
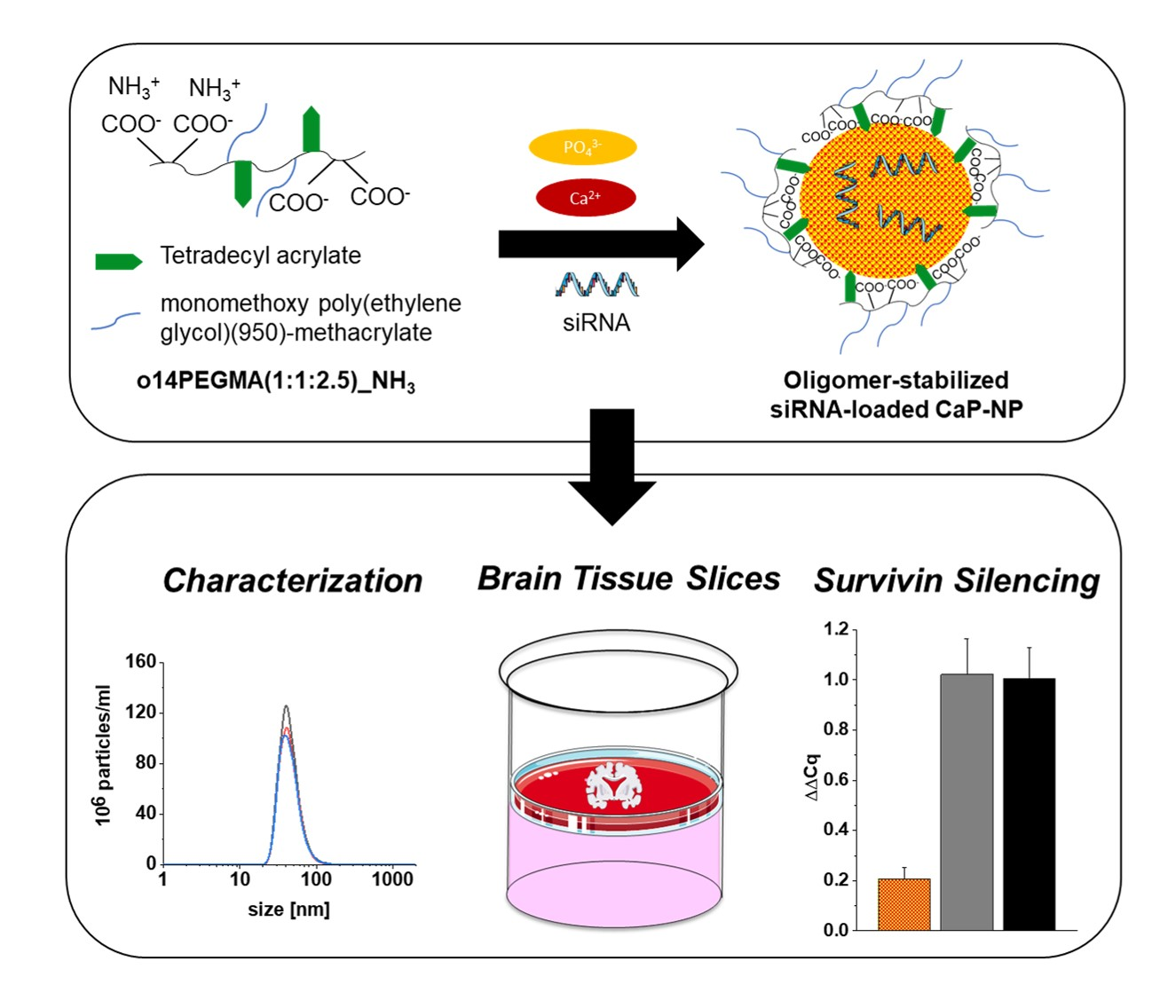
Amphiphilic Anionic Oligomer-Stabilized Calcium Phosphate Nanoparticles with Prospects in siRNA Delivery via Convection-Enhanced Delivery

 , , , , , , , , and
, , , , , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
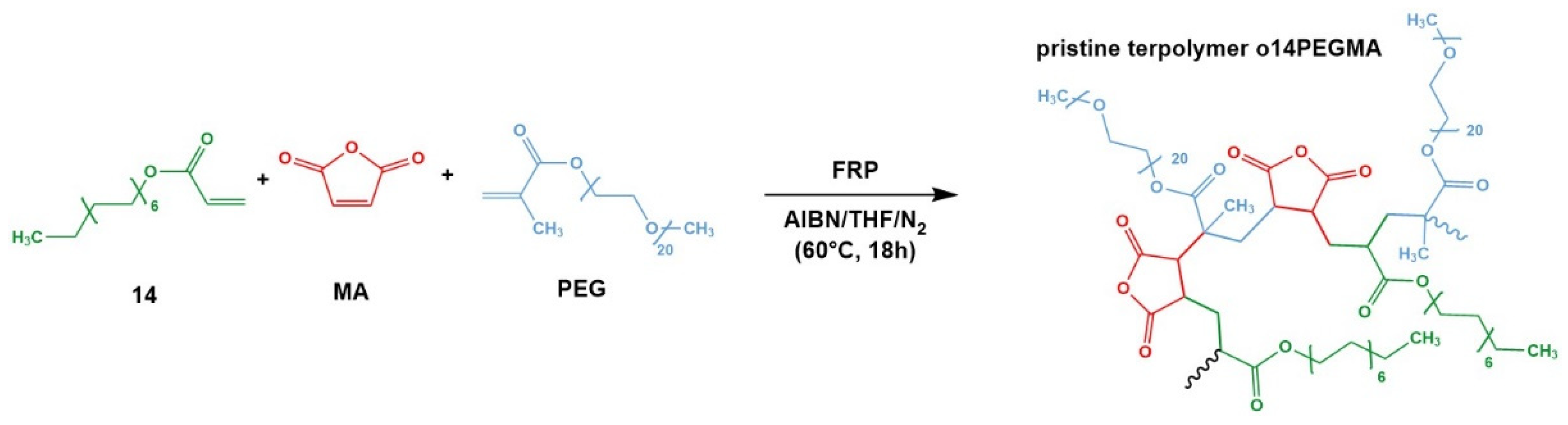
2.2. Synthesis and Characterization of the Amphiphilic Terpolymer o14PEGMA(1:1:2.5)_NH3
2.3. Covalent Derivatization of the Oligomer with Fluorescent Dye Cy5
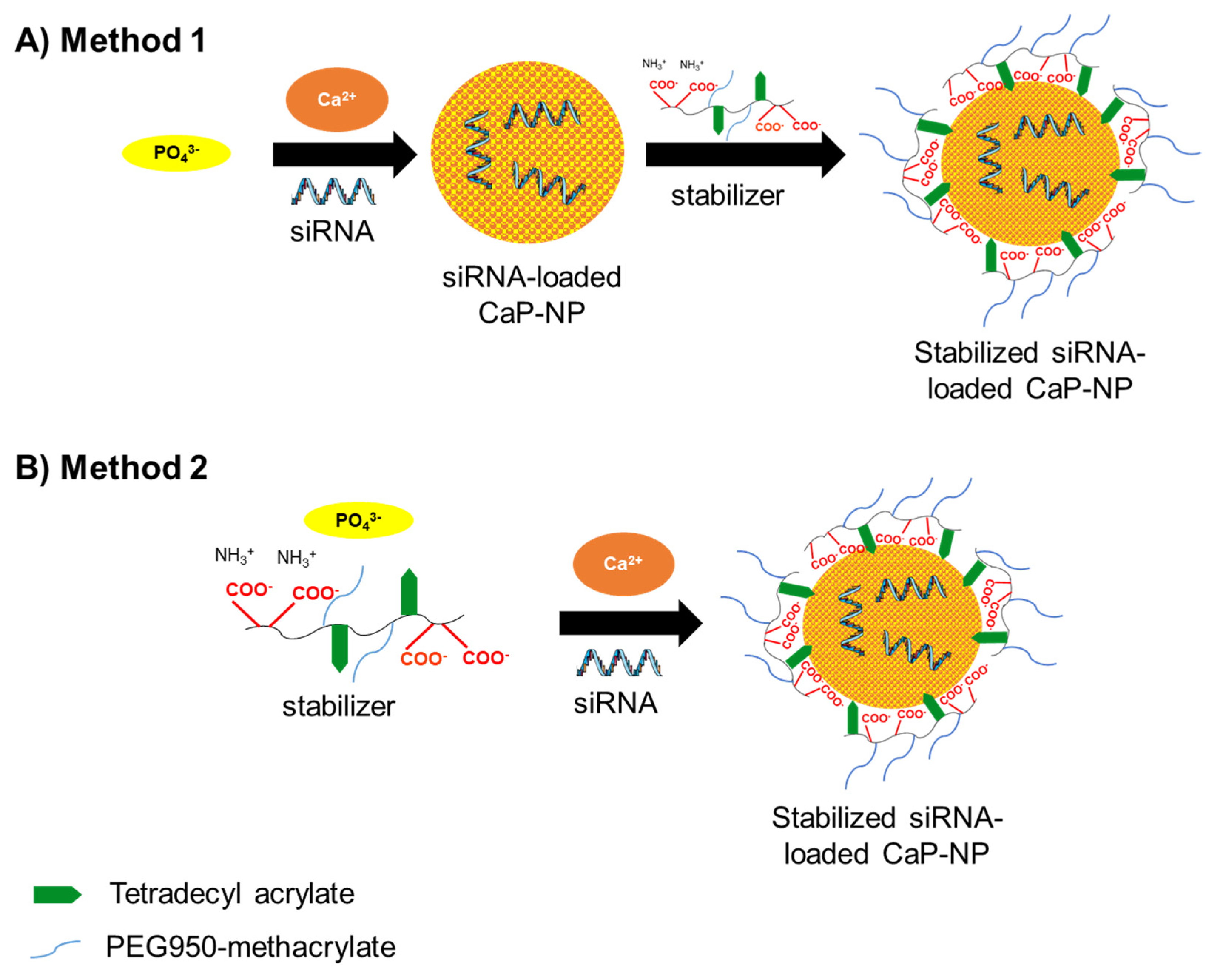
2.4. Preparation of CaP-NPs
Concentration of the CaP-NPs
2.5. Particle Size and Zeta Potential Measurements
2.6. Determination of the siRNA Binding Capacity
2.7. Determination of the Oligomer Binding Capacity
2.8. Cell Culture
2.9. Cellular Uptake of CaP-NPs
2.10. BIRC5 siRNA Transfection Experiments
2.10.1. Determination of the BIRC5 Gene Expression Levels
2.10.2. Determination of the Cell Viability
2.10.3. Caspase Assay and Annexin V Staining
2.11. In Vitro Cytocompatibility Testing
2.11.1. Determination of the Metabolic Activity
2.11.2. Live/Dead Assay
2.12. Tissue Slice Cultures
2.12.1. Preparation and Treatment
2.12.2. Microscopic Analysis
2.13. In Vivo Distribution of the CaP-NPs by CED
2.13.1. CaP-NP Preparation for CED
2.13.2. Convection-Enhanced Delivery
2.13.3. In Vivo Biodistribution
2.14. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterization of the Terpolymer o14PEGMA(1:1:2.5)_NH3
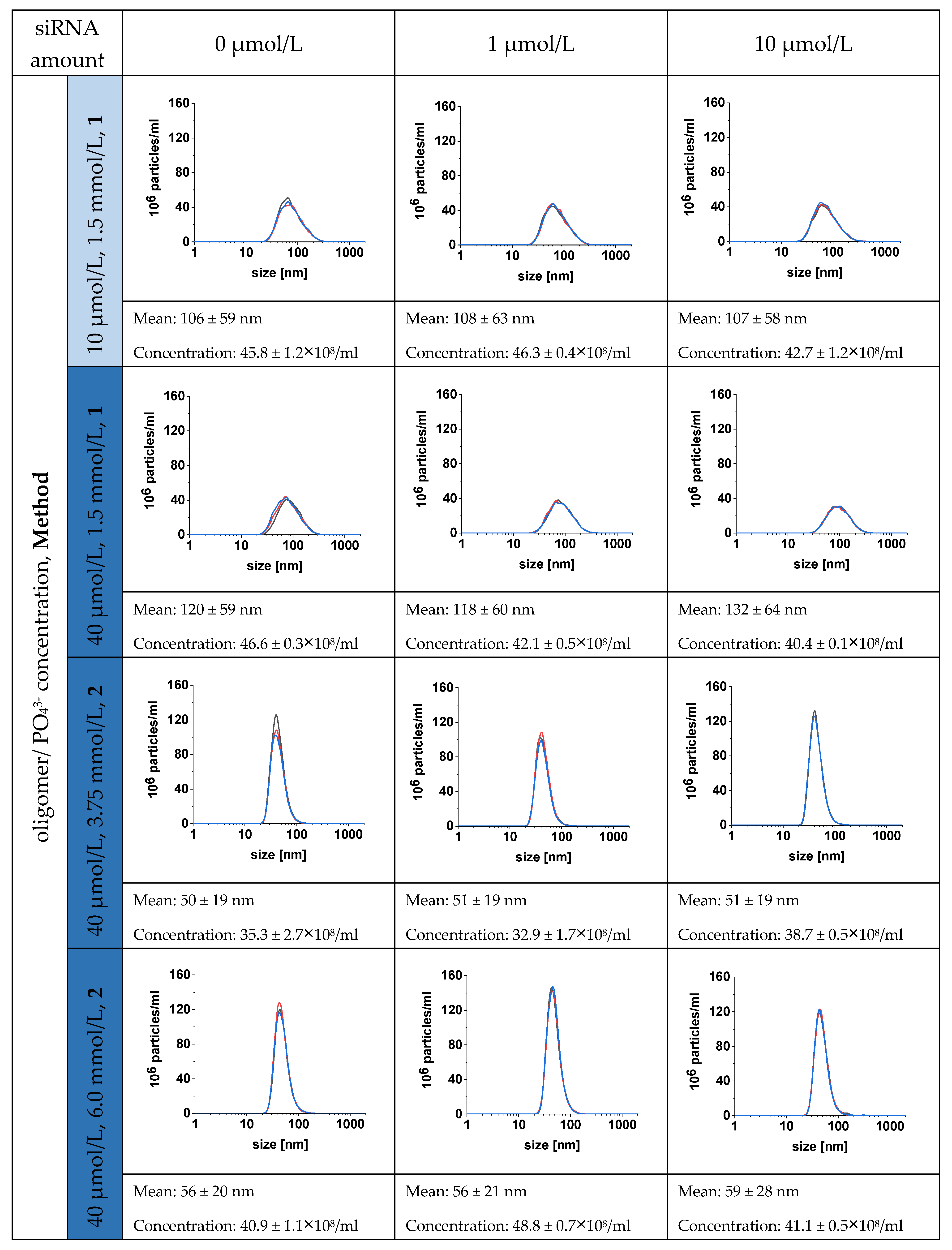
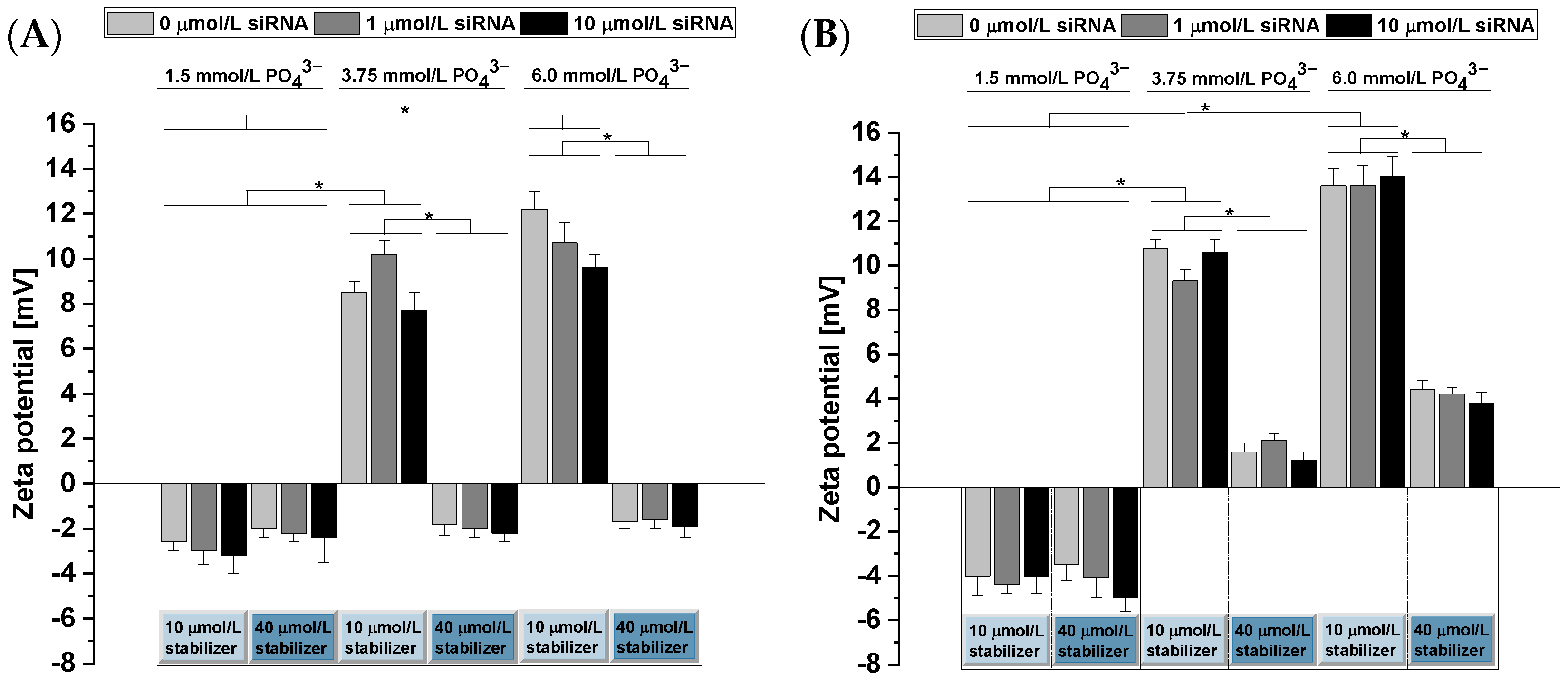
3.2. Characterization of the CaP-NPs
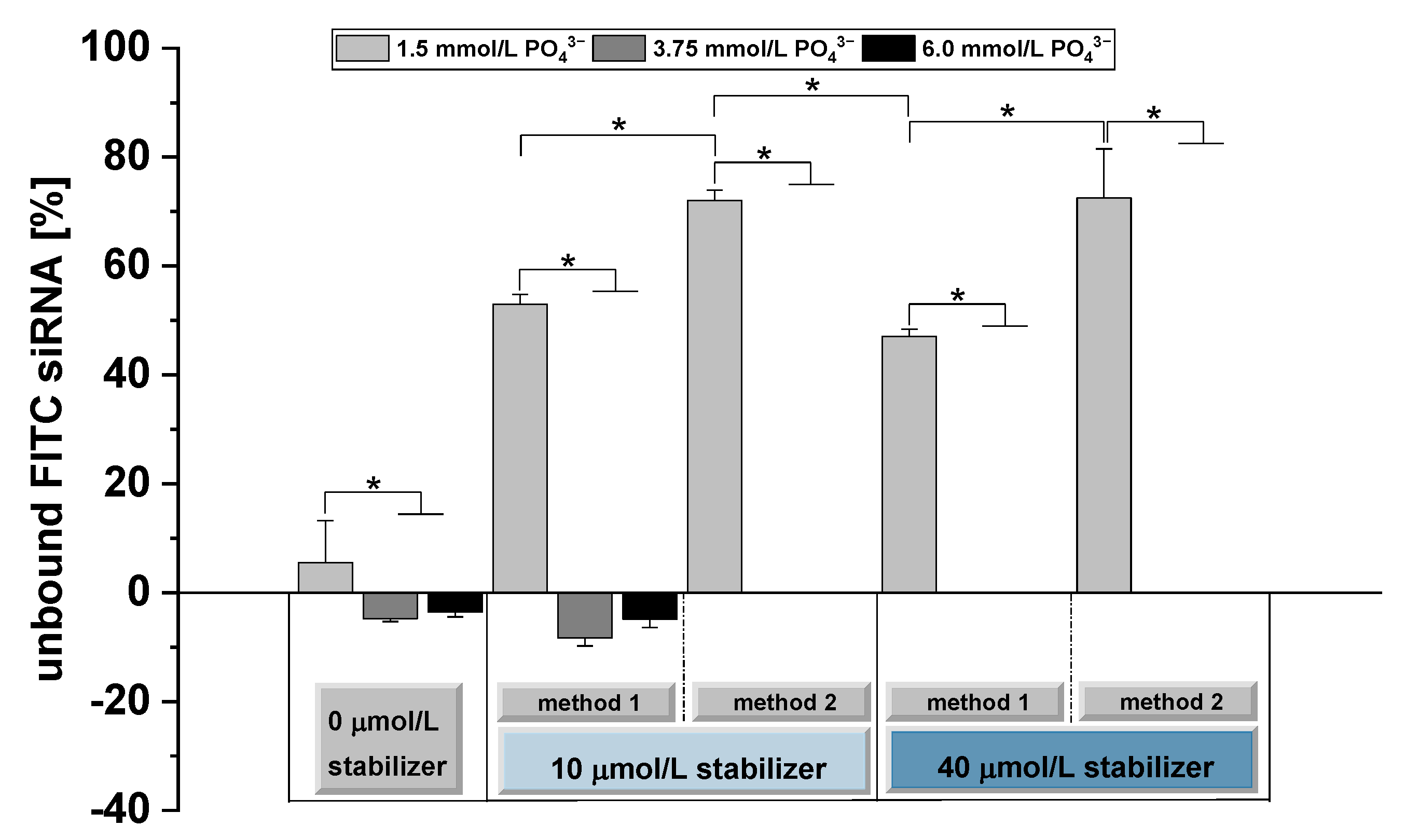
3.3. Determination of the siRNA Binding Capacity
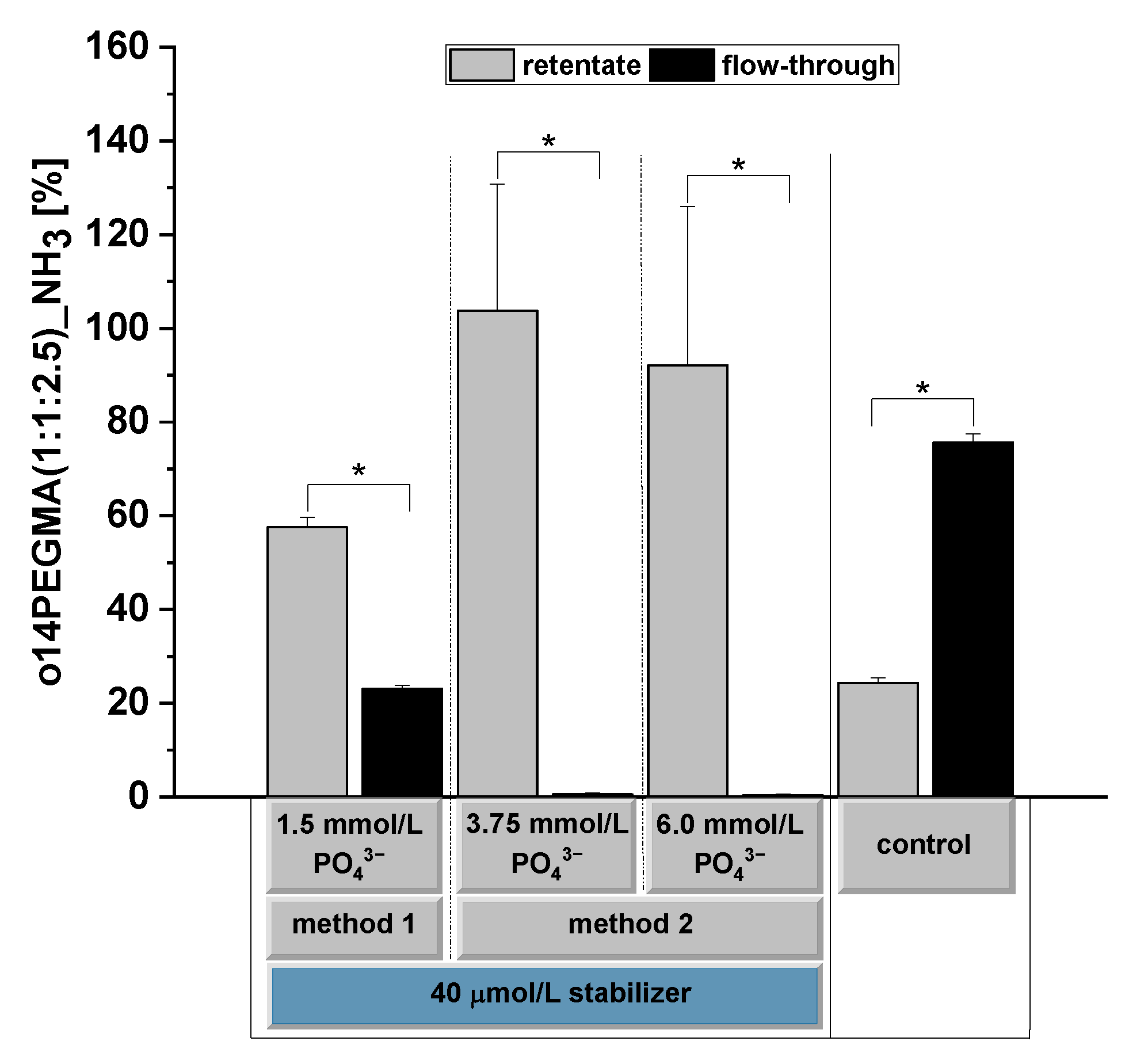
3.4. Determination of the Oligomer Binding Capacity
3.5. Determination of the Serum Stability
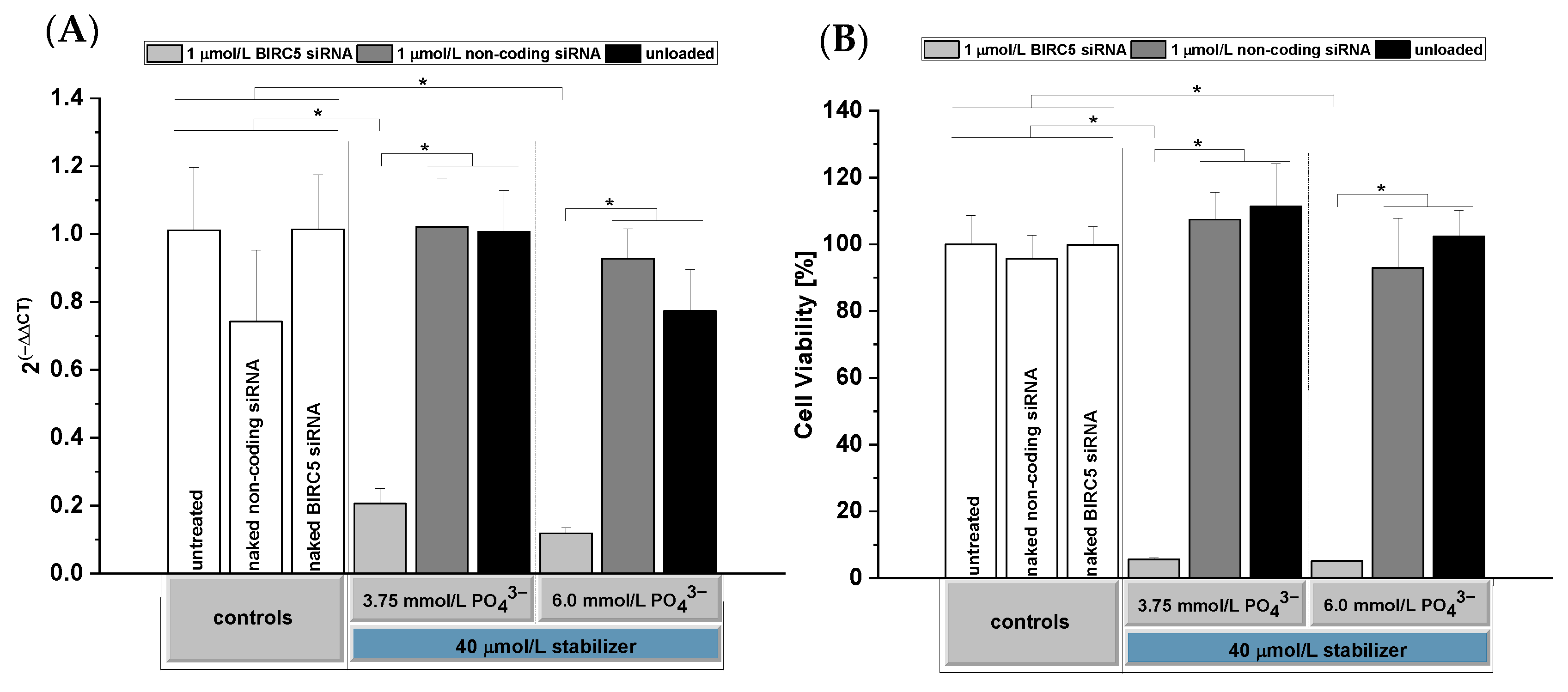
3.6. Determination of In Vitro Silencing
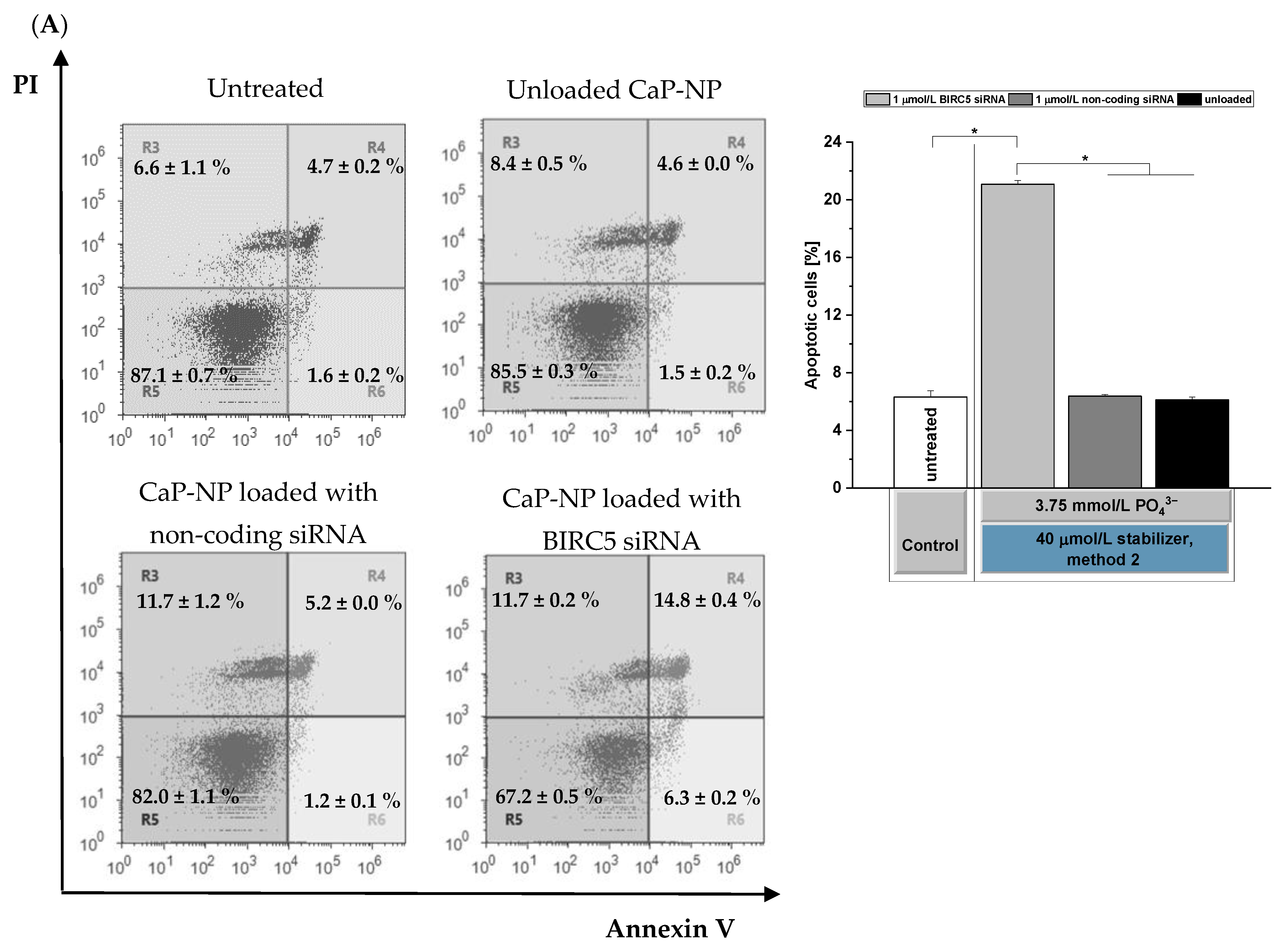
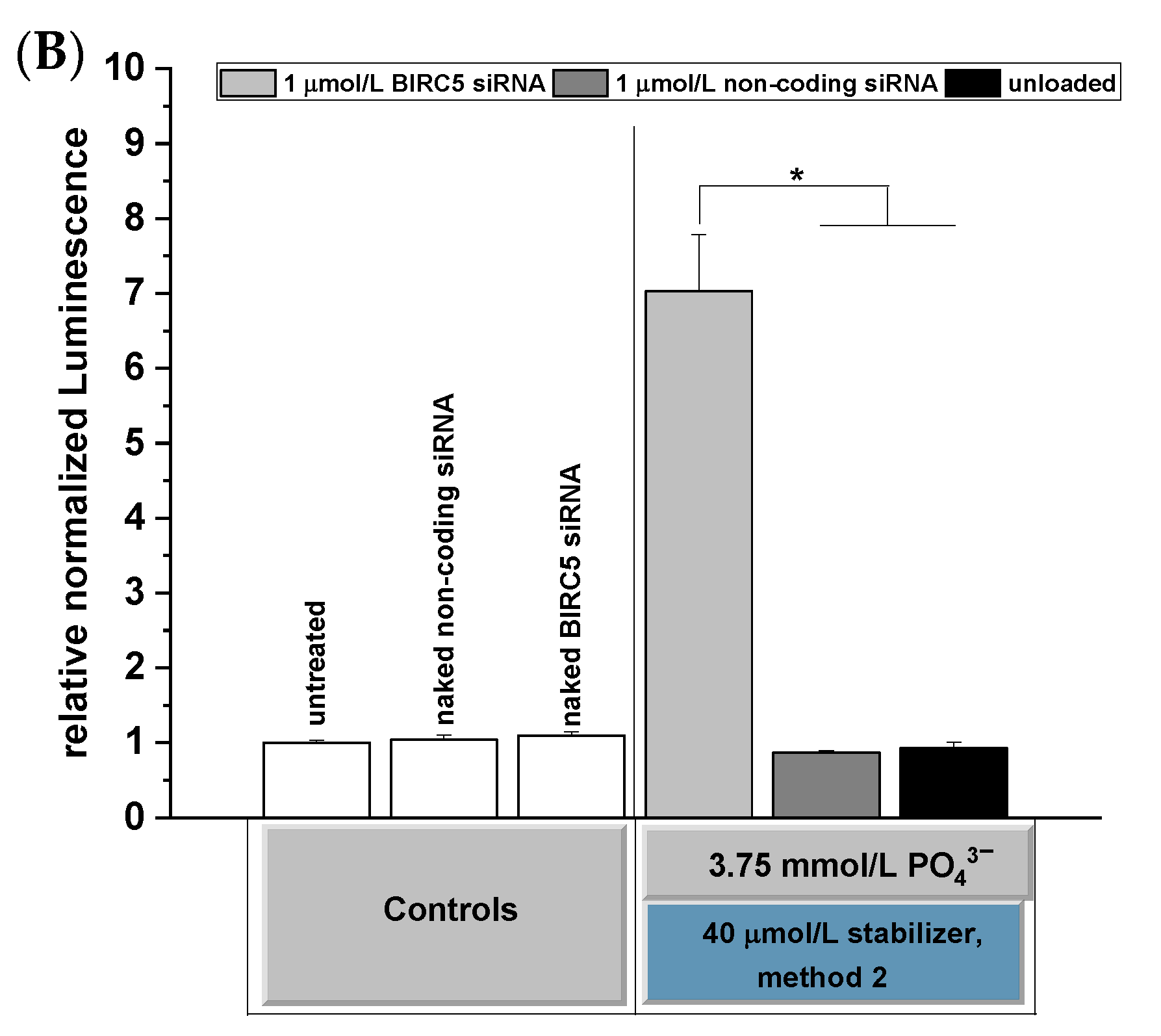
3.7. Effects of BIRC5 Silencing on Apoptosis Induction
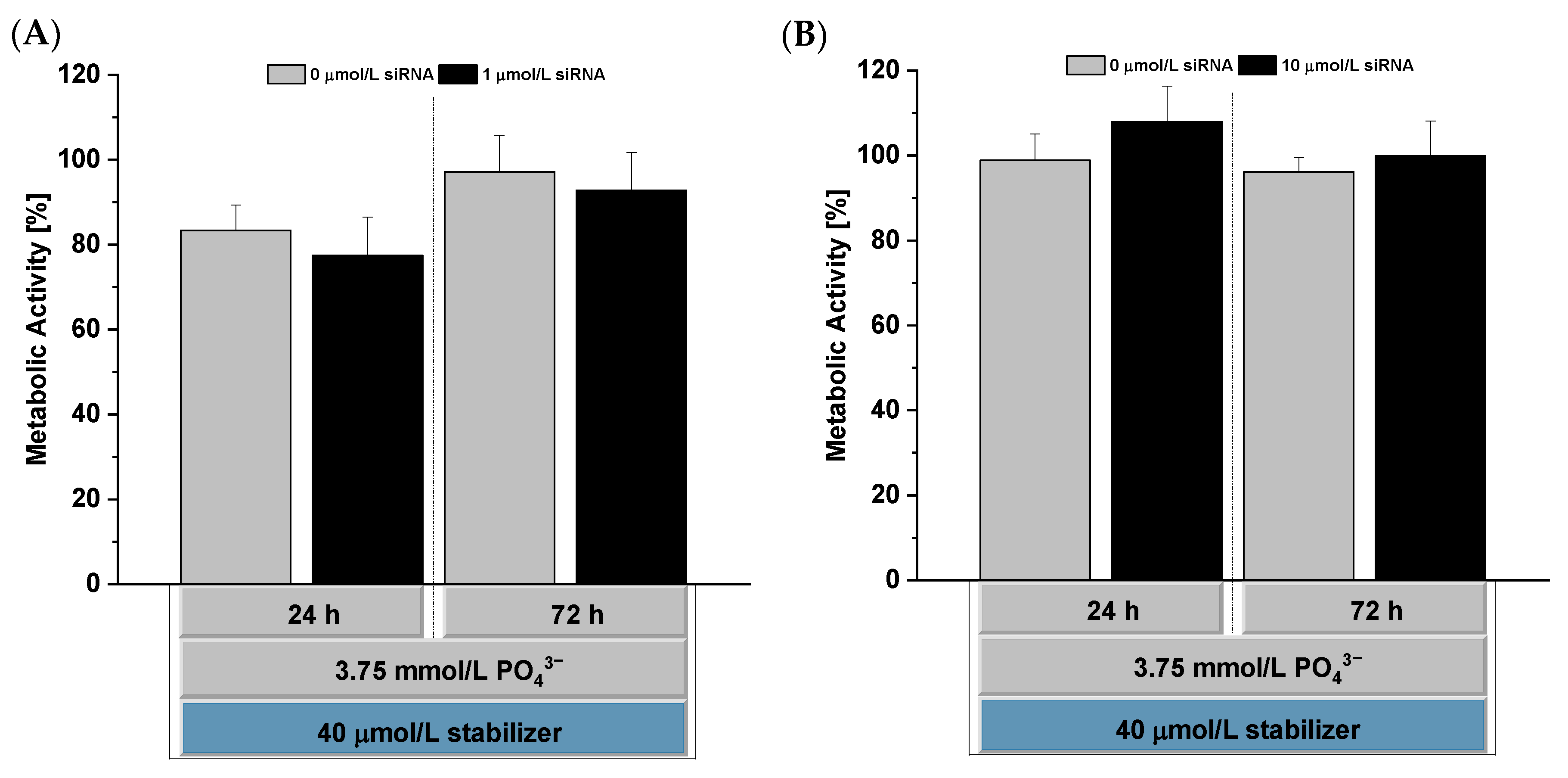
3.8. In Vitro Cytocompatibility Testing
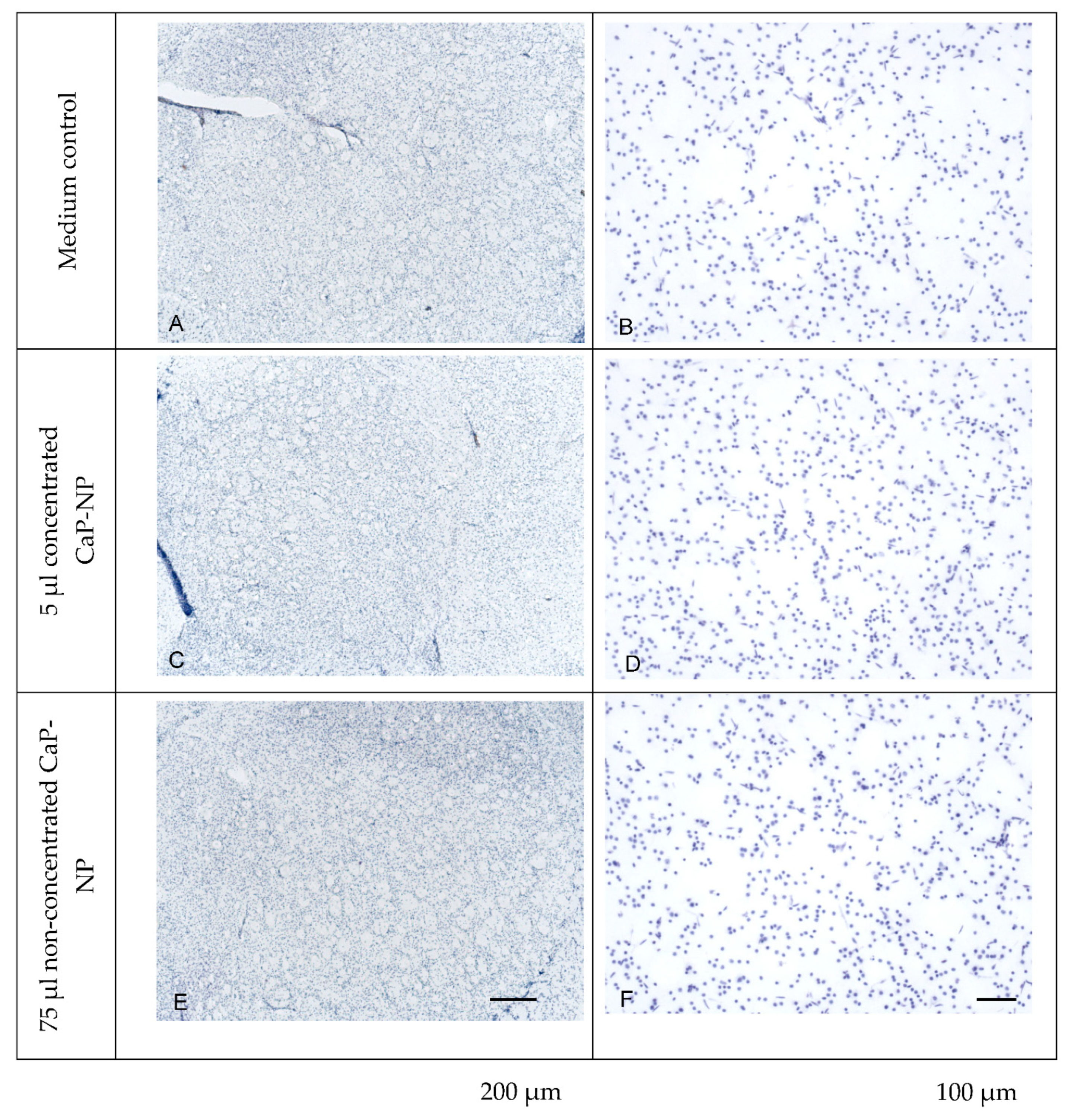
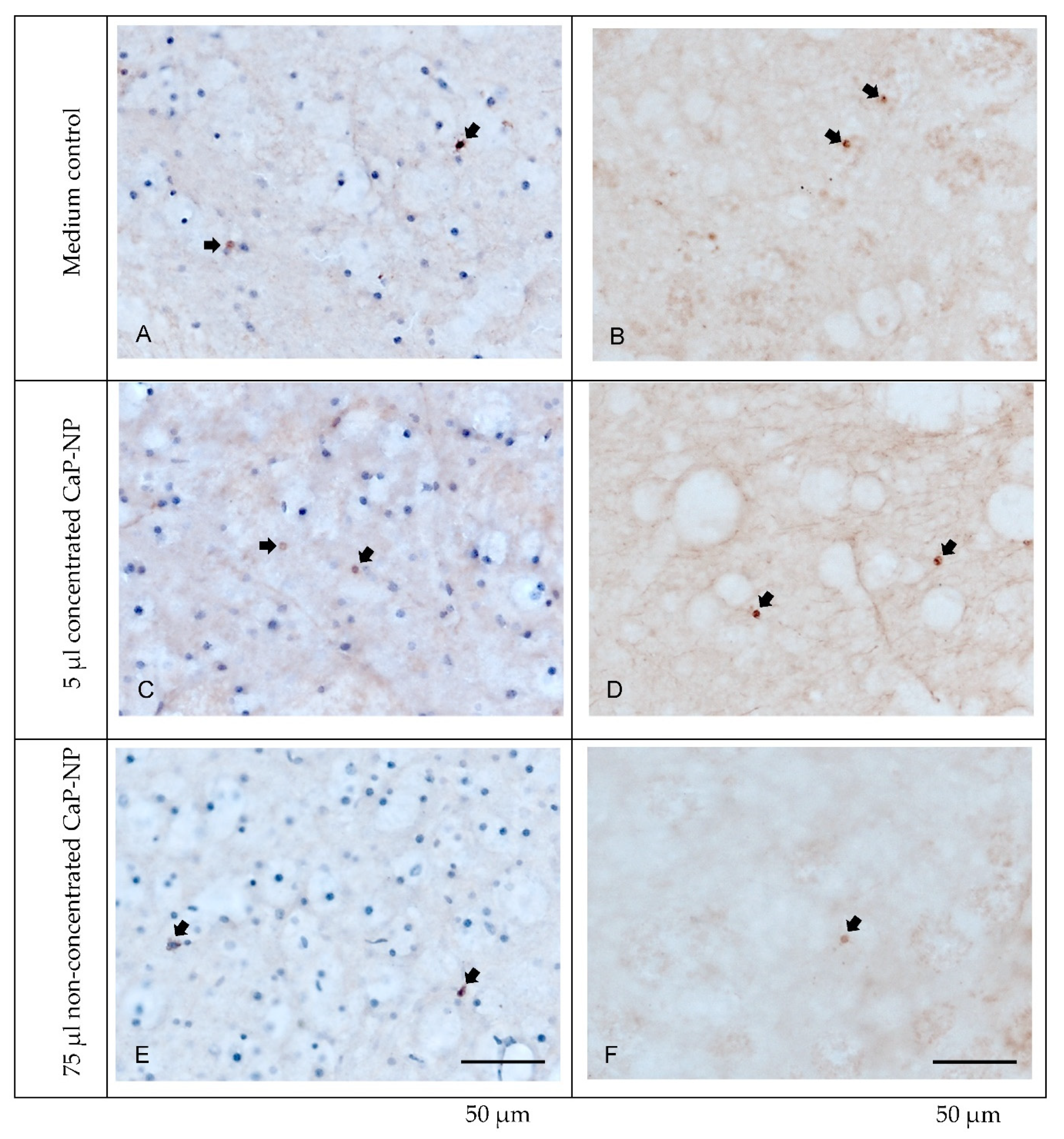
3.9. Absence of Adverse Effects of CaP-NPs in Organotypic Brain Slice Cultures
3.10. Preliminary In Vivo Distribution Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.M.; Sonabend, A.M.; Bruce, J.N. Convection-Enhanced Delivery. Neurotherapeutics 2017, 14, 358–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saucier-Sawyer, J.K.; Seo, Y.-E.; Gaudin, A.; Quijano, E.; Song, E.; Sawyer, A.J.; Deng, Y.; Huttner, A.; Saltzman, W.M. Distribution of polymer nanoparticles by convection-enhanced delivery to brain tumors. J. Control. Release 2016, 232, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendruschk, S.; Wiedemuth, R.; Aigner, A.; Töpfer, K.; Cartellieri, M.; Martin, D.; Kirsch, M.; Ikonomidou, C.; Schackert, G.; Temme, A. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro Oncol. 2011, 13, 1074–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef] [PubMed]
- Miao, G.-Y.; Lu, Q.-M.; Zhang, X.-L. Downregulation of survivin by RNAi inhibits growth of human gastric carcinoma cells. World J. Gastroenterol. 2007, 13, 1170–1174. [Google Scholar] [CrossRef] [Green Version]
- Ohka, F.; Natsume, A.; Wakabayashi, T. Current trends in targeted therapies for glioblastoma multiforme. Neurol. Res. Int. 2012, 2012, 878425. [Google Scholar] [CrossRef]
- Montazeri Aliabadi, H.; Landry, B.; Mahdipoor, P.; Uludağ, H. Induction of apoptosis by survivin silencing through siRNA delivery in a human breast cancer cell line. Mol. Pharm. 2011, 8, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, R.A.; Stamm, N.B.; Marcusson, E.; Sandusky, G.; Iversen, P.; Patel, B.K.R. Antisense inhibition of survivin expression as a cancer therapeutic. Mol. Cancer 2011, 10, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liang, H.; Cao, W.; Xu, R.; Ju, X.L. Downregulation of survivin by siRNA inhibits invasion and promotes apoptosis in neuroblastoma SH-SY5Y cells. Braz. J. Med. Biol. Res. 2014, 47, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupenthal, J.; Baehr, C.; Kiermayer, S.; Zeuzem, S.; Piiper, A. Inhibition of RNAse A family enzymes prevents degradation and loss of silencing activity of siRNAs in serum. Biochem. Pharmacol. 2006, 71, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Tatiparti, K.; Sau, S.; Kashaw, S.K.; Iyer, A.K. siRNA Delivery Strategies: A Comprehensive Review of Recent Developments. Nanomaterials 2017, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L.-S. Delivery of siRNA therapeutics: Barriers and carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V.; Epple, M. Biological and Medical Significance of Calcium Phosphates. Angew. Chem. Int. Ed. 2002, 41, 3130–3146. [Google Scholar] [CrossRef]
- Graham, F.L.; van der Eb, A.J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973, 52, 456–467. [Google Scholar] [CrossRef]
- C Joshi, J.; Bhardwaj, A.; Roy, I.; Gulati, K.; Ray, A. Experimental Studies on the Systemic Toxicity and Biodistribution of Synthesized Calcium Phosphate Nanoparticles after Oral Administration in Rats. Pharm. Nanotechnol. 2016, 4, 202–212. [Google Scholar] [CrossRef]
- Neuhaus, B.; Tosun, B.; Rotan, O.; Frede, A.; Westendorf, A.M.; Epple, M. Nanoparticles as transfection agents: A comprehensive study with ten different cell lines. RSC Adv. 2016, 6, 18102–18112. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Jin, W.; Wang, Y.-N.; Pan, H.; Sun, Z.; Tang, R. Effect of the aggregation state of amorphous calcium phosphate on hydroxyapatite nucleation kinetics. RSC Adv. 2017, 7, 25497–25503. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.; Schallhorn, A.; Wurm, F.M. Transfecting mammalian cells: Optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 1996, 24, 596–601. [Google Scholar] [CrossRef]
- Bisso, S.; Mura, S.; Castagner, B.; Couvreur, P.; Leroux, J.-C. Dual delivery of nucleic acids and PEGylated-bisphosphonates via calcium phosphate nanoparticles. Eur. J. Pharm. Biopharm. 2019, 142, 142–152. [Google Scholar] [CrossRef]
- Giger, E.V.; Castagner, B.; Räikkönen, J.; Mönkkönen, J.; Leroux, J.-C. siRNA transfection with calcium phosphate nanoparticles stabilized with PEGylated chelators. Adv. Healthc. Mater. 2013, 2, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Andina, D.; Ge, J.; Labarre, A.; Leroux, J.-C.; Castagner, B. Characterization of Calcium Phosphate Nanoparticles Based on a PEGylated Chelator for Gene Delivery. ACS Appl. Mater. Interfaces 2017, 9, 10435–10445. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, Y.; Furukawa, S.; Kataoka, K. Block copolymer-coated calcium phosphate nanoparticles sensing intracellular environment for oligodeoxynucleotide and siRNA delivery. J. Control. Release 2004, 97, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, Y.; Furukawa, S.; Ishii, A.; Kataoka, K. Organic-inorganic hybrid-nanocarrier of siRNA constructing through the self-assembly of calcium phosphate and PEG-based block aniomer. J. Control. Release 2006, 111, 368–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.-W.; Zhou, L.-Z.; Sun, Y.; Wu, G.-J.; Gu, H.-C.; Duan, Y.-R.; Chen, F.; Zhu, Y.-J. Calcium phosphate/PLGA-mPEG hybrid porous nanospheres: A promising vector with ultrahigh gene loading and transfection efficiency. J. Mater. Chem. 2010, 20, 1161–1166. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, Y.; Xiang, J.; Piao, Y.; Zhou, Z.; Tang, J.; Liu, X.; Shen, Y. Stabilized calcium phosphate hybrid nanocomposite using a benzoxaborole-containing polymer for pH-responsive siRNA delivery. Biomater. Sci. 2018, 6, 3178–3188. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Huang, L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J. Control. Release 2012, 158, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Li, J.; Liu, F.; Huang, L. Systemic delivery of siRNA via LCP nanoparticle efficiently inhibits lung metastasis. Mol. Ther. 2012, 20, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Peng, L.; Mumper, R.J.; Huang, L. Combinational delivery of c-myc siRNA and nucleoside analogs in a single, synthetic nanocarrier for targeted cancer therapy. Biomaterials 2013, 34, 8459–8468. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Yu, B.; Yang, X.; Huo, T.; Lee, L.J.; Barth, R.F.; Lee, R.J. Lipid-coated nano-calcium-phosphate (LNCP) for gene delivery. Int. J. Pharm. 2010, 392, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Devarasu, T.; Saad, R.; Ouadi, A.; Frisch, B.; Robinet, E.; Laquerrière, P.; Voegel, J.-C.; Baumert, T.; Ogier, J.; Meyer, F. Potent calcium phosphate nanoparticle surface coating for in vitro and in vivo siRNA delivery: A step toward multifunctional nanovectors. J. Mater. Chem. B 2013, 1, 4692–4700. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Neumann, S.; Kovtun, A.; Chernousova, S.; Heumann, R.; Epple, M. An outer shell of positively charged poly(ethyleneimine) strongly increases the transfection efficiency of calcium phosphate/DNA nanoparticles. J. Mater. Sci. 2010, 45, 4952–4957. [Google Scholar] [CrossRef]
- Xu, X.-L.; Yang, H.-Y.; Ou, B.; Lin, S.-D.; Wu, H.; He, W.; Jiang, Q.-C.; Luo, B.-M.; Li, G.-P. Hydroxyapatite nanoparticles modified by branched polyethylenimine are effective non-viral vectors for siRNA transfection of hepatoma cells in vitro. Int. J. Oncol. 2015, 46, 2138–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunniffe, G.M.; O’Brien, F.J.; Partap, S.; Levingstone, T.J.; Stanton, K.T.; Dickson, G.R. The synthesis and characterization of nanophase hydroxyapatite using a novel dispersant-aided precipitation method. J. Biomed. Mater. Res. A 2010, 95, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, G.; Molino, G.; Fiorilli, S.; Vitale-Brovarone, C. Synthesis and incorporation of rod-like nano-hydroxyapatite into type I collagen matrix: A hybrid formulation for 3D printing of bone scaffolds. J. Eur. Ceram. Soc. 2020, 40, 3689–3697. [Google Scholar] [CrossRef]
- Mencía Castaño, I.; Curtin, C.M.; Duffy, G.P.; O’Brien, F.J. Next generation bone tissue engineering: Non-viral miR-133a inhibition using collagen-nanohydroxyapatite scaffolds rapidly enhances osteogenesis. Sci. Rep. 2016, 6, 27941. [Google Scholar] [CrossRef] [PubMed]
- Gillich, T.; Acikgöz, C.; Isa, L.; Schlüter, A.D.; Spencer, N.D.; Textor, M. PEG-stabilized core-shell nanoparticles: Impact of linear versus dendritic polymer shell architecture on colloidal properties and the reversibility of temperature-induced aggregation. ACS Nano 2013, 7, 316–329. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Quinsaat, J.E.Q.; Ono, T.; Maeki, M.; Tokeshi, M.; Isono, T.; Tajima, K.; Satoh, T.; Sato, S.-I.; Miura, Y.; et al. Enhanced dispersion stability of gold nanoparticles by the physisorption of cyclic poly(ethylene glycol). Nat. Commun. 2020, 11, 6089. [Google Scholar] [CrossRef]
- ISO 10993-5:2009(en), Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity. Available online: https://www.iso.org/obp/ui#iso:std:iso:10993:-5:ed-3:v1:en (accessed on 16 December 2021).
- Kascholke, C.; Loth, T.; Kohn-Polster, C.; Möller, S.; Bellstedt, P.; Schulz-Siegmund, M.; Schnabelrauch, M.; Hacker, M.C. Dual-Functional Hydrazide-Reactive and Anhydride-Containing Oligomeric Hydrogel Building Blocks. Biomacromolecules 2017, 18, 683–694. [Google Scholar] [CrossRef]
- Loth, T.; Hennig, R.; Kascholke, C.; Hötzel, R.; Hacker, M.C. Reactive and stimuli-responsive maleic anhydride containing macromers—multi-functional cross-linkers and building blocks for hydrogel fabrication. React. Funct. Polym. 2013, 73, 1480–1492. [Google Scholar] [CrossRef]
- Nawaz, H.A.; Schröck, K.; Schmid, M.; Krieghoff, J.; Maqsood, I.; Kascholke, C.; Kohn-Polster, C.; Schulz-Siegmund, M.; Hacker, M.C. Injectable oligomer-cross-linked gelatine hydrogels via anhydride-amine-conjugation. J. Mater. Chem. B 2021, 9, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Wölk, C.; Nawaz, H.A.; Maqsood, I.; Strati, F.; Brezesinski, G.; Hause, G.; Schulz-Siegmund, M.; Hacker, M.C. Amphiphilic Functionalized Oligomers: A Promising Strategy for the Postfabrication Functionalization of Liposomes. Adv. Mater. Interfaces 2020, 7, 2001168. [Google Scholar] [CrossRef]
- US2725367A—Salts of Terpolymers of Maleic Anhydride, Diisobutylene and Divinylbenzene—Google Patents. Available online: https://currentprotocols.onlinelibrary.wiley.com/doi/10.1002/0471140864.ps0404s38/otherversions (accessed on 13 October 2021).
- Bortel, E.; Stysło, M. On the chemical modifications of poly(maleic anhydride-co-isobutene) by means of hydrolysis, ammoniation or aminations. Makromol. Chem. 1990, 191, 2653–2662. [Google Scholar] [CrossRef]
- Phillips, A.T.; Signs, M.W. Desalting, concentration, and buffer exchange by dialysis and ultrafiltration. Curr. Protoc. Protein Sci. 2005, 38, 4. [Google Scholar] [CrossRef]
- Heine, C.; Franke, H. Organotypic Slice Co-culture Systems to Study Axon Regeneration in the Dopaminergic System Ex Vivo. In Axon Growth and Regeneration: Methods and Protocols; Murray, A.J., Ed.; Humana Press: New York, NY, USA, 2014; pp. 97–111. ISBN 978-1-4939-0777-9. [Google Scholar]
- Sidorcenco, V.; Krahnen, L.; Schulz, M.; Remy, J.; Kögel, D.; Temme, A.; Krügel, U.; Franke, H.; Aigner, A. Glioblastoma Tissue Slice Tandem-Cultures for Quantitative Evaluation of Inhibitory Effects on Invasion and Growth. Cancers 2020, 12, 2707. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, F.; Zou, X.; Torbey, M. Comparison of unbiased estimation of neuronal number in the rat hippocampus with different staining methods. J. Neurosci. Methods 2015, 254, 73–79. [Google Scholar] [CrossRef]
- Pittella, F.; Miyata, K.; Maeda, Y.; Suma, T.; Watanabe, S.; Chen, Q.; Christie, R.J.; Osada, K.; Nishiyama, N.; Kataoka, K. Pancreatic cancer therapy by systemic administration of VEGF siRNA contained in calcium phosphate/charge-conversional polymer hybrid nanoparticles. J. Control. Release 2012, 161, 868–874. [Google Scholar] [CrossRef]
- Sokolova, V.; Prymak, O.; Meyer-Zaika, W.; Cölfen, H.; Rehage, H.; Shukla, A.; Epple, M. Synthesis and characterization of DNA-functionalized calcium phosphate nanoparticles. Mater. Werkst. 2006, 37, 441–445. [Google Scholar] [CrossRef]
- Tobin, L.A.; Xie, Y.; Tsokos, M.; Chung, S.I.; Merz, A.A.; Arnold, M.A.; Li, G.; Malech, H.L.; Kwong, K.F. Pegylated siRNA-loaded calcium phosphate nanoparticle-driven amplification of cancer cell internalization in vivo. Biomaterials 2013, 34, 2980–2990. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Ishii, A.; Nishiyama, N.; Matsumoto, S.; Ishii, T.; Yamasaki, Y.; Kataoka, K. PEGylated Calcium Phosphate Nanocomposites as Smart Environment-Sensitive Carriers for siRNA Delivery. Adv. Mater. 2009, 21, 3520–3525. [Google Scholar] [CrossRef]
- Tang, J.; Li, L.; Howard, C.B.; Mahler, S.M.; Huang, L.; Xu, Z.P. Preparation of optimized lipid-coated calcium phosphate nanoparticles for enhanced in vitro gene delivery to breast cancer cells. J. Mater. Chem. B 2015, 3, 6805–6812. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Lee, J.E.; Byun, E.; Kim, N.W.; Lee, K.; Lee, H.; Sim, S.J.; Lee, D.S.; Jeong, J.H. Target-specific delivery of siRNA by stabilized calcium phosphate nanoparticles using dopa-hyaluronic acid conjugate. J. Control. Release 2014, 192, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Pittella, F.; Zhang, M.; Lee, Y.; Kim, H.J.; Tockary, T.; Osada, K.; Ishii, T.; Miyata, K.; Nishiyama, N.; Kataoka, K. Enhanced endosomal escape of siRNA-incorporating hybrid nanoparticles from calcium phosphate and PEG-block charge-conversional polymer for efficient gene knockdown with negligible cytotoxicity. Biomaterials 2011, 32, 3106–3114. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.-C.; Tseng, Y.-C.; Mozumdar, S.; Huang, L. Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J. Control. Release 2010, 142, 416–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauchle, E.; Thude, S.; Brucker, S.Y.; Schenke-Layland, K. Cell death stages in single apoptotic and necrotic cells monitored by Raman microspectroscopy. Sci. Rep. 2014, 4, 4698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logue, S.E.; Elgendy, M.; Martin, S.J. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat. Protoc. 2009, 4, 1383–1395. [Google Scholar] [CrossRef]
- Stepanek, O.; Brdicka, T.; Angelisova, P.; Horvath, O.; Spicka, J.; Stockbauer, P.; Man, P.; Horejsi, V. Interaction of late apoptotic and necrotic cells with vitronectin. PLoS ONE 2011, 6, e19243. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stock Solution (Used for CaP-NP Preparation) | Final Concentration (in Formulation) | |
|---|---|---|
| o14PEGMA(1:1:2.5)_NH3 | 2 µmol/L | 0.7 µmol/L |
| 5 µmol/L | 1.7 µmol/L | |
| 10 µmol/L | 3.3 µmol/L | |
| 40 µmol/L | 13.3 µmol/L |
| Stock Solution (Used for Loading of CaP-NPs) | Final Amount/Well | ||
|---|---|---|---|
| non-concentrated (48-well) | BIRC5 siRNA | 20 µmol/L | 1.0 µg |
| AllStars Negative Control siRNA | 20 µmol/L | 1.0 µg | |
| AllStars Negative Control siRNA AF647 | 20 µmol/L | 1.0 µg | |
| 200 µmol/L | 10.0 µg | ||
| Concentrated (96-well) | BIRC5 siRNA | 200 µmol/L | 10.0 µg |
| AllStars Negative Control siRNA | 200 µmol/L | 10.0 µg | |
| AllStars Negative Control siRNA AF647 | 200 µmol/L | 10.0 µg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitrach, F.; Schmid, M.; Toussaint, M.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Franke, H.; Ewe, A.; Aigner, A.; Wölk, C.; Brust, P.; et al. Amphiphilic Anionic Oligomer-Stabilized Calcium Phosphate Nanoparticles with Prospects in siRNA Delivery via Convection-Enhanced Delivery. Pharmaceutics 2022, 14, 326. https://doi.org/10.3390/pharmaceutics14020326
Mitrach F, Schmid M, Toussaint M, Dukic-Stefanovic S, Deuther-Conrad W, Franke H, Ewe A, Aigner A, Wölk C, Brust P, et al. Amphiphilic Anionic Oligomer-Stabilized Calcium Phosphate Nanoparticles with Prospects in siRNA Delivery via Convection-Enhanced Delivery. Pharmaceutics. 2022; 14(2):326. https://doi.org/10.3390/pharmaceutics14020326
Chicago/Turabian StyleMitrach, Franziska, Maximilian Schmid, Magali Toussaint, Sladjana Dukic-Stefanovic, Winnie Deuther-Conrad, Heike Franke, Alexander Ewe, Achim Aigner, Christian Wölk, Peter Brust, and et al. 2022. "Amphiphilic Anionic Oligomer-Stabilized Calcium Phosphate Nanoparticles with Prospects in siRNA Delivery via Convection-Enhanced Delivery" Pharmaceutics 14, no. 2: 326. https://doi.org/10.3390/pharmaceutics14020326