TMT-Based Quantitative Proteomics Analysis Reveals Airborne PM2.5-Induced Pulmonary Fibrosis

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals and Whole-Body Inhalation
2.3. PM2.5 Sampling and Physical and Chemical Characterization
2.4. Lung Preparation and Histopathological Examination
2.5. Protein Extraction and Digestion
2.6. Tandem Mass Tags Labeling and High-Performance Liquid Chromatography Fractionation
2.7. Reversed-Phase Liquid Chromatography–Tandem Mass Spectrometry
2.8. Data Processing and Database Searching
2.9. Data Normalization and Protein Network Analysis
2.10. Quantitative Real-Time Polymerase Chain Reaction Analyses
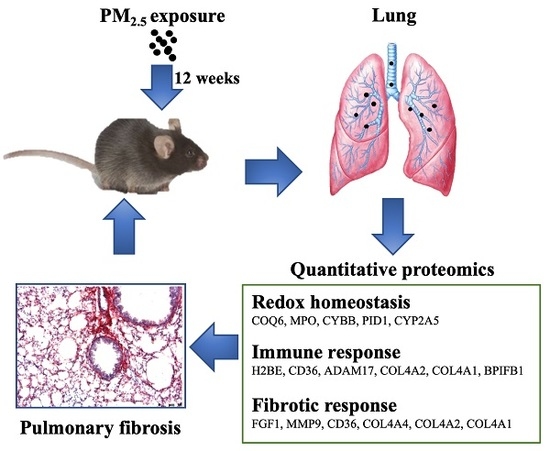
3. Results
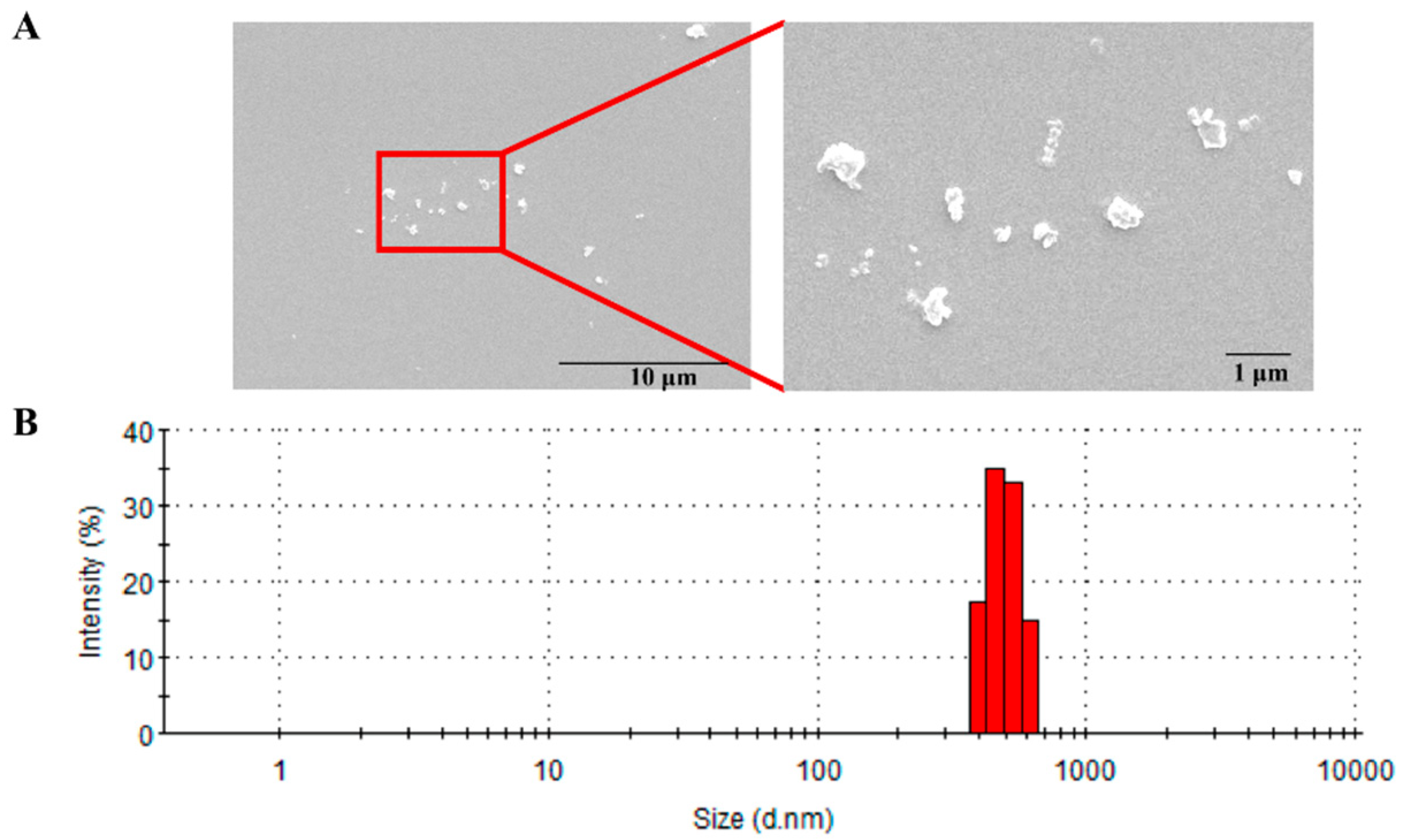
3.1. Physiochemical Characterization of PM2.5
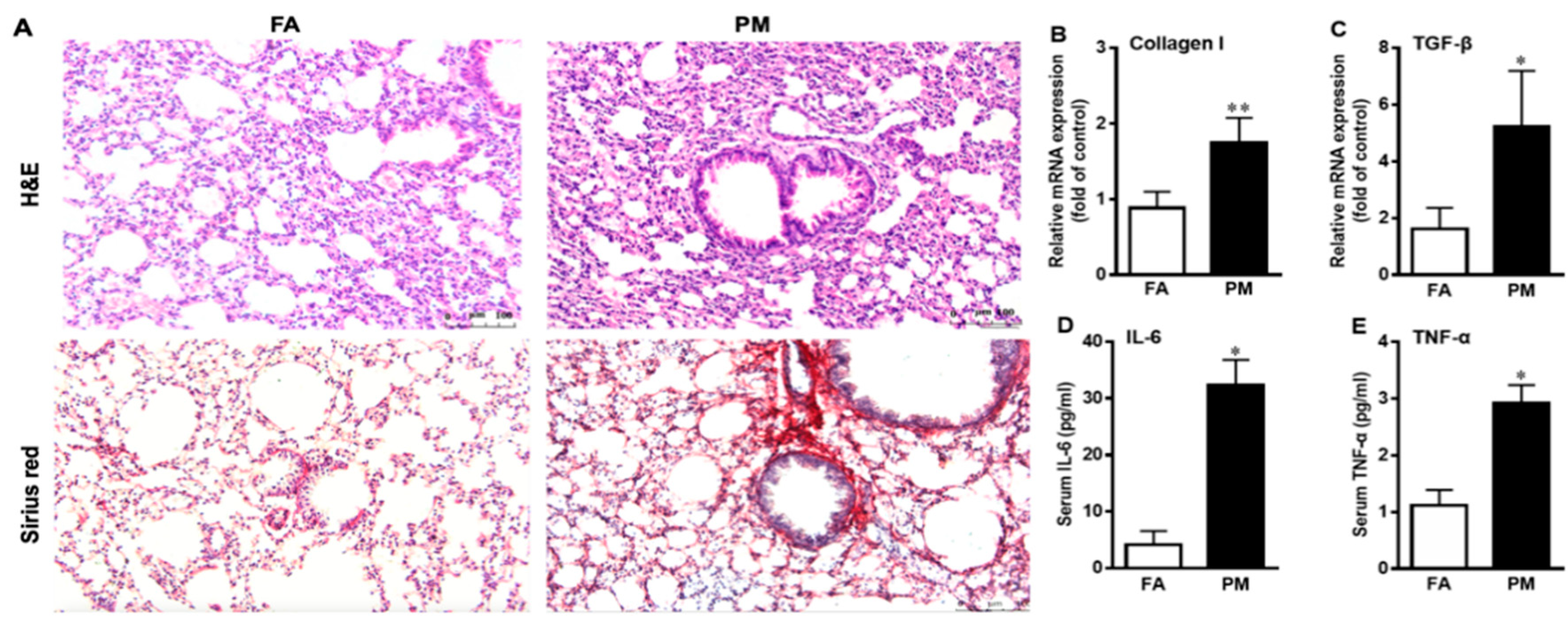
3.2. PM2.5 Exposure Induces Pulmonary Inflammation and Fibrosis
3.3. Quantitative Proteomics Analyses to Reveal PM2.5-Regulated Pulmonary Proteins
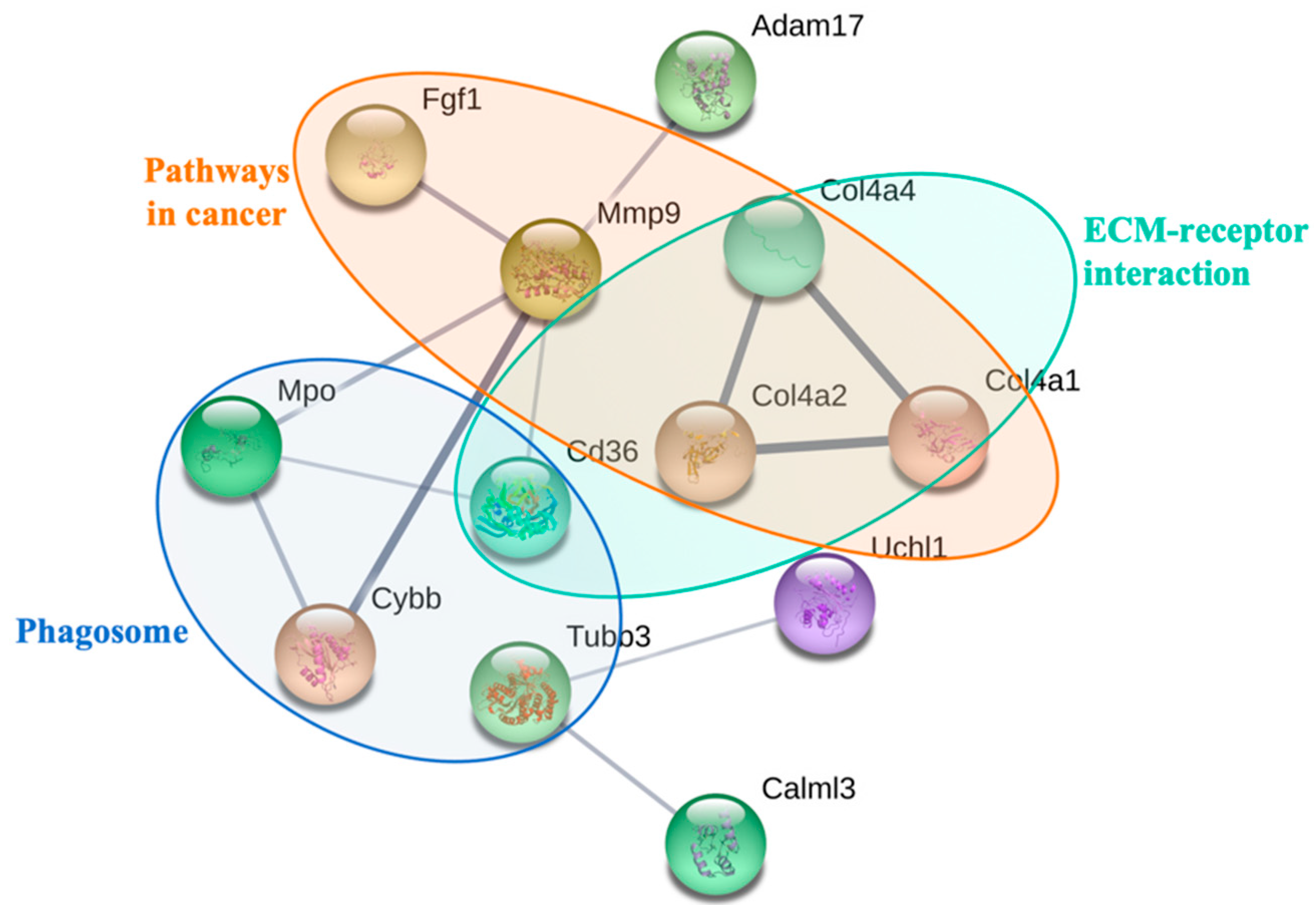
3.4. Biological Interaction of Differentially Expressed Proteins in Response to PM2.5 Exposure
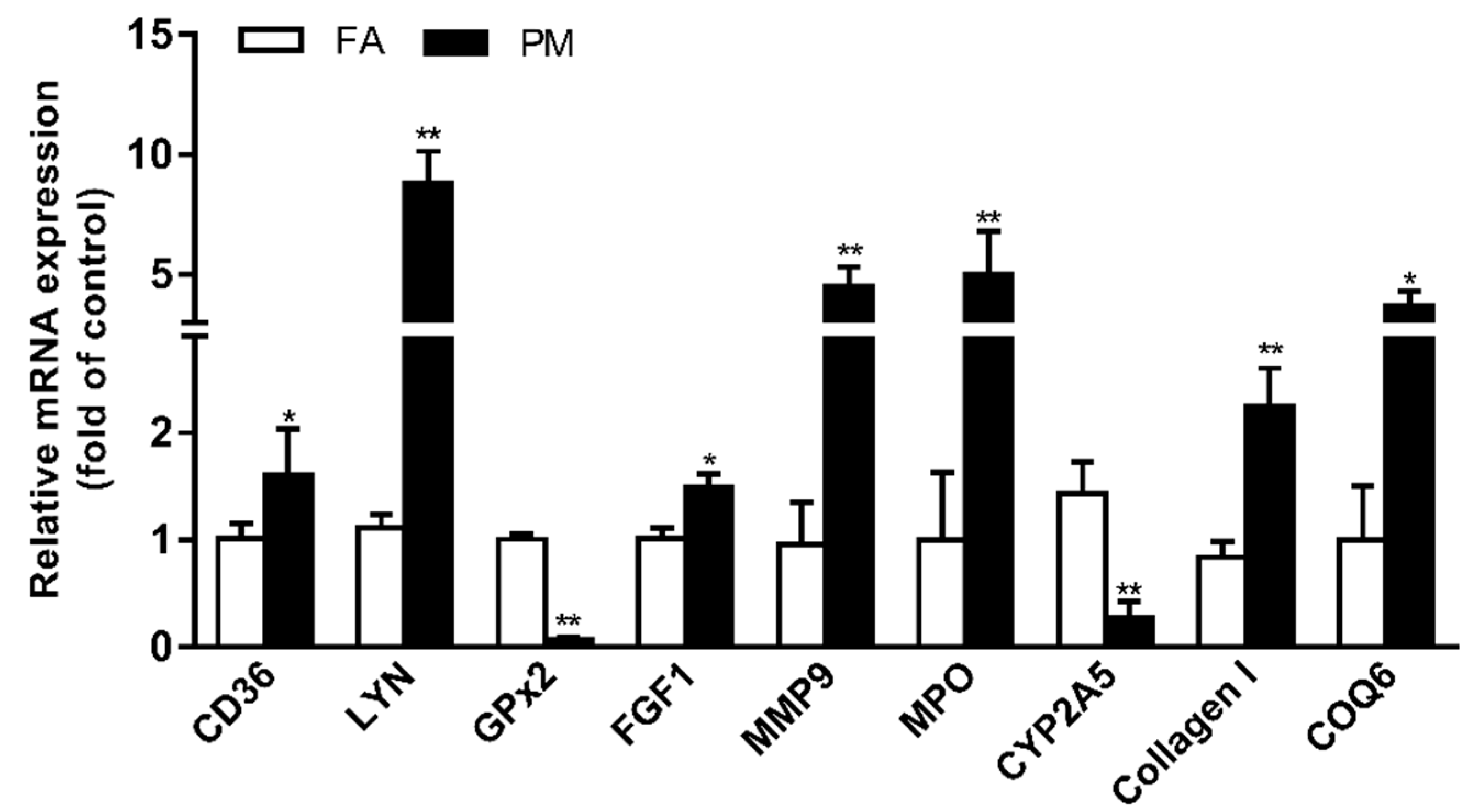
3.5. Verification of Protein Expression by RT-PCR Analysis
4. Discussion
4.1. Characterization of PM2.5
4.2. PM2.5 Activates the Phagosome Pathway
4.3. PM2.5 Alters Redox Homeostasis
4.4. Proteins Involved in PM2.5-Induced Pulmonary Inflammation and Fibrosis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Valavanidis, A.; Fiotakis, K.; Vlachogianni, T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Health Part C 2008, 26, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, N.; Liu, F.T. Pulmonary effects of diesel exhaust: Neutrophilic inflammation, oxidative injury, and asthma. Am. J. Pathol. 2011, 179, 2678–2682. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef]
- Cho, H.Y.; Reddy, S.P.; Yamamoto, M.; Kleeberger, S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB 2004, 18, 1258–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, D.R.; Magalhães, C.B.; Lopes, A.A.; Lanças, T.; Mauad, T.; Malm, O.; Valença, S.S.; Saldiva, P.H.; Faffe, D.S.; Zin, W.A. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhal. Toxicol. 2011, 23, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Kou, X.; Xie, L.; Cheng, F.; Hong, G. Effects of ambient PM2.5 on pathological injury, inflammation, oxidative stress, metabolic enzyme activity, and expression of c-fos and c-jun in lungs of rats. Environ. Sci. Pollut. Res. Int. 2015, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Rajagopalan, S.; Sun, Q.; Zhang, K. Real-world exposure of airborne particulate matter triggers oxidative stress in an animal model. Int. J. Physiol. Pathophysiol. Pharmacol. 2010, 2, 64–68. [Google Scholar] [PubMed]
- Deng, X.; Rui, W.; Zhang, F.; Ding, W. PM2.5 induces Nrf2-mediated defense mechanisms against oxidative stress by activating PIK3/AKT signaling pathway in human lung alveolar epithelial A549 cells. Cell Biol. Toxicol. 2013, 29, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.A.; Chung, J.S.; Cho, S.H.; Kim, H.J.; Yoo, Y.D. Romo1 expression contributes to oxidative stress-induced death of lung epithelial cells. Biochem. Biophys. Res. Commun. 2013, 439, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Pennathur, S.; Vivekanandan-Giri, A.; Locy, M.L.; Kulkarni, T.; Zhi, D.; Zeng, L.; Byun, J.; de Andrade, J.A.; Thannickal, V.J. Oxidative modifications of protein tyrosyl residues are increased in plasma of human subjects with interstitial lung disease. Am. J. Respir. Crit. Care Med. 2016, 193, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Steenhof, M.; Gosens, I.; Strak, M.; Godri, K.J.; Hoek, G.; Cassee, F.R.; Mudway, I.S.; Kelly, F.J.; Harrison, R.M.; Lebret, E.; et al. In vitro toxicity of particulate matter (PM) collected at different sites in the Netherlands is associated with PM composition, size fraction and oxidative potential—The RAPTES project. Part. Fibre Toxicol. 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Bo, L.; Jiang, S.; Xie, Y.; Kan, H.; Song, W.; Zhao, J. Effect of vitamin E and omega-3 fatty acids on protecting ambient PM2.5-induced inflammatory response and oxidative stress in vascular endothelial cells. PLoS ONE 2016, 11, e0152216. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zhang, F.; Wang, L.; Rui, W.; Long, F.; Zhao, Y.; Chen, D.; Ding, W. Airborne fine particulate matter induces multiple cell death pathways in human lung epithelial cells. Apoptosis 2014, 19, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Dysart, M.M.; Galvis, B.R.; Russell, A.G.; Barker, T.H. Environmental particulate (PM2.5) augments stiffness-induced alveolar epithelial cell mechanoactivation of transforming growth factor beta. PLoS ONE 2014, 9, e106821. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, X.; Tian, Z.; Jiang, R.; Chen, R.; Song, W.; Zhao, J. Preexposure to PM2.5 exacerbates acute viral myocarditis associated with Th17 cell. Int. J. Cardiol. 2013, 168, 3837–3845. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.W.; Yan, Y.; Caruso, F.; Nice, E.C. Emerging techniques in proteomics for probing nano–bio interactions. ACS Nano 2012, 6, 10438–10448. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. Proteome and proteomics: New technologies, new concepts, and new words. Electrophoresis 1998, 19, 1853–1861. [Google Scholar] [CrossRef]
- Verano-Braga, T.; Miethling-Graff, R.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Brewer, J.R.; Erdmann, H.; Kjeldsen, F. Insights into the cellular response triggered by silver nanoparticles using quantitative proteomics. ACS Nano 2014, 8, 2161–2175. [Google Scholar] [CrossRef]
- Van, U.P.; Kuhn, K.; Prinz, T.; Legner, H.; Schmid, P.; Baumann, C.; Tommassen, J. Identification of proteins of Neisseria meningitidis induced under iron-limiting conditions using the isobaric tandem mass tag (TMT) labeling approach. Proteomics 2009, 9, 1771–1781. [Google Scholar] [CrossRef]
- Huang, Q.; Zhang, J.; Peng, S.; Tian, M.; Chen, J.; Shen, H. Effects of water soluble PM2.5 extracts exposure on human lung epithelial cells (A549): A proteomic study. J. Appl. Toxicol. 2014, 34, 675–687. [Google Scholar] [CrossRef]
- Rui, W.; Guan, L.; Zhang, F.; Zhang, W.; Ding, W. PM2.5-induced oxidative stress increases adhesion molecules expression in human endothelial cells through the ERK/AKT/NF-κB-dependent pathway. J. Appl. Toxicol. 2016, 36, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Chen, H.; Yang, T.; Rui, W.; Liu, F.; Zhang, F.; Zhao, Y.; Ding, W. Direct effects of airborne PM2.5 exposure on macrophage polarizations. Biochim. Biophys. Acta 2016, 1860, 2835–2843. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zhang, F.; Rui, W.; Long, F.; Wang, L.; Feng, Z.; Chen, D.; Ding, W. PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol. In Vitro 2013, 27, 1762–1770. [Google Scholar] [CrossRef]
- Brunekreef, B.; Holgate, S.T. Air pollution and health. Lancet 2002, 360, 1233. [Google Scholar] [CrossRef]
- Voliotis, A.; Karali, I.; Kouras, A.; Samara, C. Fine and ultrafine particle doses in the respiratory tract from digital printing operations. Environ. Sci. Pollut. Res. 2017, 24, 3027–3037. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Dan, W.; Shuo, Y.; Fang, Z.; Wenjun, D. Oxidative stress induced by urban fine particles in cultured EA.hy926 cells. Hum. Exp. Toxicol. 2011, 30, 579–590. [Google Scholar] [CrossRef]
- Zhou, Q.; Liu, B.; Chen, Y.; Han, X.; Wei, X.; Zhu, Y.; Zhou, X.; Chen, J. Characterization of PAHs in size-fractionated submicron atmospheric particles and their association with the intracellular oxidative stress. Chemosphere 2017, 182, 1–7. [Google Scholar] [CrossRef]
- Hu, H.; Asweto, C.O.; Wu, J.; Shi, Y.; Feng, L.; Yang, X.; Liang, S.; Cao, L.; Duan, J.; Sun, Z. Gene expression profiles and bioinformatics analysis of human umbilical vein endothelial cells exposed to PM2.5. Chemosphere 2017, 183, 589–598. [Google Scholar] [CrossRef]
- Fujimoto, E.; Kobayashi, T.; Fujimoto, N.; Akiyama, M.; Tajima, S.; Nagai, R. AGE-modified collagens I and III induce keratinocyte terminal differentiation through AGE receptor CD36: Epidermal-dermal interaction in acquired perforating dermatosis. J. Investig. Dermatol. 2010, 130, 405–414. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Limónpacheco, J.; Gonsebatt, M.E. The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutat. Res. 2009, 674, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Jürgen, A.; Furtmüller, P.G.; Günther, R.; Christian, O. Redox properties of the couple compound I/native enzyme of myeloperoxidase and eosinophil peroxidase. Eur. J. Biochem. 2001, 268, 5142–5148. [Google Scholar] [Green Version]
- Gryglewski, R.J.; Palmer, R.M.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef]
- Ismail, A.; Leroux, V.; Smadja, M.; Gonzalez, L.; Lombard, M.; Pierrel, F.; Mellot-Draznieks, C.; Fontecave, M. Coenzyme Q Biosynthesis: Evidence for a substrate access channel in the FAD-dependent monooxygenase Coq6. PLoS Comput. Biol. 2016, 12, e1004690. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Tian, D.; Zhang, C.; Zhao, S.; Su, M. Clinicopathological and prognostic significance of GPX2 protein expression in esophageal squamous cell carcinoma. BMC Cancer 2016, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Van Eeden, S.F.; Yeung, A.; Quinlam, K.; Hogg, J.C. Systemic response to ambient particulate matter: Relevance to chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 61–67. [Google Scholar] [CrossRef]
- Yun, R.S.; Jin, S.U.; Hong, L.Y.; Wei, G.Z. Effects of environmental PM2.5 pollution on the morbidity of asthma and lung cancer. J. Lab. Med. 2002, 23, 367–378. [Google Scholar]
- Liu, L.; Poon, R.; Chen, L.; Frescura, A.-M.; Montuschi, P.; Ciabattoni, G.; Wheeler, A.; Dales, R. Acute effects of Air pollution on pulmonary function, airway inflammation, and oxidative stress in asthmatic children. Environ. Health Perspect. 2009, 117. [Google Scholar] [CrossRef]
- Miao, H.; Ichinose, T.; Kobayashi, M.; Arashidani, K.; Yoshida, S.; Nishikawa, M.; Takano, H.; Sun, G.; Shibamoto, T. Differences in allergic inflammatory responses between urban PM2.5 and fine particle derived from desert-dust in murine lungs. Toxicol. Appl. Pharmacol. 2016, 297, 41–55. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, J.; Jiang, R.; Song, W. Rat lung response to ozone and fine particulate matter (PM2.5) exposures. Environ. Toxicol. 2015, 30, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Kwapiszewska, G.; Gungl, A.; Wilhelm, J.; Marsh, L.M.; Puthenparampil, H.T.; Sinn, K.; Didiasova, M.; Klepetko, W.; Kosanovic, D.; Schermuly, R.T.; et al. Transcriptome profiling reveals the complexity of pirfenidone effects in IPF. Eur. Respir. J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Montaño, M.; Becerril, C.; Cisneros-Lira, J.; Barrera, L.; Ruíz, V.; Pardo, A.; Selman, M. Acidic fibroblast growth factor decreases α-smooth muscle actin expression and induces apoptosis in human normal lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Xing, R.G.; Chen, L.; Liu, C.R.; Miao, Z.G. PI3K/Akt signaling is involved in the pathogenesis of bleomycininduced pulmonary fibrosis via regulation of epithelialmesenchymal transition. Mol. Med. Rep. 2016, 14, 5699–5706. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, S.M. Deficiency of Psgl-1 accelerates bleomycin (BLM)-induced lung fibrosis and inflammation in mice through activating PI3K/AKT. Biochem. Biophys. Res. Commun. 2017, 491, 558–565. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Concentration (μg/mg) | |
|---|---|---|
| Metal elements | Ca | 13.76 ± 0.30 |
| K | 10.33 ± 0.03 | |
| Na | 7.96 ± 0.08 | |
| Fe | 7.06 ± 0.13 | |
| Al | 6.24 ± 0.06 | |
| Mg | 3.98 ± 0.09 | |
| Ti | 1.66 ± 0.02 | |
| Zn | 1.62 ± 0.01 | |
| Pb | 0.54 ± 0.002 | |
| Mn | 0.35 ± 0.004 | |
| Cu | 0.20 ± 0.003 | |
| V | 0.13 ± 0.002 | |
| Ba | 0.13 ± 0.001 | |
| Cr | 0.081 ± 0.001 | |
| As | 0.064 ± 0.001 | |
| Ni | 0.045 ± 0.001 | |
| Sr | 0.045 ± 0.003 | |
| Cd | 0.014 ± 0.001 | |
| Mo | 0.11 ± 0.001 | |
| Cs | 0.006 ± 0.0003 | |
| Co | 0.004 ± 0.0008 | |
| Inorganic ions | SO42− | 212.71 ± 3.67 |
| NO3− | 97.52 ± 6.81 | |
| NH4+ | 85.61 ± 0.52 | |
| Cl− | 1.85 ± 0.41 | |
| Carbon | OC | 44.09 ± 0.32 |
| EC | 25.51 ± 0.75 | |
| OC/EC | 1.72 ± 0.65 |
| Pathway | Term | Count | p-Value | Molecular Pathway |
|---|---|---|---|---|
| 1 | ECM–receptor interaction | 4 | 0.0010 | COL4A4 ↑, CD36 ↑, COL4A2 ↑, COL4A1 ↑ |
| 2 | Pathways in cancer | 6 | 0.0017 | FZD1 ↑, COL4A4 ↑, COL4A2 ↑, FGF1 ↑, MMP9 ↑, COL4A1 ↑ |
| 3 | Phagosome | 4 | 0.0072 | CYBB ↑, MPO ↑, TUBB3 ↑, CD36 ↑ |
| 4 | Small cell lung cancer | 3 | 0.0160 | COL4A4 ↑, COL4A2 ↑, COL4A1 ↑ |
| 5 | Protein digestion and absorption | 3 | 0.0175 | COL4A4 ↑, COL4A2 ↑, COL4A1 ↑ |
| 6 | Amoebiasis | 3 | 0.0297 | COL4A4 ↑, COL4A2 ↑, COL4A1 ↑ |
| 7 | PI3K–Akt signaling pathway | 4 | 0.0458 | COL4A4 ↑, COL4A2 ↑, FGF1 ↑, COL4A1 ↑ |
| Accession | Gene | PM/FARatio Average | Annotation |
|---|---|---|---|
| Redox homeostasis | |||
| Q8R1S0 | COQ6 | 1.619 | Ubiquinone biosynthesis monooxygenase |
| P11247 | MPO | 1.391 | Myeloperoxidase |
| Q61093 | CYBB | 1.306 | Cytochrome b-245 heavy chain |
| Q3UBG2 | PID1 | 1.298 | PTB-containing, cubilin and LRP1-interacting protein |
| P20852 | CYP2A5 | 0.584 | Cytochrome P450 2A5 |
| Immune response | |||
| Q64524 | H2BE | 2.130 | Histone H2B type 2-E |
| Q08857 | CD36 | 1.428 | Platelet glycoprotein 4 |
| Q9Z0F8 | ADAM17 | 1.302 | Disintegrin and metalloproteinase domain-containing protein 17 |
| P08122 | COL4A2 | 1.319 | Collagen alpha-2(IV) chain |
| P02463 | COL4A1 | 1.313 | Collagen alpha-1(IV) chain |
| Q61114 | BPIFB1 | 0.531 | BPI fold-containing family B member 1 |
| Fibrotic response | |||
| P61148 | FGF1 | 1.558 | Fibroblast growth factor 1 |
| P41245 | MMP9 | 1.434 | Matrix metalloproteinase-9 |
| Q08857 | CD36 | 1.428 | Platelet glycoprotein 4 |
| Q9QZR9 | COL4A4 | 1.362 | Collagen alpha-4(IV) chain |
| P08122 | COL4A2 | 1.319 | Collagen alpha-2(IV) chain |
| P02463 | COL4A1 | 1.313 | Collagen alpha-1(IV) chain |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Zhang, W.; Zhang, F.; Roepstorff, P.; Yang, F.; Lu, Z.; Ding, W. TMT-Based Quantitative Proteomics Analysis Reveals Airborne PM2.5-Induced Pulmonary Fibrosis. Int. J. Environ. Res. Public Health 2019, 16, 98. https://doi.org/10.3390/ijerph16010098
Liu S, Zhang W, Zhang F, Roepstorff P, Yang F, Lu Z, Ding W. TMT-Based Quantitative Proteomics Analysis Reveals Airborne PM2.5-Induced Pulmonary Fibrosis. International Journal of Environmental Research and Public Health. 2019; 16(1):98. https://doi.org/10.3390/ijerph16010098
Chicago/Turabian StyleLiu, Shan, Wei Zhang, Fang Zhang, Peter Roepstorff, Fuquan Yang, Zhongbing Lu, and Wenjun Ding. 2019. "TMT-Based Quantitative Proteomics Analysis Reveals Airborne PM2.5-Induced Pulmonary Fibrosis" International Journal of Environmental Research and Public Health 16, no. 1: 98. https://doi.org/10.3390/ijerph16010098