
Phosphodiesterase 10A Inhibition Modulates the Corticostriatal Activity and L-DOPA-Induced Dyskinesia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Stimulating and Recording Electrode Placements
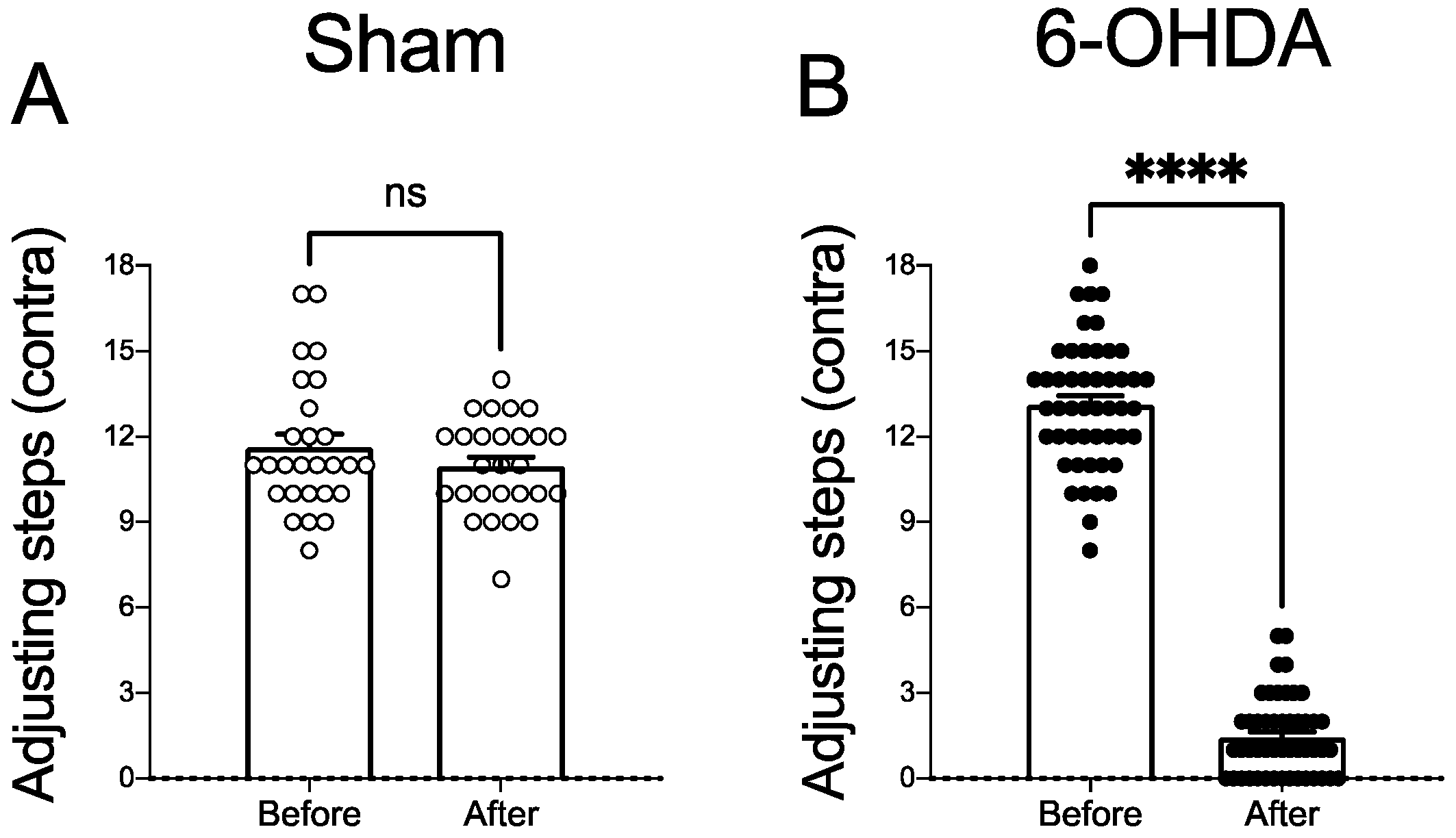
2.2. Dopaminergic Lesion Assessment
2.3. L-DOPA Facilitates Corticostriatal Transmission in Dyskinetic Animals
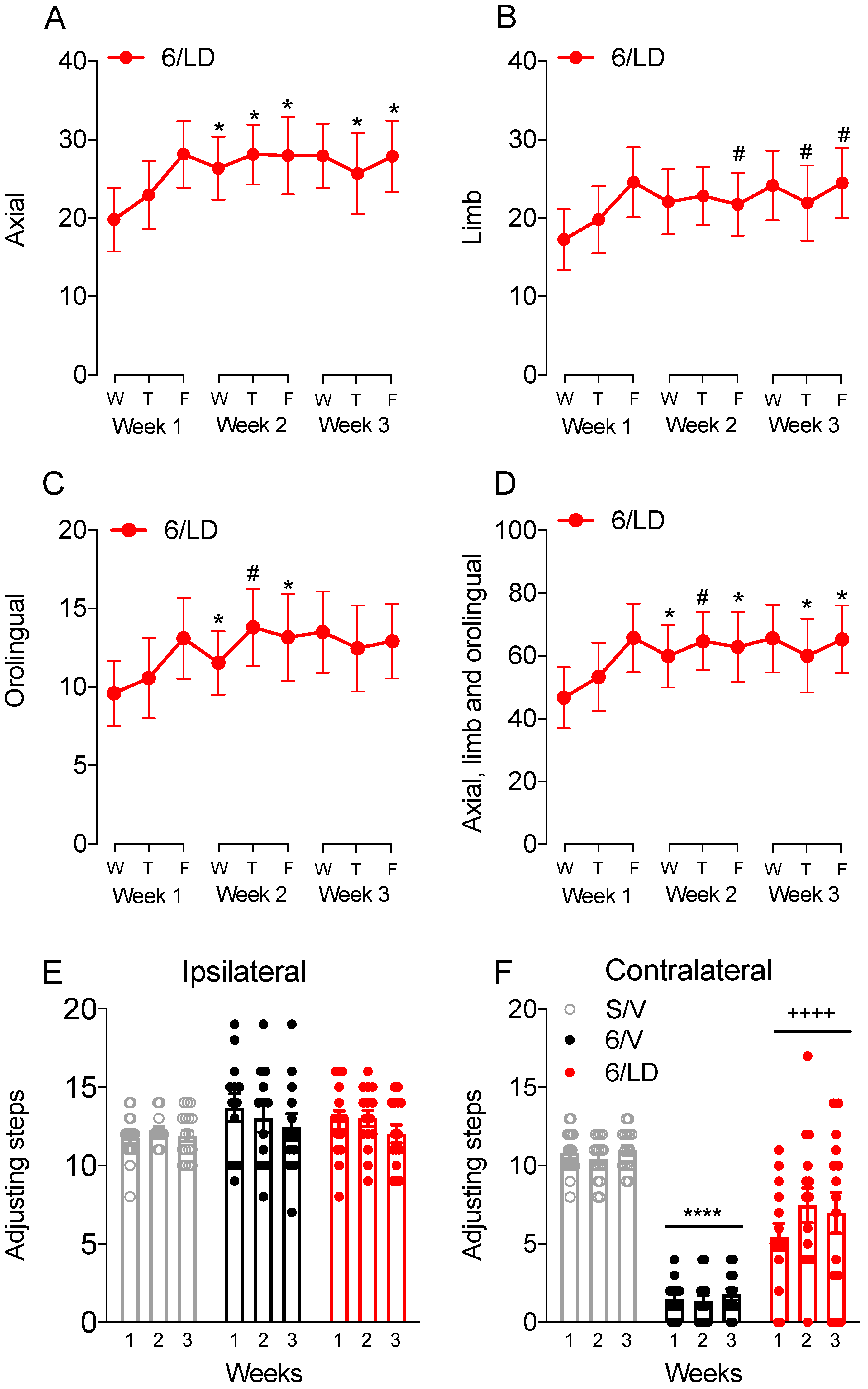
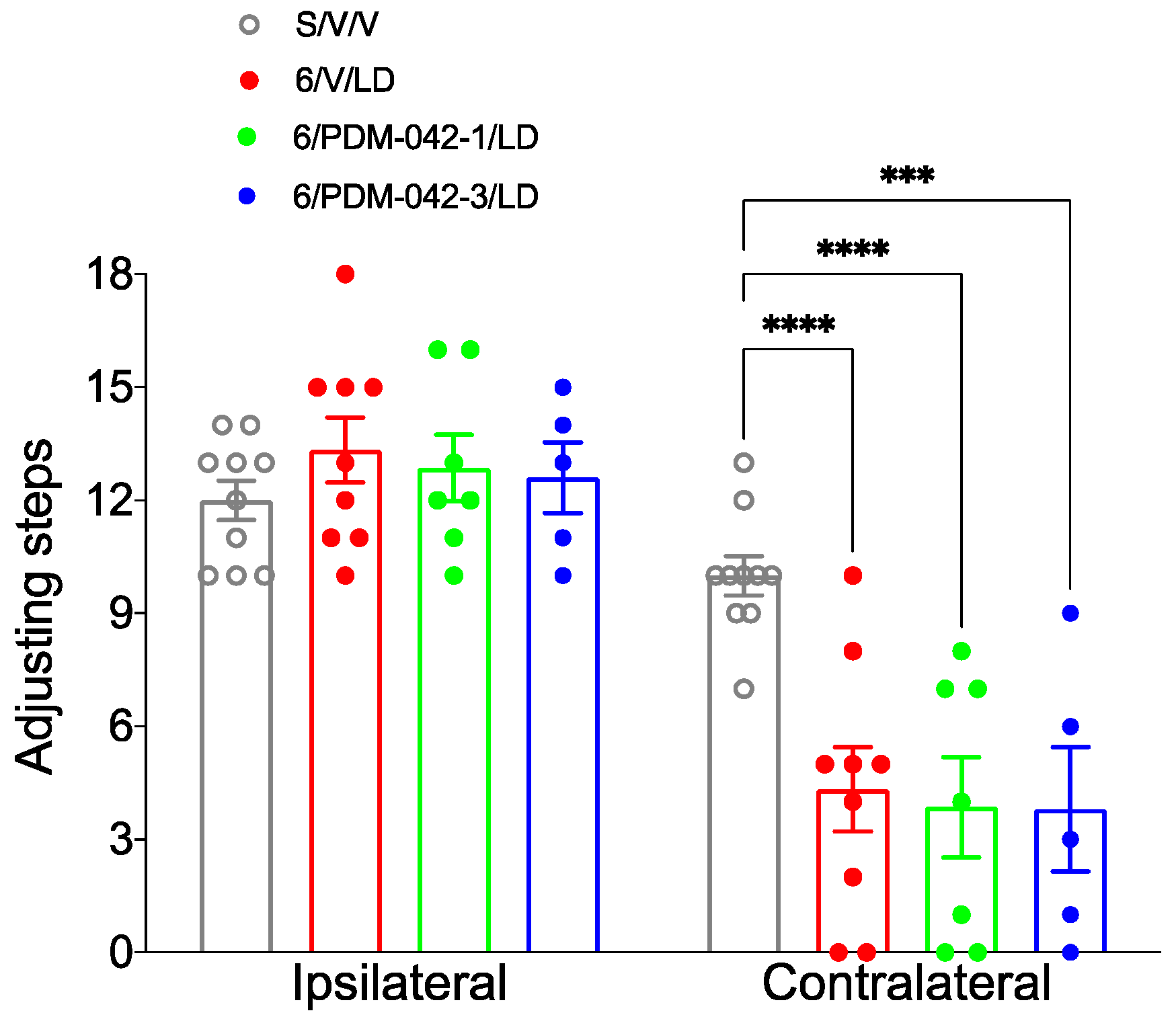
2.3.1. AIMs and Stepping Test
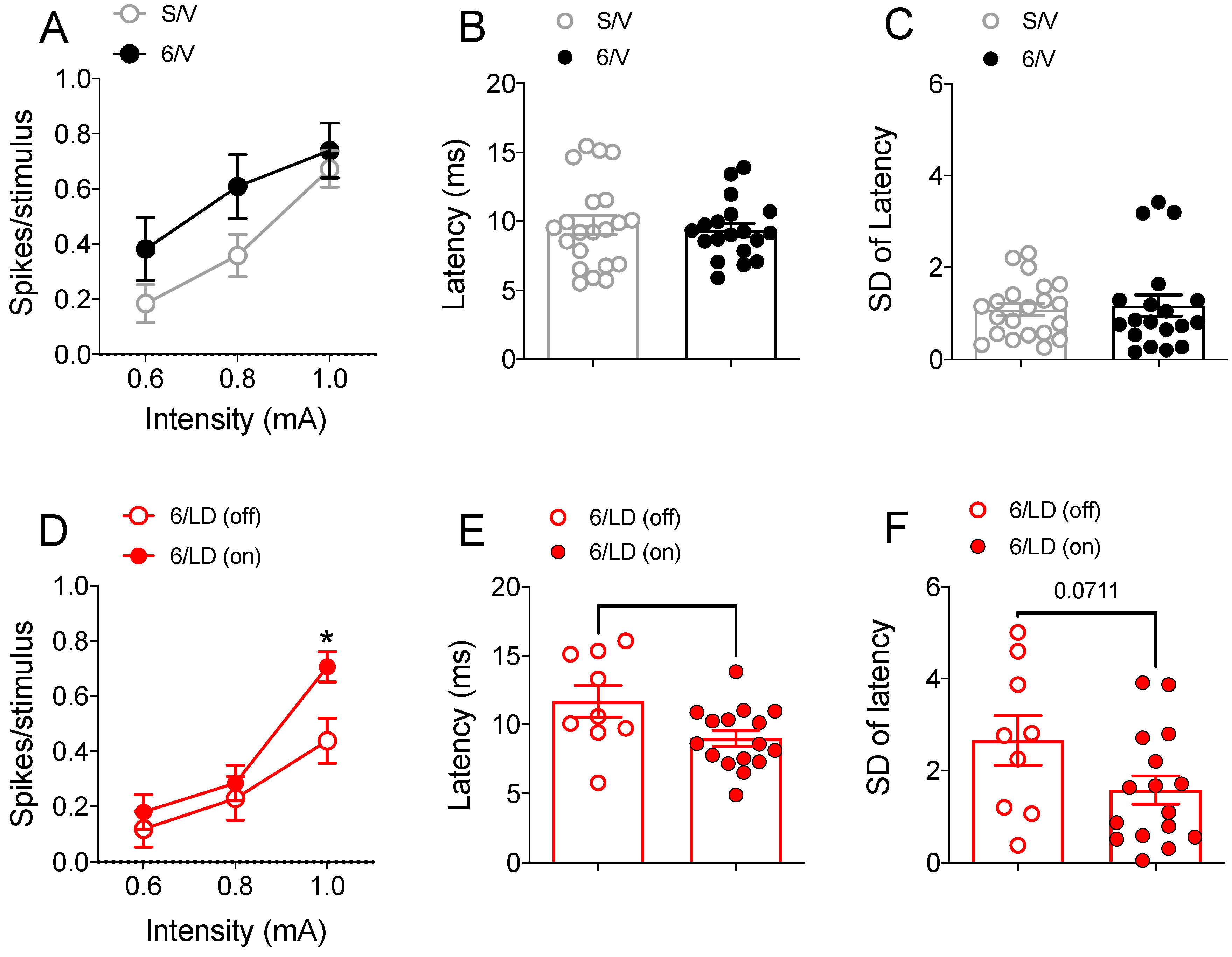
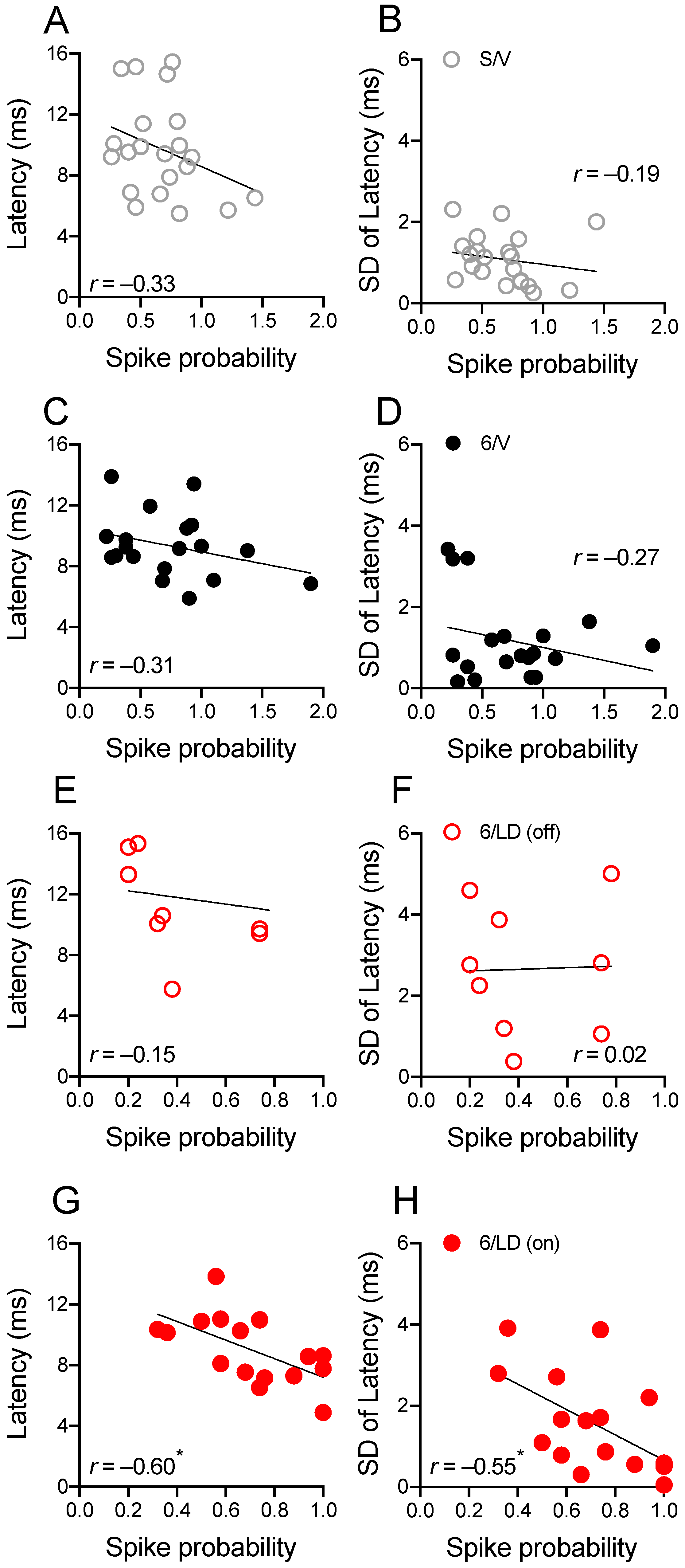
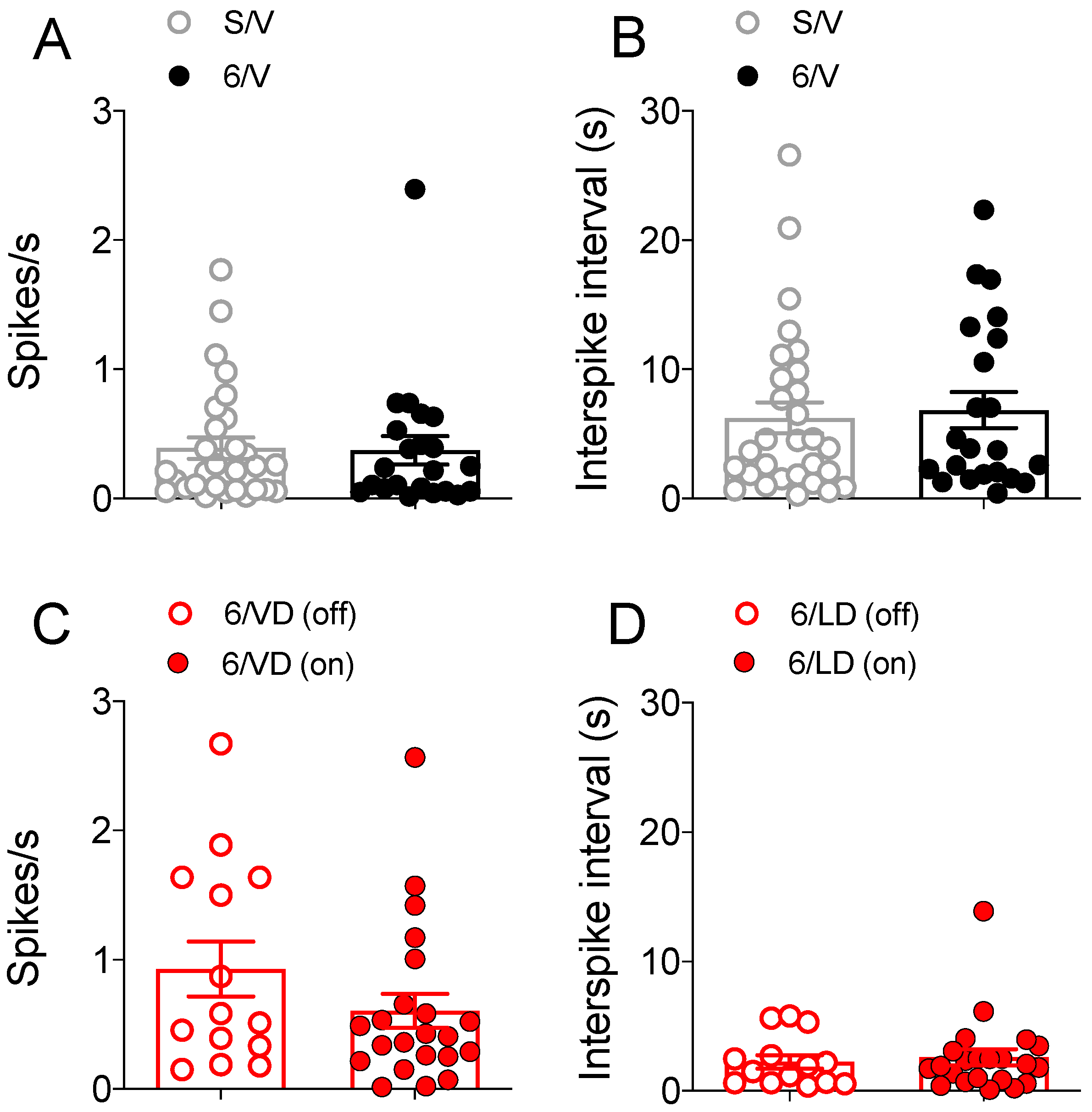
2.3.2. Electrophysiological Recordings in Cortically Responsive MSNs
2.3.3. Electrophysiological Recordings in Spontaneously Active MSNs
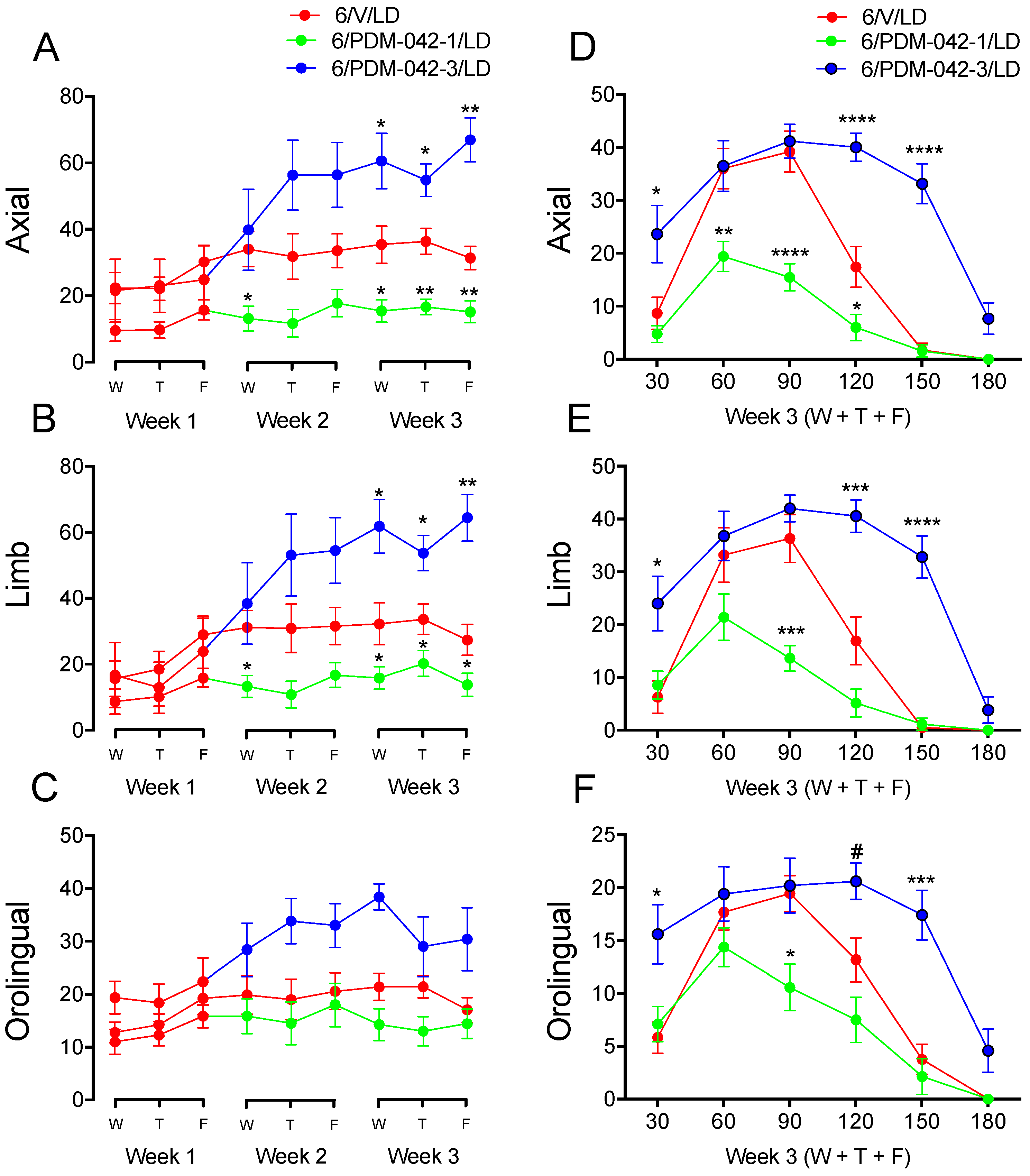
2.4. Effects of Chronic Administration of the PDE10A Inhibitor PDM-042 on AIMs
2.4.1. The Effects of PDM-042 on AIMs Are Dose-Dependent
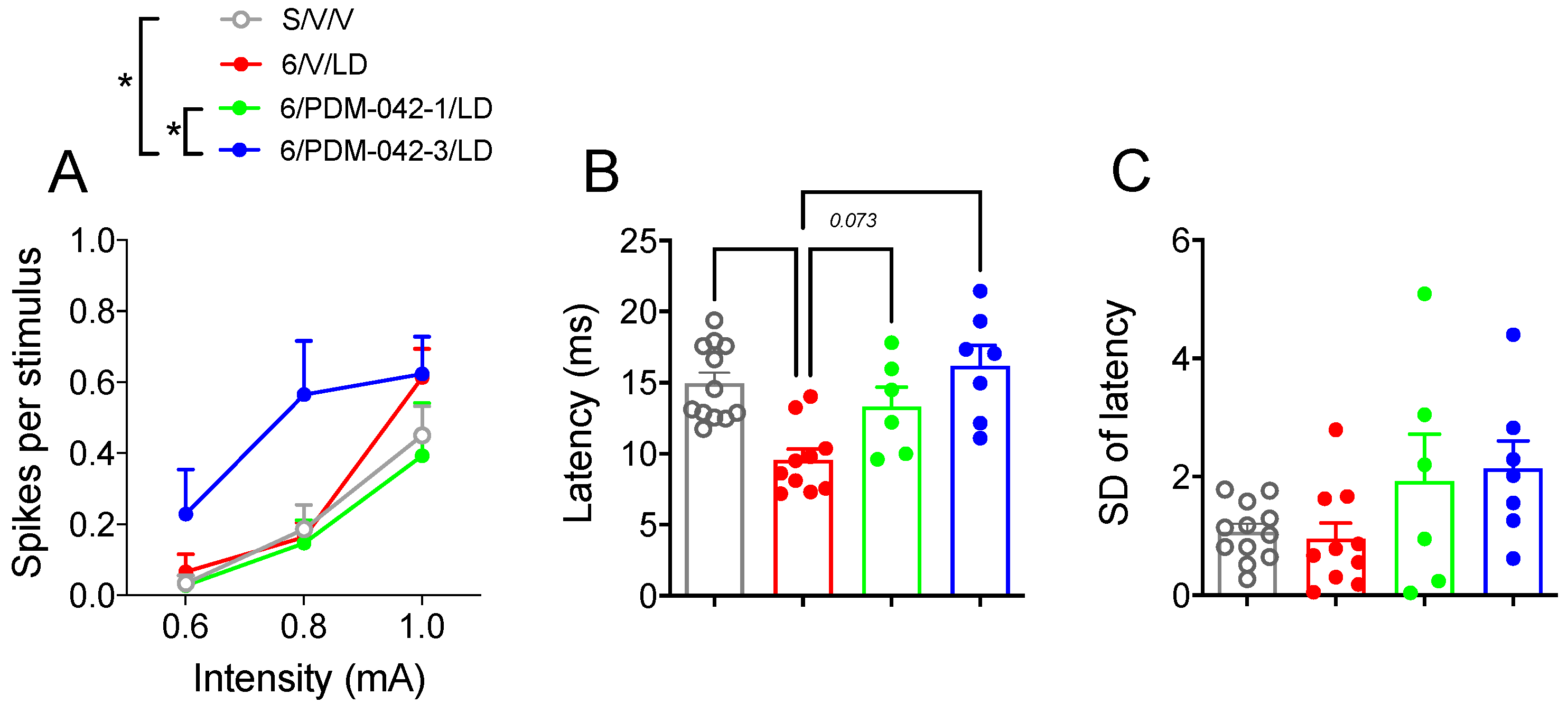
2.4.2. PDM-042 Alters Corticostriatal Activity in Dyskinetic Animals
3. Discussion
3.1. Modulation of AIMs by PDE10A Inhibition
3.2. Modulation of Striatal MSN Activity by PDE10A Inhibition
4. Materials and Methods
4.1. Animals
4.2. Drugs
4.3. 6-OHDA Lesion
4.4. Treatment Groups and Drug Administration
4.5. Stepping Test and Lesion Assessment
4.6. Abnormal Involuntary Movements (AIMs)
4.7. Electrophysiology
4.8. Tissue Processing
4.9. Tyrosine Hydroxylase Immunohistochemistry and Lesion Assessment
4.10. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tepper, J.M.; Bolam, J.P. Functional Diversity and Specificity of Neostriatal Interneurons. Curr. Opin. Neurobiol. 2004, 14, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Lanciego, J.L.; Luquin, N.; Obeso, J.A. Functional Neuroanatomy of the Basal Ganglia. Cold Spring Harb. Perspect. Med. 2012, 2, a009621. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Jenner, P. Parkinson Disease: From Pathology to Molecular Disease Mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodríguez-Oroz, M.C.; Benitez-Temino, B.; Blesa, F.J.; Guridi, J.; Marin, C.; Rodriguez, M. Functional Organization of the Basal Ganglia: Therapeutic Implications for Parkinson’s Disease. Mov. Disord. 2008, 23 (Suppl. 3), S548–S559. [Google Scholar] [CrossRef]
- Stayte, S.; Vissel, B. Advances in Non-Dopaminergic Treatments for Parkinson’s Disease. Front. Neurosci. 2014, 8, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabresi, P.; Di Filippo, M.; Ghiglieri, V.; Tambasco, N.; Picconi, B. Levodopa-Induced Dyskinesias in Patients with Parkinson’s Disease: Filling the Bench-to-Bedside Gap. Lancet Neurol. 2010, 9, 1106–1117. [Google Scholar] [CrossRef]
- DeLong, M.R. Primate Models of Movement Disorders of Basal Ganglia Origin. Trends Neurosci. 1990, 13, 281–285. [Google Scholar] [CrossRef]
- Ryan, M.B.; Bair-Marshall, C.; Nelson, A.B. Aberrant Striatal Activity in Parkinsonism and Levodopa-Induced Dyskinesia. Cell Rep. 2018, 23, 3438–3446.e5. [Google Scholar] [CrossRef]
- Cerasa, A.; Koch, G.; Donzuso, G.; Mangone, G.; Morelli, M.; Brusa, L.; Bassi, M.S.; Ponzo, V.; Picazio, S.; Passamonti, L.; et al. A Network Centred on the Inferior Frontal Cortex Is Critically Involved in Levodopa-Induced Dyskinesias. Brain 2015, 138, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Herz, D.M.; Haagensen, B.N.; Nielsen, S.H.; Madsen, K.H.; Løkkegaard, A.; Siebner, H.R. Resting-State Connectivity Predicts Levodopa-Induced Dyskinesias in Parkinson’s Disease. Mov. Disord. 2016, 31, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Herz, D.M.; Haagensen, B.N.; Christensen, M.S.; Madsen, K.H.; Rowe, J.B.; Løkkegaard, A.; Siebner, H.R. Abnormal Dopaminergic Modulation of Striato-Cortical Networks Underlies Levodopa-Induced Dyskinesias in Humans. Brain 2015, 138, 1658–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbach, D.; Conti, M.M.; Ostock, C.Y.; George, J.A.; Goldenberg, A.A.; Melikhov-Sosin, M.; Nuss, E.E.; Bishop, C. The Role of Primary Motor Cortex (M1) Glutamate and GABA Signaling in L-DOPA-Induced Dyskinesia in Parkinsonian Rats. J. Neurosci. 2016, 36, 9873–9887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erro, R.; Mencacci, N.E.; Bhatia, K.P. The Emerging Role of Phosphodiesterases in Movement Disorders. Mov. Disord. 2021, 36, 2225–2243. [Google Scholar] [CrossRef] [PubMed]
- Threlfell, S.; Sammut, S.; Menniti, F.S.; Schmidt, C.J.; West, A.R. Inhibition of Phosphodiesterase 10A Increases the Responsiveness of Striatal Projection Neurons to Cortical Stimulation. J. Pharmacol. Exp. Ther. 2009, 328, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, M.; D’Angelo, V.; Esposito, Z.; Nuccetelli, V.; Sorge, R.; Martorana, A.; Stefani, A.; Bernardi, G.; Sancesario, G. Lowered CAMP and CGMP Signalling in the Brain during Levodopa-Induced Dyskinesias in Hemiparkinsonian Rats: New Aspects in the Pathogenetic Mechanisms. Eur. J. Neurosci. 2008, 28, 941–950. [Google Scholar] [CrossRef]
- Picconi, B.; Bagetta, V.; Ghiglieri, V.; Paillè, V.; di Filippo, M.; Pendolino, V.; Tozzi, A.; Giampà, C.; Fusco, F.R.; Sgobio, C.; et al. Inhibition of Phosphodiesterases Rescues Striatal Long-Term Depression and Reduces Levodopa-Induced Dyskinesia. Brain 2011, 134, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Solís, O.; Espadas, I.; Del-Bel, E.A.; Moratalla, R. Nitric Oxide Synthase Inhibition Decreases L-DOPA-Induced Dyskinesia and the Expression of Striatal Molecular Markers in Pitx3(−/−) Aphakia Mice. Neurobiol. Dis. 2015, 73, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Padovan-Neto, F.E.; West, A.R. Regulation of Striatal Neuron Activity by Cyclic Nucleotide Signaling and Phosphodiesterase Inhibition: Implications for the Treatment of Parkinson’s Disease. Adv. Neurobiol. 2017, 17, 257–283. [Google Scholar]
- Beck, G.; Maehara, S.; Chang, P.L.; Papa, S.M. A Selective Phosphodiesterase 10A Inhibitor Reduces L -Dopa-Induced Dyskinesias in Parkinsonian Monkeys. Mov. Disord. 2018, 33, 805–814. [Google Scholar] [CrossRef]
- Arakawa, K.; Yuge, N.; Maehara, S. Ameliorative Effects of a Phosphodiesterase 10A Inhibitor, MR1916 on l -DOPA-Induced Dyskinesia in Parkinsonian Rats. Pharmacol. Rep. 2020, 72, 443–448. [Google Scholar] [CrossRef]
- Xie, W.R.; Deng, H.; Li, H.; Bowen, T.L.; Strong, J.A.; Zhang, J.M. Robust Increase of Cutaneous Sensitivity, Cytokine Production and Sympathetic Sprouting in Rats with Localized Inflammatory Irritation of the Spinal Ganglia. Neuroscience 2006, 142, 809–822. [Google Scholar] [CrossRef] [Green Version]
- Siuciak, J.A.; McCarthy, S.A.; Chapin, D.S.; Fujiwara, R.A.; James, L.C.; Williams, R.D.; Stock, J.L.; McNeish, J.D.; Strick, C.A.; Menniti, F.S.; et al. Genetic Deletion of the Striatum-Enriched Phosphodiesterase PDE10A: Evidence for Altered Striatal Function. Neuropharmacology 2006, 51, 374–385. [Google Scholar] [CrossRef] [PubMed]
- West, A.R.; Grace, A.A. The Nitric Oxide-Guanylyl Cyclase Signaling Pathway Modulates Membrane Activity States and Electrophysiological Properties of Striatal Medium Spiny Neurons Recorded in Vivo. J. Neurosci. 2004, 24, 1924–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padovan-Neto, F.E.; Sammut, S.; Chakroborty, S.; Dec, A.M.; Threlfell, S.; Campbell, P.W.; Mudrakola, V.; Harms, J.F.; Schmidt, C.J.; West, A.R. Facilitation of Corticostriatal Transmission Following Pharmacological Inhibition of Striatal Phosphodiesterase 10A: Role of Nitric Oxide-Soluble Guanylyl Cyclase-CGMP Signaling Pathways. J. Neurosci. 2015, 35, 5781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckman, P.R.A.; Blokland, A.; Bollen, E.P.P.; Prickaerts, J. Phosphodiesterase Inhibition and Modulation of Corticostriatal and Hippocampal Circuits: Clinical Overview and Translational Considerations. Neurosci. Biobehav. Rev. 2018, 87, 233–254. [Google Scholar] [CrossRef]
- Albin, R.L.; Young, A.B.; Penney, J.B. The Functional Anatomy of Basal Ganglia Disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- Alexander, G.E.; Crutcher, M.D. Functional Architecture of Basal Ganglia Circuits: Neural Substrates of Parallel Processing. Trends Neurosci. 1990, 13, 266–271. [Google Scholar] [CrossRef]
- Wahyu, I.D.; Chiken, S.; Hasegawa, T.; Sano, H.; Nambu, A. Abnormal Cortico-Basal Ganglia Neurotransmission in a Mouse Model of l-DOPA-Induced Dyskinesia. J. Neurosci. 2021, 41, 2668–2683. [Google Scholar] [CrossRef]
- Nishi, A.; Kuroiwa, M.; Miller, D.B.; O’Callaghan, J.P.; Bateup, H.S.; Shuto, T.; Sotogaku, N.; Fukuda, T.; Heintz, N.; Greengard, P.; et al. Distinct Roles of PDE4 and PDE10A in the Regulation of CAMP/PKA Signaling in the Striatum. J. Neurosci. 2008, 28, 10460–10471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polito, M.; Guiot, E.; Gangarossa, G.; Longueville, S.; Doulazmi, M.; Valjent, E.; Hervé, D.; Girault, J.A.; Paupardin-Tritsch, D.; Castro, L.R.V.; et al. Selective Effects of PDE10A Inhibitors on Striatopallidal Neurons Require Phosphatase Inhibition by DARPP-32. eNeuro 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Elsevier: Amsterdam, The Netherlands, 2007; ISBN 0080475132. [Google Scholar]
- Guimarães, R.P.; Ribeiro, D.L.; dos Santos, K.B.; Godoy, L.D.; Corrêa, M.R.; Padovan-Neto, F.E. The 6-Hydroxydopamine Rat Model of Parkinson’s Disease. J. Vis. Exp. 2021, 176. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Nikkhah, G.; Bentlage, C.; Björklund, A. Forelimb Akinesia in the Rat Parkinson Model: Differential Effects of Dopamine Agonists and Nigral Transplants as Assessed by a New Stepping Test. J. Neurosci. 1995, 15, 3863–3875. [Google Scholar] [CrossRef]
- Megens, A.A.H.P.; Hendrickx, H.M.R.; Mahieu, M.M.A.; Wellens, A.L.Y.; de Boer, P.; Vanhoof, G. PDE10A Inhibitors Stimulate or Suppress Motor Behavior Dependent on the Relative Activation State of the Direct and Indirect Striatal Output Pathways. Pharmacol. Res. Perspect. 2014, 2, e00057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.J.; Chapin, D.S.; Cianfrogna, J.; Corman, M.L.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; Lebel, L.A.; McCarthy, S.A.; Nelson, F.R.; et al. Preclinical Characterization of Selective Phosphodiesterase 10A Inhibitors: A New Therapeutic Approach to the Treatment of Schizophrenia. J. Pharmacol. Exp. Ther. 2008, 325, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, K.; Maehara, S. Combination of the phosphodiesterase 10A inhibitor, MR1916 with risperidone shows additive antipsychotic-like effects without affecting cognitive enhancement and cataleptic effects in rats. Neuropsychopharmacol Rep. 2020, 40, 190–195. [Google Scholar] [CrossRef]
- Wilson, J.M.; Ogden, A.M.L.; Loomis, S.; Gilmour, G.; Baucum, A.J.; Belecky-Adams, T.L.; Merchant, K.M. Phosphodiesterase 10A Inhibitor, MP-10 (PF-2545920), Produces Greater Induction of c-Fos in Dopamine D2 Neurons than in D1 Neurons in the Neostriatum. Neuropharmacology 2015, 99, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in Targeting Cyclic Nucleotide Phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Menniti, F.S.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for Drug Development. Nat. Rev. Drug Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef]
- Beaumont, V.; Zhong, S.; Lin, H.; Xu, W.; Bradaia, A.; Steidl, E.; Gleyzes, M.; Wadel, K.; Buisson, B.; Padovan-Neto, F.E.; et al. Phosphodiesterase 10A Inhibition Improves Cortico-Basal Ganglia Function in Huntington’s Disease Models. Neuron 2016, 92, 1220–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menniti, F.S.; Chappie, T.A.; Schmidt, C.J. PDE10A Inhibitors—Clinical Failure or Window Into Antipsychotic Drug Action? Front. Neurosci. 2021, 14, 1442. [Google Scholar] [CrossRef]
- Geerts, H.; Spiros, A.; Roberts, P. Phosphodiesterase 10 Inhibitors in Clinical Development for CNS Disorders. Expert Rev. Neurother. 2017, 17, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Cenci, M.A.; Jörntell, H.; Petersson, P. On the Neuronal Circuitry Mediating L-DOPA-Induced Dyskinesia. J. Neural Transm. 2018, 125, 1157–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belić, J.J.; Halje, P.; Richter, U.; Petersson, P.; Hellgren Kotaleski, J. Untangling Cortico-Striatal Connectivity and Cross-Frequency Coupling in L-DOPA-Induced Dyskinesia. Front. Syst. Neurosci. 2016, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupre, K.B.; Cruz, A.V.; McCoy, A.J.; Delaville, C.; Gerber, C.M.; Eyring, K.W.; Walters, J.R. Effects of L-Dopa Priming on Cortical High Beta and High Gamma Oscillatory Activity in a Rodent Model of Parkinson’s Disease. Neurobiol. Dis. 2016, 86, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-Z.; Rothwell, J.C.; Lu, C.-S.; Chuang, W.-L.; Chen, R.-S. Abnormal Bidirectional Plasticity-like Effects in Parkinson’s Disease. Brain 2011, 134, 2312–2320. [Google Scholar] [CrossRef] [Green Version]
- Guerra, A.; Suppa, A.; D’Onofrio, V.; Di Stasio, F.; Asci, F.; Fabbrini, G.; Berardelli, A. Abnormal Cortical Facilitation and L-Dopa-Induced Dyskinesia in Parkinson’s Disease. Brain Stimul. 2019, 12, 1517–1525. [Google Scholar] [CrossRef]
- Bonate, R.; Kurek, G.; Hrabak, M.; Patterson, S.; Padovan-Neto, F.; West, A.R.; Steiner, H. Phosphodiesterase 10A (PDE10A): Regulator of Dopamine Agonist-Induced Gene Expression in the Striatum. Cells 2022, 11, 2214. [Google Scholar] [CrossRef] [PubMed]
- Heiman, M.; Heilbut, A.; Francardo, V.; Kulicke, R.; Fenster, R.J.; Kolaczyk, E.D.; Mesirov, J.P.; Surmeier, D.J.; Cenci, M.A.; Greengard, P. Molecular Adaptations of Striatal Spiny Projection Neurons during Levodopa-Induced Dyskinesia. Proc. Natl. Acad. Sci. USA 2014, 111, 4578–4583. [Google Scholar] [CrossRef] [Green Version]
- Padovan-Neto, F.E.; Patterson, S.; Nivea, N.M.; Altwal, F.; Beverley, J.A.; West, A.R.; Steiner, H. Selective Regulation of 5-HT1B Serotonin Receptor Expression in the Striatum by Dopamine Depletion and Repeated L-DOPA Treatment: Relationship to L-DOPA-Induced Dyskinesias. Mol. Neurobiol. 2020, 57, 736. [Google Scholar] [CrossRef]
- Bariotto-dos-Santos, K.; Ribeiro, D.L.; Guimarães, R.P.; Padovan-Neto, F.E. Rating L-DOPA-Induced Dyskinesias in the Unilaterally 6-OHDA-Lesioned Rat Model of Parkinson’s Disease. J. Vis. Exp. 2021, 176, 1–16. [Google Scholar] [CrossRef]
- Jayasinghe, V.R.; Flores-Barrera, E.; West, A.R.; Tseng, K.Y. Frequency-Dependent Corticostriatal Disinhibition Resulting from Chronic Dopamine Depletion: Role of Local Striatal CGMP and GABA-AR Signaling. Cereb. Cortex 2017, 27, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenci, M.A.; Lee, C.S.; Björklund, A. L-DOPA-Induced Dyskinesia in the Rat Is Associated with Striatal Overexpression of Prodynorphin- and Glutamic Acid Decarboxylase MRNA. Eur. J. Neurosci. 1998, 10, 2694–2706. [Google Scholar] [CrossRef]
- Padovan-Neto, F.E.; Echeverry, M.B.; Tumas, V.; Del-Bel, E.A. Nitric Oxide Synthase Inhibition Attenuates L-DOPA-Induced Dyskinesias in a Rodent Model of Parkinson’s Disease. Neuroscience 2009, 159, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.; Kirik, D.; Björklund, A.; Cenci, M.A. L-DOPA-Induced Dyskinesia in the Intrastriatal 6-Hydroxydopamine Model of Parkinson’s Disease: Relation to Motor and Cellular Parameters of Nigrostriatal Function. Neurobiol. Dis. 2002, 10, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Sammut, S.; Threlfell, S.; West, A.R. Nitric Oxide-Soluble Guanylyl Cyclase Signaling Regulates Corticostriatal Transmission and Short-Term Synaptic Plasticity of Striatal Projection Neurons Recorded in Vivo. Neuropharmacology 2010, 58, 624–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharott, A.; Doig, N.M.; Mallet, N.; Magill, P.J. Relationships between the Firing of Identified Striatal Interneurons and Spontaneous and Driven Cortical Activities in Vivo. J. Neurosci. 2012, 32, 13221–13236. [Google Scholar] [CrossRef] [Green Version]
- Mallet, N.; Ballion, B.; Le Moine, C.; Gonon, F. Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J. Neurosci. 2006, 26, 3875–3884. [Google Scholar] [CrossRef] [Green Version]
- Notter, T.; Panzanelli, P.; Pfister, S.; Mircsof, D.; Fritschy, J.-M. A Protocol for Concurrent High-Quality Immunohistochemical and Biochemical Analyses in Adult Mouse Central Nervous System. Eur. J. Neurosci. 2014, 39, 165–175. [Google Scholar] [CrossRef]
- Tseng, K.Y.; Kargieman, L.; Gacio, S.; Riquelme, L.A.; Murer, M.G. Consequences of Partial and Severe Dopaminergic Lesion on Basal Ganglia Oscillatory Activity and Akinesia. Eur. J. Neurosci. 2005, 22, 2579–2586. [Google Scholar] [CrossRef]
- Tseng, K.Y.; Caballero, A.; Dec, A.; Cass, D.K.; Simak, N. Inhibition of Striatal Soluble Guanylyl Cyclase-CGMP Signaling Reverses Basal Ganglia Dysfunction and Akinesia in Experimental Parkinsonism. PLoS ONE 2011, 6, 27187. [Google Scholar] [CrossRef]
- Andersson, M.; Hilbertson, A.; Cenci, M.A. Striatal FosB Expression Is Causally Linked with L-DOPA-Induced Abnormal Involuntary Movements and the Associated Upregulation of Striatal Prodynorphin MRNA in a Rat Model of Parkinson’s Disease. Neurobiol. Dis. 1999, 6, 461–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenci, A.M.; Lundblad, M. Ratings of L-DOPA-Induced Dyskinesia in the Unilateral 6-OHDA Lesion Model of Parkinson’s Disease in Rats and Mice. In Current Protocols in Neuroscience; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Chapter 9; pp. 1–23. [Google Scholar] [CrossRef]
- Padovan-Neto, F.E.; Cavalcanti-Kiwiatkoviski, R.; Carolino, R.O.G.; Anselmo-Franci, J.; del Bel, E. Effects of Prolonged Neuronal Nitric Oxide Synthase Inhibition on the Development and Expression of L-DOPA-Induced Dyskinesia in 6-OHDA-Lesioned Rats. Neuropharmacology 2015, 89, 87–99. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guimarães, R.P.; Ribeiro, D.L.; Dos Santos, K.B.; Talarico, C.H.Z.; Godoy, L.D.; Padovan-Neto, F.E. Phosphodiesterase 10A Inhibition Modulates the Corticostriatal Activity and L-DOPA-Induced Dyskinesia. Pharmaceuticals 2022, 15, 947. https://doi.org/10.3390/ph15080947
Guimarães RP, Ribeiro DL, Dos Santos KB, Talarico CHZ, Godoy LD, Padovan-Neto FE. Phosphodiesterase 10A Inhibition Modulates the Corticostriatal Activity and L-DOPA-Induced Dyskinesia. Pharmaceuticals. 2022; 15(8):947. https://doi.org/10.3390/ph15080947
Chicago/Turabian StyleGuimarães, Rayanne Poletti, Danilo Leandro Ribeiro, Keila Bariotto Dos Santos, Carlos Henrique Zanello Talarico, Lívea Dornela Godoy, and Fernando E. Padovan-Neto. 2022. "Phosphodiesterase 10A Inhibition Modulates the Corticostriatal Activity and L-DOPA-Induced Dyskinesia" Pharmaceuticals 15, no. 8: 947. https://doi.org/10.3390/ph15080947