

Sulfadiazine Exerts Potential Anticancer Effect in HepG2 and MCF7 Cells by Inhibiting TNFα, IL1b, COX-1, COX-2, 5-LOX Gene Expression: Evidence from In Vitro and Computational Studies
 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Effects of Sulfadiazine on In Vitro COX-1, COX-2 and 5-LOX Activities
2.2. Effects of Sulfadiazine on Cell Viability of HepG2, MCF7, and THLE2 Cells
2.3. Effect of Sulfadiazine on the Levels of Inflammation-Related Parameters in LPS-Inflamed Cells
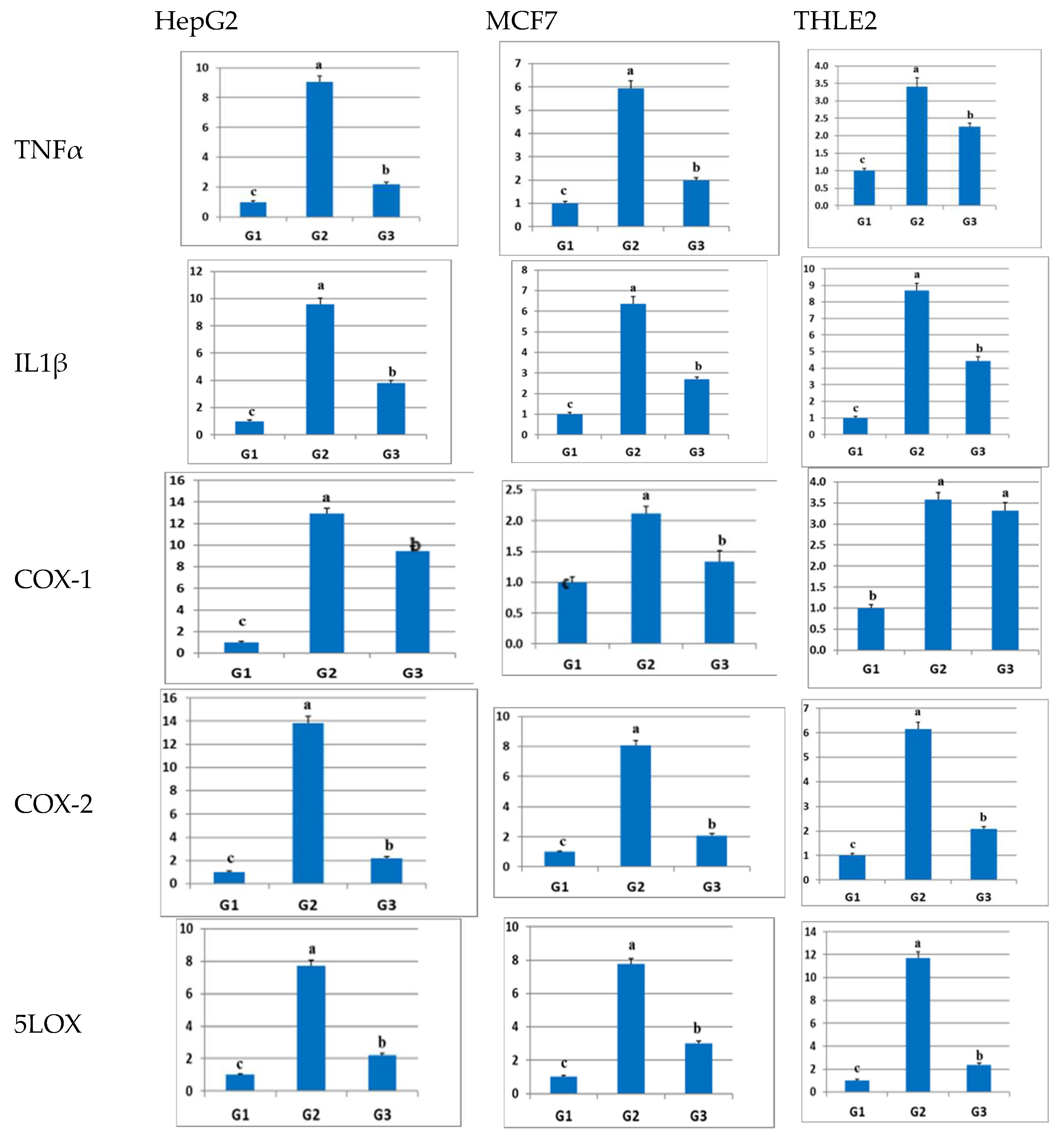
2.4. Effect of Sulfadiazine on the Expression of Inflammation-Related Genes in LPS-Inflamed Cells
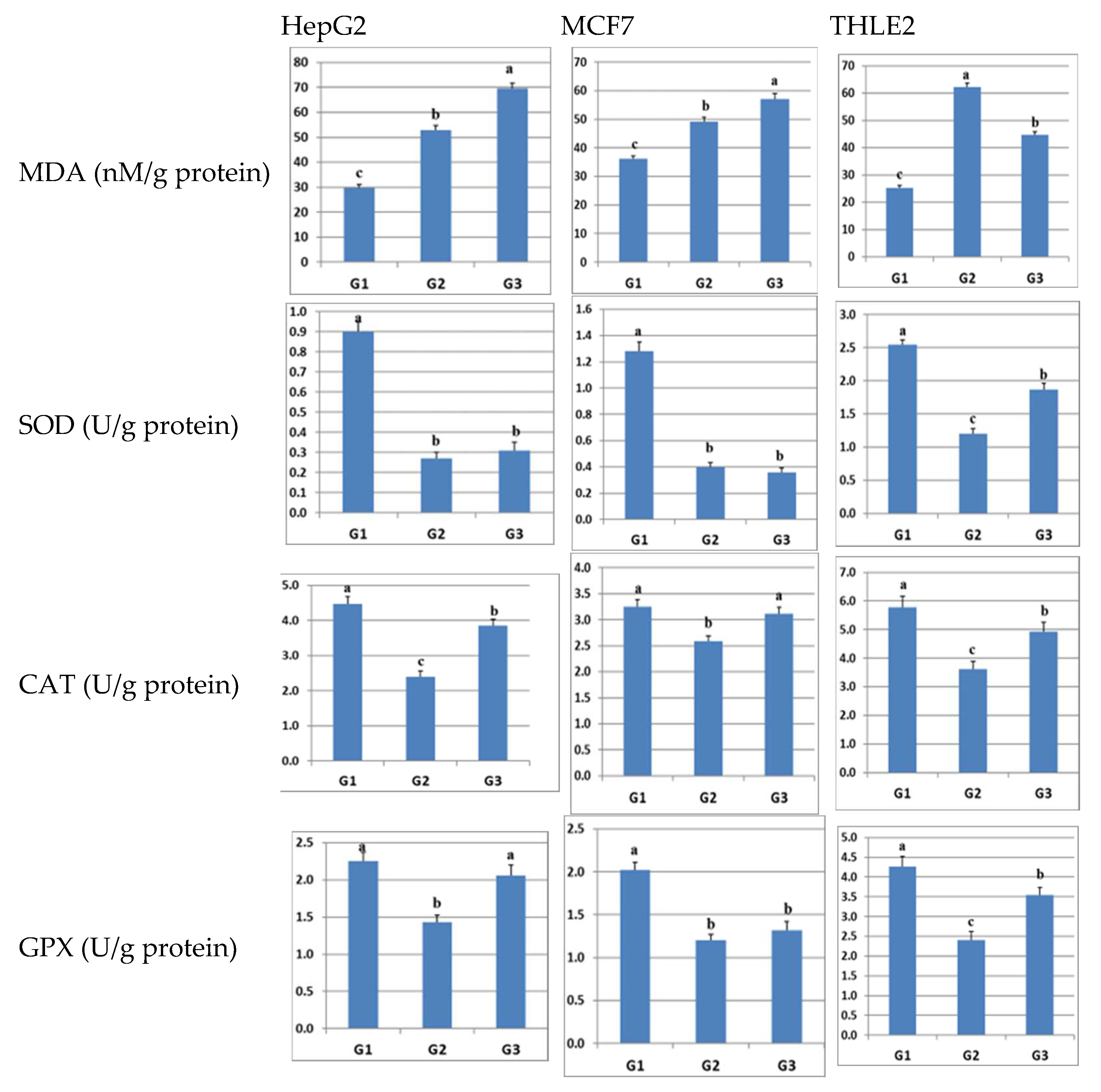
2.5. Effect of Sulfadiazine on the Levels of MDA and Antioxidant Enzymes in LPS-Inflamed Cells
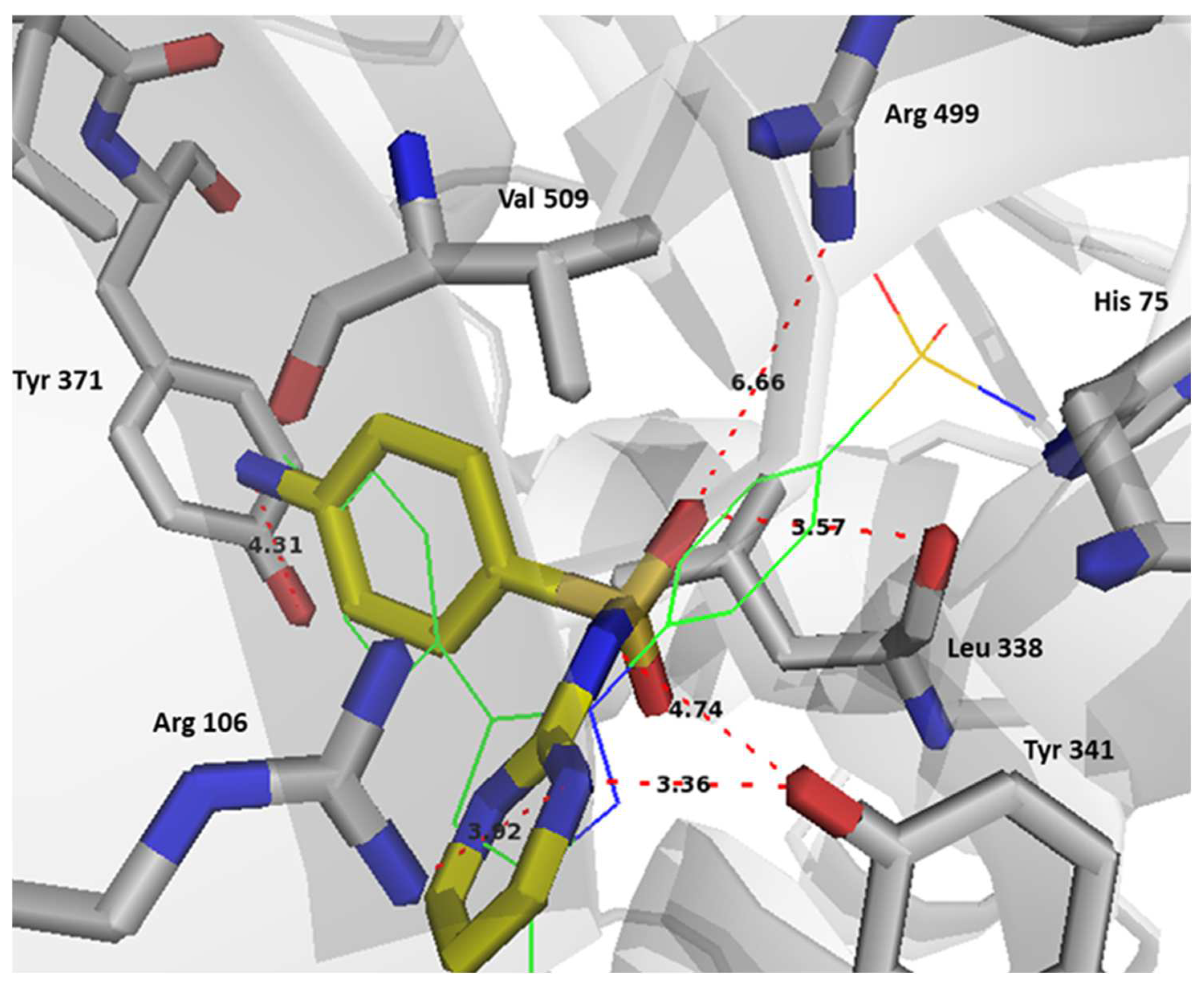
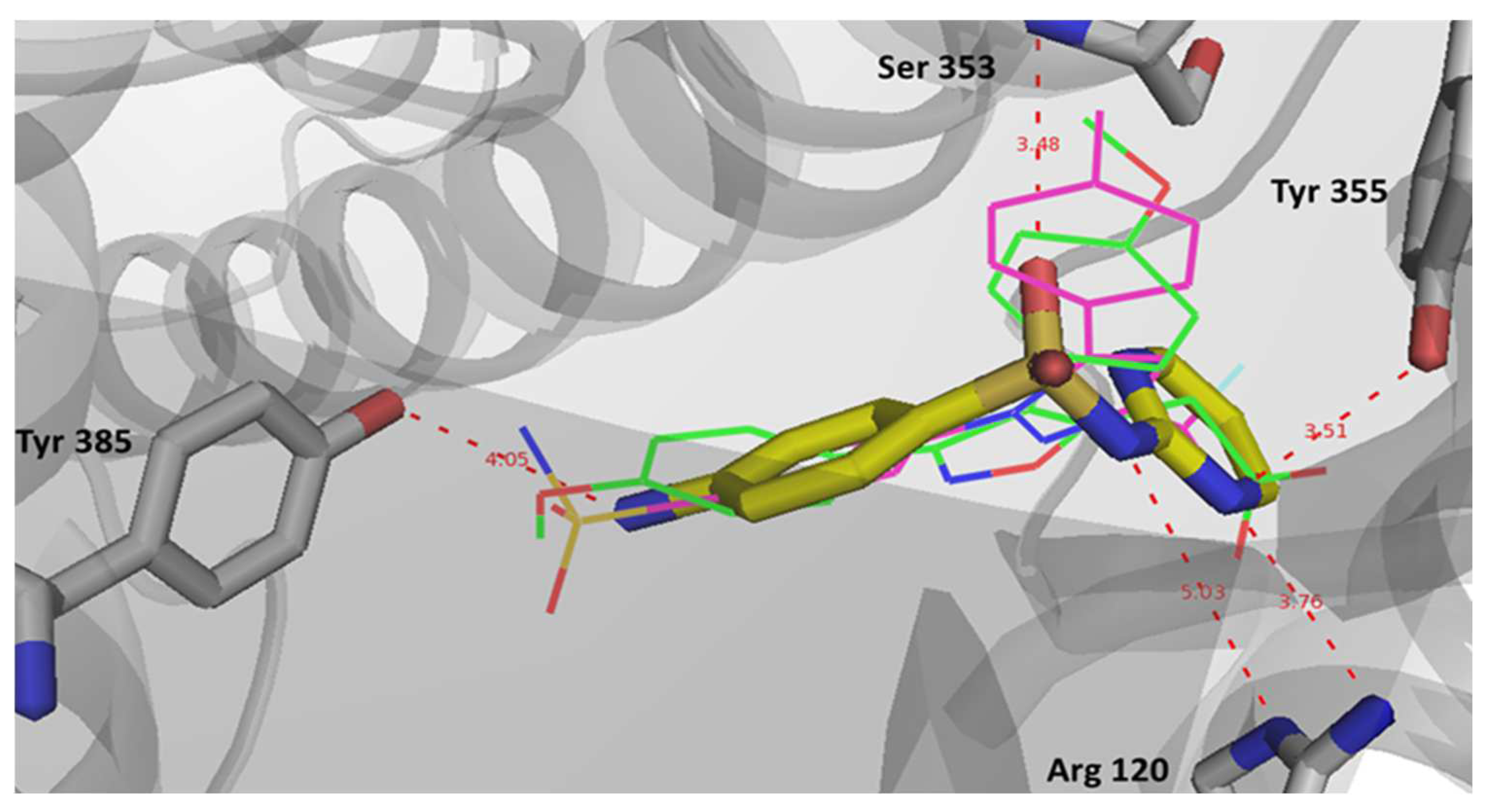
2.6. Computational Studies
3. Materials and Methods
3.1. Apparatus, Cell Lines and Chemicals
3.2. In Vitro Cyclooxygenase Inhibition Assay
3.3. Cell Viability Determination via MTT Assay
3.4. Stimulation of the Production of Inflammatory Mediators in Cell Lines
3.5. Measurement of Inflammatory Markers
3.6. Malondialdehyde Level and Activities of Antioxidant Enzymes
3.7. Quantitative Real-Time PCR Analysis
3.8. Statistical Analysis
3.9. Molecular Modeling
3.9.1. Computational Tools
3.9.2. Crystal Structures
3.9.3. Protein Preparation
3.9.4. Ligand Library Preparation
3.9.5. Binding Pocket Determination
3.9.6. Validation of Molecular Docking
3.9.7. Molecular Docking
3.9.8. Molecular Dynamics Simulation Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Nguyen, S.M.; Deppen, S.; Nguyen, G.H.; Pham, D.X.; Bui, T.D.; Tran, T.V. Projecting Cancer Incidence for 2025 in the 2 Largest Populated Cities in Vietnam. Cancer Control 2019, 26, 1073274819865274. [Google Scholar] [CrossRef] [PubMed]
- Bidram, E.; Esmaeili, Y.; Ranji-Burachaloo, H.; Al-Zaubai, N.; Zarrabi, A.; Stewart, A.; Dunstan, D.E. A concise review on cancer treatment methods and delivery systems. J. Drug Deliv. Sci. Technol. 2019, 54, 101350. [Google Scholar] [CrossRef]
- Jin, G.; Wong, S.T. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Lago, S.G.; Bahn, S. Clinical Trials and Therapeutic Rationale for Drug Repurposing in Schizophrenia. ACS Chem. Neurosci. 2019, 10, 58–78. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Gostev, M.; Knight, R.A.; Willis, A.E.; Melino, G.; Antonov, A.V. DRUGSURV: A resource for repositioning of approved and experimental drugs in oncology based on patient survival information. Cell Death Dis. 2014, 5, e1051. [Google Scholar] [CrossRef]
- Senanayake, S.L. Drug repurposing strategies for COVID-19. Future Drug Discov. 2020, 2, 10-4155. [Google Scholar] [CrossRef]
- Yusup, G.; Akutsu, Y.; Mutallip, M.; Qin, W.; Hu, X.; Komatsu-Akimoto, A.; Hoshino, I.; Hanari, N.; Mori, M.; Akanuma, N.; et al. A COX-2 inhibitor enhances the antitumor effects of chemotherapy and radiotherapy for esophageal squamous cell carcinoma. Int. J. Oncol. 2014, 44, 1146–1152. [Google Scholar] [CrossRef]
- Ristimäki, A. Cyclooxygenase 2: From inflammation to carcinogenesis. Novartis Found. Symp. 2004, 256, 215–221. [Google Scholar]
- Li, S.; Jiang, M.; Wang, L.; Yu, S. Combined chemotherapy with cyclooxygenase-2 (COX-2) inhibitors in treating human cancers: Recent advancement. Biomed. Pharmacother. 2020, 129, 110389. [Google Scholar] [CrossRef]
- Jara-Gutierrez, A.; Baladron, V. The Role of Prostaglandins in Different Types of Cancer. Cells 2021, 10, 1487. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.M.; Day, R.O. COX-2 inhibitors. Med. J. Aust. 2000, 173, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J. COX-1 and COX-2 tissue expression: Implications and predictions. J. Rheumatol. Suppl. 1997, 49, 15–19. [Google Scholar]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 2009, 17, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Saenz, A.; Atreya, C.E.; Wang, C.; Pan, B.; Dreyer, C.A.; Brunen, D.; Prahallad, A.; Munoz, D.P.; Ramms, D.J.; Burghi, V.; et al. A reversible SRC-relayed COX2 inflammatory program drives resistance to BRAF and EGFR inhibition in BRAF(V600E) colorectal tumors. Nat. Cancer 2023, 4, 240–256. [Google Scholar] [CrossRef]
- Moore, G.Y.; Pidgeon, G.P. Cross-Talk between Cancer Cells and the Tumour Microenvironment: The Role of the 5-Lipoxygenase Pathway. Int. J. Mol. Sci. 2017, 18, 236. [Google Scholar] [CrossRef]
- Ye, Y.N.; Wu, W.K.; Shin, V.Y.; Bruce, I.C.; Wong, B.C.; Cho, C.H. Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis 2005, 26, 827–834. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Lajeunesse, D.; Reboul, P.; Pelletier, J.P. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann. Rheum. Dis. 2003, 62, 501–509. [Google Scholar] [CrossRef]
- Che, X.H.; Chen, C.L.; Ye, X.L.; Weng, G.B.; Guo, X.Z.; Yu, W.Y.; Tao, J.; Chen, Y.C.; Chen, X. Dual inhibition of COX-2/5-LOX blocks colon cancer proliferation, migration and invasion in vitro. Oncol. Rep. 2016, 35, 1680–1688. [Google Scholar] [CrossRef]
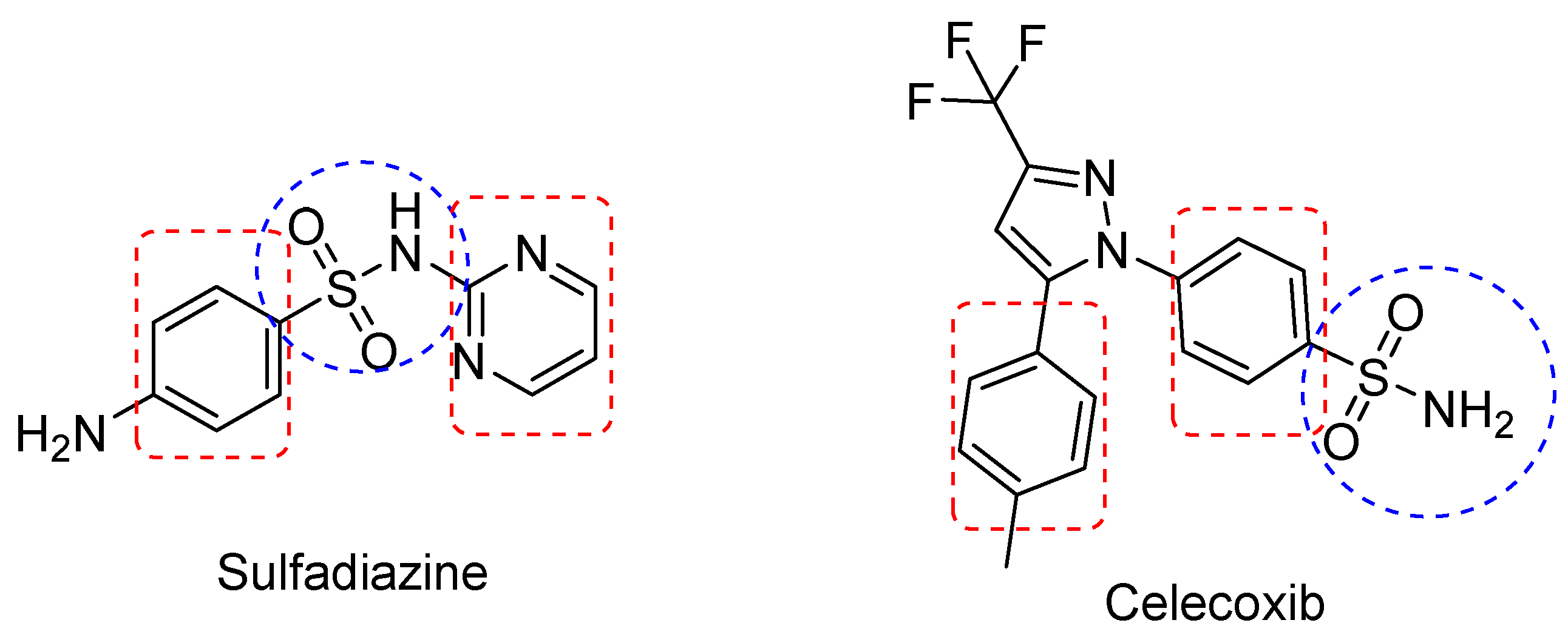
- Ovung, A.; Bhattacharyya, J. Sulfonamide drugs: Structure, antibacterial property, toxicity, and biophysical interactions. Biophys. Rev. 2021, 13, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Fang, G.; Chen, H.; Deng, X.; Tang, Z. Sulfonamide derivatives as potential anti-cancer agents and their SARs elucidation. Eur. J. Med. Chem. 2021, 226, 113837. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, A.; Sikka, A.; Yogev, O.; Herendi, L.; Balcells, C.; Ma, Y.; Poon, E.; Eckold, C.; Valbuena, G.N.; Xu, Y.; et al. Indisulam targets RNA splicing and metabolism to serve as a therapeutic strategy for high-risk neuroblastoma. Nat. Commun. 2022, 13, 1380. [Google Scholar] [CrossRef]
- Rains, C.P.; Noble, S.; Faulds, D. Sulfasalazine. A review of its pharmacological properties and therapeutic efficacy in the treatment of rheumatoid arthritis. Drugs 1995, 50, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Plosker, G.L.; Croom, K.F. Sulfasalazine: A review of its use in the management of rheumatoid arthritis. Drugs 2005, 65, 1825–1849. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Lowell, C.A. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med. 1997, 185, 1661–1670. [Google Scholar] [CrossRef]
- Granado-Serrano, A.B.; Martin, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin attenuates TNF-induced inflammation in hepatic cells by inhibiting the NF-kappaB pathway. Nutr. Cancer 2012, 64, 588–598. [Google Scholar] [CrossRef]
- Kim, H.G.; Shrestha, B.; Lim, S.Y.; Yoon, D.H.; Chang, W.C.; Shin, D.J.; Han, S.K.; Park, S.M.; Park, J.H.; Park, H.I.; et al. Cordycepin inhibits lipopolysaccharide-induced inflammation by the suppression of NF-kappaB through Akt and p38 inhibition in RAW 264.7 macrophage cells. Eur. J. Pharmacol. 2006, 545, 192–199. [Google Scholar] [CrossRef]
- Chowdhury, M.A.; Abdellatif, K.R.; Dong, Y.; Das, D.; Suresh, M.R.; Knaus, E.E. Synthesis of celecoxib analogues possessing a N-difluoromethyl-1,2-dihydropyrid-2-one 5-lipoxygenase pharmacophore: Biological evaluation as dual inhibitors of cyclooxygenases and 5-lipoxygenase with anti-inflammatory activity. J. Med. Chem. 2009, 52, 1525–1529. [Google Scholar] [CrossRef]
- Childers, M.C.; Daggett, V. Validating Molecular Dynamics Simulations against Experimental Observables in Light of Underlying Conformational Ensembles. J. Phys. Chem. B 2018, 122, 6673–6689. [Google Scholar] [CrossRef]
- Wadhwa, R.; Yadav, N.S.; Katiyar, S.P.; Yaguchi, T.; Lee, C.; Ahn, H.; Yun, C.O.; Kaul, S.C.; Sundar, D. Molecular dynamics simulations and experimental studies reveal differential permeability of withaferin-A and withanone across the model cell membrane. Sci. Rep. 2021, 11, 2352. [Google Scholar] [CrossRef]
- Adelusi, T.I.; Oyedele, A.-Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure Of Biomolecules through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Gilbert, N.C.; Bartlett, S.G.; Waight, M.T.; Neau, D.B.; Boeglin, W.E.; Brash, A.R.; Newcomer, M.E. The structure of human 5-lipoxygenase. Science 2011, 331, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Fadaly, W.A.A.; Kamel, G.M.; Elshaier, Y.; El-Magd, M.A. Design, synthesis, modeling studies and biological evaluation of thiazolidine derivatives containing pyrazole core as potential anti-diabetic PPAR-gamma agonists and anti-inflammatory COX-2 selective inhibitors. Bioorg. Chem. 2019, 82, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Roschek, B., Jr.; Fink, R.C.; Li, D.; McMichael, M.; Tower, C.M.; Smith, R.D.; Alberte, R.S. Pro-inflammatory enzymes, cyclooxygenase 1, cyclooxygenase 2, and 5-lipooxygenase, inhibited by stabilized rice bran extracts. J. Med. Food 2009, 12, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Arumanayagam, S.; Arunmani, M. Hepatoprotective and antibacterial activity of Lippia nodiflora Linn. against lipopolysaccharides on HepG2 cells. Pharmacogn. Mag. 2015, 11, 24–31. [Google Scholar] [CrossRef]
- Bisht, K.; Wagner, K.H.; Bulmer, A.C. Curcumin, resveratrol and flavonoids as anti-inflammatory, cyto- and DNA-protective dietary compounds. Toxicology 2010, 278, 88–100. [Google Scholar] [CrossRef]
- Karan, B.N.; Maity, T.K.; Pal, B.C.; Singha, T.; Jana, S. Betulinic Acid, the first lupane-type triterpenoid isolated via bioactivity-guided fractionation, and identified by spectroscopic analysis from leaves of Nyctanthes arbor-tristis: Its potential biological activities in vitro assays. Nat. Prod. Res. 2019, 33, 3287–3292. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Zhang, P.; Lin, L.; Shuai, O.; Zhang, H.M.; Liu, S.H.; Wang, J.Y. Protective effect of Bajijiasu against beta-amyloid-induced neurotoxicity in PC12 cells. Cell. Mol. Neurobiol. 2013, 33, 837–850. [Google Scholar] [CrossRef] [PubMed]
- El-Magd, M.A.; Kahilo, K.A.; Nasr, N.E.; Kamal, T.; Shukry, M.; Saleh, A.A. A potential mechanism associated with lead-induced testicular toxicity in rats. Andrologia 2017, 49, e12750. [Google Scholar] [CrossRef] [PubMed]
- Elgazar, A.A.; Selim, N.M.; Abdel-Hamid, N.M.; El-Magd, M.A.; El Hefnawy, H.M. Isolates from Alpinia officinarum Hance attenuate LPS-induced inflammation in HepG2: Evidence from in silico and in vitro studies. Phytother. Res. 2018, 32, 1273–1288. [Google Scholar] [CrossRef]
- El-Magd, M.A.; Abdo, W.S.; El-Maddaway, M.; Nasr, N.M.; Gaber, R.A.; El-Shetry, E.S.; Saleh, A.A.; Alzahrani, F.A.A.; Abdelhady, D.H. High doses of S-methylcysteine cause hypoxia-induced cardiomyocyte apoptosis accompanied by engulfment of mitochondaria by nucleus. Biomed. Pharmacother. 2017, 94, 589–597. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Alves, M.J.; Froufe, H.J.; Costa, A.F.; Santos, A.F.; Oliveira, L.G.; Osorio, S.R.; Abreu, R.M.; Pintado, M.; Ferreira, I.C. Docking studies in target proteins involved in antibacterial action mechanisms: Extending the knowledge on standard antibiotics to antimicrobial mushroom compounds. Molecules 2014, 19, 1672–1684. [Google Scholar] [CrossRef] [PubMed]
- Kwofie, S.K.; Dankwa, B.; Odame, E.A.; Agamah, F.E.; Doe, L.P.A.; Teye, J.; Agyapong, O.; Miller, W.A., 3rd; Mosi, L.; Wilson, M.D. In Silico Screening of Isocitrate Lyase for Novel Anti-Buruli Ulcer Natural Products Originating from Africa. Molecules 2018, 23, 1550. [Google Scholar] [CrossRef] [PubMed]
- Jaundoo, R.; Bohmann, J.; Gutierrez, G.E.; Klimas, N.; Broderick, G.; Craddock, T.J.A. Using a Consensus Docking Approach to Predict Adverse Drug Reactions in Combination Drug Therapies for Gulf War Illness. Int. J. Mol. Sci. 2018, 19, 3355. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | COX-1 IC50 (μM) * | COX-2 IC50 (μM) * | 5LOX IC50 (μM) * |
|---|---|---|---|
| Sulfadiazine | 18.4 | 5.27 | 19.1 |
| Celecoxib | 5.9 | 1.94 | 16.7 |
| Compound | HEPG2 IC50 (μM) | MCF7 IC50 (μM) | THLE2 IC50 (μM) |
|---|---|---|---|
| Sulfadiazine | 245.69 ± 4.1 | 215.68 ± 3.8 | 4159 ± 90.5 |
| Cisplatin | 66.92 ± 1.8 | 46.83 ± 1.3 | 2144 ± 95.3 |
| Glide Score | Selectivity COX-1/COX-2/5-LOX | |||
|---|---|---|---|---|
| COX-1 | COX-2 | 5-LOX | ||
| Sulfadiazine | −4.9 | −6.1 | −5.3 | 1:1.3:1.1 |
| Sulfamerazine | −6.1 | −5.7 | −5.3 | 1.6:1.1:1 |
| Sulfathiazole | −5.6 | −5.3 | −5.2 | 1.1:1:1 |
| Sulfameter | −5.4 | −5.7 | −5.1 | 1.1:1.1:1 |
| Sulfadimidine | −5.2 | −5.5 | −5.1 | 1:1.1:1 |
| Sulfadoxine | −5.3 | −5.3 | −4.9 | 1.1:1.1:1 |
| Sulfaguanidine | −4.8 | −5.9 | −4.8 | 1:1.2:1 |
| Sulfabenzamide | −5.6 | −7.3 | −4.4 | 1.2:1.7:1 |
| Celecoxib | −10.1 | −11.6 | −5.3 | 1.9:2.2:1 |
| Experimental IC50 (μM) * | Calculated Binding Affinity | |||||||
|---|---|---|---|---|---|---|---|---|
| COX-1 | COX-2 | 5-LOX | Selectivity COX-1/COX-2/5-LOX | COX-1 | COX-2 | 5-LOX | Selectivity COX-1/COX-2/5-LOX | |
| Sulfadiazine | 18.4 | 5.27 | 19.1 | 1:3.6:1 | −4.9 | −6.1 | −5.3 | 1:1.3:1.1 |
| Celecoxib | 5.9 | 1.94 | 16.7 | 3:8.6:1 | −10.1 | −11.6 | −5.3 | 1.9:2.2:1 |
| Gene | Forward Primer (/5 ------ /3) | Reverse Primer (/5 ------ /3) |
|---|---|---|
| TNFa | CCCAGGGACCTCTCTCTAATC | ATGGGCTACAGGCTTGTCACT |
| IL1β | ACAGATGAAGTGCTCCTTCCA | GTCGGAGATTCGTAGCTGGAT |
| Cox2 | CCCTTGGGTGTCAAAGGTAA | GCCCTCGCTTATGATCTGTC |
| COX-1 | AAGGAGATGGCAGCAGAGTT | GTGGCCGTCTTGACAATGTT |
| 5LOX | ATTGCCATCCAGCTCAACCAAACC | TGGCGATACCAAACACCTCAGACA |
| β-actin | CGACATCAGGAAGGACCTGTATGCC | GAAGATTCGTCGTGAAAGTCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomaa, M.; Gad, W.; Hussein, D.; Pottoo, F.H.; Tawfeeq, N.; Alturki, M.; Alfahad, D.; Alanazi, R.; Salama, I.; Aziz, M.; et al. Sulfadiazine Exerts Potential Anticancer Effect in HepG2 and MCF7 Cells by Inhibiting TNFα, IL1b, COX-1, COX-2, 5-LOX Gene Expression: Evidence from In Vitro and Computational Studies. Pharmaceuticals 2024, 17, 189. https://doi.org/10.3390/ph17020189
Gomaa M, Gad W, Hussein D, Pottoo FH, Tawfeeq N, Alturki M, Alfahad D, Alanazi R, Salama I, Aziz M, et al. Sulfadiazine Exerts Potential Anticancer Effect in HepG2 and MCF7 Cells by Inhibiting TNFα, IL1b, COX-1, COX-2, 5-LOX Gene Expression: Evidence from In Vitro and Computational Studies. Pharmaceuticals. 2024; 17(2):189. https://doi.org/10.3390/ph17020189
Chicago/Turabian StyleGomaa, Mohamed, Wael Gad, Dania Hussein, Faheem Hyder Pottoo, Nada Tawfeeq, Mansour Alturki, Dhay Alfahad, Razan Alanazi, Ismail Salama, Mostafa Aziz, and et al. 2024. "Sulfadiazine Exerts Potential Anticancer Effect in HepG2 and MCF7 Cells by Inhibiting TNFα, IL1b, COX-1, COX-2, 5-LOX Gene Expression: Evidence from In Vitro and Computational Studies" Pharmaceuticals 17, no. 2: 189. https://doi.org/10.3390/ph17020189