Adam, B.A.; Wang, Z.; Ballermann, B.J. The Chloride Intracellular Channel 5A Stimulates Podocyte Rac1, Protecting against Hypertension-Induced Glomerular Injury. Kidney Int. 2016, 89, 833–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolias, K.F.; Hartwig, J.H.; Ishihara, H.; Shibasaki, Y.; Cantley, L.C.; Carpenter, C.L. Type Iα Phosphatidylinositol-4-Phosphate 5-Kinase Mediates Rac-Dependent Actin Assembly. Curr. Biol. 2000, 10, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Malecka, K.A.; Szentpetery, Z.; Peterson, J.R. Synergistic Activation of P21-Activated Kinase 1 by Phosphatidylinositol 4,5-Bisphosphate and Rho GTPases. J. Biol. Chem. 2013, 288, 8887–8897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auvinen, E.; Kivi, N.; Vaheri, A. Regulation of Ezrin Localization by Rac1 and PIPK in Human Epithelial Cells. Exp. Cell Res. 2007, 313, 824–833. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-S. Advances in Studies of Tyrosine Kinase Inhibitors and Their Acquired Resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef]
- De, P.; Aske, J.C.; Dey, N. RAC1 Takes the Lead in Solid Tumors. Cells 2019, 8, 382. [Google Scholar] [CrossRef] [Green Version]
- Aashiq, M.; Silverman, D.A.; Na’ara, S.; Takahashi, H.; Amit, M. Radioiodine-Refractory Thyroid Cancer: Molecular Basis of Redifferentiation Therapies, Management, and Novel Therapies. Cancers 2019, 11, 1382. [Google Scholar] [CrossRef] [Green Version]
- Riesco-Eizaguirre, G.; Gutiérrez-Martínez, P.; García-Cabezas, M.A.; Nistal, M.; Santisteban, P. The Oncogene BRAF V600E Is Associated with a High Risk of Recurrence and Less Differentiated Papillary Thyroid Carcinoma Due to the Impairment of Na+/I− Targeting to the Membrane. Endocr. Relat. Cancer 2006, 13, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Endo, T.; Kawaguchi, A.; Ikeda, M.; Katoh, R.; Kawaoi, A.; Muramatsu, A.; Onaya, T. Increased Expression of the Sodium/Iodide Symporter in Papillary Thyroid Carcinomas. J. Clin. Investig. 1998, 101, 1296–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonacchera, M.; Viacava, P.; Agretti, P.; de Marco, G.; Perri, A.; di Cosmo, C.; de Servi, M.; Miccoli, P.; Lippi, F.; Naccarato, A.G.; et al. Benign Nonfunctioning Thyroid Adenomas Are Characterized by a Defective Targeting to Cell Membrane or a Reduced Expression of the Sodium Iodide Symporter Protein. J. Clin. Endocrinol. Metab. 2002, 87, 352–357. [Google Scholar] [CrossRef]
- Kollecker, I.; von Wasielewski, R.; Langner, C.; Müller, J.A.; Spitzweg, C.; Kreipe, H.; Brabant, G. Subcellular Distribution of the Sodium Iodide Symporter in Benign and Malignant Thyroid Tissues. Thyroid 2012, 22, 529–535. [Google Scholar] [CrossRef]
- Neumann, S.; Schuchardt, K.; Reske, A.; Reske, A.; Emmrich, P.; Paschke, R. Lack of Correlation for Sodium Iodide Symporter MRNA and Protein Expression and Analysis of Sodium Iodide Symporter Promoter Methylation in Benign Cold Thyroid Nodules. Thyroid 2004, 14, 99–111. [Google Scholar] [CrossRef]
- Trouttet-Masson, S.; Selmi-Ruby, S.; Bernier-Valentin, F.; Porra, V.; Berger-Dutrieux, N.; Decaussin, M.; Peix, J.-L.; Perrin, A.; Bournaud, C.; Orgiazzi, J.; et al. Evidence for Transcriptional and Posttranscriptional Alterations of the Sodium/Iodide Symporter Expression in Hypofunctioning Benign and Malignant Thyroid Tumors. Am. J. Pathol 2004, 165, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Kogai, T.; Sajid-Crockett, S.; Newmarch, L.S.; Liu, Y.-Y.; Brent, G.A. Phosphoinositide-3-Kinase Inhibition Induces Sodium/Iodide Symporter Expression in Rat Thyroid Cells and Human Papillary Thyroid Cancer Cells. J. Endocrinol. 2008, 199, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Amit, M.; Na’ara, S.; Francis, D.; Matanis, W.; Zolotov, S.; Eisenhaber, B.; Eisenhaber, F.; Weiler Sagie, M.; Malkin, L.; Billan, S.; et al. Post-Translational Regulation of Radioactive Iodine Therapy Response in Papillary Thyroid Carcinoma. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Read, M.L.; Lewy, G.D.; Fong, J.C.W.; Sharma, N.; Seed, R.I.; Smith, V.E.; Gentilin, E.; Warfield, A.; Eggo, M.C.; Knauf, J.A.; et al. Proto-Oncogene PBF/PTTG1IP Regulates Thyroid Cell Growth and Represses Radioiodide Treatment. Cancer Res. 2011, 71, 6153–6164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darrouzet, E.; Graslin, F.; Marcellin, D.; Tcheremisinova, I.; Marchetti, C.; Salleron, L.; Pognonec, P.; Pourcher, T. A Systematic Evaluation of Sorting Motifs in the Sodium-Iodide Symporter (NIS). Biochem. J. 2016, 473, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Khramushin, A.; Yadav, V.K.; Schueler-Furman, O.; Ivarsson, Y. Defining Binding Motifs and Dynamics of the Multi-Pocket FERM Domain from Ezrin, Radixin, Moesin and Merlin. Biochemistry 2020, in press. [Google Scholar] [CrossRef]
- Chan, C.M.; Jing, X.; Pike, L.A.; Zhou, Q.; Lim, D.-J.; Sams, S.B.; Lund, G.S.; Sharma, V.; Haugen, B.R.; Schweppe, R.E. Targeted Inhibition of Src Kinase with Dasatinib Blocks Thyroid Cancer Growth and Metastasis. Clin. Cancer Res. 2012, 18, 3580–3591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.; Tyner, J.W.; Chng, W.J.; Bi, C.; Okamoto, R.; Said, J.; Ngan, B.D.; Braunstein, G.D.; Koeffler, H.P. Effect of Dasatinib against Thyroid Cancer Cell Lines in Vitro and a Xenograft Model in Vivo. Oncol. Lett. 2012, 3, 807–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanden Borre, P.; Gunda, V.; McFadden, D.G.; Sadow, P.M.; Varmeh, S.; Bernasconi, M.; Parangi, S. Combined BRAF(V600E)- and SRC-Inhibition Induces Apoptosis, Evokes an Immune Response and Reduces Tumor Growth in an Immunocompetent Orthotopic Mouse Model of Anaplastic Thyroid Cancer. Oncotarget 2014, 5, 3996–4010. [Google Scholar] [CrossRef] [Green Version]
- Beadnell, T.C.; Mishall, K.M.; Zhou, Q.; Riffert, S.M.; Wuensch, K.E.; Kessler, B.E.; Corpuz, M.L.; Jing, X.; Kim, J.; Wang, G.; et al. The Mitogen-Activated Protein Kinase Pathway Facilitates Resistance to the Src Inhibitor Dasatinib in Thyroid Cancer. Mol. Cancer Ther. 2016, 15, 1952–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishall, K.M.; Beadnell, T.C.; Kuenzi, B.M.; Klimczak, D.M.; Superti-Furga, G.; Rix, U.; Schweppe, R.E. Sustained Activation of the AKT/MTOR and MAP Kinase Pathways Mediate Resistance to the Src Inhibitor, Dasatinib, in Thyroid Cancer. Oncotarget 2017, 8, 103014–103031. [Google Scholar] [CrossRef] [Green Version]
- Jha, V.; Macchia, M.; Tuccinardi, T.; Poli, G. Three-Dimensional Interactions Analysis of the Anticancer Target c-Src Kinase with Its Inhibitors. Cancers 2020, 12, 2327. [Google Scholar] [CrossRef] [PubMed]
- Kessler, B.E.; Mishall, K.M.; Kellett, M.D.; Clark, E.G.; Pugazhenthi, U.; Pozdeyev, N.; Kim, J.; Tan, A.C.; Schweppe, R.E. Resistance to Src Inhibition Alters the BRAF-Mutant Tumor Secretome to Promote an Invasive Phenotype and Therapeutic Escape through a FAK>p130Cas>c-Jun Signaling Axis. Oncogene 2019, 38, 2565–2579. [Google Scholar] [CrossRef] [PubMed]
- Iravani, A.; Solomon, B.; Pattison, D.A.; Jackson, P.; Ravi Kumar, A.; Kong, G.; Hofman, M.S.; Akhurst, T.; Hicks, R.J. Mitogen-Activated Protein Kinase Pathway Inhibition for Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer: An Evolving Protocol. Thyroid 2019, 29, 1634–1645. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exclusively at NIS-PM Dataset | Common to NIS-PM and NIS-WC Datasets | Exclusively at NIS-WC Dataset | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Proteins | 109 | Number of Proteins | 19 | Number of Proteins | 33 | ||||||
| GO Term | Description | Count by Term | FDR | GO Term | Description | Count by Term | FDR | GO Term | Description | Count by Term | FDR |
| GO:0007010 | cytoskeleton organization | 29 | 4.51 × 10−11 | GO:0050896 | response to stimulus | 17 | 4.00 × 10−3 | GO:0006810 | transport | 21 | 7.26 × 10−6 |
| GO:0030029 | actin filament-based process | 24 | 1.67 × 10−12 | GO:0006810 | transport | 14 | 1.90 × 10−3 | GO:0046903 | secretion | 12 | 6.47 × 10−6 |
| GO:0030036 | actin cytoskeleton organization | 21 | 3.77 × 10−11 | GO:0009888 | tissue development | 9 | 3.80 × 10−3 | GO:0002252 | immune effector process | 11 | 1.18 × 10−5 |
| GO:0097435 | supramolecular fiber organization | 17 | 3.39 × 10−8 | GO:0071453 | cellular response to oxygen levels | 4 | 4.20 × 10−3 | GO:0001775 | cell activation | 11 | 2.84 × 10−5 |
| GO:0007015 | actin filament organization | 16 | 4.51 × 10−11 | GO:0031952 | regulation of protein autophosphorylation | 3 | 4.00 × 10−3 | GO:0043312 | neutrophil degranulation | 10 | 5.22 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faria, M.; Domingues, R.; Bugalho, M.J.; Silva, A.L.; Matos, P. Analysis of NIS Plasma Membrane Interactors Discloses Key Regulation by a SRC/RAC1/PAK1/PIP5K/EZRIN Pathway with Potential Implications for Radioiodine Re-Sensitization Therapy in Thyroid Cancer. Cancers 2021, 13, 5460. https://doi.org/10.3390/cancers13215460
Faria M, Domingues R, Bugalho MJ, Silva AL, Matos P. Analysis of NIS Plasma Membrane Interactors Discloses Key Regulation by a SRC/RAC1/PAK1/PIP5K/EZRIN Pathway with Potential Implications for Radioiodine Re-Sensitization Therapy in Thyroid Cancer. Cancers. 2021; 13(21):5460. https://doi.org/10.3390/cancers13215460
Chicago/Turabian StyleFaria, Márcia, Rita Domingues, Maria João Bugalho, Ana Luísa Silva, and Paulo Matos. 2021. "Analysis of NIS Plasma Membrane Interactors Discloses Key Regulation by a SRC/RAC1/PAK1/PIP5K/EZRIN Pathway with Potential Implications for Radioiodine Re-Sensitization Therapy in Thyroid Cancer" Cancers 13, no. 21: 5460. https://doi.org/10.3390/cancers13215460