
Molecular Recognition of Methacryllysine and Crotonyllysine by the AF9 YEATS Domain
 , , and
, , and
Abstract
:1. Introduction
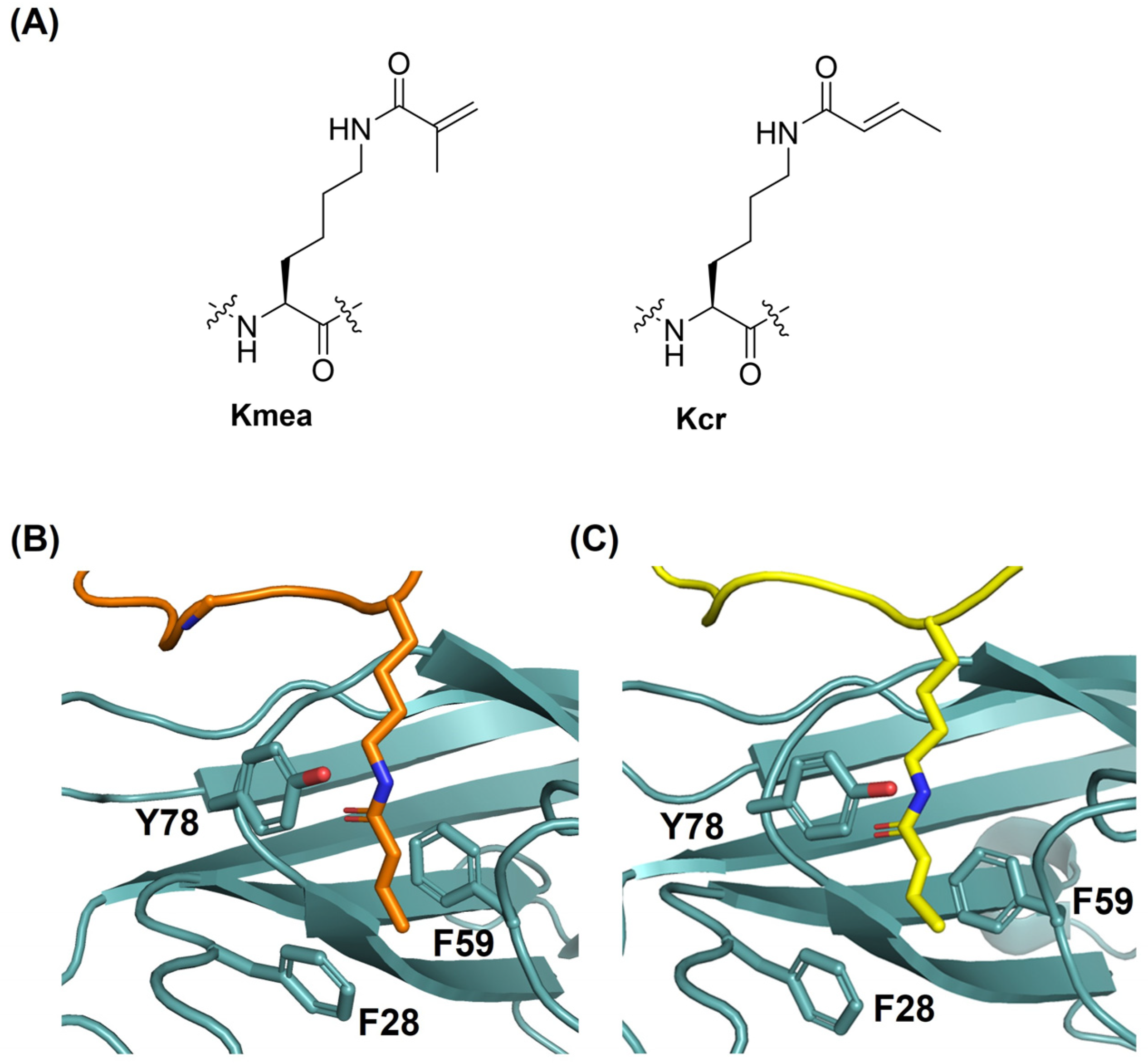
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of Histone Peptides
3.2. Expression and Purification of the AF9 YEATS Domain
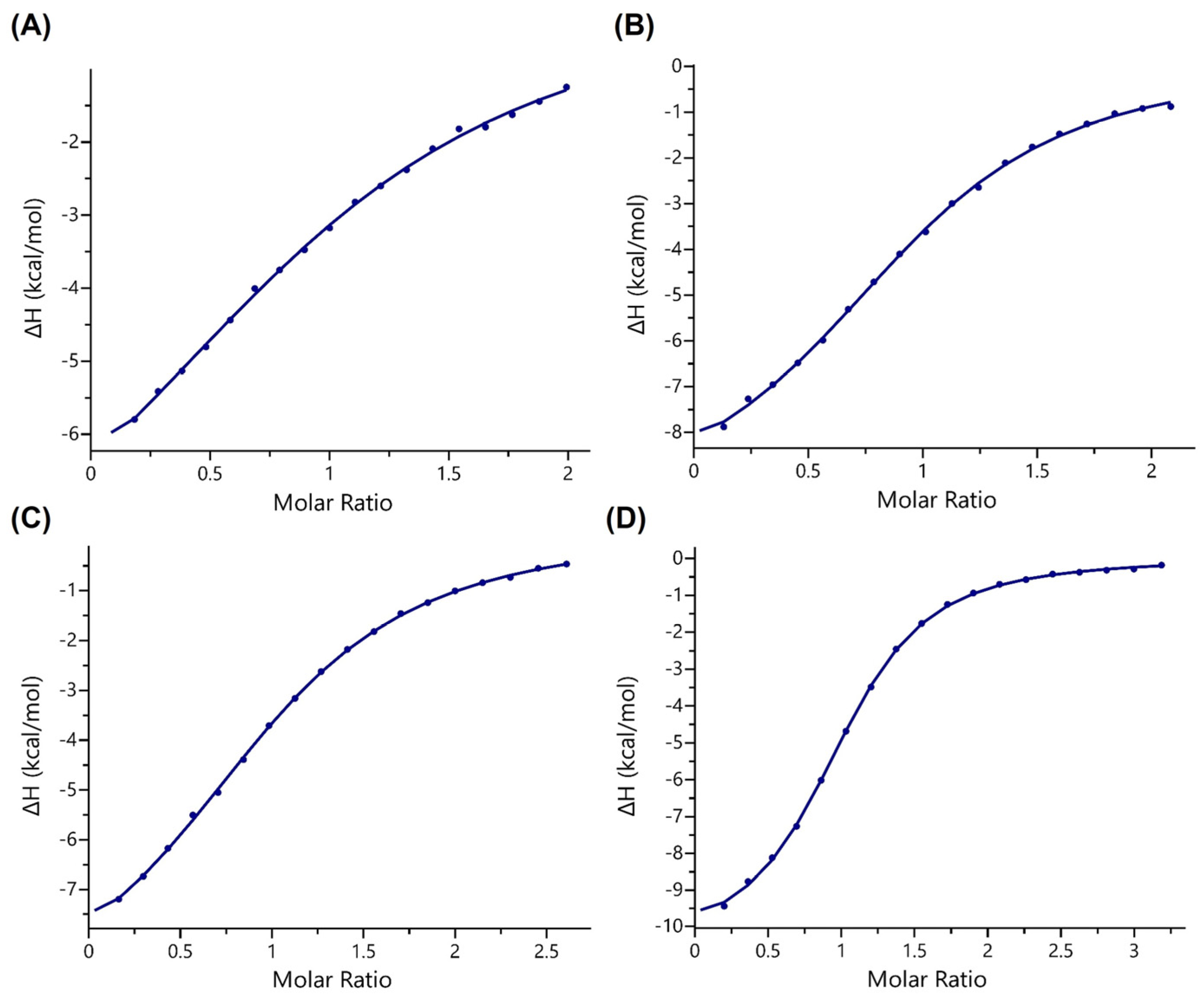
3.3. ITC Binding Assays
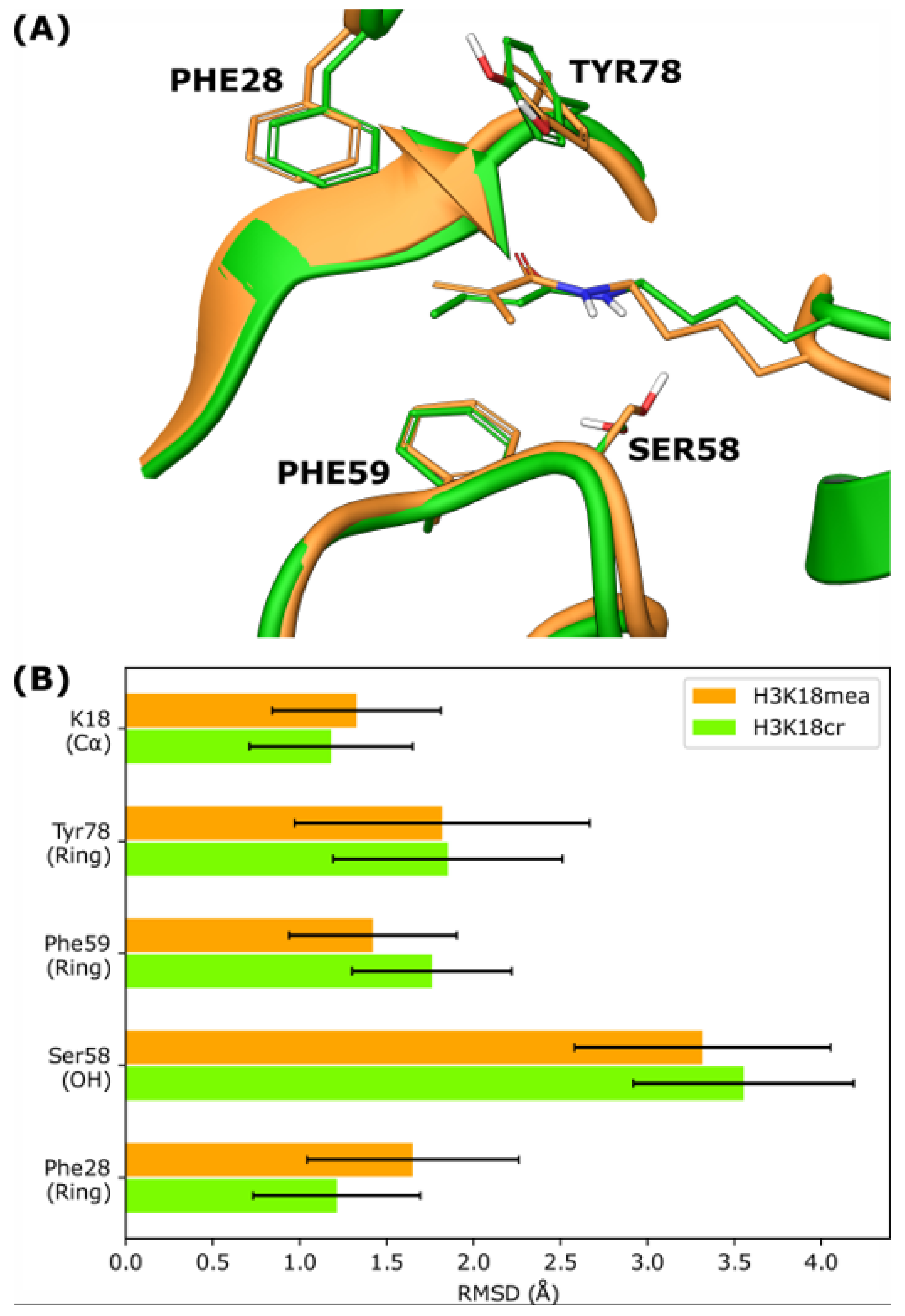
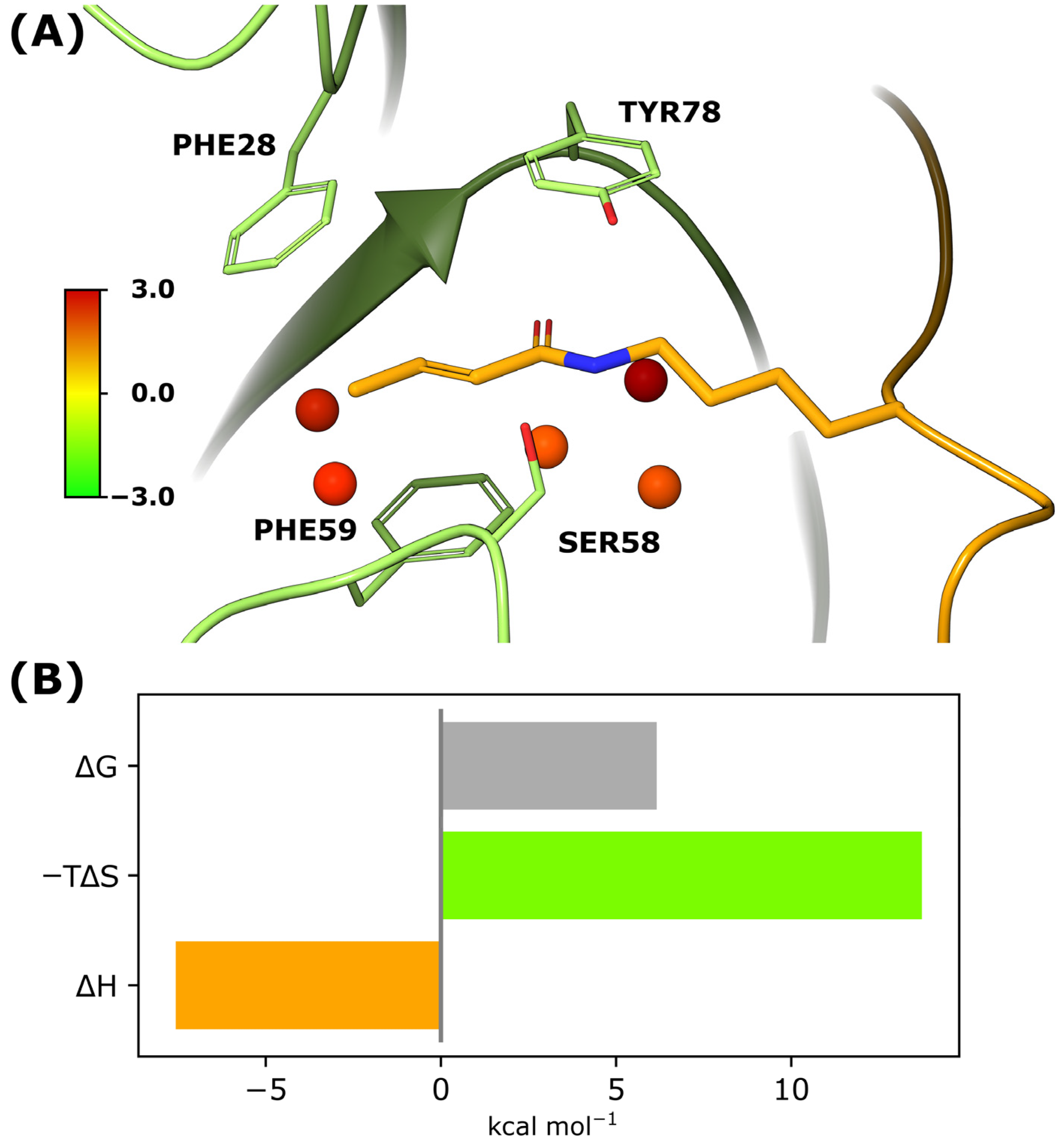
3.4. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, M.N.; Hintzen, J.C.J.; Mecinović, J. Probing lysine posttranslational modifications by unnatural amino acids. Chem. Commun. 2022, 58, 7216–7231. [Google Scholar] [CrossRef]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Zhou, X.; Taghizadeh, K.; Dong, M.; Dedon, P.C. N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Zhou, Y.; Xue, Z.; Hao, N.; Li, Y.; Guo, X.; Wang, D.; Shi, X.; Li, H. Histone benzoylation serves as an epigenetic mark for DPF and YEATS family proteins. Nucleic Acids Res. 2021, 49, 114–126. [Google Scholar] [CrossRef]
- Delaney, K.; Tan, M.; Zhu, Z.; Gao, J.; Dai, L.; Kim, S.; Ding, J.; He, M.; Halabelian, L.; Yang, L.; et al. Histone lysine methacrylation is a dynamic post-translational modification regulated by HAT1 and SIRT2. Cell Discov. 2021, 7, 122. [Google Scholar] [CrossRef]
- Sabari, B.R.; Tang, Z.; Huang, H.; Yong-Gonzalez, V.; Molina, H.; Kong, H.E.; Dai, L.; Shimada, M.; Cross, J.R.; Zhao, Y.; et al. Intracellular Crotonyl-CoA Stimulates Transcription through p300-Catalyzed Histone Crotonylation. Mol. Cell 2015, 58, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Li, C.; Lu, M.; Lu, K.; Li, H. Protein lysine crotonylation: Past, present, perspective. Cell Death Dis. 2021, 12, 703. [Google Scholar] [CrossRef]
- Liu, X.; Wei, W.; Liu, Y.; Yang, X.; Wu, J.; Zhang, Y.; Zhang, Q.; Shi, T.; Du, J.X.; Zhao, Y.; et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov. 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musselman, C.A.; Lalonde, M.-E.; Côté, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.J.; Wang, Z. Readout of epigenetic modifications. Annu. Rev. Biochem. 2013, 82, 81–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, E.M.; Huang, O.W.; Poy, F.; Oppikofer, M.; Bellon, S.F.; Tang, Y.; Cochran, A.G. A Subset of Human Bromodomains Recognizes Butyryllysine and Crotonyllysine Histone Peptide Modifications. Structure 2015, 23, 1801–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Panchenko, T.; Yang, S.; Zhao, S.; Yan, P.; Zhang, W.; Xie, W.; Li, Y.; Zhao, Y.; Allis, C.D.; et al. Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat. Chem. Biol. 2016, 12, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Li, Y.; Xiong, X.; Chen, Z.; Li, H. YEATS Domain—A Histone Acylation Reader in Health and Disease. J. Mol. Biol. 2017, 429, 1994–2002. [Google Scholar] [CrossRef]
- Li, Y.; Sabari, B.R.; Panchenko, T.; Wen, H.; Zhao, D.; Guan, H.; Wan, L.; Huang, H.; Tang, Z.; Zhao, Y.; et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol. Cell 2016, 62, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wen, H.; Xi, Y.; Tanaka, K.; Wang, H.; Peng, D.; Ren, Y.; Jin, Q.; Dent, S.Y.R.; Li, W.; et al. AF9 YEATS Domain Links Histone Acetylation to DOT1L-Mediated H3K79 Methylation. Cell 2014, 159, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zeng, L.; Zhao, C.; Ju, Y.; Konuma, T.; Zhou, M.-M. Structural Insights into Histone Crotonyl-Lysine Recognition by the AF9 YEATS Domain. Structure 2016, 24, 1606–1612. [Google Scholar] [CrossRef] [Green Version]
- Travis, C.R.; Francis, D.Y.; Williams, D.C.; Waters, M.L. Evaluation of acyllysine isostere interactions with the aromatic pocket of the AF9 YEATS domain. Protein Sci. 2023, 32, e4533. [Google Scholar] [CrossRef]
- Krone, M.W.; Travis, C.R.; Lee, G.Y.; Eckvahl, H.J.; Houk, K.N.; Waters, M.L. More Than π–π–π Stacking: Contribution of Amide−π and CH−π Interactions to Crotonyllysine Binding by the AF9 YEATS Domain. J. Am. Chem. Soc. 2020, 142, 17048–17056. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Wen, H.; Li, Y.; Lyu, J.; Xi, Y.; Hoshii, T.; Joseph, J.K.; Wang, X.; Loh, Y.-H.E.; Erb, M.A.; et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 2017, 543, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, X.-M.; Jiang, Y.; Liu, Z.; Cui, Y.; Fung, K.Y.; van der Beelen, S.H.E.; Tian, G.; Wan, L.; Shi, X.; et al. Structure-guided development of YEATS domain inhibitors by targeting π-π-π stacking. Nat. Chem. Biol. 2018, 14, 1140–1149. [Google Scholar] [CrossRef]
- Liu, Y.; Jin, R.; Lu, H.; Bian, K.; Wang, R.; Wang, L.; Gao, R.; Zhang, J.; Wu, J.; Yao, X.; et al. Fragment-Based Discovery of AF9 YEATS Domain Inhibitors. Int. J. Mol. Sci. 2022, 23, 3893. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Nguyen, C.N.; Young, T.K.; Gilson, M.K. Grid inhomogeneous solvation theory: Hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril. J. Chem. Phys. 2012, 137, 044101. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.N.; Cruz, A.; Gilson, M.K.; Kurtzman, T. Thermodynamics of Water in an Enzyme Active Site: Grid-Based Hydration Analysis of Coagulation Factor Xa. J. Chem. Theory Comput. 2014, 10, 2769–2780. [Google Scholar] [CrossRef] [Green Version]
- Maestro, S. Schrödinger, LLC.: New York, NY, USA, 2020. Available online: https://www.schrodinger.com/products/maestro (accessed on 5 April 2023).
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc 2001, 222, U403. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2019, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Molecular dynamics simulation analyses of viral fusion peptides in membranes prone to phase transition: Effects on membrane curvature, phase behavior and lipid-water interface destabilization. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; Gilson, M.K.; et al. Amber18; University of California, San Francisco: San Francisco, CA, USA, 2018. [Google Scholar]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. B 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Lazaridis, T. Inhomogeneous Fluid Approach to Solvation Thermodynamics. 1. Theory. J. Phys. Chem. B 1998, 102, 3531–3541. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Yoshida, N.; Hirata, F. Placevent: An algorithm for prediction of explicit solvent atom distribution—Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Kd (μM) | N (Stoichiometry) |
|---|---|---|
| H3K18mea | 114 ± 17 | 0.99 ± 0.03 |
| H3K18cr | 21 ± 2 | 0.98 ± 0.03 |
| H3K9mea | 59 ± 0.3 | 1.02 ± 0.01 |
| H3K9cr | 11 ± 0.8 | 0.98 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilgin, N.; Moesgaard, L.; Rahman, M.M.; Türkmen, V.A.; Kongsted, J.; Mecinović, J. Molecular Recognition of Methacryllysine and Crotonyllysine by the AF9 YEATS Domain. Int. J. Mol. Sci. 2023, 24, 7002. https://doi.org/10.3390/ijms24087002
Bilgin N, Moesgaard L, Rahman MM, Türkmen VA, Kongsted J, Mecinović J. Molecular Recognition of Methacryllysine and Crotonyllysine by the AF9 YEATS Domain. International Journal of Molecular Sciences. 2023; 24(8):7002. https://doi.org/10.3390/ijms24087002
Chicago/Turabian StyleBilgin, Nurgül, Laust Moesgaard, Mohammad M. Rahman, Vildan A. Türkmen, Jacob Kongsted, and Jasmin Mecinović. 2023. "Molecular Recognition of Methacryllysine and Crotonyllysine by the AF9 YEATS Domain" International Journal of Molecular Sciences 24, no. 8: 7002. https://doi.org/10.3390/ijms24087002