
Multi-Omics Approaches for Revealing the Epigenetic Regulation of Histone H3.1 during Spermatogonial Stem Cell Differentiation In Vitro
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
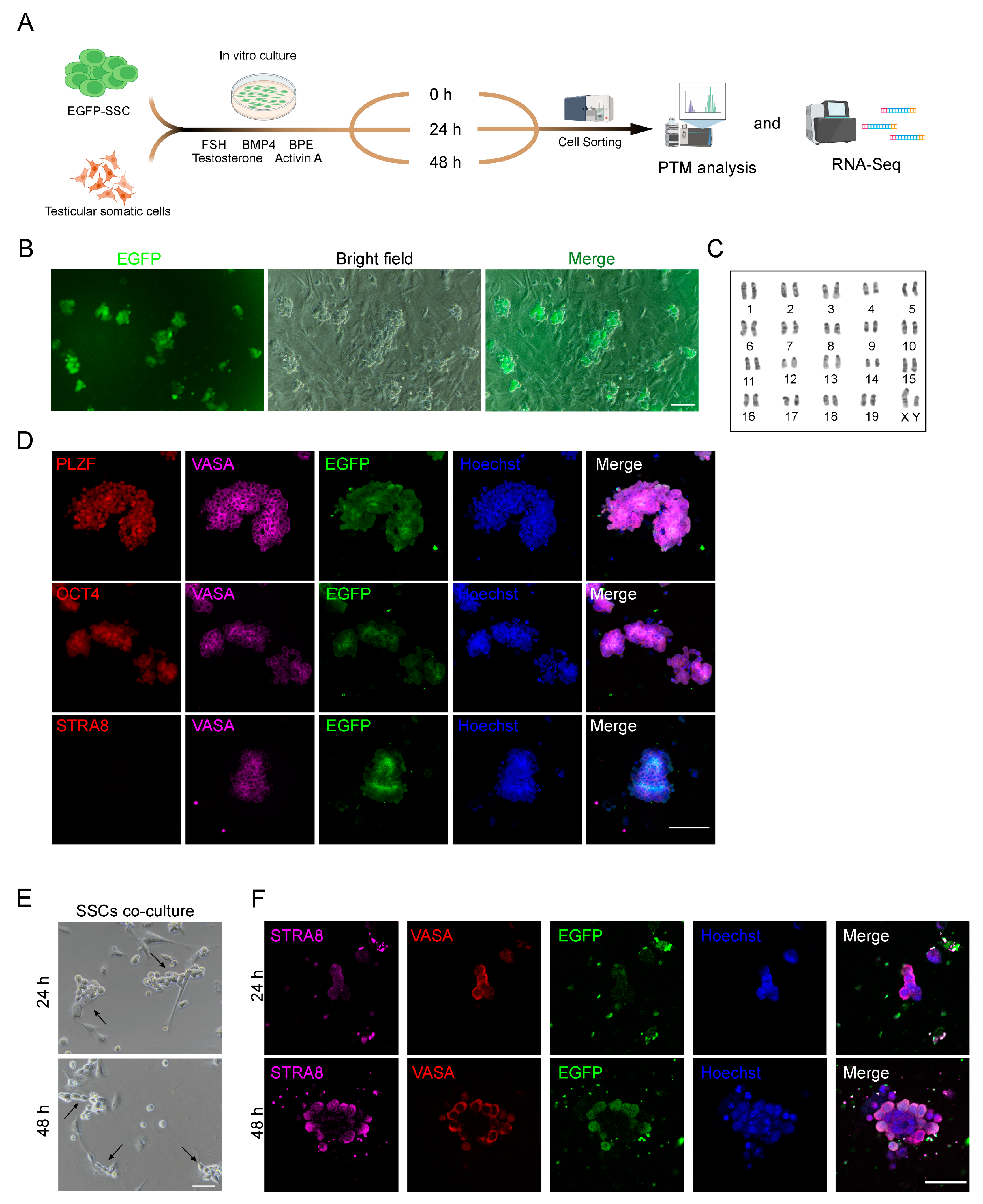
2.1. Induction of SSC Differentiation In Vitro
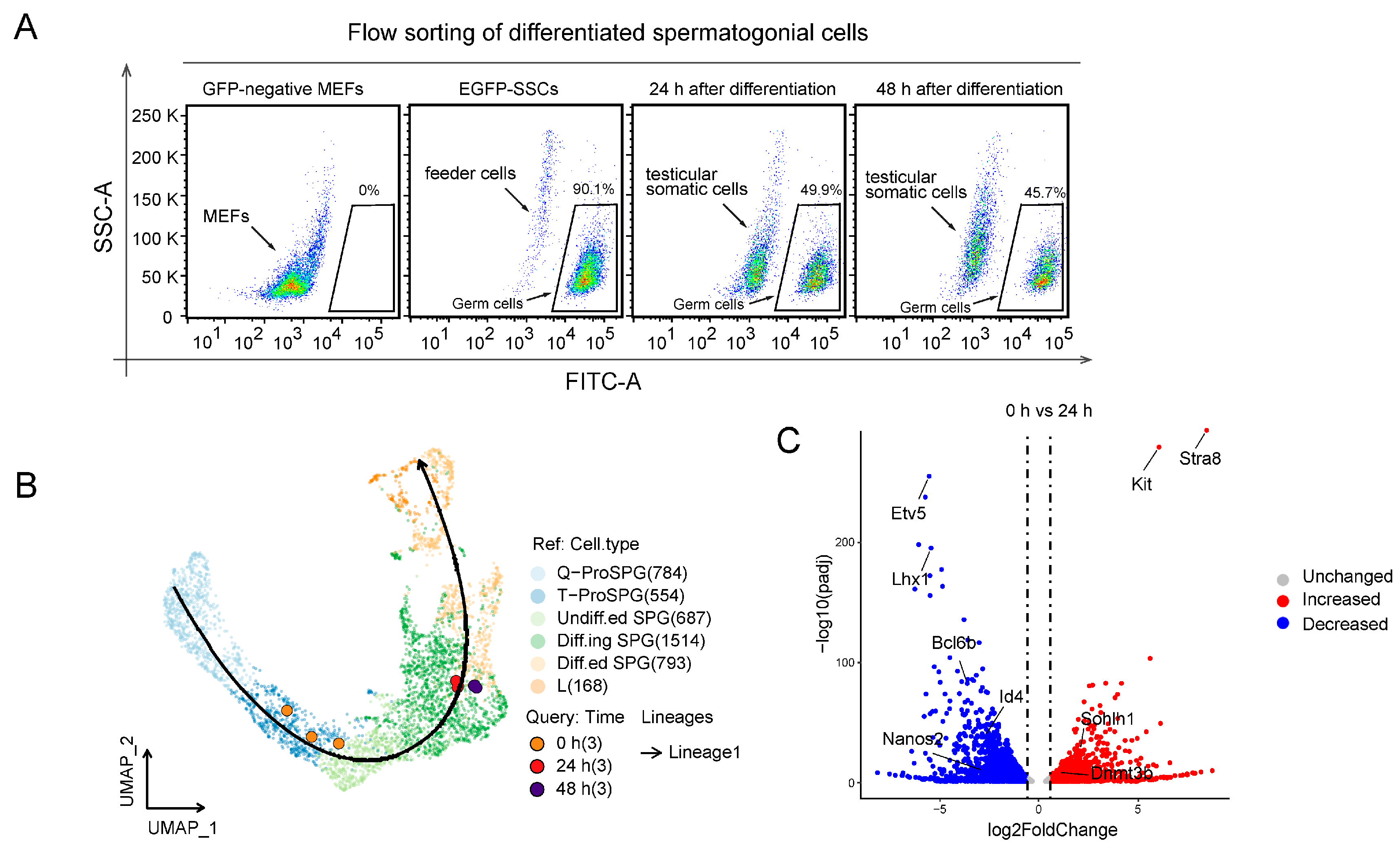
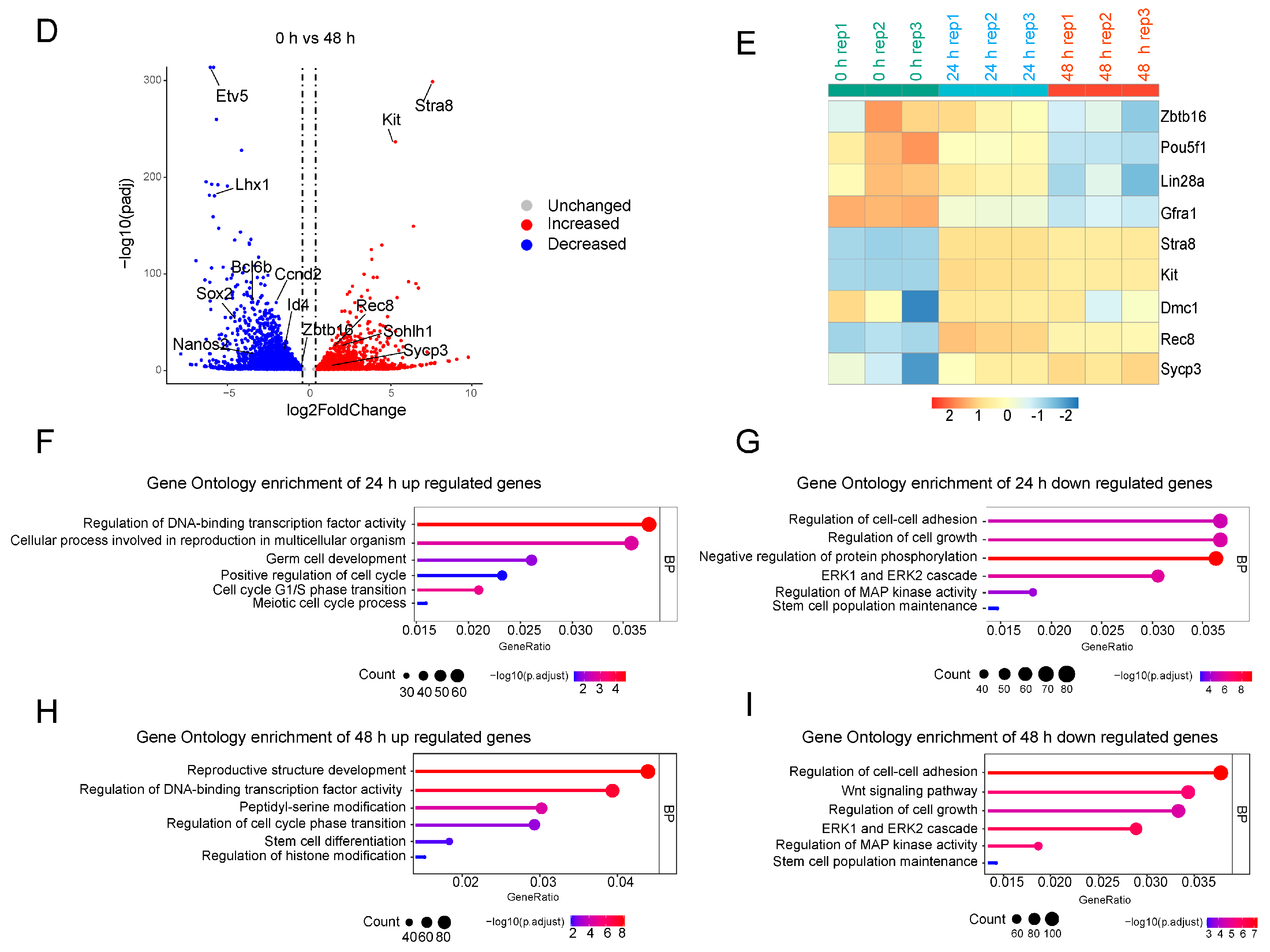
2.2. RNA-seq Analysis of SSCs Differentiation In Vitro
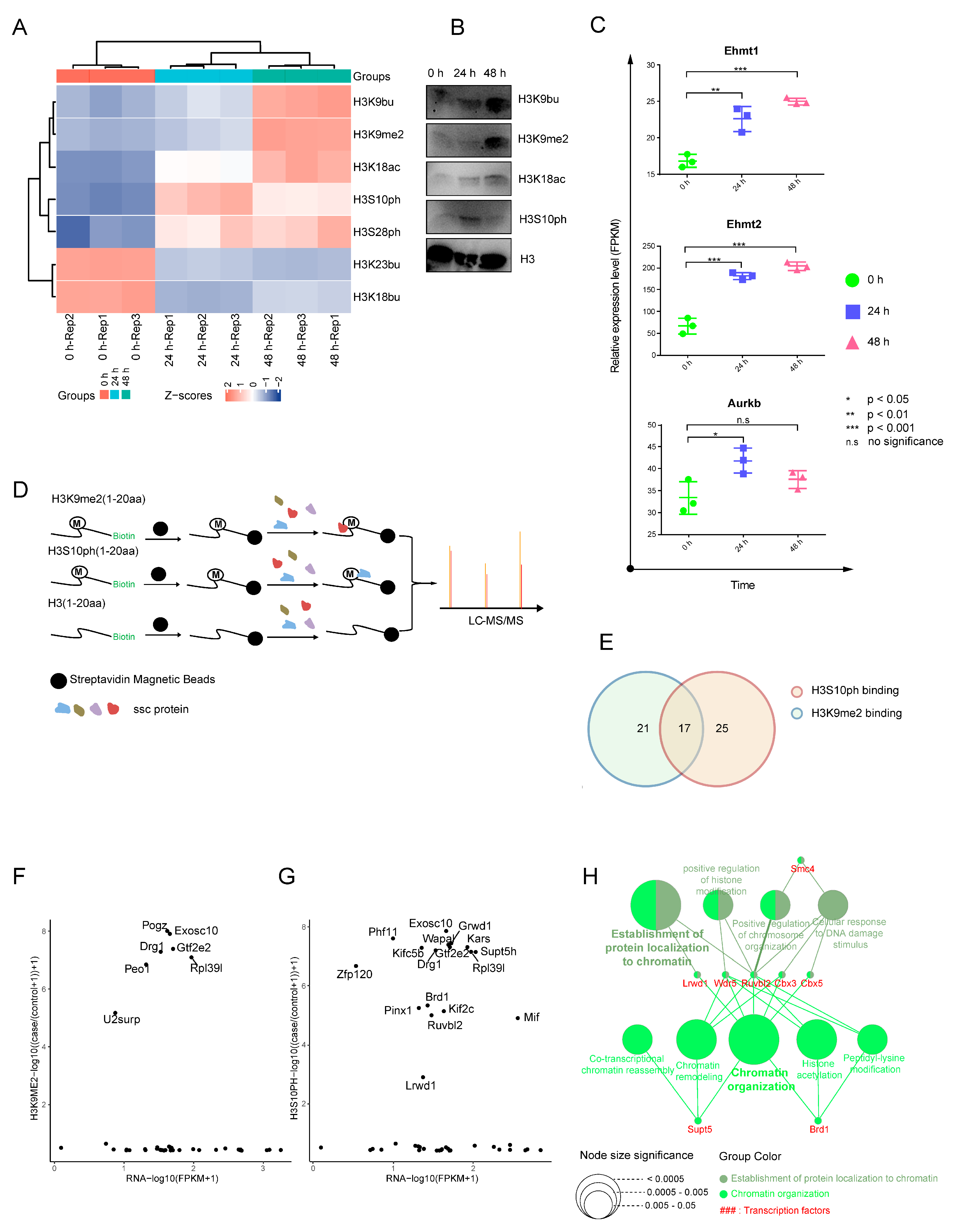
2.3. Dynamic Landscape of Histone H3 Modifications during SSC Differentiation
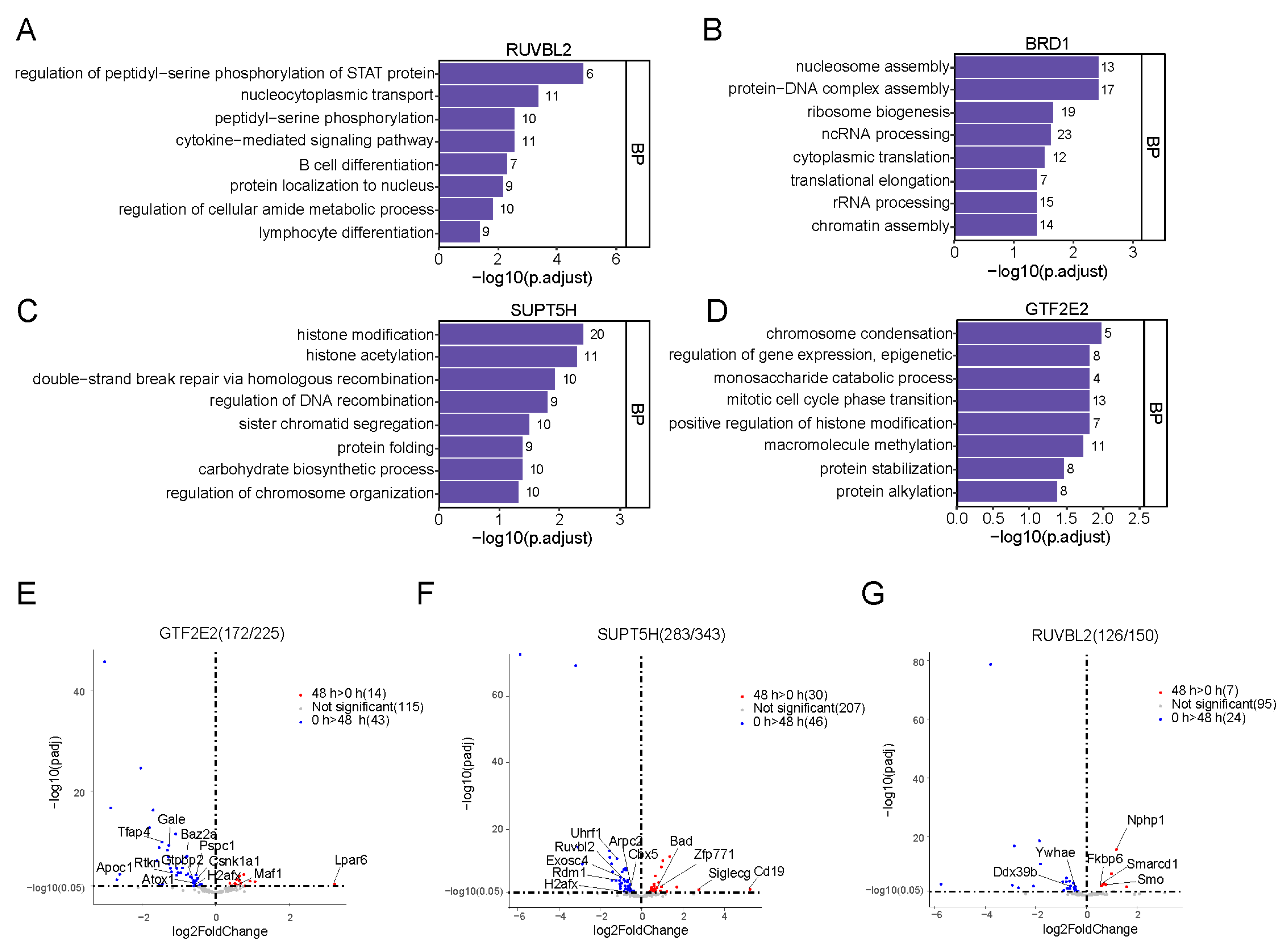
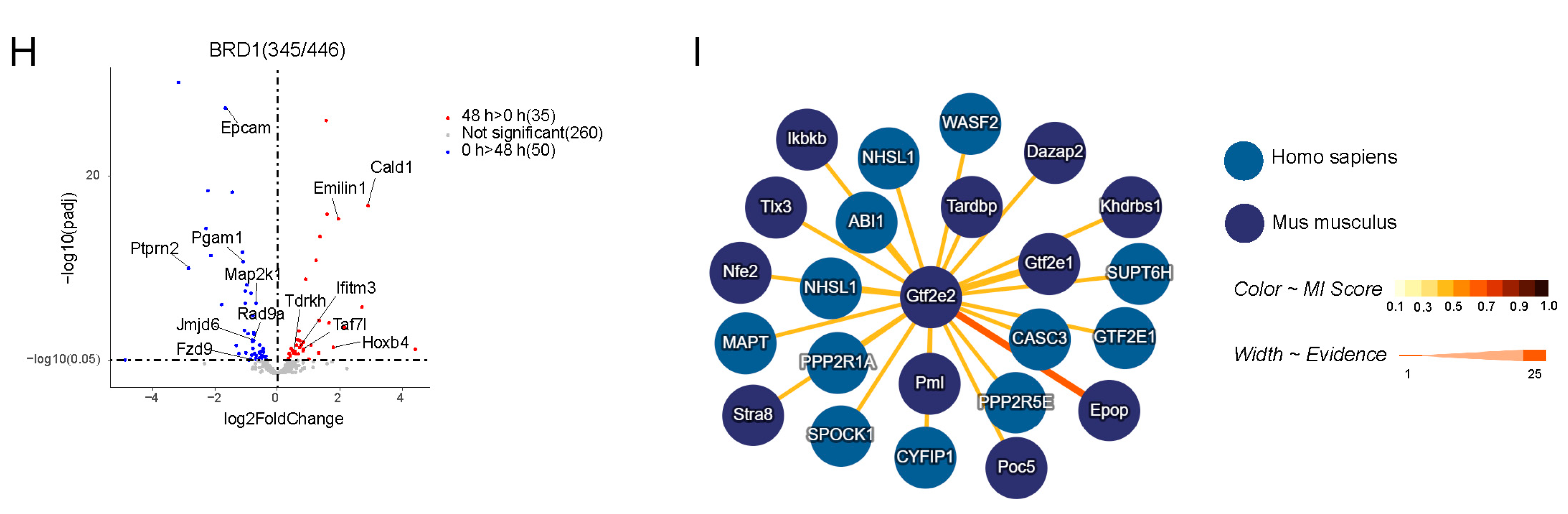
2.4. Synthetic Biotin–Labeled Peptides Enriched in Proteins That Interacted with HMs
3. Discussion
4. Materials and Methods
4.1. SSC Culture and Differentiation
4.2. Immunostaining
4.3. Karyotype
4.4. Flow Cytometry Analysis
4.5. Western Blotting
4.6. Histone Acid Extraction
4.7. Biotinylated H3K9me2 and H3S10ph Peptides Pull down Analysis
4.8. LC-MS/MS Analysis and Data Processing
4.9. Chemical Derivatization of Histones and Quantification by Parallel Reaction Monitoring
4.10. RNA-seq and Data Analysis
4.11. Differentiation Trajectory Analysis
4.12. Data Availability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Rooij, D.G.; Russell, L.D. All you wanted to know about spermatogonia but were afraid to ask. J. Androl. 2000, 21, 776–798. [Google Scholar] [PubMed]
- Legrand, J.M.D.; Chan, A.-L.; La, H.M.; Rossello, F.J.; Änkö, M.-L.; Fuller-Pace, F.V.; Hobbs, R.M. DDX5 plays essential transcriptional and post-transcriptional roles in the maintenance and function of spermatogonia. Nat. Commun. 2019, 10, 2278. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, M.; Yuan, Y.; Wang, X.; Fu, R.; Wan, H.; Xie, M.; Liu, M.; Guo, X.; Zheng, Y.; et al. Complete Meiosis from Embryonic Stem Cell-Derived Germ Cells in Vitro. Cell Stem Cell 2016, 18, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, S.-I.; Kobayashi, Y.; Shirakawa, T.; Watanabe, K.; Mizoguchi, K.; Hoshi, I.; Nakajima, K.; Nakabayashi, J.; Singh, S.; Dahl, A.; et al. Kmt2b conveys monovalent and bivalent H3K4me3 in mouse spermatogonial stem cells at germline and embryonic promoters. Development 2018, 145, dev169102. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Suen, H.C.; Luk, A.C.S.; Yang, L.; Lee, A.W.T.; Qi, H.; Lee, T.-L. Transcriptomic and epigenomic profiling of young and aged spermatogonial stem cells reveals molecular targets regulating differentiation. PLoS Genet. 2021, 17, e1009369. [Google Scholar] [CrossRef]
- Blanpain, C.; Lowry, W.E.; Geoghegan, A.; Polak, L.; Fuchs, E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 2004, 118, 635–648. [Google Scholar] [CrossRef]
- Gan, H.; Cai, T.; Lin, X.; Wu, Y.; Wang, X.; Yang, F.; Han, C. Integrative proteomic and transcriptomic analyses reveal multiple post-transcriptional regulatory mechanisms of mouse spermatogenesis. Mol. Cell. Proteom. 2013, 12, 1144–1157. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, Y.; Gao, Y.; Lin, Z.; Yang, S.; Wang, T.; Wang, Q.; Xie, N.; Hua, R.; Liu, M.; et al. Single-cell RNA-seq uncovers dynamic processes and critical regulators in mouse spermatogenesis. Cell Res. 2018, 28, 879–896. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, P.; Wan, C.; Huang, Y.; Cui, M.; Yang, X.; Hu, Y.; Zheng, Y.; Dong, J.; Wang, M.; et al. Cell-fate transition and determination analysis of mouse male germ cells throughout development. Nat. Commun. 2021, 12, 6839. [Google Scholar] [CrossRef]
- Ernst, C.; Eling, N.; Martinez-Jimenez, C.P.; Marioni, J.C.; Odom, D.T. Staged developmental mapping and X chromosome transcriptional dynamics during mouse spermatogenesis. Nat. Commun. 2019, 10, 1251. [Google Scholar] [CrossRef]
- Chen, S.-R.; Liu, Y.-X. Regulation of spermatogonial stem cell self-renewal and spermatocyte meiosis by Sertoli cell signaling. Reproduction 2015, 149, R159–R167. [Google Scholar] [CrossRef]
- Yang, F.; Whelan, E.C.; Guan, X.; Deng, B.; Wang, S.; Sun, J.; Avarbock, M.R.; Wu, X.; Brinster, R.L. FGF9 promotes mouse spermatogonial stem cell proliferation mediated by p38 MAPK signalling. Cell Prolif. 2021, 54, e12933. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Wang, Y.; Allis, C.D. Binary switches and modification cassettes in histone biology and beyond. Nature 2003, 425, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Qin, Q.; Wu, Q.; Sun, H.; Zheng, R.; Zang, C.; Zhu, M.; Wu, J.; Shi, X.; Taing, L.; et al. Cistrome Data Browser: A data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017, 45, D658–D662. [Google Scholar] [CrossRef]
- Cha, N.; Jia, B.; He, Y.; Luan, W.; Bao, W.; Han, X.; Gao, W.; Gao, Y. MicroRNA-124 suppresses the invasion and proliferation of breast cancer cells by targeting TFAP4. Oncol. Lett. 2021, 21, 271. [Google Scholar] [CrossRef]
- Tong, L.; Cao, W.; Sheng, J.; Zhu, E.; Yu, Y.; Zhong, T.; Chen, Y.; Wang, L. RDM1 plays an oncogenic role in human ovarian carcinoma cells. Artif. Cells Nanomed. Biotechnol. 2020, 48, 885–892. [Google Scholar] [CrossRef]
- He, C.; Li, A.; Lai, Q.; Ding, J.; Yan, Q.; Liu, S.; Li, Q. The DDX39B/FUT3/TGFβR-I axis promotes tumor metastasis and EMT in colorectal cancer. Cell Death Dis. 2021, 12, 74. [Google Scholar] [CrossRef]
- An, X.; Li, T.; Chen, N.; Wang, H.; Wang, X.; Ma, Y. PGAM1 regulates the glycolytic metabolism of SCs in tibetan sheep and its influence on the development of SCs. Gene 2021, 804, 145897. [Google Scholar] [CrossRef]
- Chen, K.; Zhu, L.; Guo, L.; Pan, Y.-B.; Feng, D.-F. Maf1 regulates dendritic morphogenesis and influences learning and memory. Cell Death Dis. 2020, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Qi, X.; Gao, X.; Li, J.; Qin, Y.; Yin, Y.; Gao, F.; Feng, T.; Wu, S.; Du, X. A Genome-Wide CRISPR Screen Identifies Factors Regulating Pluripotency Exit in Mouse Embryonic Stem Cells. Cells 2022, 11, 2289. [Google Scholar] [CrossRef] [PubMed]
- Alajem, A.; Biran, A.; Harikumar, A.; Sailaja, B.S.; Aaronson, Y.; Livyatan, I.; Nissim-Rafinia, M.; Sommer, A.G.; Mostoslavsky, G.; Gerbasi, V.R.; et al. Differential association of chromatin proteins identifies BAF60a/SMARCD1 as a regulator of embryonic stem cell differentiation. Cell Rep. 2015, 10, 2019–2031. [Google Scholar] [CrossRef]
- Lei, D.; Yang, W.-T.; Zheng, P.-S. HOXB4 inhibits the proliferation and tumorigenesis of cervical cancer cells by downregulating the activity of Wnt/β-catenin signaling pathway. Cell Death Dis. 2021, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef]
- An, J.; Zhang, X.; Qin, J.; Wan, Y.; Hu, Y.; Liu, T.; Li, J.; Dong, W.; Du, E.; Pan, C.; et al. The histone methyltransferase ESET is required for the survival of spermatogonial stem/progenitor cells in mice. Cell Death Dis. 2014, 5, e1196. [Google Scholar] [CrossRef]
- Chen, T.; Dent, S.Y.R. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef]
- Tachibana, M.; Nozaki, M.; Takeda, N.; Shinkai, Y. Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J. 2007, 26, 3346–3359. [Google Scholar] [CrossRef]
- Di Giacomo, M.; Comazzetto, S.; Sampath, S.C.; Sampath, S.C.; O’Carroll, D. G9a co-suppresses LINE1 elements in spermatogonia. Epigenetics Chromatin 2014, 7, 24. [Google Scholar] [CrossRef]
- Yu, L.; Ham, K.; Gao, X.; Castro, L.; Yan, Y.; Kissling, G.E.; Tucker, C.J.; Flagler, N.; Dong, R.; Archer, T.K.; et al. Epigenetic regulation of transcription factor promoter regions by low-dose genistein through mitogen-activated protein kinase and mitogen-and-stress activated kinase 1 nongenomic signaling. Cell Commun. Signal. 2016, 14, 18. [Google Scholar] [CrossRef]
- Wellard, S.R.; Schindler, K.; Jordan, P.W. Aurora B and C kinases regulate chromosome desynapsis and segregation during mouse and human spermatogenesis. J Cell Sci. 2020, 133, jcs248831. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Aréchaga, J.M.; Avarbock, M.R.; Brinster, R.L. Transplantation of testis germinal cells into mouse seminiferous tubules. Int. J. Dev. Biol. 1997, 41, 111–122. [Google Scholar] [PubMed]
- Bellvé, A.R.; Cavicchia, J.C.; Millette, C.F.; O’Brien, D.A.; Bhatnagar, Y.M.; Dym, M. Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J. Cell Biol. 1977, 74, 68–85. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Li, H.; Wang, M.; Zhang, X.; Ren, J.; Yuan, Y.; Sha, J.; Guo, X. Multi-Omics Approaches for Revealing the Epigenetic Regulation of Histone H3.1 during Spermatogonial Stem Cell Differentiation In Vitro. Int. J. Mol. Sci. 2023, 24, 3314. https://doi.org/10.3390/ijms24043314
Liu L, Li H, Wang M, Zhang X, Ren J, Yuan Y, Sha J, Guo X. Multi-Omics Approaches for Revealing the Epigenetic Regulation of Histone H3.1 during Spermatogonial Stem Cell Differentiation In Vitro. International Journal of Molecular Sciences. 2023; 24(4):3314. https://doi.org/10.3390/ijms24043314
Chicago/Turabian StyleLiu, Li, Haojie Li, Mengjie Wang, Xiangzheng Zhang, Jie Ren, Yan Yuan, Jiahao Sha, and Xuejiang Guo. 2023. "Multi-Omics Approaches for Revealing the Epigenetic Regulation of Histone H3.1 during Spermatogonial Stem Cell Differentiation In Vitro" International Journal of Molecular Sciences 24, no. 4: 3314. https://doi.org/10.3390/ijms24043314