
Paricalcitol Improves the Angiopoietin/Tie-2 and VEGF/VEGFR2 Signaling Pathways in Adriamycin-Induced Nephropathy
 , , , and
, , , and
Abstract
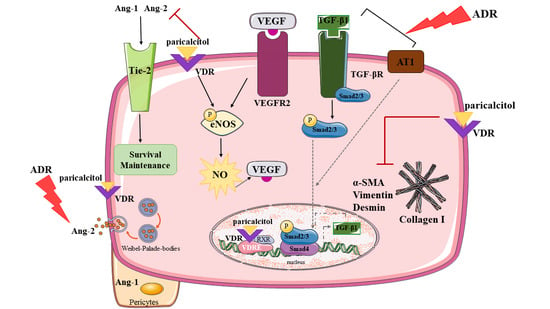
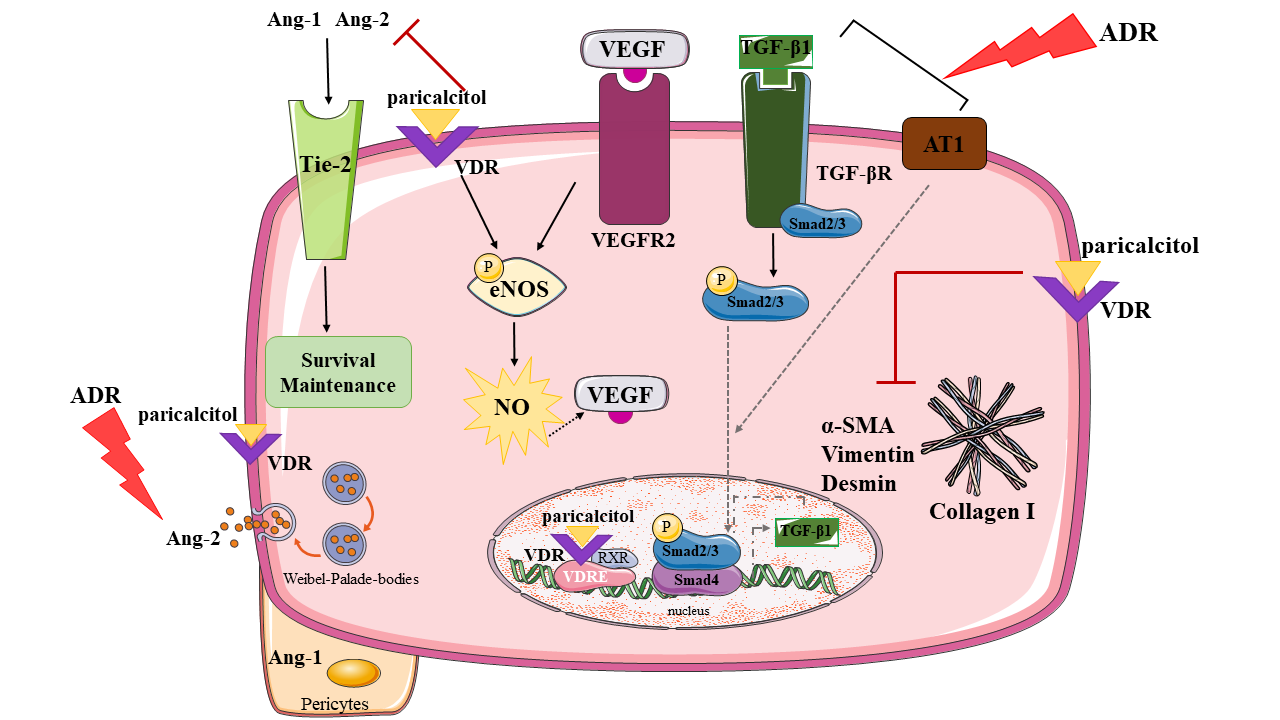
:
1. Introduction
2. Materials and Methods
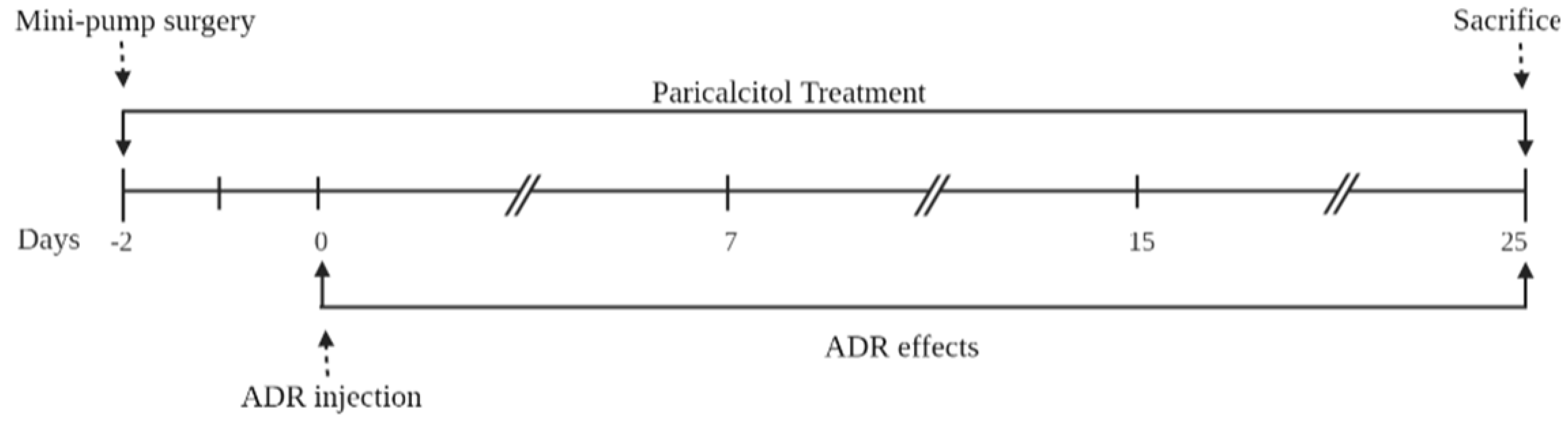
2.1. Animals and Experimental Design
2.2. Evaluation of Renal Function
2.3. Serum 25 Hydroxyvitamin D (25 OHD) Levels, Serum Parathyroid Hormone (PTH), Plasma Calcium (PCa), and Phosphorus (PP) Levels Measurement
2.4. NO in Urine, Plasma, and Renal Tissue Measurement
2.5. TGF-β1 Measurement
2.6. Histological Analysis
2.7. Immunohistochemical Analysis
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
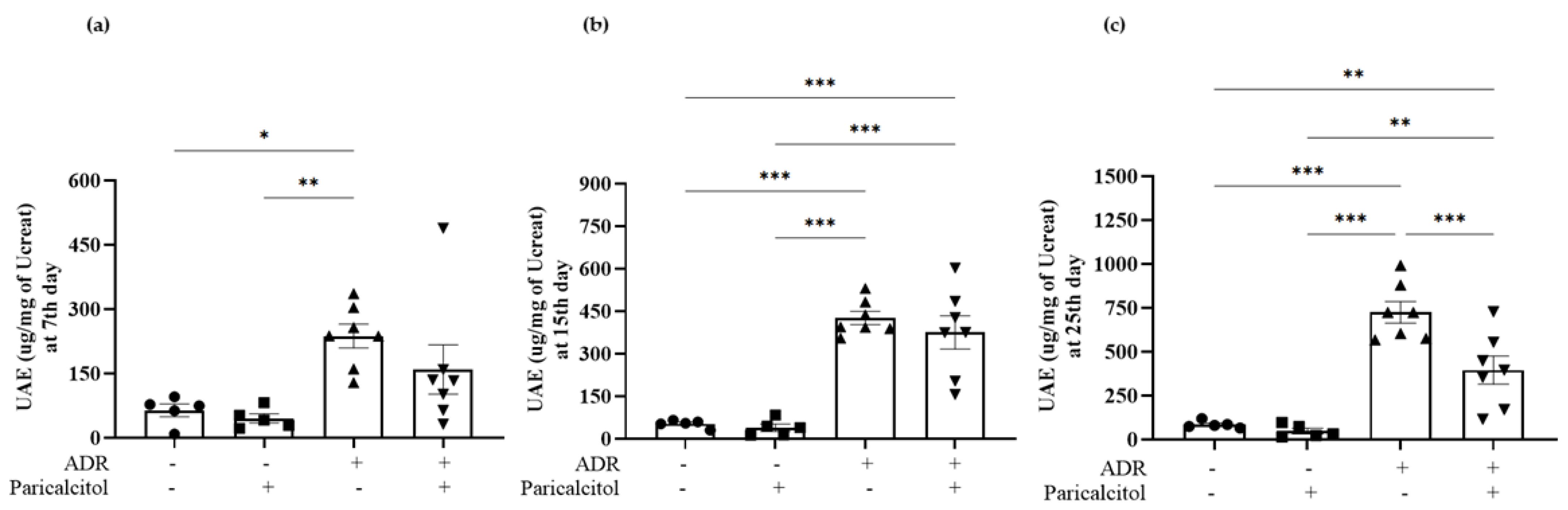
3.1. Paricalcitol Improved ADR-Induced Kidney Dysfunction
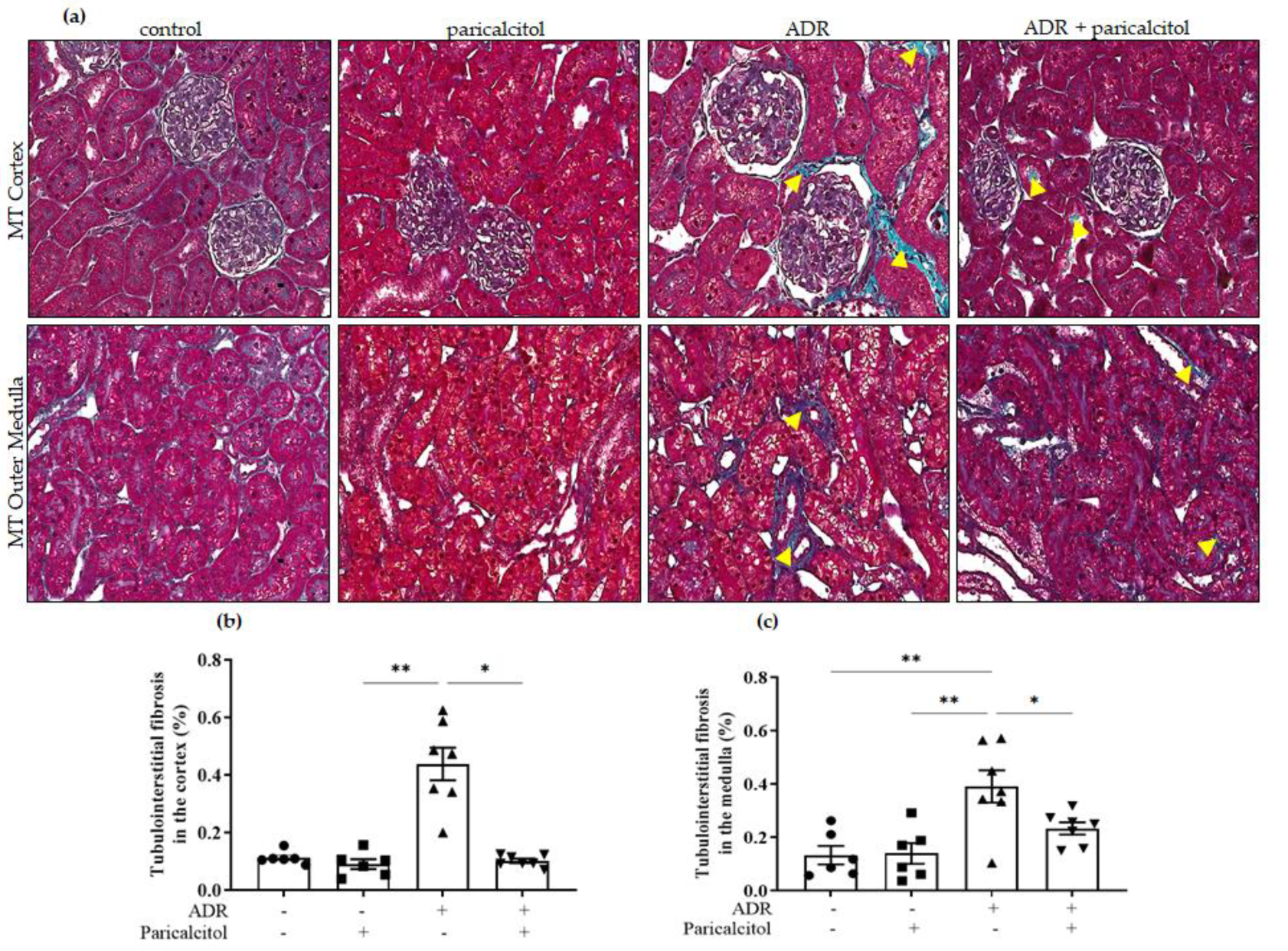
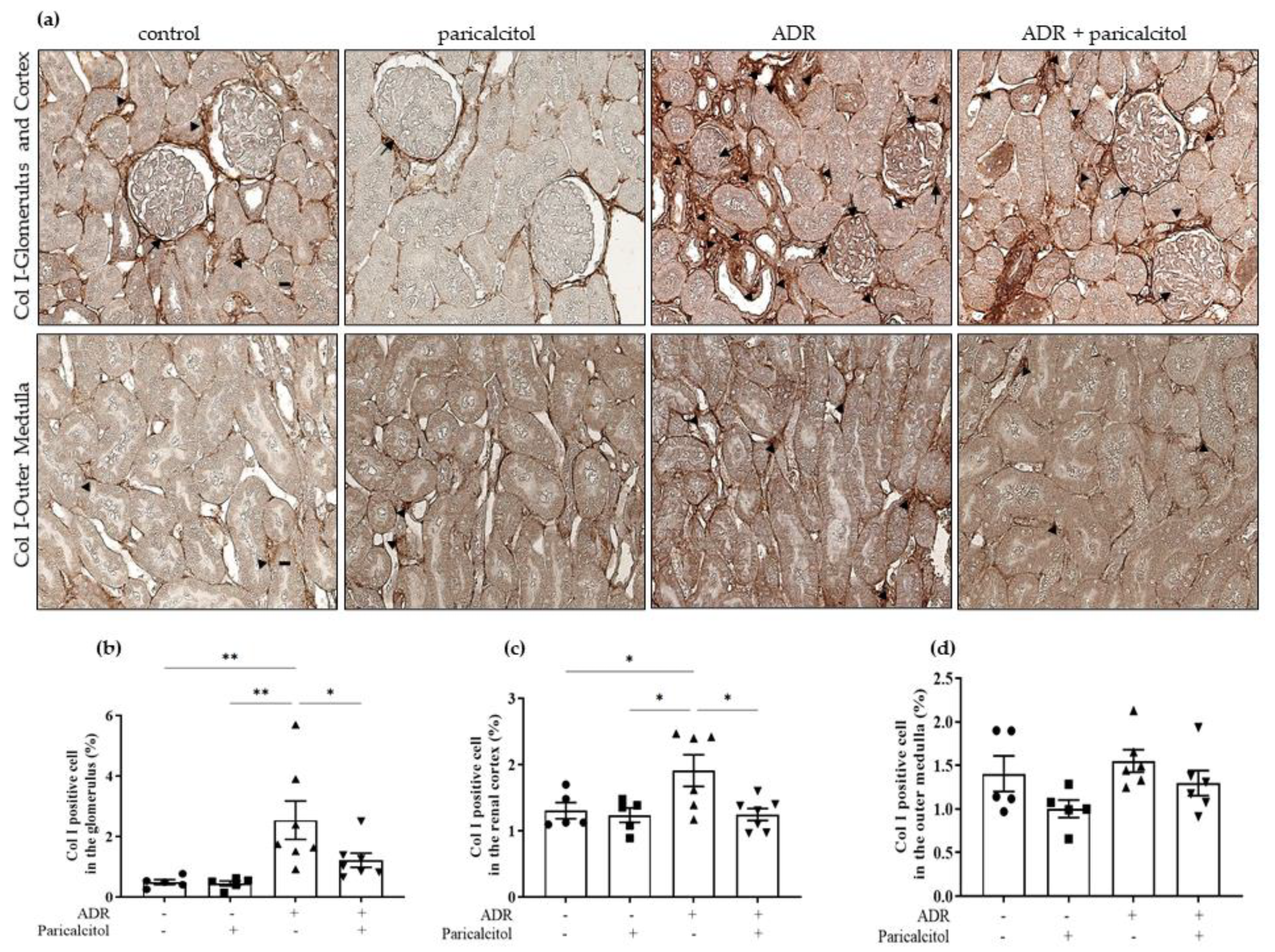
3.2. Paricalcitol Attenuated ADR-Induced Renal Structure Injury
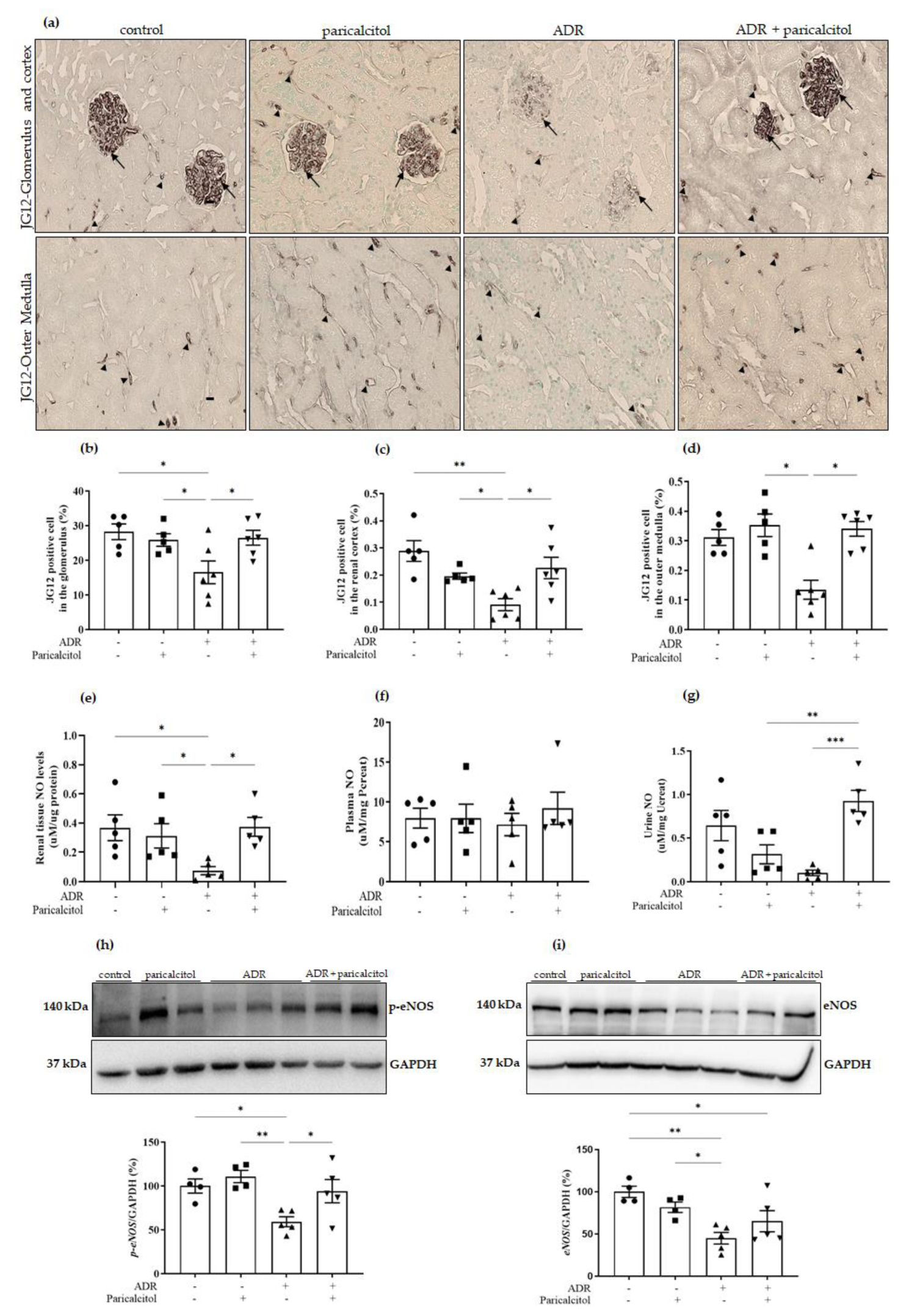
3.3. Paricalcitol Treatment Improved Endothelium Structure and Function
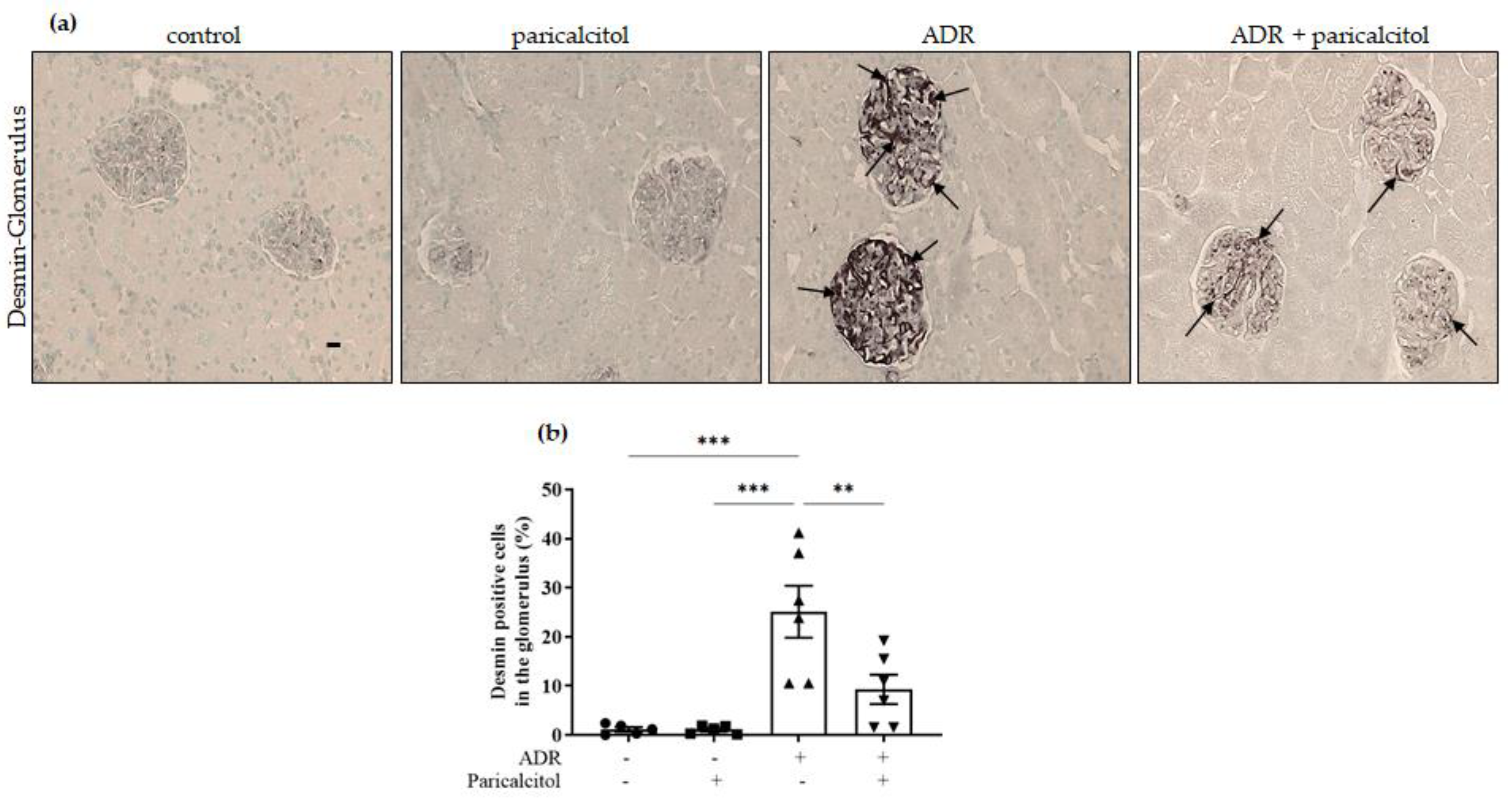
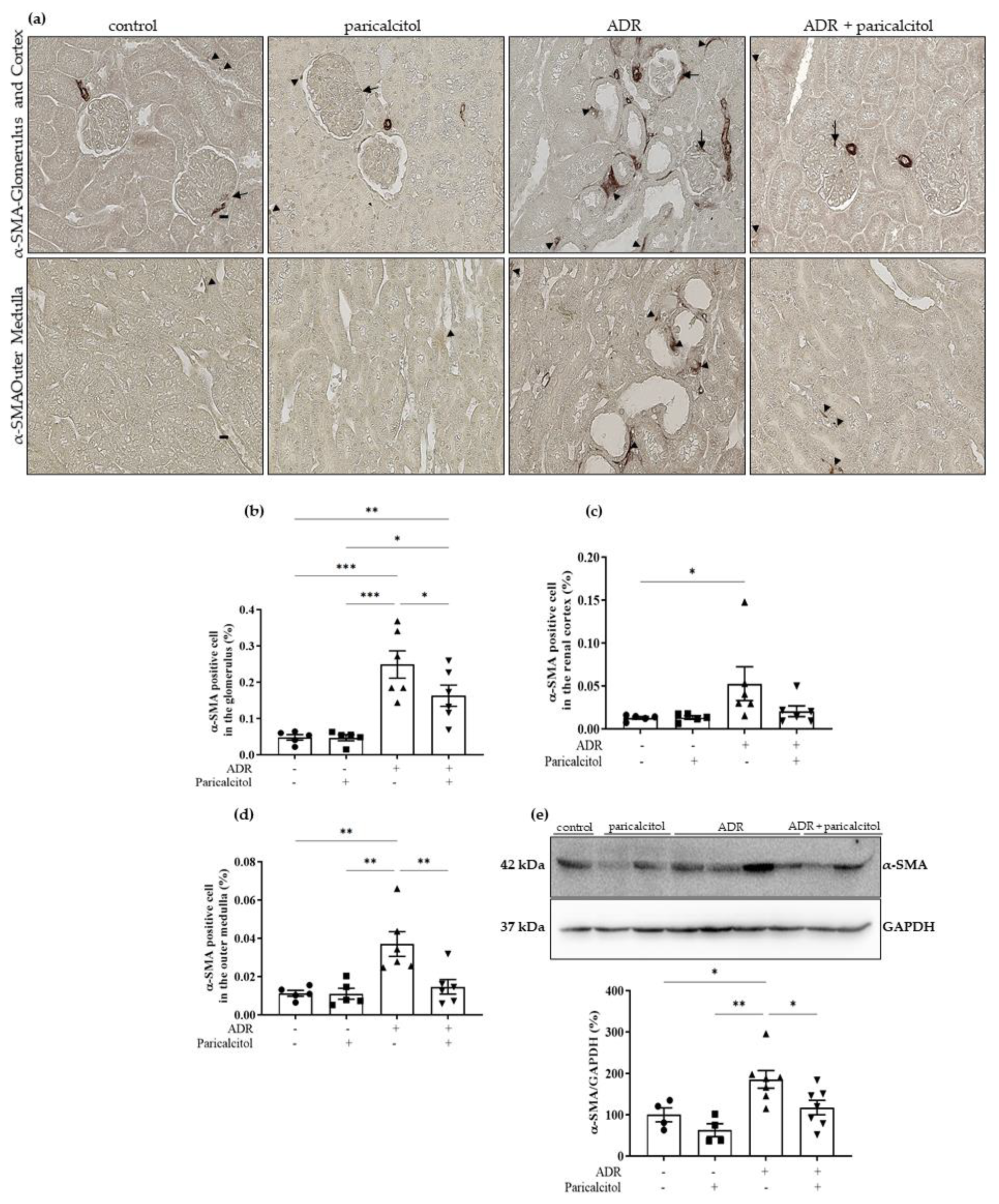
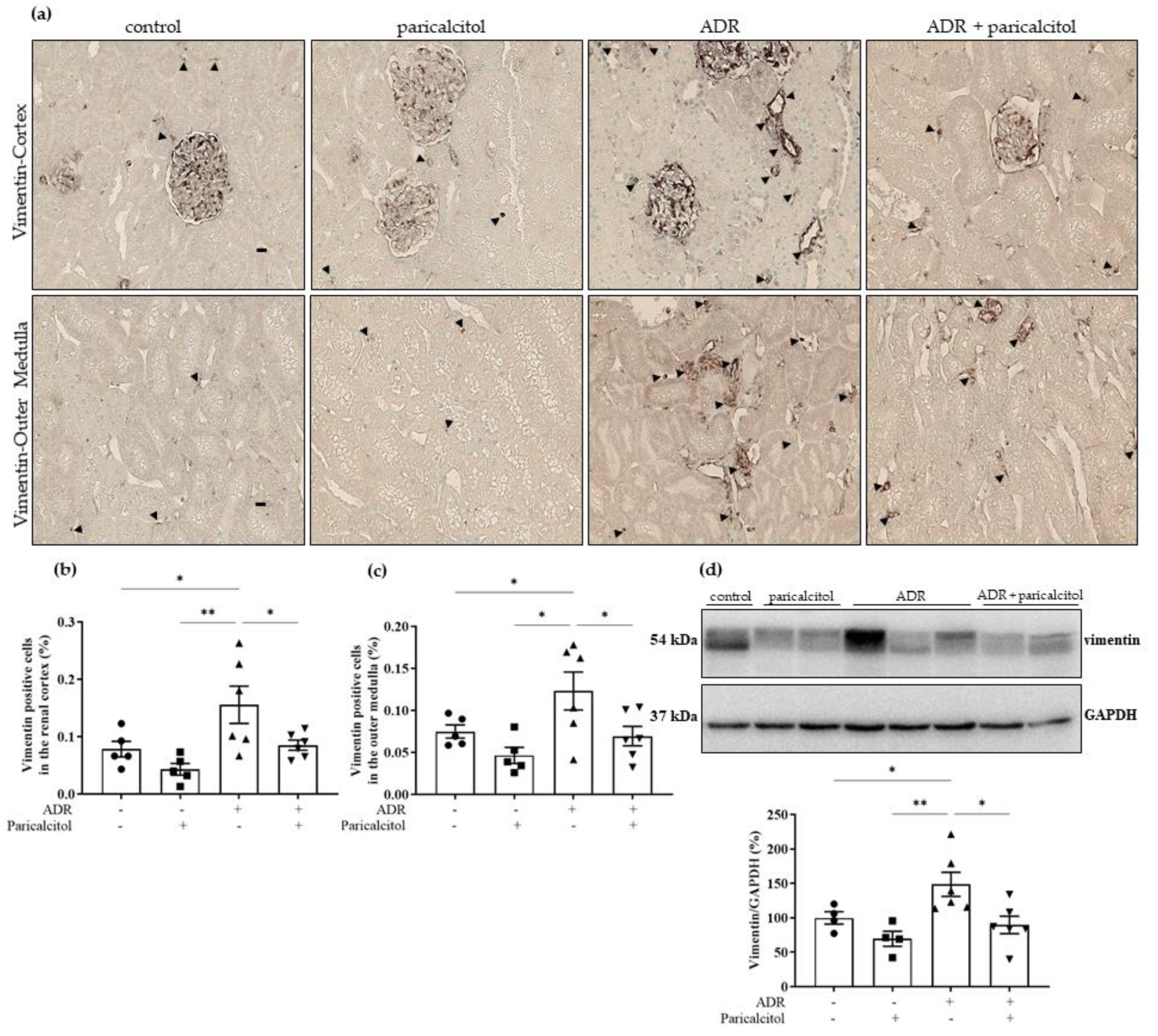
3.4. Paricalcitol Treatment Attenuated the Expression of Mesenchymal Markers
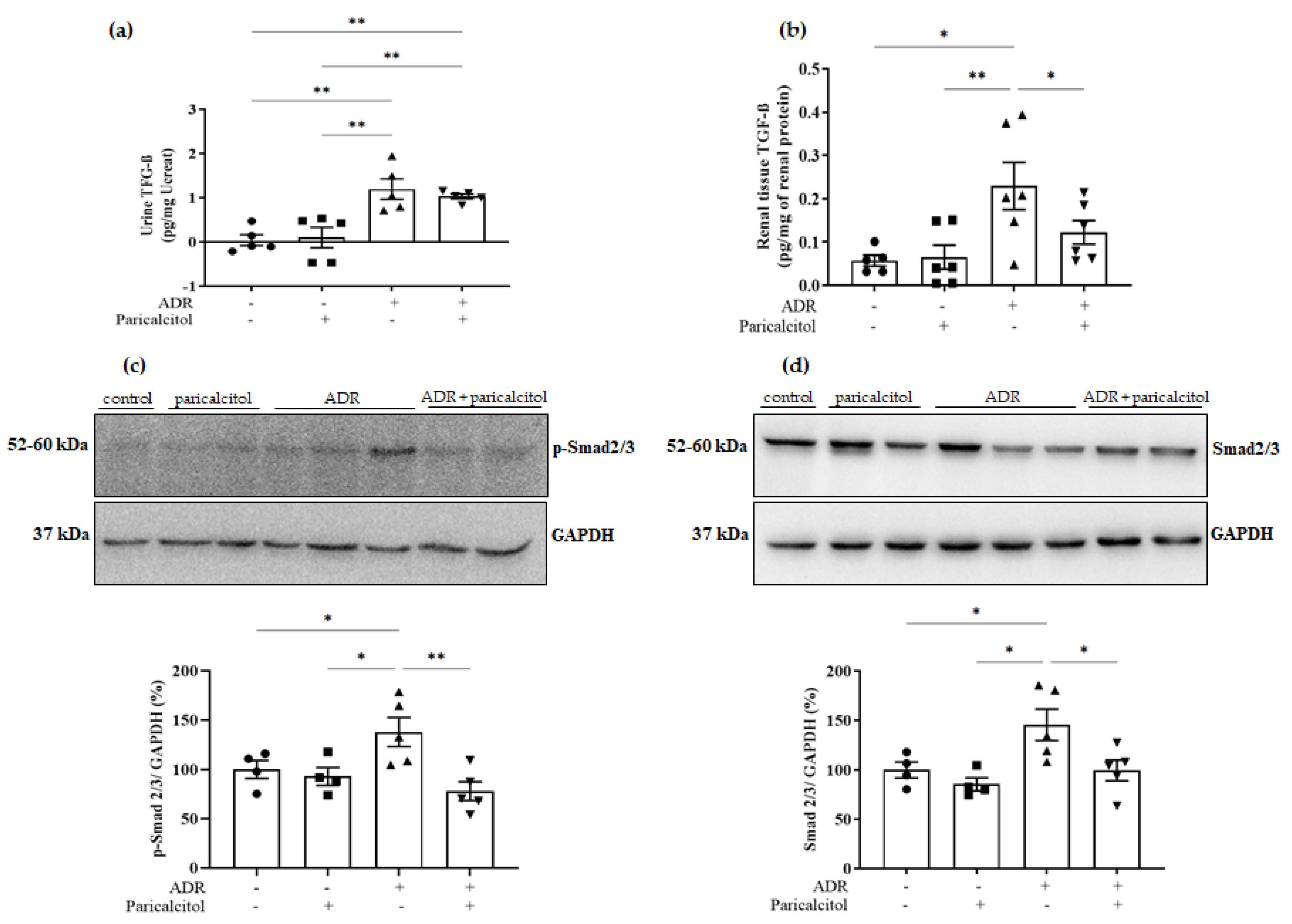
3.5. Paricalcitol Modulated TGF-β1 through the Smads Pathway
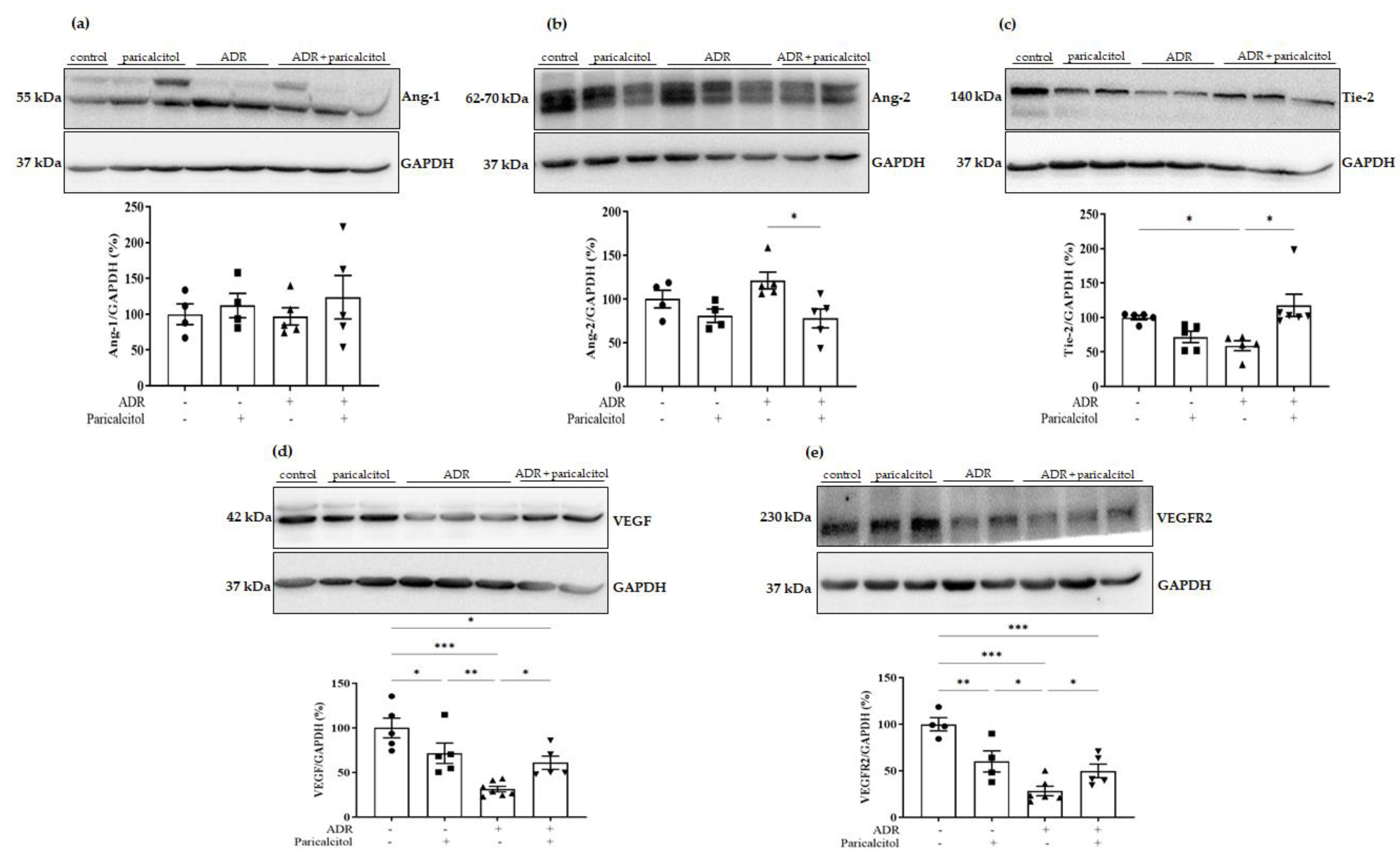
3.6. Paricalcitol Treatment Attenuated Imbalance in Pro- and Anti-Angiogenic Factors
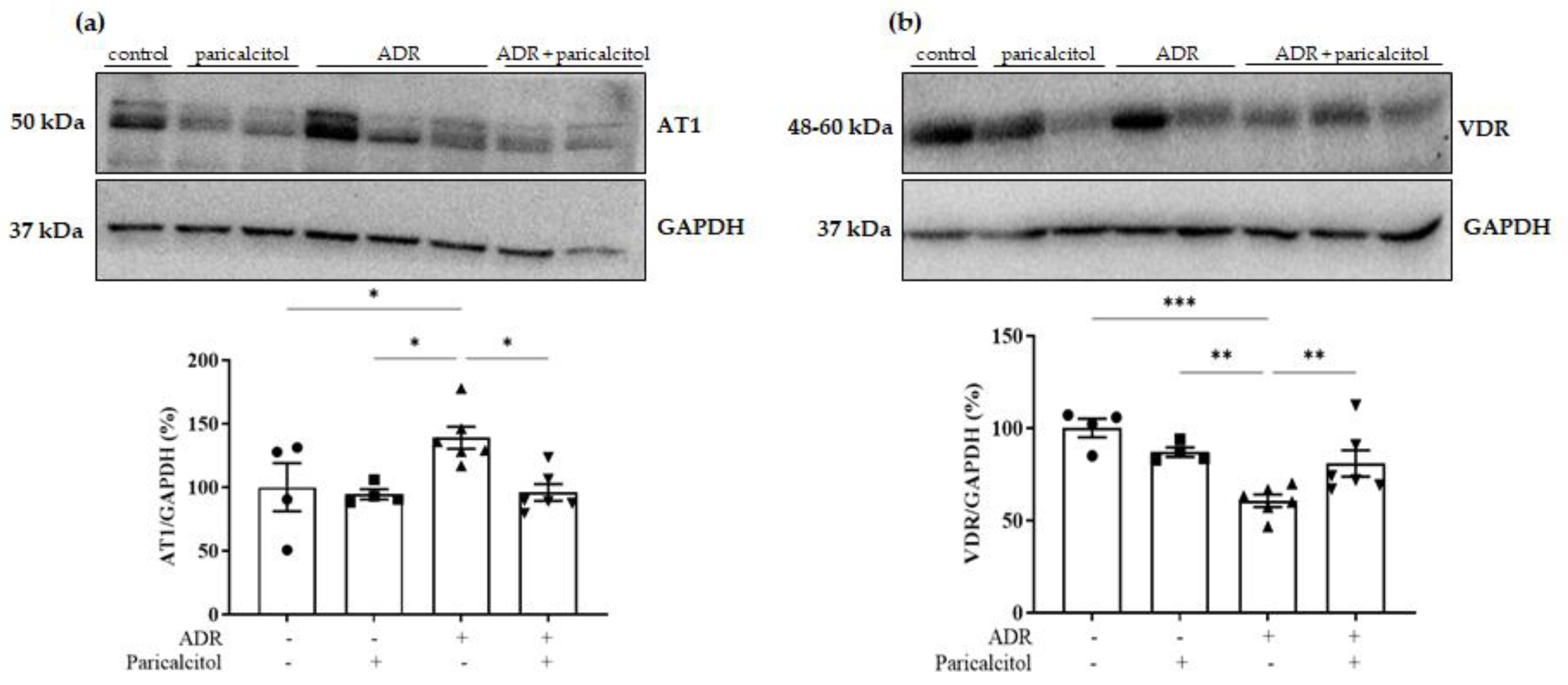
3.7. Paricalcitol Treatment Modulated AT1 and VDR Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The Master Regulator of Fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and Function of Myofibroblasts in Kidney Fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.B.Y.; Qu, X.; Caruana, G.; Li, J. The Origin of Renal Fibroblasts/Myofibroblasts and the Signals That Trigger Fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 8361945. [Google Scholar] [CrossRef] [PubMed]
- Ebefors, K.; Wiener, R.J.; Yu, L.; Azeloglu, E.U.; Yi, Z.; Jia, F.; Zhang, W.; Baron, M.H.; He, J.C.; Haraldsson, B.; et al. Endothelin Receptor–A Mediates Degradation of the Glomerular Endothelial Surface Layer via Pathologic Crosstalk between Activated Podocytes and Glomerular Endothelial Cells. Kidney Int. 2019, 96, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.B.Y.; Qu, X.; Zhang, X.; Caruana, G.; Bertram, J.F.; Li, J. Glomerular Endothelial Cell Injury and Damage Precedes that of Podocytes in Adriamycin-Induced Nephropathy. PLoS ONE 2013, 8, e55027. [Google Scholar] [CrossRef] [Green Version]
- Simic, I.; Tabatabaeifar, M.; Schaefer, F. Animal Models of Nephrotic Syndrome. Pediatr. Nephrol. 2013, 28, 2079–2088. [Google Scholar] [CrossRef]
- Bryant, C.; Cianciolo, R.; Govindarajan, R.; Agrawal, S. Adriamycin-Induced Nephropathy Is Robust in N and Modest in J Substrain of C57BL/6. Front. Cell Dev. Biol. 2022, 10, 924751. [Google Scholar] [CrossRef]
- Anderson, C.E.; Hamm, L.L.; Batuman, G.; Kumbala, D.R.; Chen, C.-S.; Kallu, S.G.; Siriki, R.; Gadde, S.; Kleinpeter, M.A.; Krane, N.K.; et al. The Association of Angiogenic Factors and Chronic Kidney Disease. BMC Nephrol. 2018, 19, 117. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Kamboj, K.; Yadav, A.K.; Kaur, P.; Kumar, V.; Jha, V. Cholecalciferol Supplementation and Angiogenic Markers in Chronic Kidney Disease. PLoS ONE 2022, 17, e0268946. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Almeida, L.; Della Coletta Francescato, H.; Antunes-Rodrigues, J.; Jose Albuquerque de Paula, F.; Giovanni Alves da Silva, C.; Silva Costa, R.; Machado Coimbra, T. Imbalance of Pro- and Anti-Angiogenic Factors Due to Maternal Vitamin D Deficiency Causes Renal Microvasculature Alterations Affecting the Adult Kidney Function. Nutrients 2019, 11, 1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnudi, L.; Benedetti, S.; Woolf, A.S.; Long, D.A. Vascular Growth Factors Play Critical Roles in Kidney Glomeruli. Clin. Sci. 2015, 129, 1225–1236. [Google Scholar] [CrossRef]
- He, H.; Wang, L.; Qiao, Y.; Zhou, Q.; Li, H.; Chen, S.; Yin, D.; Huang, Q.; He, M. Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/ENOS/NO Pathway. Front. Pharmacol. 2020, 10, 1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salanova Villanueva, L.; Gil Giraldo, Y.; Santos Sánchez-Rey, B.; Aguilera Peralta, A. Efecto Regulador de Paricalcitol Sobre Parámetros Inflamatorios, Fibróticos y Anticalcificantes En El Paciente Con Enfermedad Renal Crónica. Más Allá de La Regulación de La Enfermedad Óseo-Mineral. Nefrología 2020, 40, 171–179. [Google Scholar] [CrossRef]
- Martínez-Arias, L.; Panizo, S.; Alonso-Montes, C.; Martín-Vírgala, J.; Martín-Carro, B.; Fernández-Villabrille, S.; García Gil-Albert, C.; Palomo-Antequera, C.; Fernández-Martín, J.L.; Ruiz-Torres, M.P.; et al. Effects of Calcitriol and Paricalcitol on Renal Fibrosis in CKD. Nephrol. Dial. Transplant. 2021, 36, 793–803. [Google Scholar] [CrossRef]
- Gembillo, G.; Siligato, R.; Amatruda, M.; Conti, G.; Santoro, D. Vitamin D and Glomerulonephritis. Medicina 2021, 57, 186. [Google Scholar] [CrossRef]
- Demir, F.; Demir, M.; Aygun, H. Evaluation of the Protective Effect of Paricalcitol and Vitamin D 3 at Doxorubicin Nephrotoxicity in Rats with 99m Technetium-Dimercaptosuccinic Acid Renal Scintigraphy and Biochemical Methods. Hum. Exp. Toxicol. 2021, 40, 274–283. [Google Scholar] [CrossRef]
- Martin-Romero, A.; Perelló-Martínez, J.; Hidalgo-Santiago, J.C.; Michan-Doña, A.; Bosco López Sáez, J.; Gómez-Fernández, P. Efecto de la Administración de Diferentes Formas de Vitamina D en la Presión Arterial y Rigidez Aórticas, y su Implicación en la Reducción de la Albuminuria en la Enfermedad Renal Crónica. Clín. Investig. Arterioscler. 2022, 34, 311–321. [Google Scholar] [CrossRef]
- Wylie-Sears, J.; Levine, R.A.; Bischoff, J. Losartan Inhibits Endothelial-to-Mesenchymal Transformation in Mitral Valve Endothelial Cells by Blocking Transforming Growth Factor-β-Induced Phosphorylation of ERK. Biochem. Biophys. Res. Commun. 2014, 446, 870–875. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.-H.; Yook, J.-M.; Oh, S.-H.; Jeon, S.-J.; Noh, H.W.; Jung, H.-Y.; Choi, J.-Y.; Cho, J.-H.; Kim, C.-D.; Kim, Y.-L.; et al. Paricalcitol Improves Hypoxia-Induced and TGF-Β1-Induced Injury in Kidney Pericytes. Int. J. Mol. Sci. 2021, 22, 9751. [Google Scholar] [CrossRef]
- Wu-Wong, J.R.; Li, X.; Chen, Y. Different Vitamin D Receptor Agonists Exhibit Differential Effects on Endothelial Function and Aortic Gene Expression in 5/6 Nephrectomized Rats. J. Steroid Biochem. Mol. Biol. 2015, 148, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Miguel, P.; Valdivielso, J.M.; Medrano-Andrés, D.; Román-García, P.; Cano-Peñalver, J.L.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; López-Ongil, S. The Active Form of Vitamin D, Calcitriol, Induces a Complex Dual Upregulation of Endothelin and Nitric Oxide in Cultured Endothelial Cells. Am. J. Physiol. Metab. 2014, 307, E1085–E1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maimaitiyiming, H.; Zhou, Q.; Wang, S. Thrombospondin 1 Deficiency Ameliorates the Development of Adriamycin-Induced Proteinuric Kidney Disease. PLoS ONE 2016, 11, e0156144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Li, H.; Zhang, C.; Li, Z.; Wang, Q.; Guo, J.; Luo, C.; Wang, Y. Urinary Angiopoietin-2 Is Associated with Albuminuria in Patients with Type 2 Diabetes Mellitus. Int. J. Endocrinol. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.-C.; Lai, T.-S.; Chiang, C.-K.; Chen, Y.-M.; Wu, M.-S.; Chu, T.-S.; Wu, K.-D.; Lin, S.-L. Angiopoietin-2 Is Associated with Albuminuria and Microinflammation in Chronic Kidney Disease. PLoS ONE 2013, 8, e54668. [Google Scholar] [CrossRef] [Green Version]
- Arfian, N.; Kusuma, M.H.H.; Anggorowati, N.; Nugroho, D.B.; Jeffilano, A.; Suzuki, Y.; Ikeda, K.; Emoto, N. Vitamin D Upregulates Endothelin-1, ETBR, ENOS MRNA Expression and Attenuates Vascular Remodelling and Ischemia in Kidney Fibrosis Model in Mice. Physiol. Res. 2018, 67 (Suppl. 1), S137–S147. [Google Scholar] [CrossRef]
- Deluque, A.L.; de Almeida, L.F.; Francescato, H.D.C.; da Silva, C.G.A.; Costa, R.S.; Antunes-Rodrigues, J.; Coimbra, T.M. Effect of Calcitriol on the Renal Microvasculature Differentiation Disturbances Induced by AT1 Blockade During Nephrogenesis in Rats. Front. Med. 2020, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Faleiros, C.M.; Francescato, H.D.C.; Papoti, M.; Chaves, L.; Silva, C.G.A.; Costa, R.S.; Coimbra, T.M. Effects of Previous Physical Training on Adriamycin Nephropathy and Its Relationship with Endothelial Lesions and Angiogenesis in the Renal Cortex. Life Sci. 2017, 169, 43–51. [Google Scholar] [CrossRef]
- Chang-Chien, C.; Chuang, G.-T.; Tsai, I.-J.; Chiang, B.-L.; Yang, Y.-H. A Large Retrospective Review of Persistent Proteinuria in Children. J. Formos. Med. Assoc. 2018, 117, 711–719. [Google Scholar] [CrossRef]
- Han, X.; Dai, R.; Zhai, Y.; Shen, Q.; Sun, L.; Liu, H.; Xu, H. Anti-Proteinuria Effect of Antibody against ANGPTL3 Coil-Coiled Domain on Adriamycin-Induced Nephropathy in Mice. Biochem. Biophys. Res. Commun. 2019, 516, 812–818. [Google Scholar] [CrossRef]
- Zoja, C.; Abbate, M.; Remuzzi, G. Progression of Renal Injury toward Interstitial Inflammation and Glomerular Sclerosis Is Dependent on Abnormal Protein Filtration. Nephrol. Dial. Transplant. 2015, 30, 706–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Li, Q.; Zhen, J.; Xu, Y.; Sun, S. Simvastatin Ameliorates Renal Lipidosis through the Suppression of Renal CXCL16 Expression in Mice with Adriamycin-Induced Nephropathy. Int. J. Clin. Exp. Pathol. 2015, 8, 15696–15707. [Google Scholar] [PubMed]
- Silveira, K.D.; Barroso, L.C.; Vieira, A.T.; Cisalpino, D.; Lima, C.X.; Bader, M.; Arantes, R.M.E.; dos Santos, R.A.S.; Simões-e-Silva, A.C.; Teixeira, M.M. Beneficial Effects of the Activation of the Angiotensin-(1–7) Mas Receptor in a Murine Model of Adriamycin-Induced Nephropathy. PLoS ONE 2013, 8, e66082. [Google Scholar] [CrossRef] [Green Version]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Jiang, X.; Wen, D.; Zhang, Y.; Mao, J.; Ni, L.; Chen, J. Vitamin D Receptor/Vitamin D Response Element Directly Modulate Nestin Transcription to Ameliorate PAN-Induced Podocyte Morphological Changes. Nephron 2022, 146, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Garsen, M.; Sonneveld, R.; Rops, A.L.; Huntink, S.; van Kuppevelt, T.H.; Rabelink, T.J.; Hoenderop, J.G.; Berden, J.H.; Nijenhuis, T.; van der Vlag, J. Vitamin D Attenuates Proteinuria by Inhibition of Heparanase Expression in the Podocyte. J. Pathol. 2015, 237, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Ito, I.; Waku, T.; Aoki, M.; Abe, R.; Nagai, Y.; Watanabe, T.; Nakajima, Y.; Ohkido, I.; Yokoyama, K.; Miyachi, H.; et al. A Nonclassical Vitamin D Receptor Pathway Suppresses Renal Fibrosis. J. Clin. Investig. 2013, 123, 4579–4594. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Zhen, X.; Liu, J.; Ren, X.; Hu, Z.; Zhou, Z.; Zhu, F.; Ding, K.; Nie, J. The Antihelmenthic Phosphate Niclosamide Impedes Renal Fibrosis by Inhibiting Homeodomain-Interacting Protein Kinase 2 Expression. Kidney Int. 2017, 92, 612–624. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhou, J.; Zhang, H.; Zhou, H.; Tang, H.; Lei, C.; Ye, C.; You, C.; Chen, Y.; Wang, Y.; et al. Rhodojaponin II Attenuates Kidney Injury by Regulating TGF-Β1/Smad Pathway in Mice with Adriamycin Nephropathy. J. Ethnopharmacol. 2019, 243, 112078. [Google Scholar] [CrossRef]
- Samarakoon, R.; Overstreet, J.M.; Higgins, S.P.; Higgins, P.J. TGF-Β1 → SMAD/P53/USF2 → PAI-1 Transcriptional Axis in Ureteral Obstruction-Induced Renal Fibrosis. Cell Tissue Res. 2012, 347, 117–128. [Google Scholar] [CrossRef]
- van Meeteren, L.A.; ten Dijke, P. Regulation of Endothelial Cell Plasticity by TGF-β. Cell Tissue Res. 2012, 347, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-H.; Zhang, Y.; Wang, X.; Fan, X.-F.; Zhang, Y.; Li, X.; Gong, Y.; Han, L.-P. SIRT1 Activation Attenuates Cardiac Fibrosis by Endothelial-to-Mesenchymal Transition. Biomed. Pharmacother. 2019, 118, 109227. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.; ten Dijke, P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.-Z.; Lin, C.-J.; Hang, C.-l.; Chen, W.-Y. Calcitriol Attenuates Doxorubicin-Induced Cardiac Dysfunction and Inhibits Endothelial-to-Mesenchymal Transition in Mice. Cells 2019, 8, 865. [Google Scholar] [CrossRef] [Green Version]
- Lundwall, K.; Jörneskog, G.; Jacobson, S.H.; Spaak, J. Paricalcitol, Microvascular and Endothelial Function in Non-Diabetic Chronic Kidney Disease: A Randomized Trial. Am. J. Nephrol. 2015, 42, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Curatola, G.; Panuccio, V.; Tripepi, R.; Pizzini, P.; Versace, M.; Bolignano, D.; Cutrupi, S.; Politi, R.; Tripepi, G.; et al. Paricalcitol and Endothelial Function in Chronic Kidney Disease Trial. Hypertension 2014, 64, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial Dysfunction and Vascular Disease—A 30th Anniversary Update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Liu, H.; Zhang, J.; Huang, J.; Zhu, C.; Lu, Y.; Shi, Y.; Wang, Y. Ursolic Acid Prevents Doxorubicin-Induced Cardiac Toxicity in Mice through ENOS Activation and Inhibition of ENOS Uncoupling. J. Cell. Mol. Med. 2019, 23, 2174–2183. [Google Scholar] [CrossRef]
- Andrukhova, O.; Slavic, S.; Zeitz, U.; Riesen, S.C.; Heppelmann, M.S.; Ambrisko, T.D.; Markovic, M.; Kuebler, W.M.; Erben, R.G. Vitamin D Is a Regulator of Endothelial Nitric Oxide Synthase and Arterial Stiffness in Mice. Mol. Endocrinol. 2014, 28, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.; He, J.C. Glomerular Endothelial Cell Injury and Cross Talk in Diabetic Kidney Disease. Am. J. Physiol. Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef]
- Miao, C.; Zhu, X.; Wei, X.; Long, M.; Jiang, L.; Li, C.; Jin, D.; Du, Y. Pro- and Anti-Fibrotic Effects of Vascular Endothelial Growth Factor in Chronic Kidney Diseases. Ren. Fail. 2022, 44, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Haidar, M.; Placzko, S.; Niendorf, R.; Darashchonak, N.; Hubel, C.A.; von Versen-Höynck, F. Vitamin D Improves the Angiogenic Properties of Endothelial Progenitor Cells. Am. J. Physiol. Cell Physiol. 2012, 303, C954–C962. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Gu, B.; Gu, Y.; Groome, L.J.; Sun, J.; Wang, Y. Activation of Vitamin D Receptor Promotes VEGF and CuZn-SOD Expression in Endothelial Cells. J. Steroid Biochem. Mol. Biol. 2014, 140, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loganathan, K.; Salem Said, E.; Winterrowd, E.; Orebrand, M.; He, L.; Vanlandewijck, M.; Betsholtz, C.; Quaggin, S.E.; Jeansson, M. Angiopoietin-1 Deficiency Increases Renal Capillary Rarefaction and Tubulointerstitial Fibrosis in Mice. PLoS ONE 2018, 13, e0189433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Manson, S.R.; Lee, H.; Kim, Y.; Liu, T.; Guo, Q.; Geminiani, J.J.; Austin, P.F.; Chen, Y.M. Tubular Overexpression of Angiopoietin-1 Attenuates Renal Fibrosis. PLoS ONE 2016, 11, e0158908. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.-C.; Lee, M.-C.; Chiang, C.-K.; Liu, J.-S.; Lai, T.-S.; Chiang, W.-C.; Chen, Y.-M.; Chu, T.-S. Angiopoietin-2 Is Associated with Metabolic Syndrome in Chronic Kidney Disease. J. Formos. Med. Assoc. 2021, 120, 2113–2119. [Google Scholar] [CrossRef]
- Mansour, S.G.; Bhatraju, P.K.; Coca, S.G.; Obeid, W.; Wilson, F.P.; Stanaway, I.B.; Jia, Y.; Thiessen-Philbrook, H.; Go, A.S.; Ikizler, T.A.; et al. Angiopoietins as Prognostic Markers for Future Kidney Disease and Heart Failure Events after Acute Kidney Injury. J. Am. Soc. Nephrol. 2022, 33, 613–627. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Paricalcitol | ADR | ADR + Paricalcitol | |
|---|---|---|---|---|
| Body Weight (g) | 402 ± 21.70 | 451 ± 17.30 | 392 ± 11.90 # | 354 ± 12.70 ### |
| 25 OHD (ng/mL) | 29 ± 3.28 | 34 ± 1.96 | 28 ± 2.19 | 31 ± 1.39 |
| PTH (pg/mg) | 177 ± 22.40 | 180 ± 36.40 | 288 ± 29.50 | 270 ± 35.70 |
| PCa (mg/dL) | 9.1 ± 0.27 | 9.4 ± 0.5 | 9.1 ± 0.33 | 9.5 ± 0.24 |
| Pp (mg/dL) | 7.0 ± 0.46 | 6.7 ± 0.33 | 7.1 ± 0.23 | 7.6 ± 0.20 |
| Urine volume (mL 100 g−1 24 h−1) | 5.1 ± 1.57 | 10.4 ± 2.24 | 8.3 ± 1.26 | 9.5 ± 1.03 |
| GFR (ml min−1 100 g−1) | 0.3 ± 0.04 | 0.4 ± 0.03 | 0.2 ± 0.02 # | 0.4 ± 0.01 $ |
| FENa (%) | 0.3 ± 0.02 | 0.2 ± 0.02 | 0.3 ± 0.03 # | 0.2 ± 0.01 $ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deluque, A.L.; Oliveira, B.M.; Souza, C.S.; Maciel, A.L.D.; Francescato, H.D.C.; Giovanini, C.; de Almeida, L.F.; de Paula, F.J.A.; Costa, R.S.; Antunes-Rodrigues, J.; et al. Paricalcitol Improves the Angiopoietin/Tie-2 and VEGF/VEGFR2 Signaling Pathways in Adriamycin-Induced Nephropathy. Nutrients 2022, 14, 5316. https://doi.org/10.3390/nu14245316
Deluque AL, Oliveira BM, Souza CS, Maciel ALD, Francescato HDC, Giovanini C, de Almeida LF, de Paula FJA, Costa RS, Antunes-Rodrigues J, et al. Paricalcitol Improves the Angiopoietin/Tie-2 and VEGF/VEGFR2 Signaling Pathways in Adriamycin-Induced Nephropathy. Nutrients. 2022; 14(24):5316. https://doi.org/10.3390/nu14245316
Chicago/Turabian StyleDeluque, Amanda Lima, Beatriz Magalhães Oliveira, Cláudia Silva Souza, Ana Lívia Dias Maciel, Heloísa Della Coletta Francescato, Cleonice Giovanini, Lucas Ferreira de Almeida, Francisco José Albuquerque de Paula, Roberto Silva Costa, José Antunes-Rodrigues, and et al. 2022. "Paricalcitol Improves the Angiopoietin/Tie-2 and VEGF/VEGFR2 Signaling Pathways in Adriamycin-Induced Nephropathy" Nutrients 14, no. 24: 5316. https://doi.org/10.3390/nu14245316