
Bovine Pancreatic RNase A: An Insight into the Mechanism of Antitumor Activity In Vitro and In Vivo

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Mice
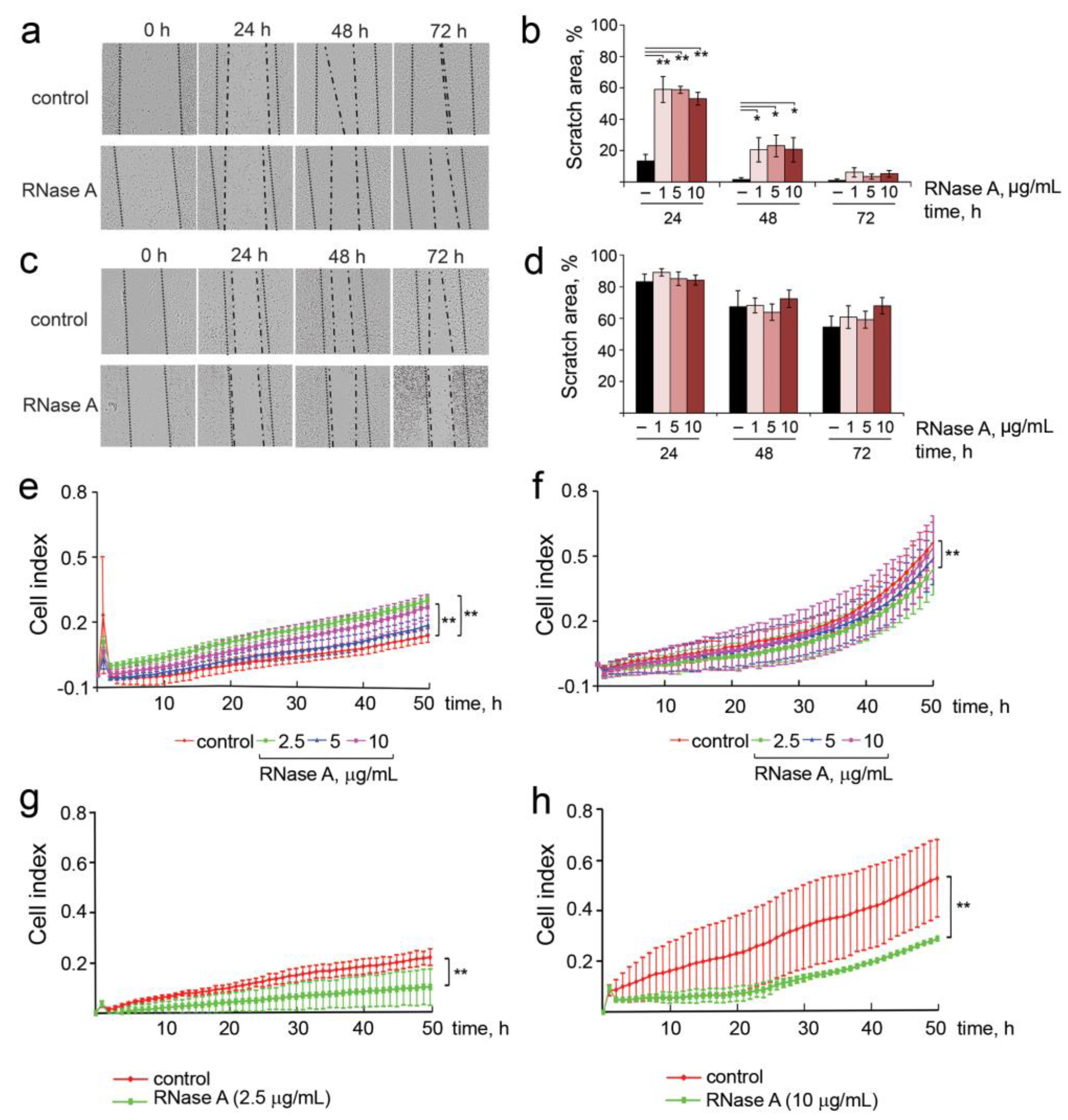
2.3. Scratch Assay
2.4. Influence of RNase A on the Migration Activity, Motility, Adhesion, Invasion and Colony Formation of B16 and HeLa Cells
2.5. Conjugation of RNase A with Biotin
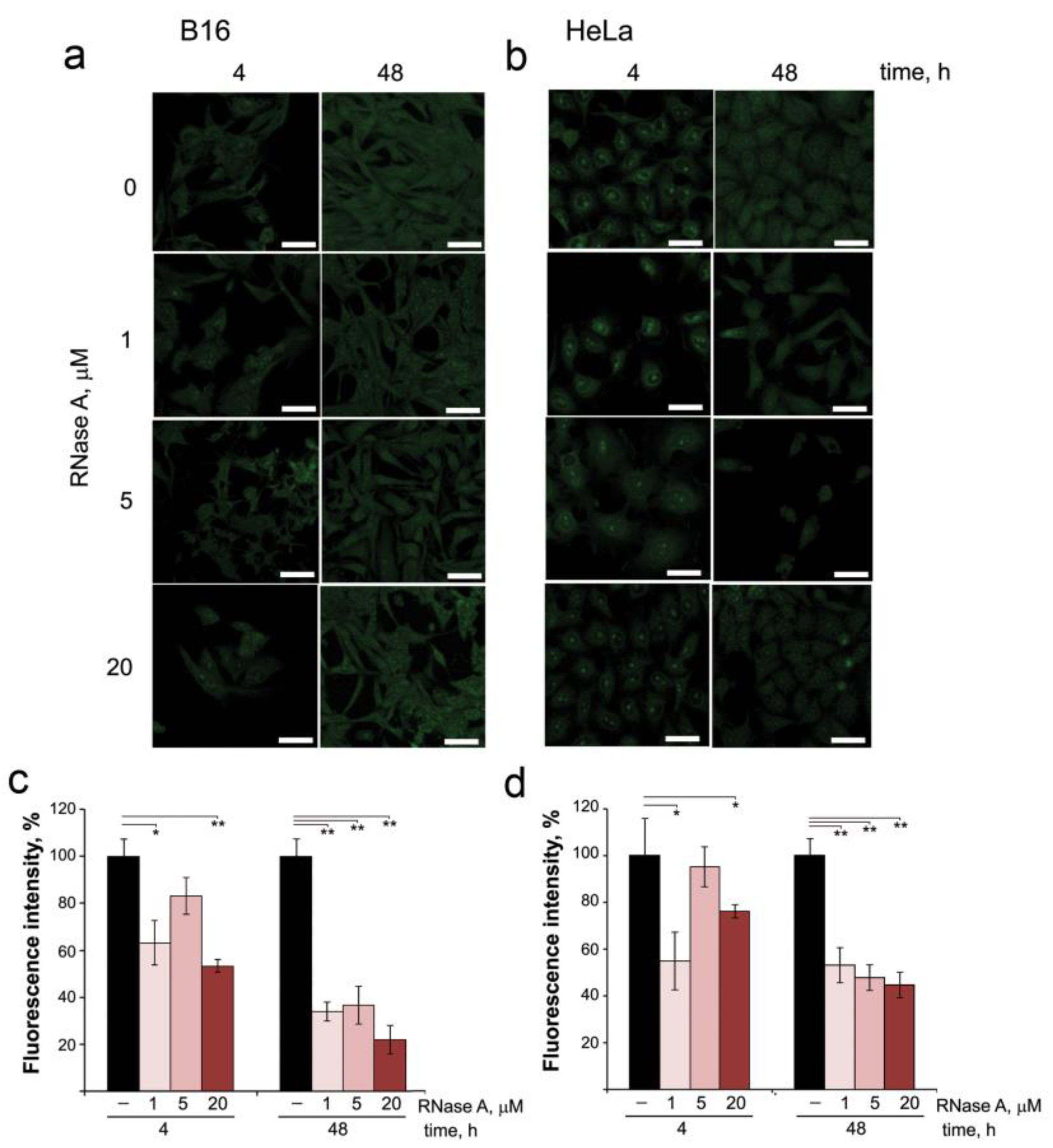
2.6. RNA Visualization by Fluorescence Microscopy
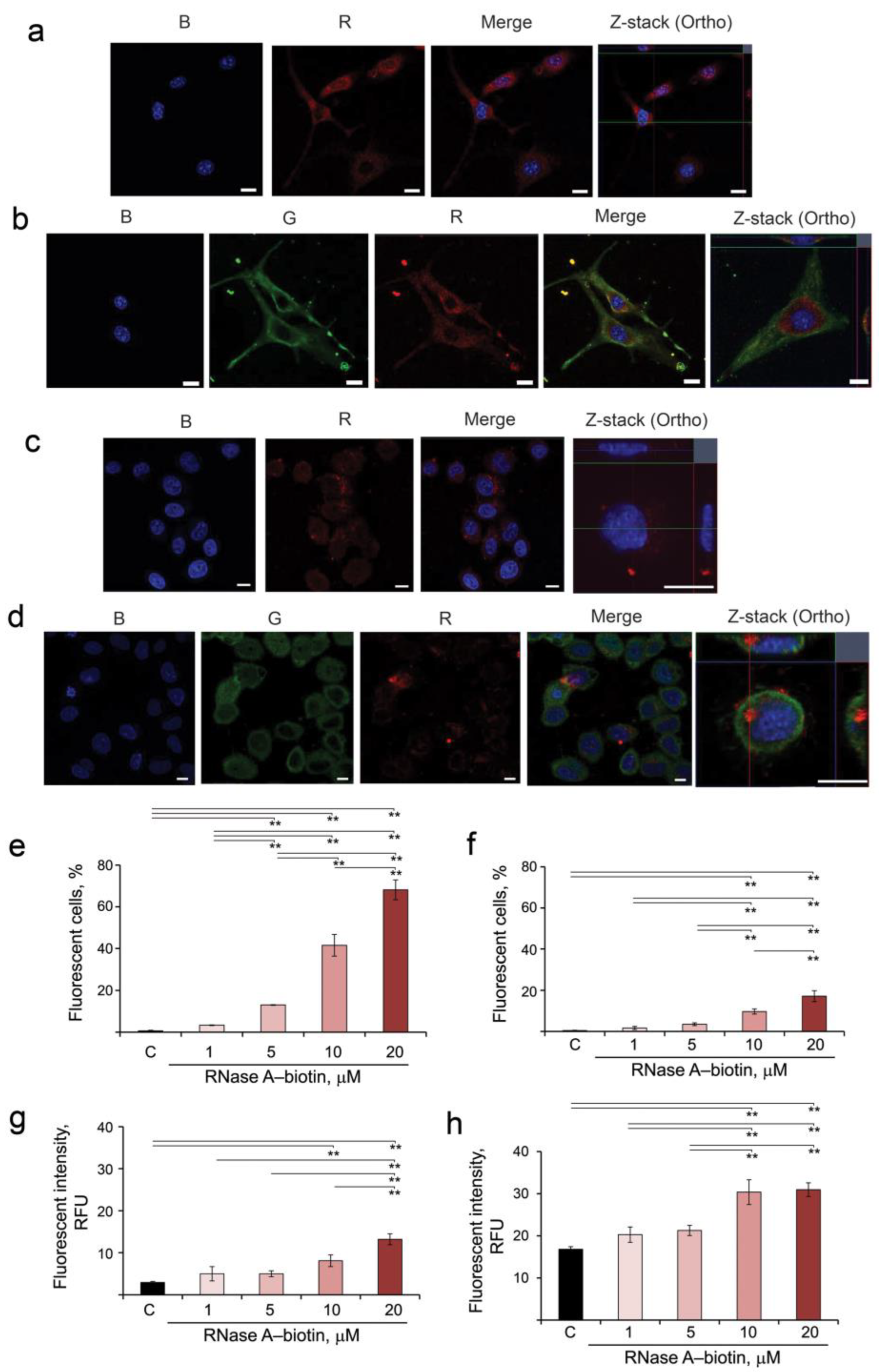
2.7. Analysis of Accumulation and Intracellular Localization of RNase A–Biotin Conjugate in HeLa and B16 Cells by Confocal Microscopy
2.8. Analysis of RNase A–Biotin Conjugate Accumulation into B16 and HeLa Cells by Flow Cytometry
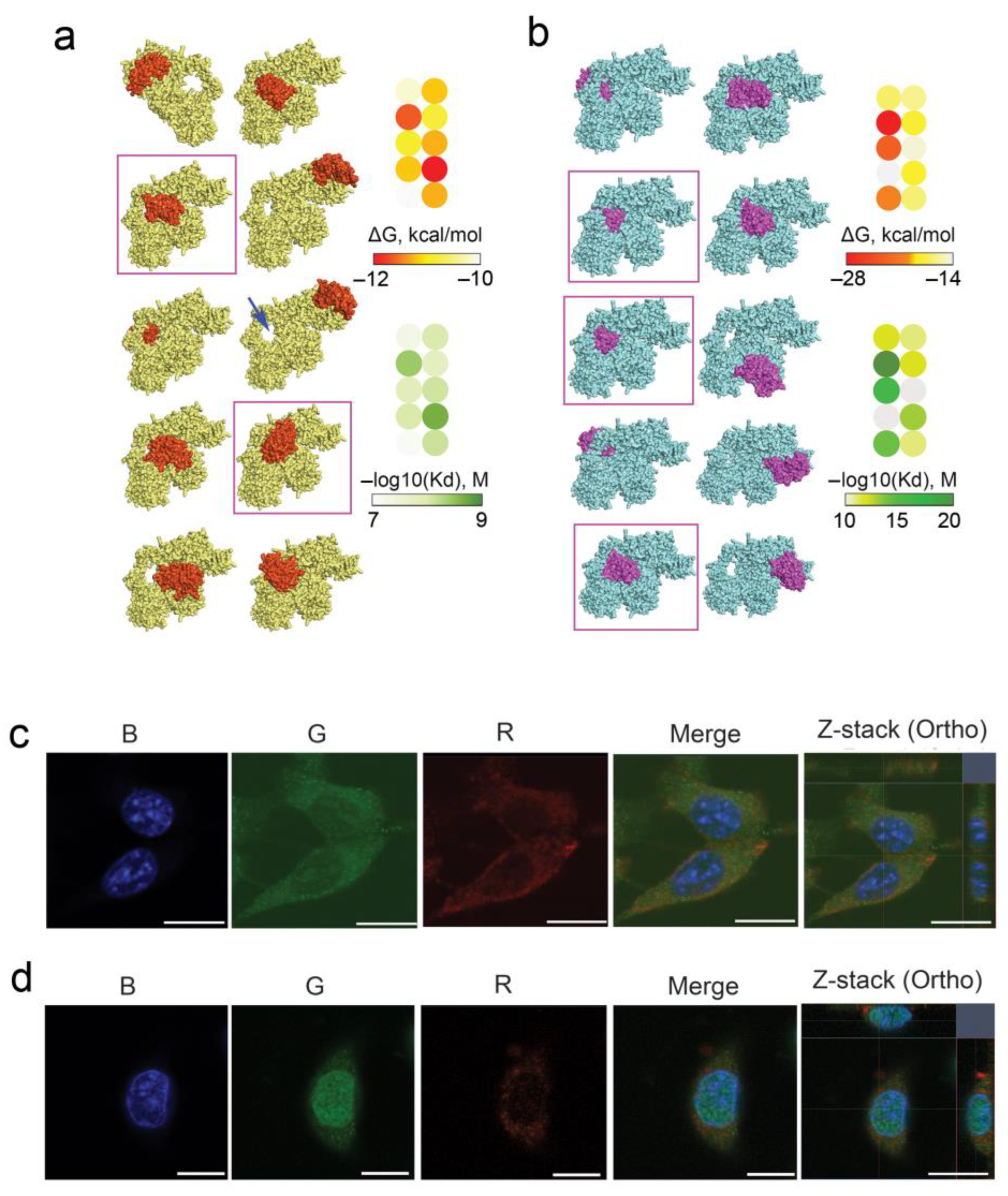
2.9. Co-Localization of RNase A–Biotin Conjugate with Ku70/Ku80 in B16 and HeLa Cells
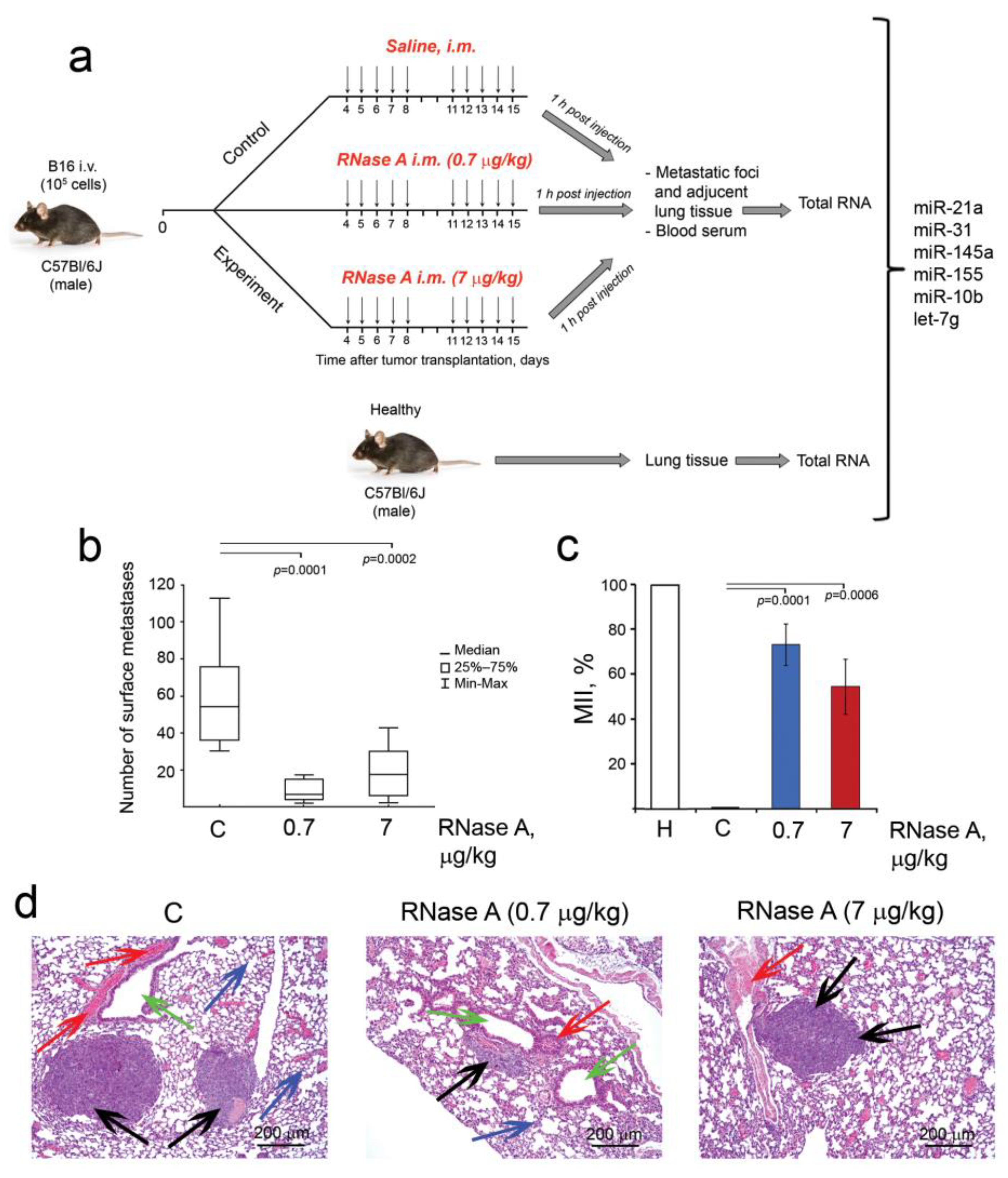
2.10. B16 Implantation and Design of the Animal Experiment
2.11. Sample Processing and RNA Extraction
2.12. RT-qPCR
2.13. Histology and Immunohistochemistry
2.14. Molecular Modeling of RNase A Interaction with Ku70/Ku80
2.15. RNase-A-Susceptible miRNA Target Prediction and Functional Analysis
2.16. Statistics
3. Results
3.1. Intracellular Accumulation and Localization of RNase A–Biotin Conjugate and Its Interaction with RI in B16 and HeLa Cells
3.2. Interaction of RNase A with Ku70/Ku80 Heterodimer: Molecular Modeling and Intracellular Behavior
3.3. The Effect of RNase A on Intracellular RNAs
3.4. The Effect of RNase A on the Migration Activity, Adhesion, Invasion and Colony Formation of B16 and HeLa Cells
3.5. The Influence of RNase A on Metastasis of Melanoma B16 In Vivo
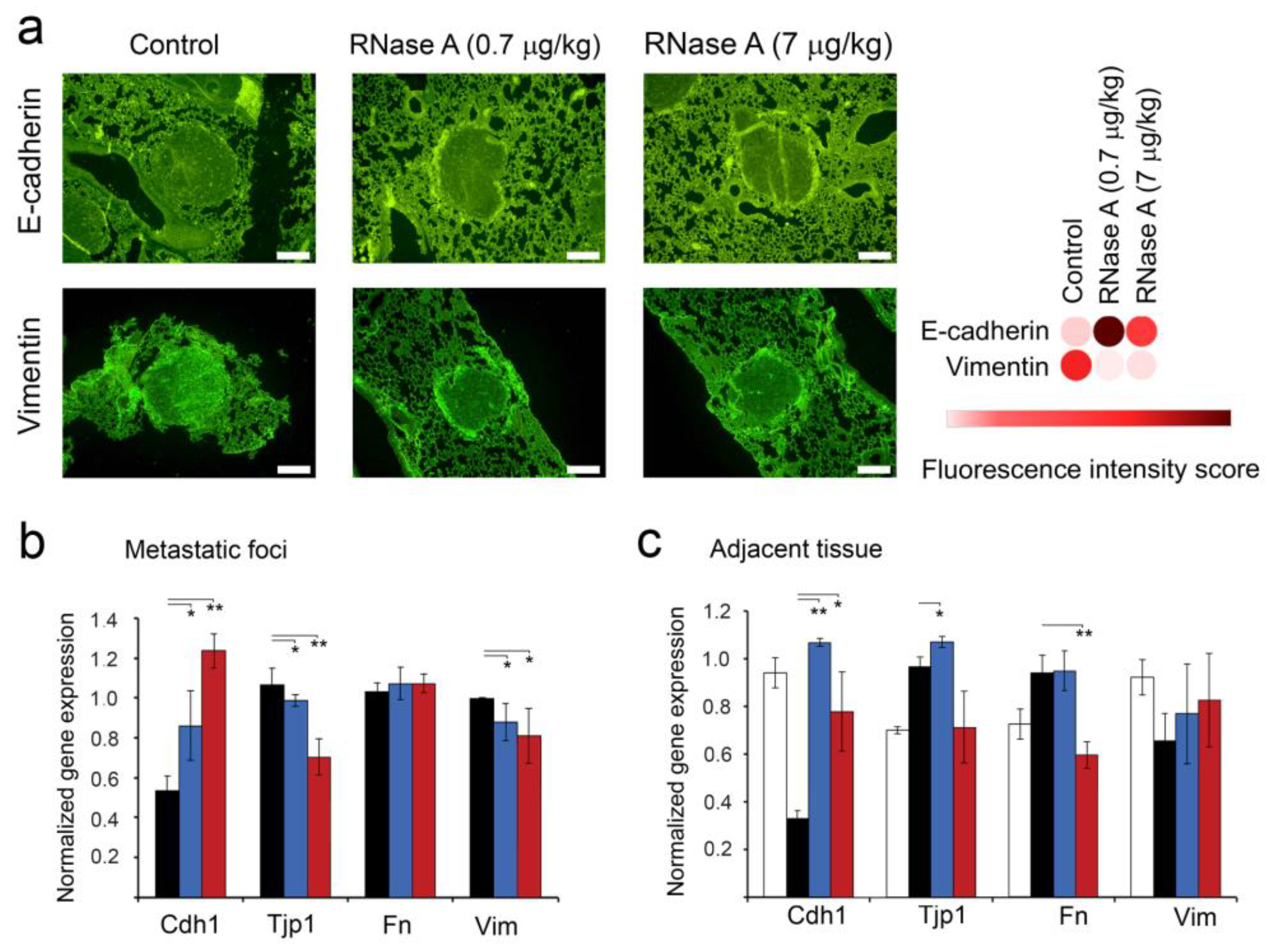
3.6. The Effect of RNase A on the Expression of EMT Markers in Metastatic Foci and Adjacent Lung Tissue of B16 Melanoma-Bearing Mice
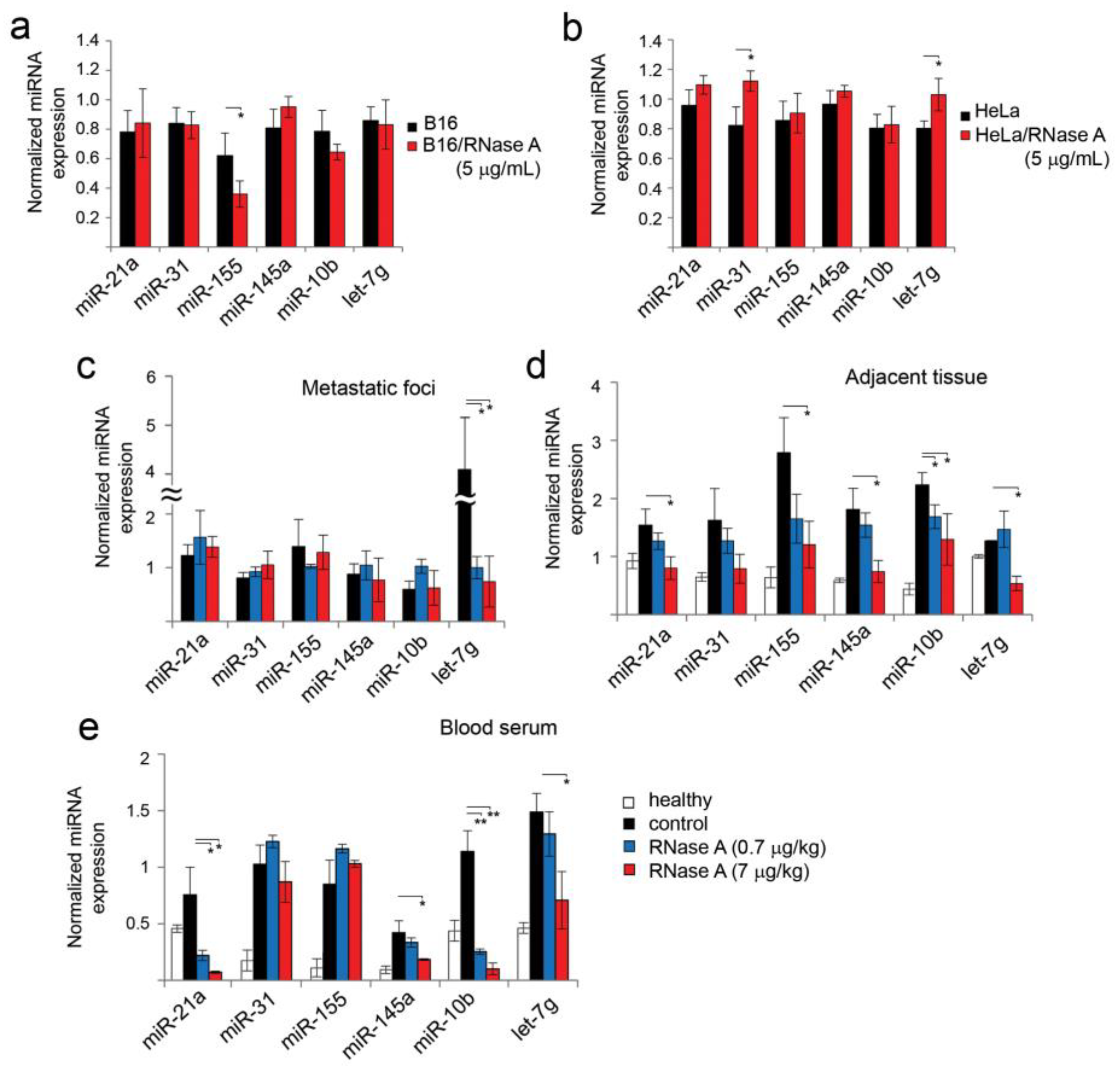
3.7. Alteration of miRNA Expression in B16 and HeLa Cells In Vitro and in B16 Metastatic Foci and Adjacent Lung Tissues In Vivo under the Action of RNase A
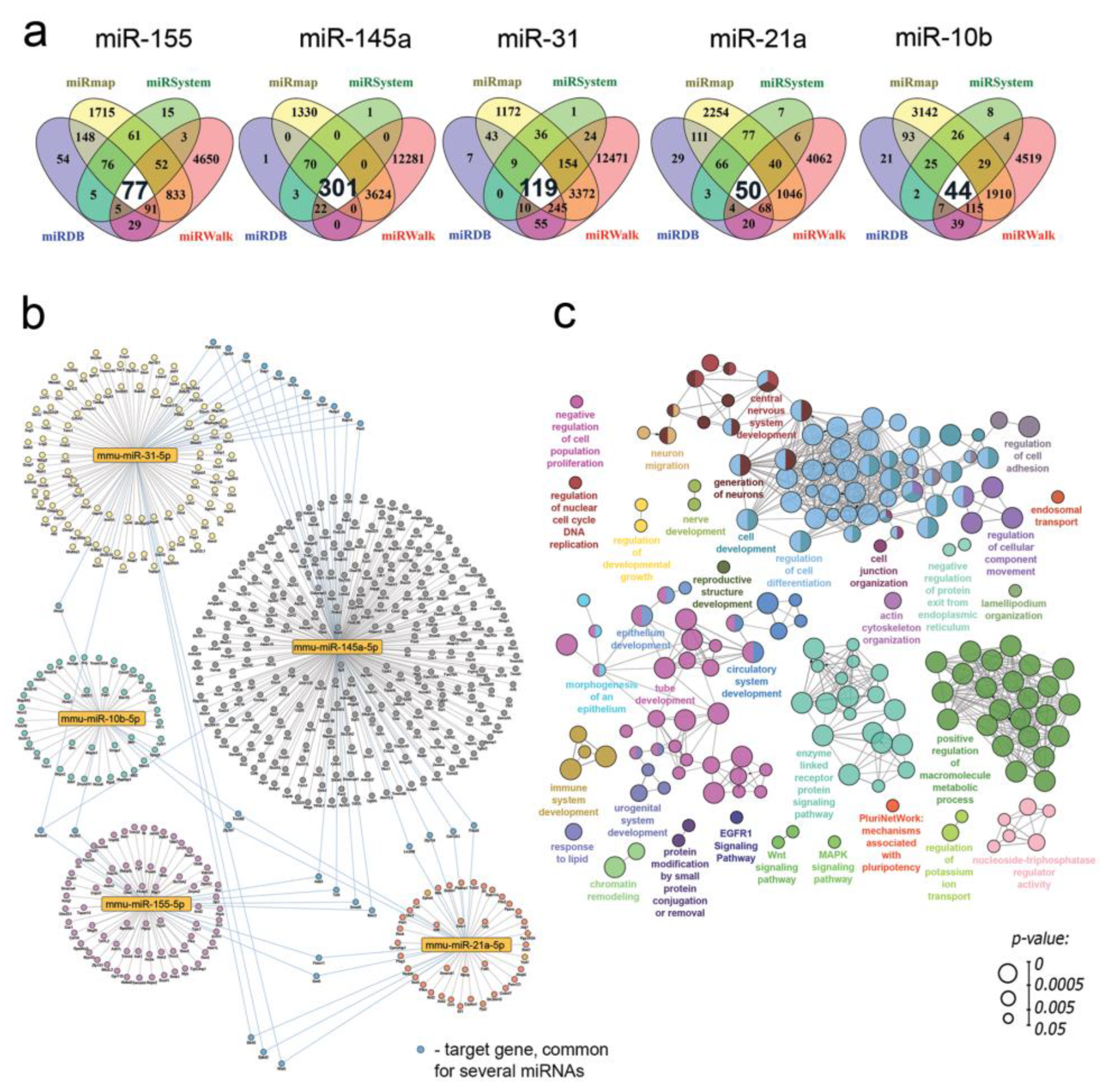
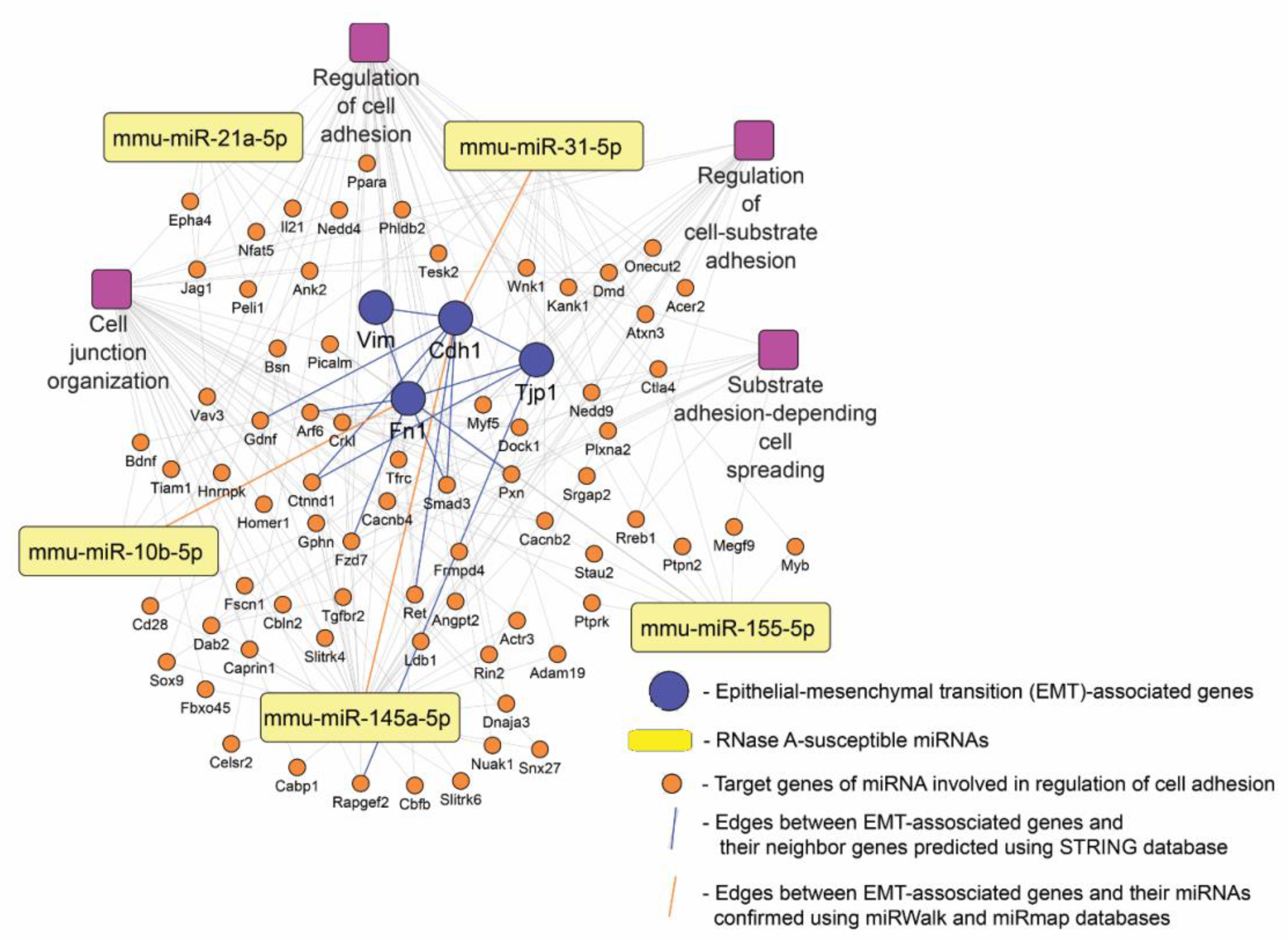
3.8. RNase-A-Susceptible miRNA: Target Prediction and Functional Analysis
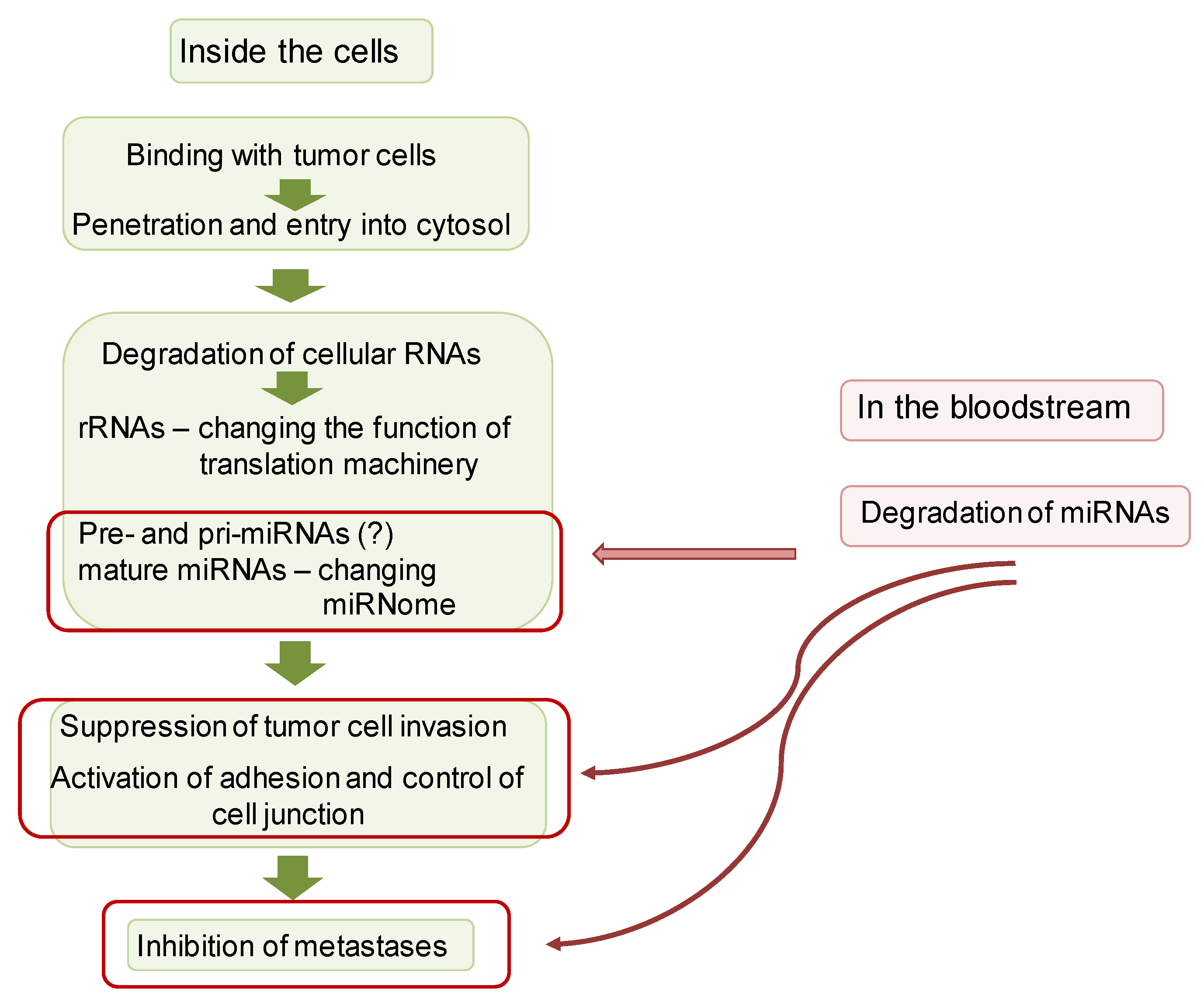
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, J.E.; Raines, R.T. Ribonucleases as novel chemotherapeutics: The ranpirnase example. BioDrugs 2008, 22, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Poučková, P.; Zadinová, M.; Hloušková, D.; Strohalm, J.; Plocová, D.; Špunda, M.; Olejár, T.; Zitko, M.; Matoušek, J.; Ulbrich, K.; et al. Polymer-conjugated bovine pancreatic and seminal ribonucleases inhibit growth of human tumors in nude mice. J. Control. Release 2004, 95, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Raines, R.T. Cytotoxicity of bovine seminal ribonuclease: Monomer versus dimer. Biochemistry 2005, 44, 15760–15767. [Google Scholar] [CrossRef] [PubMed]
- Lacadena, J.; Álvarez-García, E.; Carreras-Sangrà, N.; Herrero-Galán, E.; Alegre-Cebollada, J.; García-Ortega, L.; Oñaderra, M.; Gavilanes, J.G.; Martínez Del Pozo, Á. Fungal ribotoxins: Molecular dissection of a family of natural killers. FEMS Microbiol. Rev. 2007, 31, 212–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, T.I.; Kunwar, S.; Pastan, I. Studies on the activity of barnase toxins in vitro and in vivo. Bioconjug. Chem. 1996, 7, 23–29. [Google Scholar] [CrossRef]
- Makarov, A.A.; Kolchinsky, A.; Ilinskaya, O.N. Binase and other microbial RNases as potential anticancer agents. BioEssays 2008, 30, 781–790. [Google Scholar] [CrossRef]
- Mironova, N.L.; Petrushanko, I.Y.; Patutina, O.A.; Sen’kova, A.V.; Simonenko, O.V.; Mitkevich, V.A.; Markov, O.V.; Zenkova, M.A.; Makarov, A.A. Ribonuclease binase inhibits primary tumor growth and metastases via apoptosis induction in tumor cells. Cell Cycle 2013, 12, 2120–2131. [Google Scholar] [CrossRef]
- Raines, R.T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1065. [Google Scholar] [CrossRef]
- Ledoux, L. Action of ribonuclease on neoplastic growth II. Action on landschütz ascites cells in vitro. Biochim. Biophys. Acta 1956, 20, 369–377. [Google Scholar] [CrossRef]
- Ledoux, L.; Revell, S.H. Action of ribonuclease on neoplastic growth. I. Chemical aspects of normal tumour growth: The landschütz ascites tumour. Biochim. Biophys. Acta 1955, 18, 416–426. [Google Scholar] [CrossRef]
- Ledoux, L. Action of ribonuclease on two solid tumours in vivo. Nature 1955, 176, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, L. Action of ribonuclease on certain ascites tumours. Nature 1955, 175, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrowicz, J. Intracutaneous ribonuclease in chronic myelocytic leukæmia. Lancet 1958, 272, 420. [Google Scholar] [CrossRef]
- Telford, I.R.; Kemp, J.F.; Taylor, E.F.; Yeaman, M.W. Effect of ribonuclease on survival of ascites tumor bearing mice. Proc. Soc. Exp. Biol. Med. 1959, 100, 829–831. [Google Scholar] [CrossRef]
- De Lamirande, G. Action of deoxyribonuclease and ribonuclease on the growth of Ehrlich ascites carcinoma in mice. Nature 1961, 192, 52–54. [Google Scholar] [CrossRef]
- Leland, P.A.; Raines, R.T. Cancer chemotherapy—Ribonucleases to the rescue. Chem. Biol. 2001, 8, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.J.; McCoy, J.G.; Bingman, C.A.; Phillips, G.N.; Raines, R.T. Inhibition of human pancreatic ribonuclease by the human ribonuclease inhibitor protein. J. Mol. Biol. 2007, 368, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Naddeo, M.; Vitagliano, L.; Russo, A.; Gotte, G.; D’Alessio, G.; Sorrentino, S. Interactions of the cytotoxic RNase A dimers with the cytosolic ribonuclease inhibitor. FEBS Lett. 2005, 579, 2663–2668. [Google Scholar] [CrossRef] [Green Version]
- Shklyaeva, O.A.; Mironova, N.L.; Malkova, E.M.; Taranov, O.S.; Ryabchikova, E.I.; Zenkova, M.A.; Vlasov, V.V. Cancer-suppressive effect of RNase A and DNase I. Dokl. Biochem. Biophys. 2008, 420, 108–111. [Google Scholar] [CrossRef]
- Patutina, O.; Mironova, N.; Ryabchikova, E.; Popova, N.; Nikolin, V.; Kaledin, V.; Vlassov, V.; Zenkova, M. Inhibition of metastasis development by daily administration of ultralow doses of RNase A and DNase I. Biochimie 2011, 93, 689–696. [Google Scholar] [CrossRef]
- Dalmay, T.; Edwards, D.R. MicroRNAs and the hallmarks of cancer. Oncogene 2006, 25, 6170–6175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussen, B.M.; Hidayat, H.J.; Salihi, A.; Sabir, D.K.; Taheri, M.; Ghafouri-Fard, S. MicroRNA: A signature for cancer progression. Biomed. Pharmacother. 2021, 138, 111528. [Google Scholar] [CrossRef] [PubMed]
- Mironova, N.; Patutina, O.; Brenner, E.; Kurilshikov, A.; Vlassov, V.; Zenkova, M. MicroRNA drop in the bloodstream and microRNA boost in the tumour caused by treatment with ribonuclease A leads to an attenuation of tumour malignancy. PLoS ONE 2013, 8, e83482. [Google Scholar] [CrossRef] [PubMed]
- Mironova, N.; Patutina, O.; Brenner, E.; Kurilshikov, A.; Vlassov, V.; Zenkova, M. The systemic tumor response to RNase A treatment affects the expression of genes involved in maintaining cell malignancy. Oncotarget 2017, 8, 78796–78810. [Google Scholar] [CrossRef] [Green Version]
- Koike, M.; Ikuta, T.; Miyasaka, T.; Shiomi, T. Ku80 can translocate to the nucleus independent of the translocation of Ku70 using its own nuclear localization signal. Oncogene 1999, 18, 7495–7505. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Gottlieb, T.M.; Jackson, S.P. The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993, 72, 131–142. [Google Scholar] [CrossRef]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.-W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [Green Version]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, J.F.; Knudsen, K.E. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarski, W.; Khan, S.A. Lupus autoantigen Ku protein binds HIV-1 TAR RNA in vitro. Biochem. Biophys. Res. Commun. 1993, 196, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Monferran, S.; Muller, C.; Mourey, L.; Frit, P.; Salles, B. The membrane-associated form of the DNA repair protein Ku is involved in cell adhesion to fibronectin. J. Mol. Biol. 2004, 337, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Ginis, I.; Faller, D.V. Hypoxia affects tumor cell invasiveness in vitro: The role of hypoxia-activated ligand HAL1/13 (Ku86 autoantigen). Cancer Lett. 2000, 154, 163–174. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.H.; Ko, J.H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.W.; Lee, J.; Chinnathambi, A.; Zayed, M.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef]
- Markov, A.V.; Odarenko, K.V.; Sen’kova, A.V.; Salomatina, O.V.; Salakhutdinov, N.F.; Zenkova, M.A. Cyano enone-bearing triterpenoid soloxolone methyl inhibits epithelial-mesenchymal transition of human lung adenocarcinoma cells in vitro and metastasis of murine melanoma in vivo. Molecules 2020, 25, 5925. [Google Scholar] [CrossRef]
- Mitkevich, V.A.; Tchurikov, N.A.; Zelenikhin, P.V.; Petrushanko, I.Y.; Makarov, A.A.; Ilinskaya, O.N. Binase cleaves cellular noncoding RNAs and affects coding mRNAs. FEBS J. 2010, 277, 186–196. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Markov, O.V.; Mironova, N.L.; Shmendel, E.V.; Serikov, R.N.; Morozova, N.G.; Maslov, M.A.; Vlassov, V.V.; Zenkova, M.A. Multicomponent mannose-containing liposomes efficiently deliver RNA in murine immature dendritic cells and provide productive anti-tumour response in murine melanoma model. J. Control. Release 2015, 213, 45–56. [Google Scholar] [CrossRef]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef]
- Varkonyi-Gasic, E.; Wu, R.; Wood, M.; Walton, E.F.; Hellens, R.P. Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 2007, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Lynch, E.M.; Moreland, R.B.; Ginis, I.; Perrine, S.P.; Faller, D.V. Hypoxia-activated ligand HAL-1/13 is lupus autoantigen Ku80 and mediates lymphoid cell adhesion in vitro. Am. J. Physiol.-Cell Physiol. 2001, 280, C897–C911. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Podar, K.; Kraeft, S.K.; Wang, F.; Young, G.; Lin, B.; Gupta, D.; Lan, B.C.; Anderson, K.C. Translocation of Ku86/Ku70 to the multiple myeloma cell membrane: Functional implications. Exp. Hematol. 2002, 30, 212–220. [Google Scholar] [CrossRef]
- Teoh, G.; Urashima, M.; Greenfield, E.A.; Nguyen, K.A.; Lee, J.F.; Chauhan, D.; Ogata, A.; Treon, S.P.; Anderson, K.C. The 86-kD subunit of Ku autoantigen mediates homotypic and heterotypic adhesion of multiple myeloma cells. J. Clin. Investig. 1998, 101, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, B.S.; Allaway, G.P.; Srinivasappa, J.; Notkins, A.L. Cell surface expression of the 70-kD component of Ku, a DNA-binding nuclear autoantigen. J. Clin. Investig. 1990, 86, 1301–1305. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.; Paupert, J.; Monferran, S.; Salles, B. The double life of the Ku protein: Facing the DNA breaks and the extracellular environment. Cell Cycle 2005, 4, 438–441. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Mohamed, I.S.E.; Sen’kova, A.V.; Nadyrova, A.I.; Savin, I.A.; Markov, A.V.; Mitkevich, V.A.; Makarov, A.A.; Ilinskaya, O.N.; Mironova, N.L.; Zenkova, M.A. Antitumour activity of the ribonuclease binase from bacillus pumilus in the RLS40 tumour model is associated with the reorganisation of the miRNA network and reversion of cancer-related cascades to normal functioning. Biomolecules 2020, 10, 1509. [Google Scholar] [CrossRef]
- Chao, T.Y.; Lavis, L.D.; Raines, R.T. Cellular uptake of ribonuclease A relies on anionic glycans. Biochemistry 2010, 49, 10666–10673. [Google Scholar] [CrossRef] [Green Version]
- Chao, T.Y.; Raines, R.T. Mechanism of ribonuclease A endocytosis: Analogies to cell-penetrating peptides. Biochemistry 2011, 50, 8374–8382. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Kim, T.H.; Choi, J.H.; Lee, K.C.; Park, K.D.; Lee, S.J.; Kuh, H.J. Evaluation of interstitial protein delivery in multicellular layers model. Arch. Pharm. Res. 2012, 35, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Kurten, E.L.; Raines, R.T. Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids Res. 2003, 31, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Rutkoski, T.; Raines, R. Evasion of ribonuclease inhibitor as a determinant of ribonuclease cytotoxicity. Curr. Pharm. Biotechnol. 2008, 9, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.S.; Shapiro, R.; Vallee, B.L. Tight-binding inhibition of angiogenin and ribonuclease A by placental ribonuclease inhibitor. Biochemistry 1989, 28, 225–230. [Google Scholar] [CrossRef]
- Hoang, T.T.; Raines, R.T. Molecular basis for the autonomous promotion of cell proliferation by angiogenin. Nucleic Acids Res. 2017, 45, 818–831. [Google Scholar] [CrossRef]
- Makarov, A.A.; Ilinskaya, O.N. Cytotoxic ribonucleases: Molecular weapons and their targets. FEBS Lett. 2003, 540, 15–20. [Google Scholar] [CrossRef]
- Ilinskaya, O.N.; Dreyer, F.; Mitkevich, V.A.; Shaw, K.L.; Pace, C.N.; Makarov, A.A. Changing the net charge from negative to positive makes ribonuclease Sa cytotoxic. Protein Sci. 2002, 11, 2522–2525. [Google Scholar]
- Ilinskaya, O.N.; Zelenikhin, P.V.; Petrushanko, I.Y.; Mitkevich, V.A.; Prassolov, V.S.; Makarov, A.A. Binase induces apoptosis of transformed myeloid cells and does not induce T-cell immune response. Biochem. Biophys. Res. Commun. 2007, 361, 1000–1005. [Google Scholar] [CrossRef]
- Ilinskaya, O.; Decker, K.; Koschinski, A.; Dreyer, F.; Repp, H. Bacillus intermedius ribonuclease as inhibitor of cell proliferation and membrane current. Toxicology 2001, 156, 101–107. [Google Scholar] [CrossRef]
- Zhang, S.; Schlott, B.; Görlach, M.; Grosse, F. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res. 2004, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; De Paola, E.; Paronetto, M.P. The RNA helicase A in malignant transformation. Oncotarget 2016, 7, 28711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baulida, J. Epithelial-to-mesenchymal transition transcription factors in cancer-associated fibroblasts. Mol. Oncol. 2017, 11, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [PubMed]
- Vella, L.J. The emerging role of exosomes in epithelial-mesenchymal-transition in cancer. Front. Oncol. 2014, 4, 361. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Handra-Luca, A.; Hong, S.M.; Walter, K.; Wolfgang, C.; Hruban, R.; Goggins, M. Tumour epithelial vimentin expression and outcome of pancreatic ductal adenocarcinomas. Br. J. Cancer 2011, 104, 1296–1302. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Cells w/o Any Treatment | Cells Treated with RNase A | ||
|---|---|---|---|---|
| Absolute Expression Level, a.u. (2) | Fold Changes in miRNA Level (3) | |||
| B16 | HeLa | B16 | HeLa | |
| mir-21a | 0.7 | 0.9 | n.a. | n.a. |
| mir-145a | 0.8 | 0.95 | n.a. | n.a. |
| mir-31 | 0.75 | 0.86 | n.a. | n.a. |
| mir-10b | 0.8 | 0.78 | 1.1↓–1.5↓ | n.a. |
| let-7g | 0.75 | 0.8 | n.a. | n.a. |
| miR-155 | 0.56 | 1.0 | 1.6–2.0↓↓ | n.a. |
| U6 snRNA (1) | 0.86 | 0.82 | n.a. | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, I.S.E.; Sen’kova, A.V.; Markov, O.V.; Markov, A.V.; Savin, I.A.; Zenkova, M.A.; Mironova, N.L. Bovine Pancreatic RNase A: An Insight into the Mechanism of Antitumor Activity In Vitro and In Vivo. Pharmaceutics 2022, 14, 1173. https://doi.org/10.3390/pharmaceutics14061173
Mohamed ISE, Sen’kova AV, Markov OV, Markov AV, Savin IA, Zenkova MA, Mironova NL. Bovine Pancreatic RNase A: An Insight into the Mechanism of Antitumor Activity In Vitro and In Vivo. Pharmaceutics. 2022; 14(6):1173. https://doi.org/10.3390/pharmaceutics14061173
Chicago/Turabian StyleMohamed, Islam Saber Ead, Aleksandra V. Sen’kova, Oleg V. Markov, Andrey V. Markov, Innokenty A. Savin, Marina A. Zenkova, and Nadezhda L. Mironova. 2022. "Bovine Pancreatic RNase A: An Insight into the Mechanism of Antitumor Activity In Vitro and In Vivo" Pharmaceutics 14, no. 6: 1173. https://doi.org/10.3390/pharmaceutics14061173