
First Characterization of the Transcriptome of Lung Fibroblasts of SSc Patients and Healthy Donors of African Ancestry

Abstract
:1. Introduction
2. Results
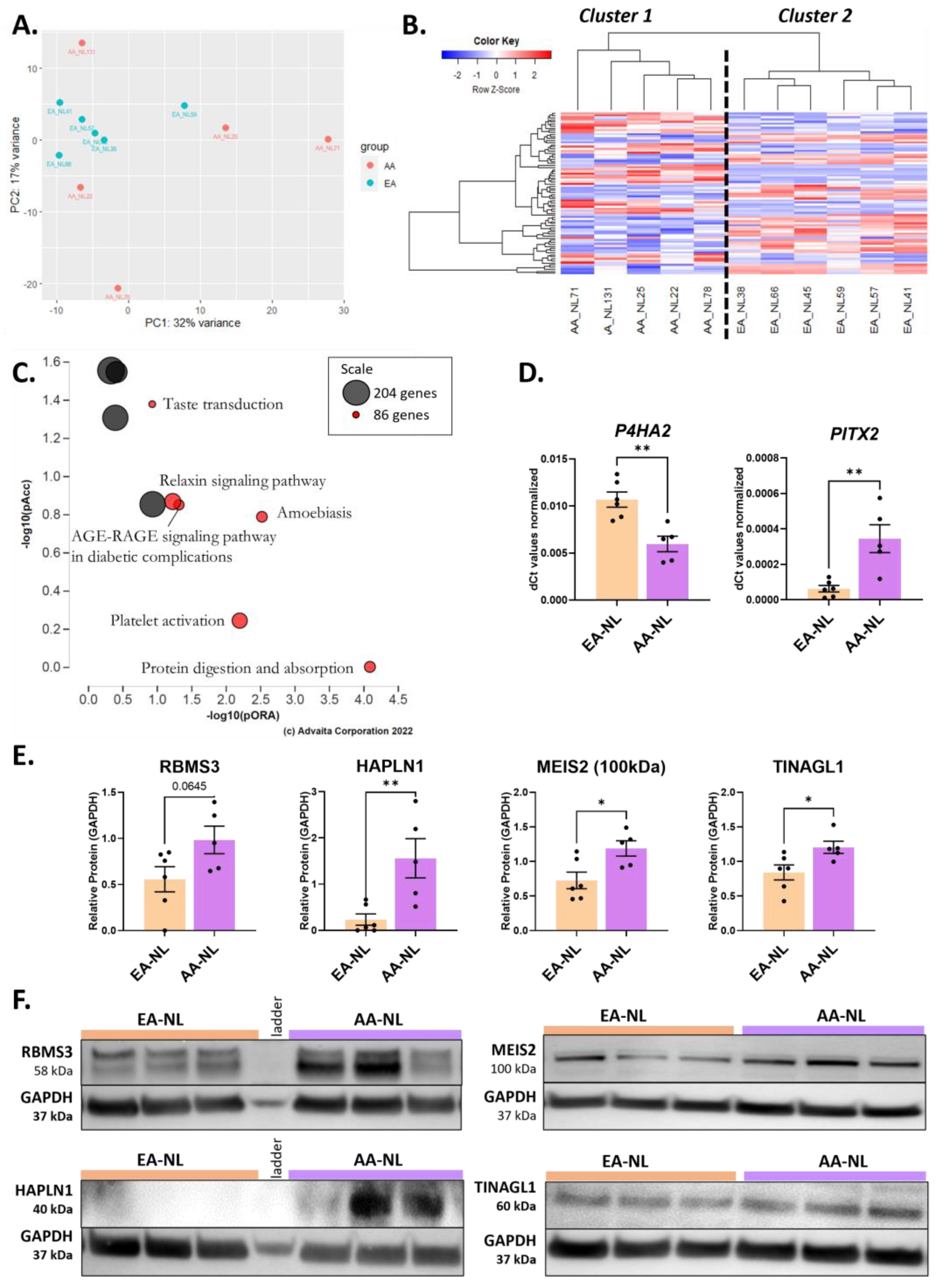
2.1. Perturbation of ECM Organization, Regulation of Myoblast Proliferation, and Histone Methyltransferase Activity in AA-NL Fibroblasts
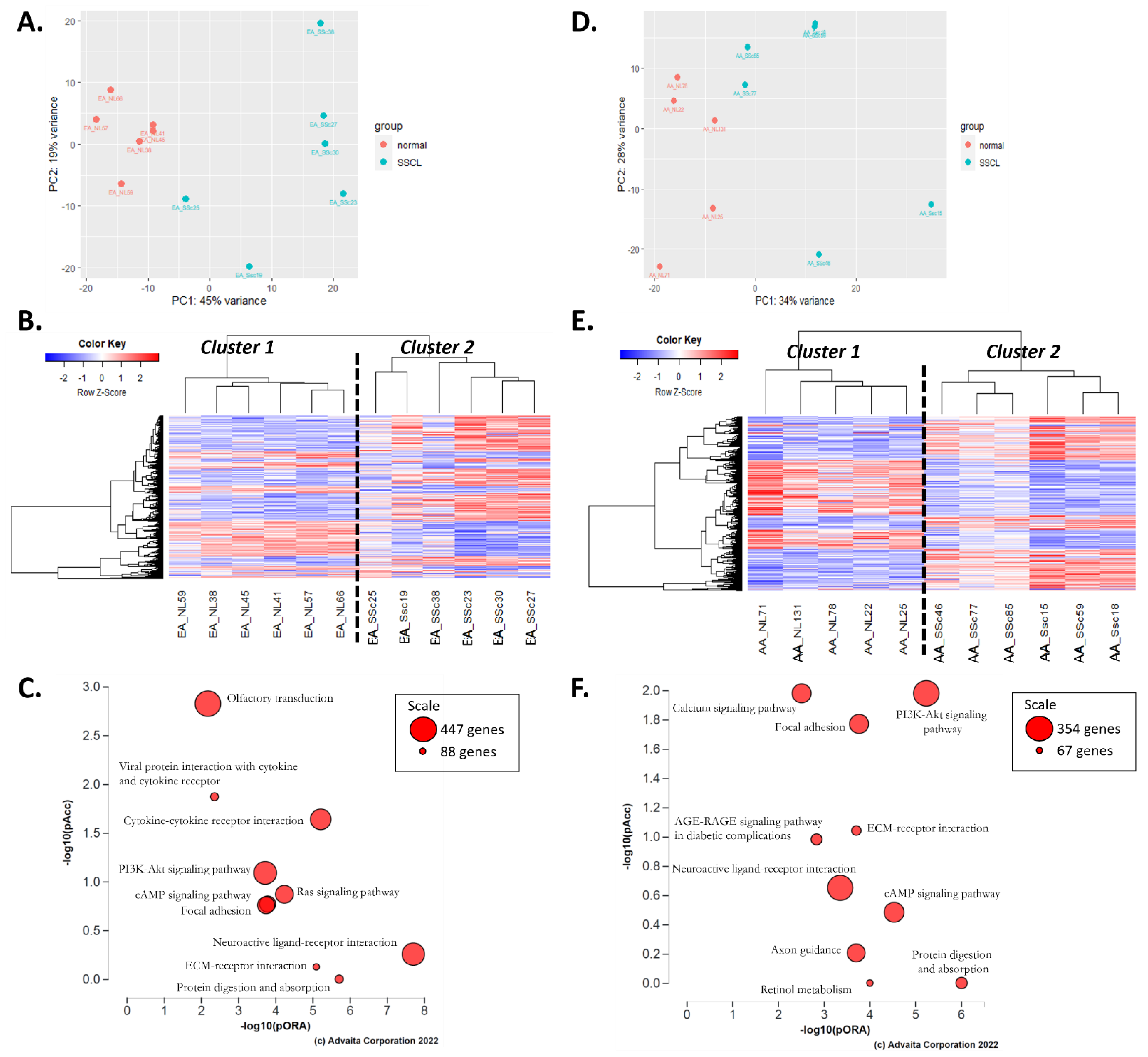
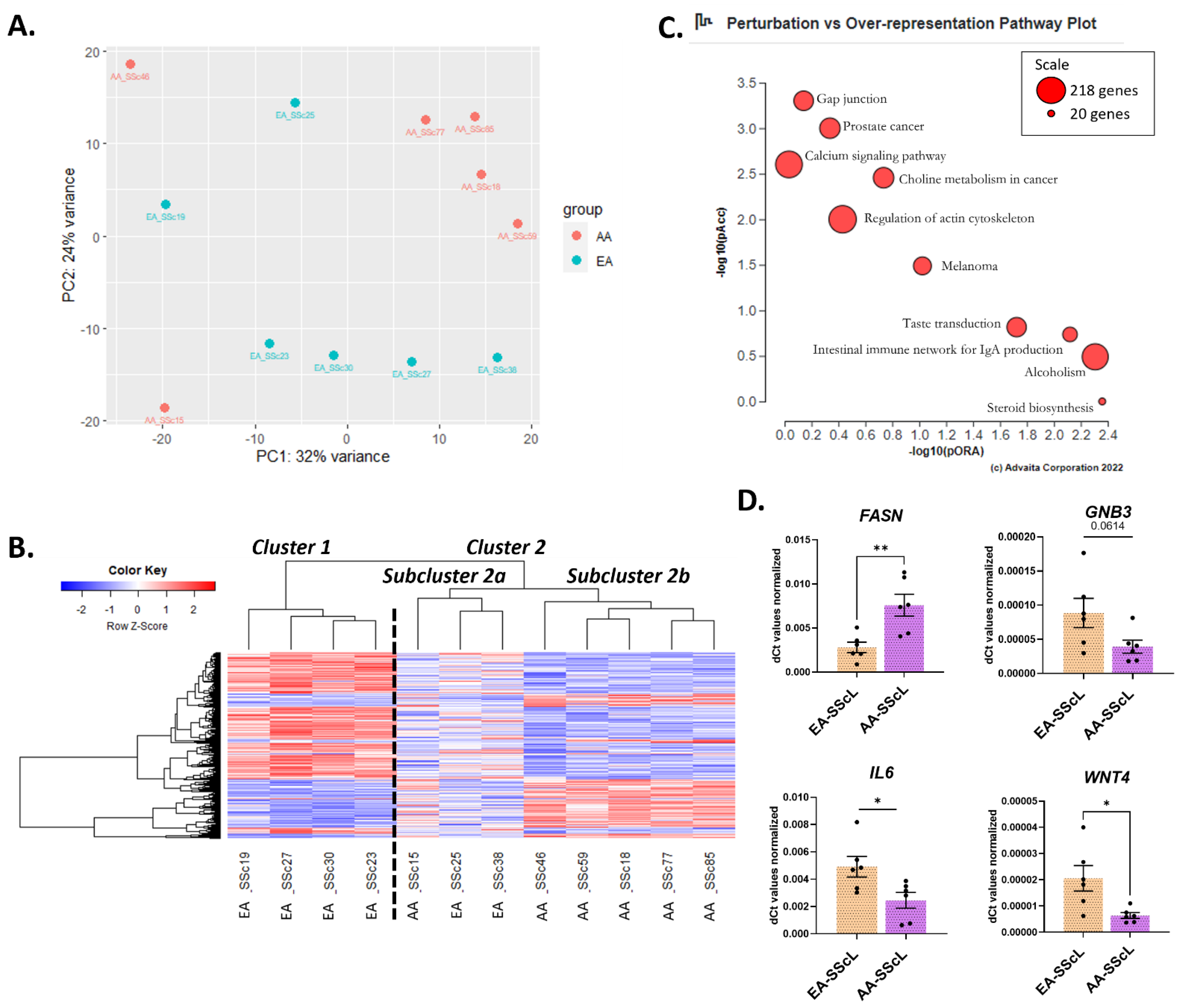
2.2. Comparison of Disease Mechanisms in Fibroblasts of AA and EA Patients with SSc
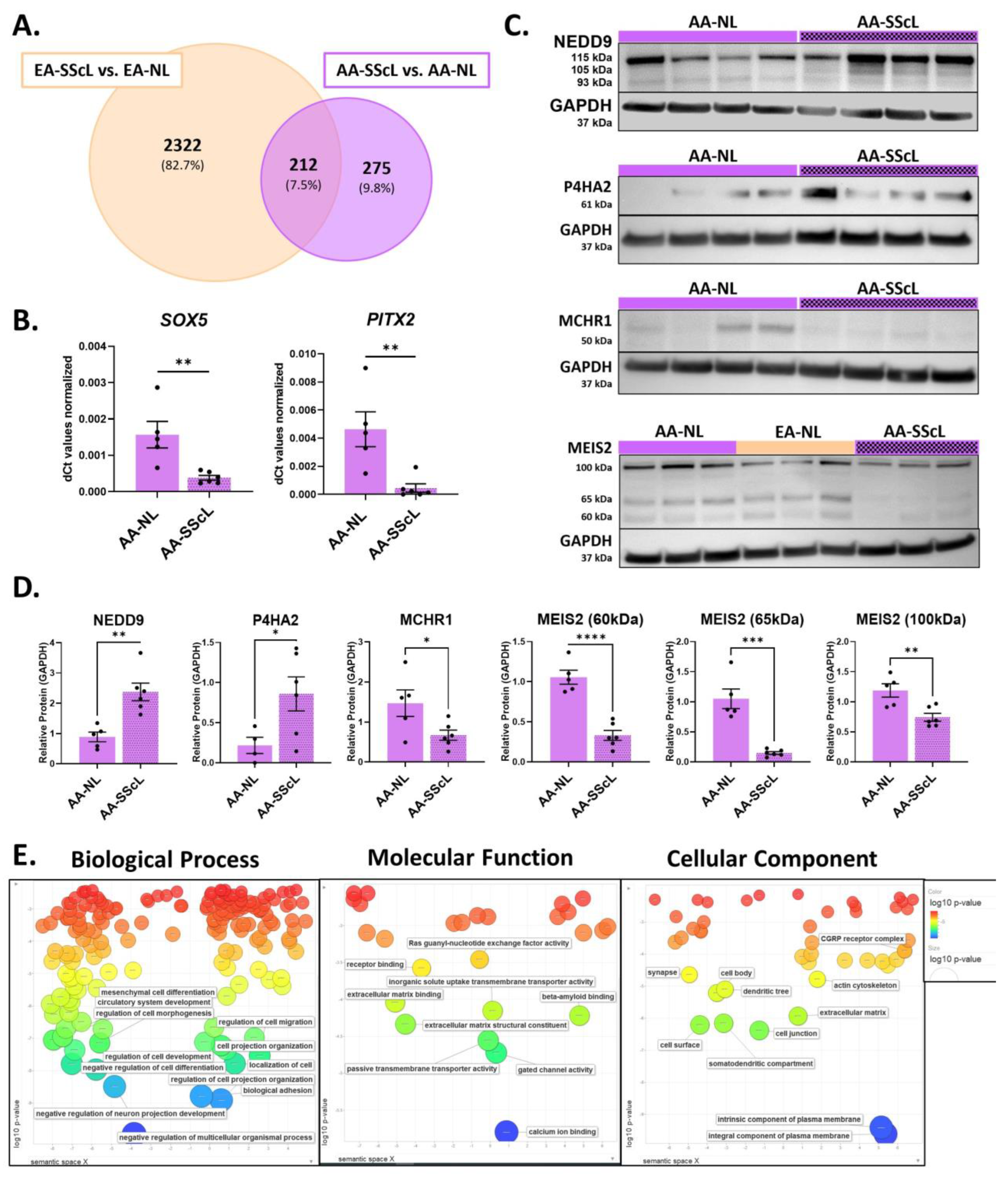
2.3. AA-SScL vs. EA-SScL: What Are the Differences in Disease State?
3. Discussion
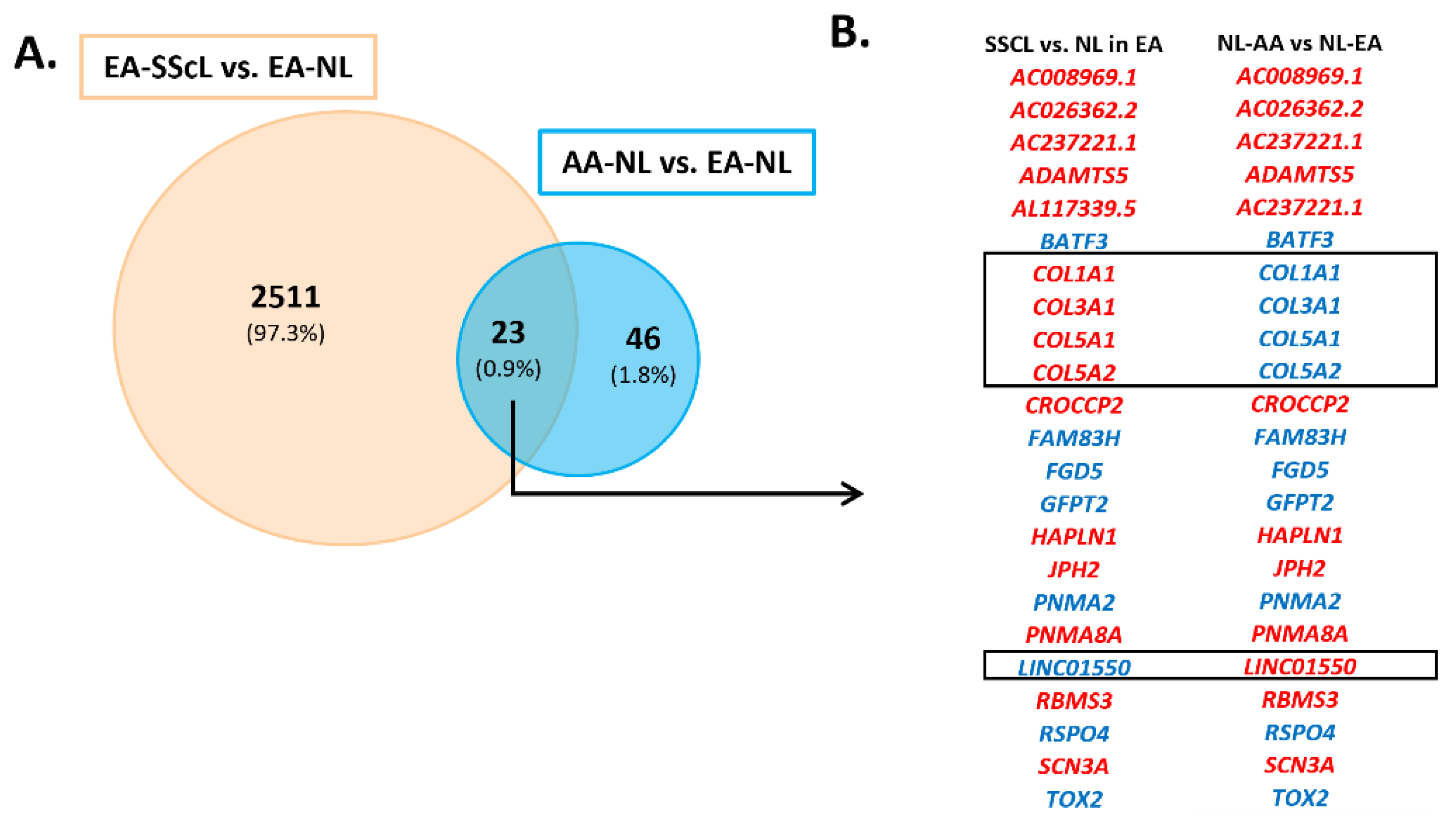
3.1. The Unique Transcriptome of AA-NL Fibroblasts (AA-NL vs. EA-NL)
3.2. The Transcriptome of AA-NL Fibroblasts Has Disease-Like Features
3.3. Disease Mechanisms Unique to AA-SScL
3.4. The Unique Transcriptome of AA-SScL Fibroblasts (AA-SScL vs. EA-SScL)
4. Materials and Methods
4.1. Patient Information and Cell Culture of the Primary Human Pulmonary Fibroblasts
4.2. RNA Extraction and Preparation
4.3. RNA Sequencing & Differential Expression Analysis
4.4. Functional Enrichment and Impact Analysis
4.5. Immunoblotting
4.6. cDNA and qPCR
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cottin, V.; Brown, K.K. Interstitial lung disease associated with systemic sclerosis (SSc-ILD). Respir. Res. 2019, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Tyndall, A.J.; Bannert, B.; Vonk, M.; Airò, P.; Cozzi, F.; Carreira, P.E.; Bancel, D.F.; Allanore, Y.; Müller-Ladner, U.; Distler, O.; et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 2010, 69, 1809–1815. [Google Scholar] [CrossRef]
- Highland, K.B.; Distler, O.; Kuwana, M.; Allanore, Y.; Assassi, S.; Azuma, A.; Bourdin, A.; Denton, C.P.; Distler, J.H.; Hoffmann-Vold, A.M. Efficacy and safety of nintedanib in patients with systemic sclerosis-associated interstitial lung disease treated with mycophenolate: A subgroup analysis of the SENSCIS trial. Lancet Respir. Med. 2021, 9, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Lin, C.J.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Reveille, J.D. Ethnicity and race and systemic sclerosis: How it affects susceptibility, severity, antibody genetics, and clinical manifestations. Curr. Rheumatol. Rep. 2003, 5, 160–167. [Google Scholar] [CrossRef]
- Gelber, A.C.; Manno, R.L.; Shah, A.A.; Woods, A.; Le, E.N.; Boin, F.; Hummers, L.K.; Wigley, F.M. Race and Association With Disease Manifestations and Mortality in Scleroderma. Medicine 2013, 92, 191–205. [Google Scholar] [CrossRef]
- Mayes, M.D.; Lacey, J.V., Jr.; Beebe-Dimmer, J.; Gillespie, B.W.; Cooper, B.; Laing, T.J.; Schottenfeld, D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2003, 48, 2246–2255. [Google Scholar] [CrossRef]
- Nietert, P.J.; Mitchell, H.C.; Bolster, M.B.; Shaftman, S.R.; Tilley, B.C.; Silver, R.M. Racial variation in clinical and immunological manifestations of systemic sclerosis. J. Rheumatol. 2006, 33, 263–268. [Google Scholar]
- Moore, D.F.; Kramer, E.; Eltaraboulsi, R.; Steen, V.D. Increased morbidity and mortality of scleroderma in African Americans compared to non–African Americans. Arthritis Care Res. 2019, 71, 1154–1163. [Google Scholar] [CrossRef]
- Nietert, P.J.; Silver, R.M.; Mitchell, H.C.; Shaftman, S.R.; Tilley, B.C. Demographic and clinical factors associated with in-hospital death among patients with systemic sclerosis. J. Rheumatol. 2005, 32, 1888–1892. [Google Scholar]
- Silver, R.M.; Bogatkevich, G.; Tourkina, E.; Nietert, P.J.; Hoffman, S. Racial differences between blacks and whites with systemic sclerosis. Curr. Opin. Rheumatol. 2012, 24, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Garrett, S.M.; Baker Frost, D.; Feghali-Bostwick, C. The mighty fibroblast and its utility in scleroderma research. J. scleroderma and related disorders 2017, 2, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Akter, T.; Silver, R.M.; Bogatkevich, G.S. Recent advances in understanding the pathogenesis of scleroderma-interstitial lung disease. Curr. Rheumatol. Rep. 2014, 16, 411. [Google Scholar] [CrossRef] [PubMed]
- Castelino, F.V.; Varga, J. Emerging cellular and molecular targets in fibrosis: Implications for scleroderma pathogenesis and targeted therapy. Curr. Opin. Rheumatol. 2014, 26, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Carrino, D.A.; Lauer, M.E.; Vasanji, A.; Wylie, J.D.; Nelson, C.M.; Apte, S.S. Pericellular versican regulates the fibroblast-myofibroblast transition: A role for ADAMTS5 protease-mediated proteolysis. J. Biol. Chem. 2011, 286, 34298–34310. [Google Scholar] [CrossRef] [PubMed]
- Velasco, J.; Li, J.; DiPietro, L.; Stepp, M.A.; Sandy, J.D.; Plaas, A. Adamts5 deletion blocks murine dermal repair through CD44-mediated aggrecan accumulation and modulation of transforming growth factor β1 (TGFβ1) signaling. J. Biol. Chem. 2011, 286, 26016–26027. [Google Scholar] [CrossRef]
- Lam, A.P.; Flozak, A.S.; Russell, S.; Wei, J.; Jain, M.; Mutlu, G.M.; Budinger, G.S.; Feghali-Bostwick, C.A.; Varga, J.; Gottardi, C.J. Nuclear β-catenin is increased in systemic sclerosis pulmonary fibrosis and promotes lung fibroblast migration and proliferation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 915–922. [Google Scholar] [CrossRef]
- Zhang, X.; MacDonald, B.T.; Gao, H.; Shamashkin, M.; Coyle, A.J.; Martinez, R.V.; He, X. Characterization of Tiki, a new family of Wnt-specific metalloproteases. J. Biol. Chem. 2016, 291, 2435–2443. [Google Scholar] [CrossRef]
- Wei, J.; Fang, F.; Lam, A.P.; Sargent, J.L.; Hamburg, E.; Hinchcliff, M.E.; Gottardi, C.J.; Atit, R.; Whitfield, M.L.; Varga, J. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012, 64, 2734–2745. [Google Scholar] [CrossRef]
- Åhrman, E.; Hallgren, O.; Malmström, L.; Hedström, U.; Malmström, A.; Bjermer, L.; Zhou, X.-H.; Westergren-Thorsson, G.; Malmström, J. Quantitative proteomic characterization of the lung extracellular matrix in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. J. Proteom. 2018, 189, 23–33. [Google Scholar] [CrossRef]
- Sato, M.; Nagano, T. Involvement of filamin A and filamin A-interacting protein (FILIP) in controlling the start and cell shape of radially migrating cortical neurons. Anat. Sci. Int. 2005, 80, 19–29. [Google Scholar] [CrossRef]
- Mezawa, M.; Pinto, V.I.; Kazembe, M.P.; Lee, W.S.; McCulloch, C.A. Filamin A regulates the organization and remodeling of the pericellular collagen matrix. FASEB J. 2016, 30, 3613–3627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Jiang, Z.; Wang, J.; Yang, N.; Qi, Q.; Zhou, W.; Feng, Z.; Li, W.; Zhang, Q.; Huang, B. Epithelial membrane protein 1 promotes glioblastoma progression through the PI3K/AKT/mTOR signaling pathway. Oncol. Rep. 2019, 42, 605–614. [Google Scholar] [CrossRef]
- Wulf, P.; Suter, U. Embryonic expression of epithelial membrane protein 1 in early neurons. Dev. Brain Res. 1999, 116, 169–180. [Google Scholar] [CrossRef]
- Taylor, V.; Welcher, A.A.; Amgen, E.P.; Suter, U. Epithelial membrane protein-1, peripheral myelin protein 22, and lens membrane protein 20 define a novel gene family. J. Biol. Chem. 1995, 270, 28824–28833. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Wang, A.; Meisgen, F.; Pivarcsi, A.; Sonkoly, E.; Ståhle, M.; Landén, N.X. MicroRNA-31 promotes skin wound healing by enhancing keratinocyte proliferation and migration. J. Investig. Dermatol. 2015, 135, 1676–1685. [Google Scholar] [CrossRef]
- Akaiwa, M.; Fukui, E.; Matsumoto, H. Tubulointerstitial nephritis antigen-like 1 deficiency alleviates age-dependent depressed ovulation associated with ovarian collagen deposition in mice. Reprod. Med. Biol. 2020, 19, 50–57. [Google Scholar] [CrossRef]
- Bergmeier, V.; Etich, J.; Pitzler, L.; Frie, C.; Koch, M.; Fischer, M.; Rappl, G.; Abken, H.; Tomasek, J.J.; Brachvogel, B. Identification of a myofibroblast-specific expression signature in skin wounds. Matrix Biol. 2018, 65, 59–74. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.; Ceribelli, A.; Cavaciocchi, F.; Crotti, C.; Massarotti, M.; Belloli, L.; Marasini, B.; Isailovic, N.; Generali, E.; Selmi, C. Nailfold videocapillaroscopy and serum VEGF levels in scleroderma are associated with internal organ involvement. Autoimmun. Highlights 2016, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Dong, Z.; Gu, H.; Guo, Z.; Yu, Z. TINAGL1 promotes hepatocellular carcinogenesis through the activation of TGF-β signaling-medicated VEGF expression. Cancer Manag. Res. 2019, 11, 767. [Google Scholar] [CrossRef]
- Distler, O.; Distler, J.H.W.; Scheid, A.; Acker, T.; Hirth, A.; Rethage, J.; Michel, B.A.; Gay, R.E.; Muüller-Ladner, U.; Matucci-Cerinic, M. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ. Res. 2004, 95, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Hjalt, T.A.; Amendt, B.A.; Murray, J.C. PITX2 regulates procollagen lysyl hydroxylase (PLOD) gene expression: Implications for the pathology of Rieger syndrome. J. Cell Biol. 2001, 152, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Cai, G.; Baugh, L.M.; Liu, Z.; Smith, A.; Watson, M.; Popovich, D.; Zhang, T.; Stawski, L.S.; Trojanowska, M. Systemic Sclerosis Dermal Fibroblasts Induce Cutaneous Fibrosis Through Lysyl Oxidase–like 4: New Evidence From Three-Dimensional Skin-like Tissues. Arthritis Rheumatol. 2020, 72, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Schweickert, A.; Campione, M.; Steinbeisser, H.; Blum, M. Pitx2 isoforms: Involvement of Pitx2c but not Pitx2a or Pitx2b in vertebrate left–right asymmetry. Mech. Dev. 2000, 90, 41–51. [Google Scholar] [CrossRef]
- Shiratori, H.; Sakuma, R.; Watanabe, M.; Hashiguchi, H.; Mochida, K.; Sakai, Y.; Nishino, J.; Saijoh, Y.; Whitman, M.; Hamada, H. Two-step regulation of left–right asymmetric expression of Pitx2: Initiation by nodal signaling and maintenance by Nkx2. Mol. Cell 2001, 7, 137–149. [Google Scholar] [CrossRef]
- Schayek, H.; Bentov, I.; Jacob-Hirsch, J.; Yeung, C.; Khanna, C.; Helman, L.J.; Plymate, S.; Werner, H. Global methylation analysis identifies PITX2 as an upstream regulator of the androgen receptor and IGF-I receptor genes in prostate cancer. Horm. Metab. Res. 2012, 44, 511–519. [Google Scholar] [CrossRef]
- Liu, H.; Wu, X.; Gan, C.; Wang, L.; Wang, G.; Yue, L.; Liu, Z.; Wei, W.; Su, X.; Zhang, Q. A novel multikinase inhibitor SKLB-YTH-60 ameliorates inflammation and fibrosis in bleomycin-induced lung fibrosis mouse models. Cell Prolif. 2021, 54, e13081. [Google Scholar] [CrossRef]
- Machon, O.; Masek, J.; Machonova, O.; Krauss, S.; Kozmik, Z. Meis2 is essential for cranial and cardiac neural crest development. BMC Dev. Biol. 2015, 15, 1–16. [Google Scholar] [CrossRef]
- Fernandes-Silva, H.; Vaz-Cunha, P.; Barbosa, V.B.; Silva-Gonçalves, C.; Correia-Pinto, J.; Moura, R.S. Retinoic acid regulates avian lung branching through a molecular network. Cell. Mol. Life Sci. 2017, 74, 4599–4619. [Google Scholar] [CrossRef]
- Wan, Z.; Chai, R.; Yuan, H.; Chen, B.; Dong, Q.; Zheng, B.; Mou, X.; Pan, W.; Tu, Y.; Yang, Q. MEIS2 promotes cell migration and invasion in colorectal cancer. Oncol. Rep. 2019, 42, 213–223. [Google Scholar] [CrossRef]
- Bjerke, G.A.; Hyman-Walsh, C.; Wotton, D. Cooperative transcriptional activation by Klf4, Meis2, and Pbx1. Mol. Cell. Biol. 2011, 31, 3723–3733. [Google Scholar] [CrossRef]
- Chandran, R.R.; Xie, Y.; Gallardo-Vara, E.; Adams, T.; Garcia-Milian, R.; Kabir, I.; Sheikh, A.Q.; Kaminski, N.; Martin, K.A.; Herzog, E.L. Distinct roles of KLF4 in mesenchymal cell subtypes during lung fibrogenesis. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Malaab, M.; Renaud, L.; Takamura, N.; Zimmerman, K.D.; da Silveira, W.A.; Ramos, P.S.; Haddad, S.; Peters-Golden, M.; Penke, L.R.; Wolf, B. Antifibrotic factor KLF4 is repressed by the miR-10/TFAP2A/TBX5 axis in dermal fibroblasts: Insights from twins discordant for systemic sclerosis. Ann. Rheum. Dis. 2022, 81, 268–277. [Google Scholar] [CrossRef]
- Wojcik, A.J.; Skaflen, M.D.; Srinivasan, S.; Hedrick, C.C. A critical role for ABCG1 in macrophage inflammation and lung homeostasis. J. Immunol. 2008, 180, 4273–4282. [Google Scholar] [CrossRef] [PubMed]
- Baldán, Á.; Tarr, P.; Vales, C.S.; Frank, J.; Shimotake, T.K.; Hawgood, S.; Edwards, P.A. Deletion of the transmembrane transporter ABCG1 results in progressive pulmonary lipidosis. J. Biol. Chem. 2006, 281, 29401–29410. [Google Scholar] [CrossRef] [PubMed]
- McPeek, M.; Malur, A.; Tokarz, D.A.; Lertpiriyapong, K.; Gowdy, K.M.; Murray, A.G.; Wingard, C.J.; Fessler, M.B.; Barna, B.P.; Thomassen, M.J. Alveolar Macrophage ABCG1 Deficiency Promotes Pulmonary Granulomatous Inflammation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 332–340. [Google Scholar] [CrossRef]
- Romero, F.; Shah, D.; Duong, M.; Penn, R.B.; Fessler, M.B.; Madenspacher, J.; Stafstrom, W.; Kavuru, M.; Lu, B.; Kallen, C.B. A pneumocyte–macrophage paracrine lipid axis drives the lung toward fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Kloss, M.; Decker, P.; Baltz, K.M.; Baessler, T.; Jung, G.; Rammensee, H.-G.; Steinle, A.; Krusch, M.; Salih, H.R. Interaction of monocytes with NK cells upon Toll-like receptor-induced expression of the NKG2D ligand MICA. J. Immunol. 2008, 181, 6711–6719. [Google Scholar] [CrossRef]
- El Sharkawy, R.; Bayoumi, A.; Metwally, M.; Mangia, A.; Berg, T.; Romero-Gomez, M.; Abate, M.L.; Irving, W.L.; Sheridan, D.; Dore, G.J. A variant in the MICA gene is associated with liver fibrosis progression in chronic hepatitis C through TGF-β1 dependent mechanisms. Sci. Rep. 2019, 9, 1439. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef]
- Darling, T.K.; Lamb, T.J. Emerging Roles for Eph Receptors and Ephrin Ligands in Immunity. Front. Immunol. 2019, 10, 1473. [Google Scholar] [CrossRef]
- Mimche, P.N.; Brady, L.M.; Bray, C.F.; Lee, C.M.; Thapa, M.; King, T.P.; Quicke, K.; McDermott, C.D.; Mimche, S.M.; Grakoui, A. The receptor tyrosine kinase EphB2 promotes hepatic fibrosis in mice. Hepatology 2015, 62, 900–914. [Google Scholar] [CrossRef] [Green Version]
- Lagares, D.; Ghassemi-Kakroodi, P.; Tremblay, C.; Santos, A.; Probst, C.K.; Franklin, A.; Santos, D.M.; Grasberger, P.; Ahluwalia, N.; Montesi, S.B. ADAM10-mediated ephrin-B2 shedding promotes myofibroblast activation and organ fibrosis. Nat. Med. 2017, 23, 1405. [Google Scholar] [CrossRef]
- Abraham, D.J.; Varga, J. Scleroderma: From cell and molecular mechanisms to disease models. Trends Immunol. 2005, 26, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Mechanisms in the loss of capillaries in systemic sclerosis: Angiogenesis versus vasculogenesis. J. Cell. Mol. Med. 2010, 14, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- NicAmhlaoibh, R.; Shtivelman, E. Metastasis suppressor CC3 inhibits angiogenic properties of tumor cells in vitro. Oncogene 2001, 20, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Plantier, L.; Renaud, H.; Respaud, R.; Marchand-Adam, S.; Crestani, B. Transcriptome of cultured lung fibroblasts in idiopathic pulmonary fibrosis: Meta-analysis of publically available microarray datasets reveals repression of inflammation and immunity pathways. Int. J. Mol. Sci. 2016, 17, 2091. [Google Scholar] [CrossRef]
- Xu, K.; Chong, D.C.; Rankin, S.A.; Zorn, A.M.; Cleaver, O. Rasip1 is required for endothelial cell motility, angiogenesis and vessel formation. Dev. Biol. 2009, 329, 269–279. [Google Scholar] [CrossRef]
- Koo, Y.; Barry, D.M.; Xu, K.; Tanigaki, K.; Davis, G.E.; Mineo, C.; Cleaver, O. Rasip1 is essential to blood vessel stability and angiogenic blood vessel growth. Angiogenesis 2016, 19, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Bei, Y.; Hua-Huy, T.; Nicco, C.; Duong-Quy, S.; Le-Dong, N.-N.; Tiev, K.-P.; Chéreau, C.; Batteux, F.; Dinh-Xuan, A.T. RhoA/Rho-kinase activation promotes lung fibrosis in an animal model of systemic sclerosis. Exp. Lung Res. 2016, 42, 44–55. [Google Scholar] [CrossRef]
- Zhuan, B.; Lu, Y.; Chen, Q.; Zhao, X.; Li, P.; Yuan, Q.; Yang, Z. Overexpression of the long noncoding RNA TRHDE-AS1 inhibits the progression of lung cancer via the miRNA-103/KLF4 axis. J. Cell. Biochem. 2019, 120, 17616–17624. [Google Scholar] [CrossRef] [PubMed]
- Bower, K.E.; Fritz, J.M.; McGuire, K.L. Transcriptional repression of MMP-1 by p21SNFT and reduced in vitro invasiveness of hepatocarcinoma cells. Oncogene 2004, 23, 8805–8814. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.; Dinakarpandian, D.; Yoshida, N.; Lauer-Fields, J.L.; Fields, G.B.; Visse, R.; Nagase, H. Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. 2004, 23, 3020–3030. [Google Scholar] [CrossRef]
- Dees, C.; Schlottmann, I.; Funke, R.; Distler, A.; Palumbo-Zerr, K.; Zerr, P.; Lin, N.-Y.; Beyer, C.; Distler, O.; Schett, G. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann. Rheum. Dis. 2014, 73, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.; Estivill, X.; Ramsay, M.; Tikly, M. Dysregulation of the Wnt signaling pathway in South African patients with diffuse systemic sclerosis. Clin. Rheumatol. 2019, 38, 933–938. [Google Scholar] [CrossRef]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H. Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Evanko, S.P.; Gooden, M.D.; Kang, I.; Chan, C.K.; Vernon, R.B.; Wight, T.N. A Role for HAPLN1 During Phenotypic Modulation of Human Lung Fibroblasts In Vitro. J. Histochem. Cytochem. 2020, 68, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Tyler, A.; Mahoney, J.M.; Carter, G.W. Genetic interactions affect lung function in patients with systemic sclerosis. G3 Genes Genomes Genet. 2020, 10, 151–163. [Google Scholar] [CrossRef]
- Chen, J.; Kwong, D.L.-W.; Zhu, C.-L.; Chen, L.-L.; Dong, S.-S.; Zhang, L.-Y.; Tian, J.; Qi, C.-B.; Cao, T.-T.; Wong, A.M.G.; et al. RBMS3 at 3p24 Inhibits Nasopharyngeal Carcinoma Development via Inhibiting Cell Proliferation, Angiogenesis, and Inducing Apoptosis. PLoS ONE 2012, 7, e44636. [Google Scholar] [CrossRef]
- Farhan, M.A.; Azad, A.K.; Touret, N.; Murray, A.G. FGD5 regulates VEGF receptor-2 coupling to PI3 kinase and receptor recycling. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2301–2310. [Google Scholar] [CrossRef]
- Kurogane, Y.; Miyata, M.; Kubo, Y.; Nagamatsu, Y.; Kundu, R.K.; Uemura, A.; Ishida, T.; Quertermous, T.; Hirata, K.-I.; Rikitake, Y. FGD5 mediates proangiogenic action of vascular endothelial growth factor in human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 988–996. [Google Scholar] [CrossRef]
- Kohl, P. Heterogeneous cell coupling in the heart: An electrophysiological role for fibroblasts. Circ. Res. 2003, 93, 381–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez, C.; Benamer, N.; Morley, G.E. The cardiac fibroblast: Functional and electrophysiological considerations in healthy and diseased hearts. J. Cardiovasc. Pharmacol. 2011, 57, 380. [Google Scholar] [CrossRef]
- Dolmetsch, R.E.; Xu, K.; Lewis, R.S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998, 392, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.J.; Mukherjee, S.; Ask, K. Calcium Homeostasis and Ionic Mechanisms in Pulmonary Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2015, 53, 135–148. [Google Scholar] [CrossRef]
- Mahling, R.; Ben-Johny, M. A bridge from the endoplasmic reticulum to the plasma membrane comes into view. Proc. Natl. Acad. Sci. USA 2022, 119, e2202254119. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Wang, Y.; Chen, B.; Wang, Y.; Yuan, J.; Zhang, L.; Hall, D.; Wu, J.; Shi, Y.; Zhu, Q. EC coupling structural protein junctophilin-2 encodes a stress-adaptive transcription regulator. Science 2018, 362, eaan3303. [Google Scholar] [CrossRef]
- Li, G.-R.; Sun, H.-Y.; Chen, J.-B.; Zhou, Y.; Tse, H.-F.; Lau, C.-P. Characterization of multiple ion channels in cultured human cardiac fibroblasts. PLoS ONE 2009, 4, e7307. [Google Scholar] [CrossRef]
- Xu, W.; Deng, M.; Meng, X.; Sun, X.; Tao, X.; Wang, D.; Zhang, S.; Zhen, Y.; Liu, X.; Liu, M. The alterations in molecular markers and signaling pathways in chronic thromboembolic pulmonary hypertension, a study with transcriptome sequencing and bioinformatic analysis. Front. Cardiovasc. Med. 2022, 9, 961305. [Google Scholar] [CrossRef]
- Naranjo, M.; Hassoun, P.M. Systemic sclerosis-associated pulmonary hypertension: Spectrum and impact. Diagnostics 2021, 11, 911. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, W.; Liu, D.; Luo, H. The role of metabolism in the pathogenesis of systemic sclerosis. Metabolism 2019, 93, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kolm-Litty, V.; Sauer, U.; Nerlich, A.; Lehmann, R.; Schleicher, E. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. J. Clin. Investig. 1998, 101, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Mutlu, G.M. Metabolic requirements of pulmonary fibrosis: Role of fibroblast metabolism. FEBS J. 2021, 288, 6331–6352. [Google Scholar] [CrossRef]
- Tran, D.H.; May, H.I.; Li, Q.; Luo, X.; Huang, J.; Zhang, G.; Niewold, E.; Wang, X.; Gillette, T.G.; Deng, Y. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat. Commun. 2020, 11, 1771. [Google Scholar] [CrossRef] [PubMed]
- Asbun, J.; Villarreal, F.J. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 47, 693–700. [Google Scholar] [CrossRef]
- Weigert, C.; Brodbeck, K.; Lehmann, R.; Häring, H.U.; Schleicher, E.D. Overexpression of glutamine: Fructose-6-phosphate-amidotransferase induces transforming growth factor-β1 synthesis in NIH-3T3 fibroblasts. FEBS Lett. 2001, 488, 95–99. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Xia, W.; Lei, D.; Voorhees, J.J.; Fisher, G.J. Age-dependent alterations of decorin glycosaminoglycans in human skin. Sci. Rep. 2013, 3, 2422. [Google Scholar] [CrossRef]
- Kuroda, K.; Shinkai, H. Gene expression of types I and III collagen, decorin, matrix metalloproteinases and tissue inhibitors of metalloproteinases in skin fibroblasts from patients with systemic sclerosis. Arch. Dermatol. Res. 1997, 289, 567–572. [Google Scholar] [CrossRef]
- Abdel-Wahab, N.; Wicks, S.J.; Mason, R.M.; Chantry, A. Decorin suppresses transforming growth factor-β-induced expression of plasminogen activator inhibitor-1 in human mesangial cells through a mechanism that involves Ca2+-dependent phosphorylation of Smad2 at serine-240. Biochem. J. 2002, 362, 643–649. [Google Scholar] [CrossRef]
- Baghy, K.; Iozzo, R.V.; Kovalszky, I. Decorin–TGFβ axis in hepatic fibrosis and cirrhosis. J. Histochem. Cytochem. 2012, 60, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Kehlet, S.N.; Bager, C.; Willumsen, N.; Dasgupta, B.; Brodmerkel, C.; Curran, M.; Brix, S.; Leeming, D.; Karsdal, M. Cathepsin-S degraded decorin are elevated in fibrotic lung disorders–development and biological validation of a new serum biomarker. BMC Pulm. Med. 2017, 17, 110. [Google Scholar] [CrossRef]
- Merline, R.; Moreth, K.; Beckmann, J.; Nastase, M.V.; Zeng-Brouwers, J.; Tralhão, J.G.; Lemarchand, P.; Pfeilschifter, J.; Schaefer, R.M.; Iozzo, R.V. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci. Signal. 2011, 4, ra75. [Google Scholar] [CrossRef] [Green Version]
- Moreth, K.; Iozzo, R.V.; Schaefer, L. Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 2012, 11, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Loewen, M.S.; Walner, D.L.; Caldarelli, D.D. Improved airway healing using transforming growth factor beta-3 in a rabbit model. Wound Repair Regen. 2001, 9, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Occleston, N.L.; O’Kane, S.; Laverty, H.G.; Cooper, M.; Fairlamb, D.; Mason, T.; Bush, J.A.; Ferguson, M.W. Discovery and development of avotermin (recombinant human transforming growth factor beta 3): A new class of prophylactic therapeutic for the improvement of scarring. Wound Repair Regen. 2011, 19, s38–s48. [Google Scholar] [CrossRef]
- Yu, L.; Border, W.A.; Huang, Y.; Noble, N.A. TGF-β isoforms in renal fibrogenesis. Kidney Int. 2003, 64, 844–856. [Google Scholar] [CrossRef]
- Vellanki, R.N.; Zhang, L.; Guney, M.A.; Rocheleau, J.V.; Gannon, M.; Volchuk, A. OASIS/CREB3L1 induces expression of genes involved in extracellular matrix production but not classical endoplasmic reticulum stress response genes in pancreatic β-cells. Endocrinology 2010, 151, 4146–4157. [Google Scholar] [CrossRef]
- Garrett, S.M.; Hsu, E.; Thomas, J.M.; Pilewski, J.M.; Feghali-Bostwick, C. Insulin-like growth factor (IGF)-II-mediated fibrosis in pathogenic lung conditions. PLoS ONE 2019, 14, e0225422. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.; Feghali-Bostwick, C.A. Insulin-like growth factor-II is increased in systemic sclerosis-associated pulmonary fibrosis and contributes to the fibrotic process via Jun N-terminal kinase-and phosphatidylinositol-3 kinase-dependent pathways. Am. J. Pathol. 2008, 172, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Kelley, K.; Melichian, D.S.; Tamaki, Z.; Fang, F.; Su, Y.; Feng, G.; Pope, R.M.; Budinger, G.S.; Mutlu, G.M. Toll-like receptor 4 signaling augments transforming growth factor-β responses: A novel mechanism for maintaining and amplifying fibrosis in scleroderma. Am. J. Pathol. 2013, 182, 192–205. [Google Scholar] [CrossRef]
- Bradbury, P.; Mahmassani, M.; Zhong, J.; Turner, K.; Paul, A.; Verrills, N.M.; O’Neill, G.M. PP2A phosphatase suppresses function of the mesenchymal invasion regulator NEDD9. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Law, S.F.; Zhang, Y.-Z.; Klein-Szanto, A.J.; Golemis, E.A. Cell cycle-regulated processing of HEF1 to multiple protein forms differentially targeted to multiple subcellular compartments. Mol. Cell. Biol. 1998, 18, 3540–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samokhin, A.O.; Stephens, T.; Wertheim, B.M.; Wang, R.-S.; Vargas, S.O.; Yung, L.-M.; Cao, M.; Brown, M.; Arons, E.; Dieffenbach, P.B. NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Sci. Transl. Med. 2018, 10, eaap7294. [Google Scholar] [CrossRef]
- Kuivaniemi, H.; Tromp, G. Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Xu, Y.; Xu, X.; Keene, D.R.; Gurusiddappa, S.; Liang, X.; Wary, K.K.; Hoöoök, M. A novel binding site in collagen type III for integrins α1β1 and α2β1. J. Biol. Chem. 2005, 280, 32512–32520. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Ning, J.; Jiang, T.; Kang, A.; Li, L.; Pang, Y.; Zhang, B.; Huang, X.; Wang, Q. The proteasome-dependent degradation of ALKBH5 regulates ECM deposition in PM2. 5 exposure-induced pulmonary fibrosis of mice. J. Hazard. Mater. 2022, 432, 128655. [Google Scholar] [CrossRef]
- Mishra, S.; Shah, M.I.; Kumar, S.U.; Kumar, D.T.; Gopalakrishnan, C.; Al-Subaie, A.M.; Magesh, R.; Doss, C.G.P.; Kamaraj, B. Network analysis of transcriptomics data for the prediction and prioritization of membrane-associated biomarkers for idiopathic pulmonary fibrosis (IPF) by bioinformatics approach. Adv. Protein Chem. Struct. Biol. 2021, 123, 241–273. [Google Scholar]
- Smits, P.; Li, P.; Mandel, J.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; De Crombrugghe, B.; Lefebvre, V. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev. Cell 2001, 1, 277–290. [Google Scholar] [CrossRef]
- Hersh, C.P.; Silverman, E.K.; Gascon, J.; Bhattacharya, S.; Klanderman, B.J.; Litonjua, A.A.; Lefebvre, V.; Sparrow, D.; Reilly, J.J.; Anderson, W.H. SOX5 is a candidate gene for chronic obstructive pulmonary disease susceptibility and is necessary for lung development. Am. J. Respir. Crit. Care Med. 2011, 183, 1482–1489. [Google Scholar] [CrossRef]
- Xu, L.; Zheng, L.; Wang, Z.; Li, C.; Li, S.; Xia, X.; Zhang, P.; Li, L.; Zhang, L. TNF-α-Induced SOX5 upregulation is involved in the osteogenic differentiation of human bone marrow mesenchymal stem cells through KLF4 signal pathway. Mol. Cells 2018, 41, 575. [Google Scholar]
- Shi, Y.; Wu, Q.; Xuan, W.; Feng, X.; Wang, F.; Tsao, B.P.; Zhang, M.; Tan, W. Transcription factor SOX5 promotes the migration and invasion of fibroblast-like synoviocytes in part by regulating MMP-9 expression in collagen-induced arthritis. Front. Immunol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Kamen, D.L. Vitamin D in lupus: New kid on the block? Bull. NYU Hosp. Jt. Dis. 2010, 68, 218. [Google Scholar]
- Prabhu, A.V.; Luu, W.; Li, D.; Sharpe, L.J.; Brown, A.J. DHCR7: A vital enzyme switch between cholesterol and vitamin D production. Prog. Lipid Res. 2016, 64, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Eslick, G.D.; Nowson, C.; Smith, C.; Bensoussan, A. Use of calcium or calcium in combination with vitamin D supplementation to prevent fractures and bone loss in people aged 50 years and older: A meta-analysis. Lancet 2007, 370, 657–666. [Google Scholar] [CrossRef]
- Patwardhan, V.G.; Khadilkar, A.V.; Chiplonkar, S.A.; Mughal, Z.M.; Khadilkar, V.V. Varying relationship between 25-hydroxy-vitamin D, high density lipoprotein cholesterol, and serum 7-dehydrocholesterol reductase with sunlight exposure. J. Clin. Lipidol. 2015, 9, 652–657. [Google Scholar] [CrossRef]
- Illescas-Montes, R.; Melguizo-Rodríguez, L.; Ruiz, C.; Costela-Ruiz, V.J. Vitamin D and autoimmune diseases. Life Sci. 2019, 233, 116744. [Google Scholar] [CrossRef] [PubMed]
- Ginde, A.A.; Liu, M.C.; Camargo, C.A. Demographic differences and trends of vitamin D insufficiency in the US population, 1988–2004. Arch. Intern. Med. 2009, 169, 626–632. [Google Scholar] [CrossRef]
- Shoenfeld, N.; Amital, H.; Shoenfeld, Y. The effect of melanism and vitamin D synthesis on the incidence of autoimmune disease. Nat. Clin. Pract. Rheumatol. 2009, 5, 99–105. [Google Scholar] [CrossRef]
- Terao, M.; Yang, L.; Matsumura, S.; Yutani, M.; Murota, H.; Katayama, I. A vitamin D analog inhibits Th2 cytokine-and TGFβ-induced periostin production in fibroblasts: A potential role for vitamin D in skin sclerosis. Derm. Endocrinol. 2015, 7, e1010983. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, T.; Yao, L.; Xing, Y.; Zhao, X.; Fu, J.; Xue, X. Chronic vitamin D deficiency induces lung fibrosis through activation of the renin-angiotensin system. Sci. Rep. 2017, 7, 3312. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, Y.; Xue, L.; Li, B.; Zhang, Z. 1α,25-dihydroxyvitamin D3 Attenuates TGF-β-Induced Pro-Fibrotic Effects in Human Lung Epithelial Cells through Inhibition of Epithelial–Mesenchymal Transition. Nutrients 2017, 9, 980. [Google Scholar] [CrossRef]
- Ramirez, A.M.; Wongtrakool, C.; Welch, T.; Steinmeyer, A.; Zügel, U.; Roman, J. Vitamin D inhibition of pro-fibrotic effects of transforming growth factor β1 in lung fibroblasts and epithelial cells. J. Steroid Biochem. Mol. Biol. 2010, 118, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Tzilas, V.; Bouros, E.; Barbayianni, I.; Karampitsakos, T.; Kourtidou, S.; Ntassiou, M.; Ninou, I.; Aidinis, V.; Bouros, D.; Tzouvelekis, A. Vitamin D prevents experimental lung fibrosis and predicts survival in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2019, 55, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Potter, J.A.; Price, G.W.; Cliff, C.L.; Green, C.R.; Squires, P.E.; Hills, C.E. Collagen I modifies connexin-43 hemichannel activity via integrin α2β1 binding in TGFβ1-evoked renal tubular epithelial cells. Int. J. Mol. Sci. 2021, 22, 3644. [Google Scholar] [CrossRef] [PubMed]
- Colasante, G.; Lignani, G.; Rubio, A.; Medrihan, L.; Yekhlef, L.; Sessa, A.; Massimino, L.; Giannelli, S.G.; Sacchetti, S.; Caiazzo, M. Rapid conversion of fibroblasts into functional forebrain GABAergic interneurons by direct genetic reprogramming. Cell Stem Cell 2015, 17, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Bhat, R.; Axtell, R.; Mitra, A.; Miranda, M.; Lock, C.; Tsien, R.W.; Steinman, L. Inhibitory role for GABA in autoimmune inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2580–2585. [Google Scholar] [CrossRef]
- Reyes-García, M.G.; Hernández-Hernández, F.; Hernández-Téllez, B.; García-Tamayo, F. GABA (A) receptor subunits RNA expression in mice peritoneal macrophages modulate their IL-6/IL-12 production. J. Neuroimmunol. 2007, 188, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; O’Reilly, S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin. Exp. Immunol. 2019, 195, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Feghali, C.; Bost, K.; Boulware, D.; Levy, L. Mechanisms of pathogenesis in scleroderma. I. Overproduction of interleukin 6 by fibroblasts cultured from affected skin sites of patients with scleroderma. J. Rheumatol. 1992, 19, 1207–1211. [Google Scholar]
- Luo, Y.; Balle, T. GABAA receptors as targets for anaesthetics and analgesics and promising candidates to help treat coronavirus infections: A mini-review. Basic Clin. Pharmacol. Toxicol. 2022, 131, 443–451. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhang, W.; Roehrl, M.W.; Roehrl, V.B.; Roehrl, M.H. An autoantigen atlas from human lung HFL1 cells offers clues to neurological and diverse autoimmune manifestations of COVID-19. BioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Lykke-Andersen, J. mRNA surveillance: The perfect persist. J Cell Sci 2002, 115, 3033–3038. [Google Scholar] [CrossRef]
- Amin, B.J.H.; Kakamad, F.H.; Ahmed, G.S.; Ahmed, S.F.; Abdulla, B.A.; Mikael, T.M.; Salih, R.Q.; Salh, A.M.; Hussein, D.A. Post COVID-19 pulmonary fibrosis; a meta-analysis study. Ann. Med. Surg. 2022, 77, 103590. [Google Scholar] [CrossRef]
- Domsic, R.T.; Rodriguez-Reyna, T.; Lucas, M.; Fertig, N.; Medsger, T.A., Jr. Skin thickness progression rate: A predictor of mortality and early internal organ involvement in diffuse scleroderma. Ann Rheum Dis 2011, 70, 104–109. [Google Scholar] [CrossRef]
- Nguyen, X.-X.; Renaud, L.; Feghali-Bostwick, C. Identification of Impacted Pathways and Transcriptomic Markers as Potential Mediators of Pulmonary Fibrosis in Transgenic Mice Expressing Human IGFBP5. Int. J. Mol. Sci. 2021, 22, 12609. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A systems biology approach for pathway level analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef]
- Khatri, P.; Draghici, S.; Tarca, A.L.; Hassan, S.S.; Romero, R. A system biology approach for the steady-state analysis of gene signaling networks. In Proceedings of the Iberoamerican Congress on Pattern Recognition, Valparaiso, Chile, 13–16 November 2007; pp. 32–41. [Google Scholar]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.-s.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Gerarduzzi, C. Emerging roles of matricellular proteins in systemic sclerosis. Int. J. Mol. Sci. 2020, 21, 4776. [Google Scholar] [CrossRef]
- Colletti, M.; Galardi, A.; De Santis, M.; Guidelli, G.M.; Di Giannatale, A.; Di Luigi, L.; Antinozzi, C. Exosomes in systemic sclerosis: Messengers between immune, vascular and fibrotic components? Int. J. Mol. Sci. 2019, 20, 4337. [Google Scholar] [CrossRef] [PubMed]
- Mouawad, J.E.; Sharma, S.; Renaud, L.; Pilewski, J.M.; Nadig, S.N.; Feghali-Bostwick, C. Reduced Cathepsin L expression and secretion into the extracellular milieu contribute to lung fibrosis in systemic sclerosis. Rheumatology 2022, keac411. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enrichment | Hit in Query List |
|---|---|
| Driven by downregulated DEGs | Downregulated DEGs entered |
| PW- Beta1 integrin cell surface interactions (2.06 × 10−8) PW- Integrin signaling pathway (2.17 × 10−6) PW- Integrins in angiogenesis (2.53 × 10−6) | COL3A1, COL5A1, COL5A2, LAMC1, COL1A1 |
| PW- Focal adhesion (5.13 × 10−6) | COL3A1, COL5A1, COL5A2, LAMC1, COL1A1 |
| PW- Collagen biosynthesis and modifying enzymes (2.79 × 10−8) PW- Collagen formation (1.17 × 10−7) PW- ECM-receptor interaction (7.03 × 10−8) BP- ECM organization (2.01 × 10−4) PW- Genes encoding collagen proteins (3.06 × 10−7) PW- Collagen chain trimerization (4.01 × 10−7) PW- Assembly of collagen fibrils and other multimeric structures (1.09 × 10−6) BP- Collagen fibril organization (1.29 × 10−6) MF- ECM structural constituent conferring tensile strength (3.86 × 10−7) PW- Ensemble of genes encoding core ECM including ECM glycoproteins, collagens, and proteoglycans (1.08 × 10−6) PW- Extracellular matrix organization (1.72 × 10−6) BP- Extracellular structure organization (5.99 × 10−5) PW- Ensemble of genes encoding ECM and ECM-associated proteins (2.28 × 10−4) | COL3A1, COL5A1, COL5A2, P4HA2, COL1A1, LAMC1, RSPO4, ABCG1 |
| BP- Negative regulation of endodermal cell differentiation (1.12 × 10−5) BP- Skin development (3.57 × 10−4) | COL3A1, PTGS1, COL1A1, COL5A1, COL5A2 |
| PW- Syndecan-1-mediated signaling events (3.68 × 10−7) | COL3A1, COL5A1, COL5A2, COL1A1 |
| MF- Platelet-derived growth factor binding (7.81 × 10−7) PW- Platelet activation (5.70 × 10−4) | COL3A1, COL5A1, COL1A1, PTGS1 |
| PW- COX reactions (1.36 × 10−3) | PTGS1 |
| Driven by upregulated DEGs | Upregulated DEGs entered |
| MF- Histone methyltransferase activity (H3-K9 specific) (4.52 × 10−5) MF- Histone-lysine N-methyltransferase activity (8.34 × 10−4) | PRDM8, PRDM16 |
| MF- Extracellular matrix binding (1.56 × 10−3) | TINAGL1, ADAMTS5 |
| MF- N-acetyllactosaminide beta-1,6-N-acetylglucosaminyltransferase activity (2.80 × 10−3) | GCNT2 |
| MF- Activating transcription factor binding (3.22 × 10−3) | PRDM16, PITX2 |
| MF- mRNA 3′-UTR binding (3.50 × 10−3) | RBMS3, RBM38 |
| MF- L27 domain binding (3.74 × 10−3) | LIN7A |
| BP- Positive regulation of myoblast proliferation (4.13 × 10−5) | MEIS2, PITX2 |
| BP- Sensory organ development (1.84 × 10−4) | PRDM16, MEIS2, LIN7A, PITX2, EPHB1 |
| BP- Central nervous system projection neuron axonogenesis (2.81 × 10−4) | PRDM8, EPHB1 |
| Enrichment | Hit in Query List |
|---|---|
| PW: ECM organization (6.66 × 10−7) PW: Ensemble of genes encoding ECM and ECM-associated proteins (1.00 × 10−4) | ANXA5, ASPN, BMP5, CLEC14A, CLEC2B, COL1A2, COL6A6, EGFL7, FBLN1, FBLN2, ITIH5, KDR, LAMA2, MXRA5, NID1, OMD, P4HA2, S100A4, SEMA4D, SFRP2, SPARC, ADAMTS15, HPSE2, ITGA10, ITGB2, JAM2, LRP4, P4HA3, PARM1, SEMA3C, SEMA3D, TGFB3 |
| MF: Calcium ion binding (1.61 × 10−6) | AIF1L, ANXA5, ASPH, ASPN, CD248, DLL4, EGFL7, FBLN1, FBLN2, FKBP9, FSTL5, NID1, PCDHGA4, PRRG3, RCN3, SPARC, SWAP70, S100A4, CAPS, CRACR2B, LRP4, PLCD4, PLN, PLS1, PRRG4 |
| BP: Calcium ion transport (3.35 × 10−6) | ANXA5, ASPH, CACHD1, CLIC2, LHCGR, PANX1, TRPV4, CALCRL, CLDN16, CACNA1A, CRACR2B, CRH, DRD1, HES1, HOMER2, MCHR1, PLN, RAMP1, TRPC5 |
| BP: Cell adhesion (1.42 × 10−9) | MYADM, ABAT, SWAP70, CYTH3, CD55, CD83, ACVRL1, NEDD9, BMP5, FBLN1, FBLN2, ACKR3, PKP2, NID1, KDR, GPNMB, SEMA4D, SFRP2, COL6A6, PCDHGA4, CARMIL1, LAMA2, XG, APOD, OMD, PDPN, TRPV4, DUSP10, EGFL7, PALLD, HLA-DPA1, MCAM, ITGB2, PGM5, JAM2, CADM1, NTM, HES1, SDK2, PODXL, ITGA10, ST6GAL1, NLGN1, CLDN16, EPHA4 |
| BP: Regulation of cell development (1.62 × 10−8) | ATF5, BMP5, CARMIL1, DLL4, DUSP10, EPHB2, FBLN1, KDR, METRN, MME, MYADM, NEDD9, PDPN, SEMA4D, TRPV4, CACNA1A, EPHA4, EPOR, HES1, KALRN, LRP4, NGEF, NLGN1, NTM, OSTN, RAPGEF4, SEMA3C, SEMA3D, SOX11, SOX5, ST6GAL1, STX1B, TRPC5, XYLT1, ZNF536 |
| MF: Gated channel activity (1.90 × 10−5) MF: Passive transmembrane transporter activity (2.78 × 10−5) | ASIC2, ASPH, CACHD1, CLCN4, CLIC2, EPHB2, GRIK2, HCN2, PANX1, PDPN, PKP2, SHROOM2, TRPV4, ANO5, AQP1, CACNA1A, CRH, KCNA4, KCNMB4, NLGN1, PLN, TRPC5 |
| PW: Insulin secretion (0.044) PW: Maturity onset diabetes of the young (0.007) PW: Type II diabetes mellitus (0.028) | ADCY1, SLC2A1, CREB3L1, RAPGEF4, CAMK2B, KCNMB4 HHEX, HES1 MAPK10, IRS2, CACNA1A, SOCS2 |
| PW: TGF-beta signaling pathway (0.031) | BMP5, GREM1, DCN, FBN1, PITX2, TGFB3, ACVR2B |
| PW: Viral myocarditis (0.040) | LAMA2, CD55,HLA-DPA1, ITGB2, CXADR |
| PW: Proteoglycans in cancer (0.012) | IGF2, KDR, TLR4, TWIST2, COL1A1, COL1A2, DCN, LUM, HPSE2, CAMK2B |
| Enrichment | Hit in Query List |
|---|---|
| PW: Cholesterol biosynthetic (1.34 × 10−6) PW: Steroid biosynthetic (1.92 × 10−6) PW: Steroid biosynthesis (0.01558) PW: Activation of gene expression by SREBF (SREBP) (1.07 × 10−4) | DHCR7, FASN, FDPS, LSS, MVD, NSDHL |
| BP: Organic hydroxy compound metabolic process (1.66 × 10−6) BP: Alcohol metabolic process (7.24 × 10−6) | DHCR7, FASN, FDPS, LSS, MVD, MYOF, NPC1, NPC2, NSDHL, SRD5A1, ADH6, ASAH2, CFTR, DRD4, GNB3, GRK3, MAOA, MC1R, P2RY1, SNCAIP, WNT4 |
| PW: Ensemble of genes encoding ECM and ECM-associated proteins (5.11 × 10−4) | ANXA4, CSTB, CTSK, FGF5, OGN, PLXDC1, SPARCL1, TIMP2, ADAM32, ANGPTL1, CCL8, CHAD, COL5A3, COL7A1, COL11A2, CRISPLD1, CTSW, IL6, IL15, OGFOD2, PDGFB, SEMA3D, VWDE, WNT4 |
| PW: Neutrophil degranulation (9.41 × 10−4) BP: Neutrophil activation involved in immune response (2.68 × 10−4) | ABCA13, CD59, CSTB, DNASE1L1, GM2A, NPC2, PTPRN2, TIMP2, VAT1 |
| PW: Beta1 integrin cell surface interactions (9.00 × 10−4) | ITGA8, CD14, COL7A1, COL11A2, VCAM1 |
| PW: Gap junction (0.003218) PW: Calcium signaling (0.016369) | PDGFB |
| PW: GABAergic synapse (0.04595) PW: Neuroactive ligand-receptor interaction (0.029797) | GNB3, GABBR1, DRD4, P2RY1, CALCRL, MC1R, RXFP1 |
| PW: Regulation of actin cytoskeleton (0.024435) | PDGFB, MYH1, ITGA8, FGF5 |
| PW: Hematopoietic cell lineage (0.038564) PW: Intestinal immune network for IgA production (0.010566) | HLA-DPA1, IL6, IL15, CD14,CD59 |
| PW: mRNA surveillance pathway (0.041045) | PABPC4L, PABPC1L, DDX39B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renaud, L.; Waldrep, K.M.; da Silveira, W.A.; Pilewski, J.M.; Feghali-Bostwick, C.A. First Characterization of the Transcriptome of Lung Fibroblasts of SSc Patients and Healthy Donors of African Ancestry. Int. J. Mol. Sci. 2023, 24, 3645. https://doi.org/10.3390/ijms24043645
Renaud L, Waldrep KM, da Silveira WA, Pilewski JM, Feghali-Bostwick CA. First Characterization of the Transcriptome of Lung Fibroblasts of SSc Patients and Healthy Donors of African Ancestry. International Journal of Molecular Sciences. 2023; 24(4):3645. https://doi.org/10.3390/ijms24043645
Chicago/Turabian StyleRenaud, Ludivine, Kristy M. Waldrep, Willian A. da Silveira, Joseph M. Pilewski, and Carol A. Feghali-Bostwick. 2023. "First Characterization of the Transcriptome of Lung Fibroblasts of SSc Patients and Healthy Donors of African Ancestry" International Journal of Molecular Sciences 24, no. 4: 3645. https://doi.org/10.3390/ijms24043645