
Modification of Ischemia/Reperfusion-Induced Alterations in Subcellular Organelles by Ischemic Preconditioning


Abstract
:1. Introduction
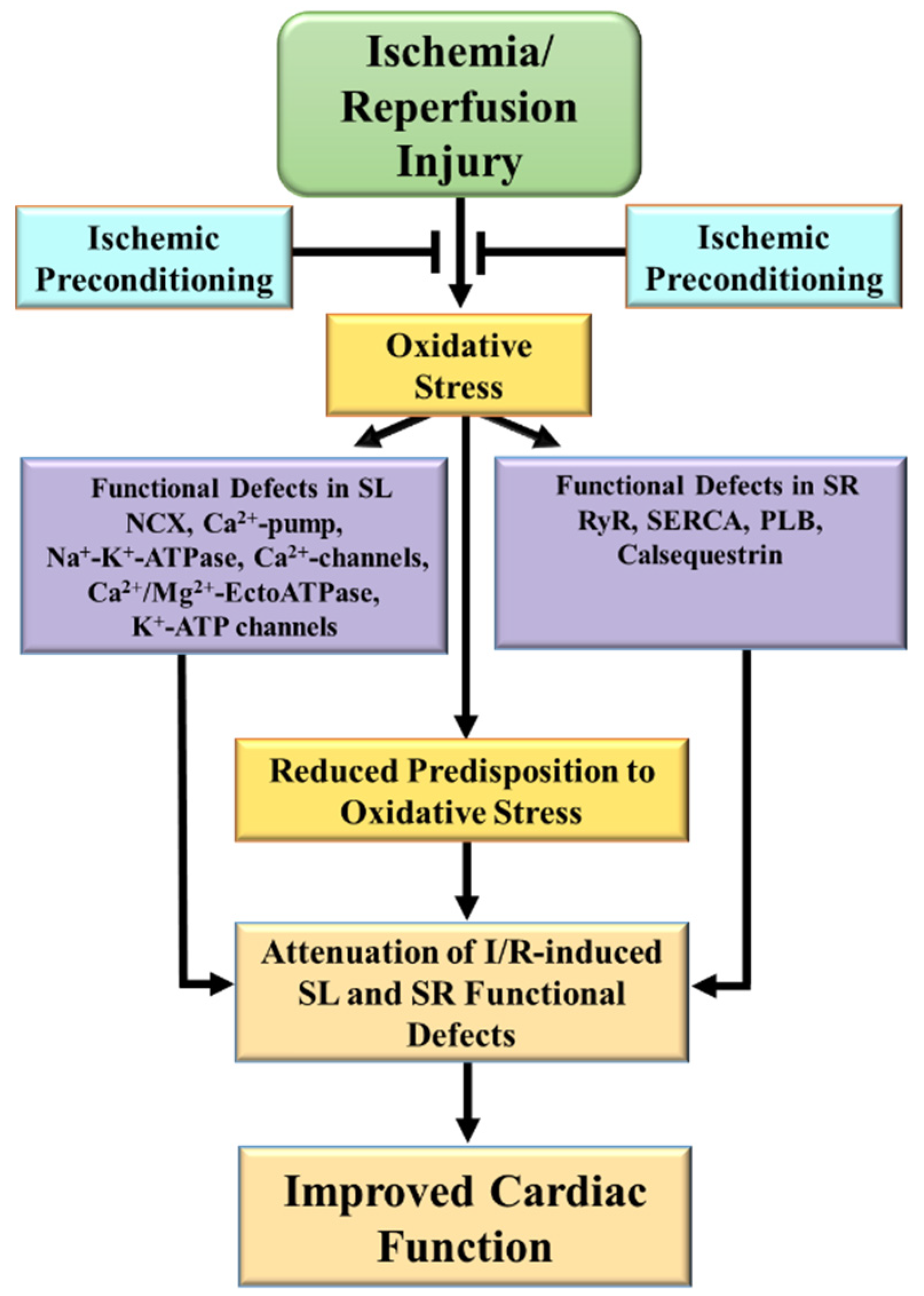
2. Alterations in Cardiac SL and SR Ca2+-Transporting Activities Due to I/R
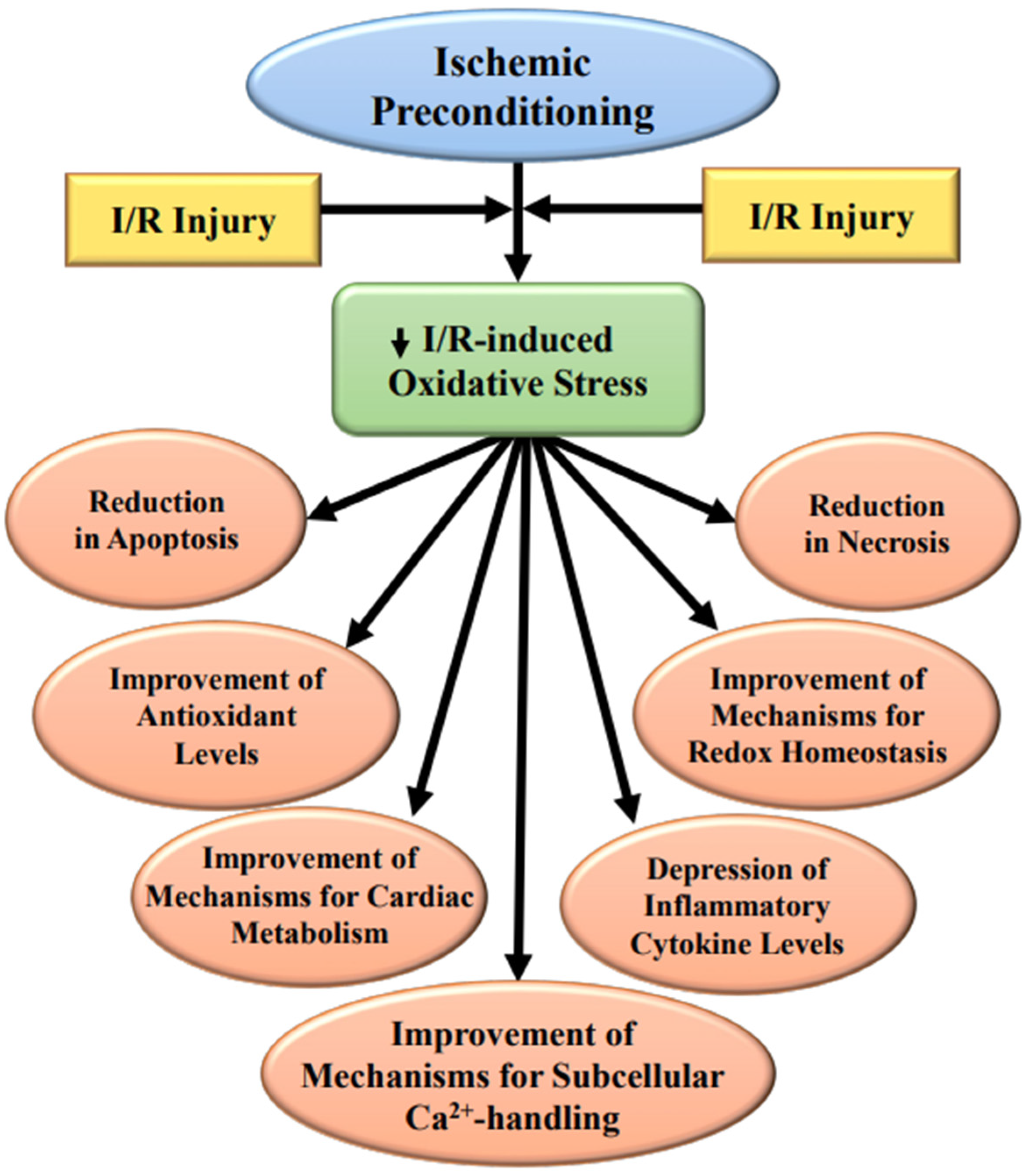
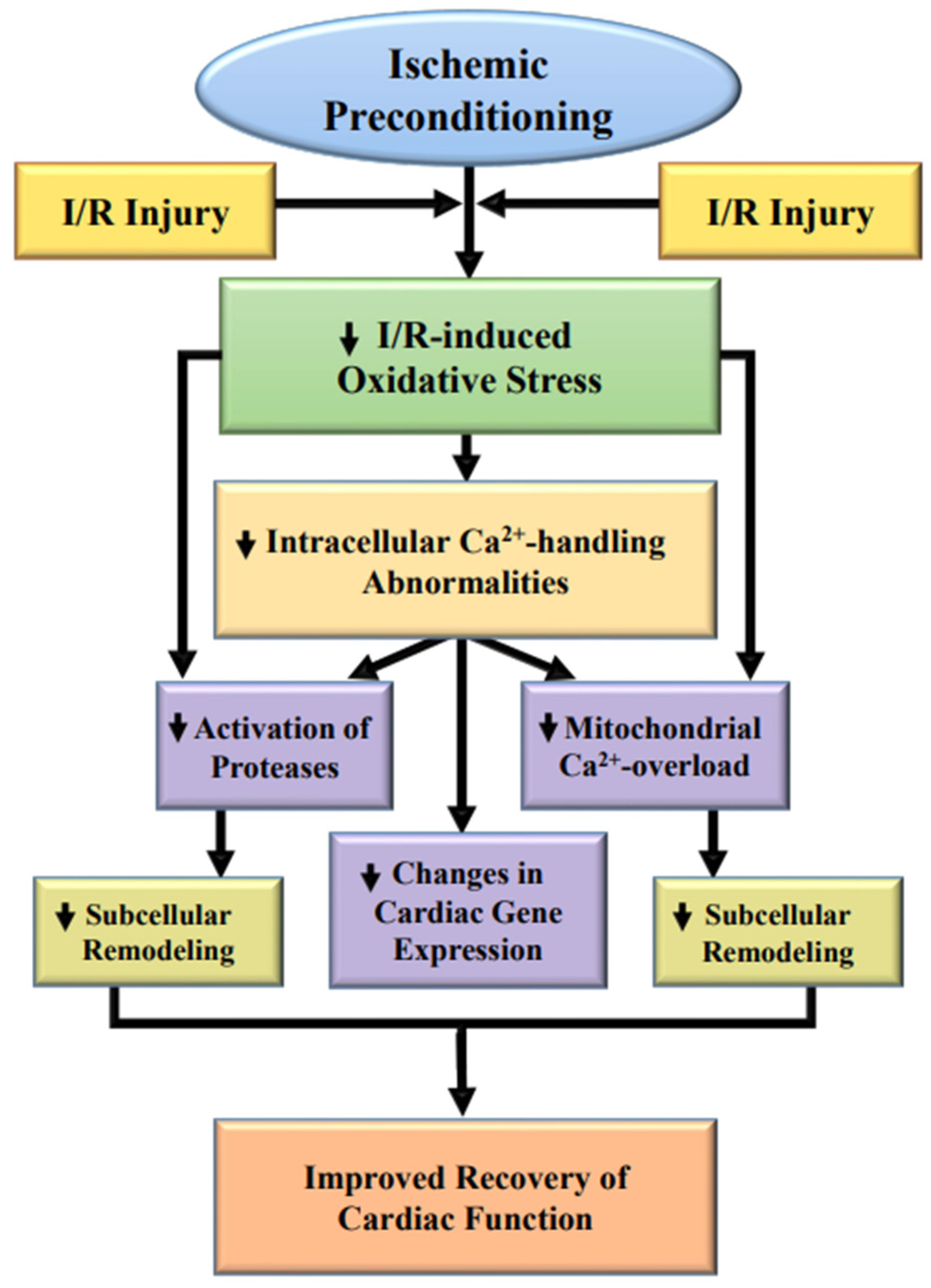
3. Subcellular Modification Due to Ischemic Preconditioning
3.1. Protection of the SL Defects
3.2. Protection for the SR Defects
3.3. Protection of the Mitochondrial Defects
3.4. Evidence for Attenuation of I/R-Induced SL and SR Defects by IP
4. Modification of I/R-Induced Defects in Signal Transduction
4.1. Role of Inflammatory Cytokines in IP-Induced Cardioprotection
4.2. Impact of IP on the Activation of Proteolysis
4.3. Role of Nrf2 Signal Transduction in IP-Induced Cardioprotection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jennings, R.B.; Reimer, K.A. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 1991, 42, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Hearse, D.J.; Bolli, R. Reperfusion induced injury: Manifestations, mechanisms, and clinical relevance. Cardiovasc. Res. 1992, 26, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Marban, E. Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 1999, 79, 609–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Golfman, L.; Takeda, S.; Takeda, N.; Nagano, M. Evidence for the role of oxidative stress in acute ischemic heart disease: A brief review. Can. J. Cardiol. 1999, 15, 587–593. [Google Scholar]
- Piper, H.M.; Meuter, K.; Schafer, C. Cellular mechanisms of ischemia-reperfusion injury. Ann. Thorac. Surg. 2003, 75, S644–S648. [Google Scholar] [CrossRef]
- Marczin, N.; El-Habashi, N.; Hoare, G.S.; Bundy, R.E.; Yacoub, M. Antioxidants in myocardial ischemia-reperfusion injury: Therapeutic potential and basic mechanisms. Arch. Biochem. Biophys. 2003, 420, 222–236. [Google Scholar] [CrossRef]
- Kim, S.J.; Depre, C.; Vatner, S.F. Novel mechanisms mediating stunned myocardium. Heart Fail. Rev. 2003, 8, 143–153. [Google Scholar] [CrossRef]
- Simonovic, N.; Jakovljevic, V.; Jeremic, J.; Finderle, Z.; Srejovic, I.; Nikolic Turnic, T.; Milosavljevic, I.; Zivkovic, V. Comparative effects of calcium and potassium channel modulators on ischemia/reperfusion injury in the isolated rat heart. Mol. Cell. Biochem. 2019, 450, 175–185. [Google Scholar] [CrossRef]
- Huang, Z.; Li, H.; Guo, F.; Jia, Q.; Zhang, Y.; Liu, X.; Shi, G. Egr-1, the potential target of calcium channel blockers in cardioprotection with ischemia/reperfusion injury in rats. Cell Physiol. Biochem. 2009, 24, 17–24. [Google Scholar] [CrossRef]
- Karmazyn, M. NHE-1: Still a viable therapeutic target. J. Mol. Cell. Cardiol. 2013, 61, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Kandilci, H.B.; Richards, M.A.; Fournier, M.; Şimşek, G.; Chung, Y.J.; Lakhal-Littleton, S.; Swietach, P. Cardiomyocyte Na+/H+ exchanger-1 activity is reduced in hypoxia. Front. Cardiovasc. Med. 2021, 7, 617038. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Nakao, K.; Shibata, Y.; Sone, T.; Takayama, T.; Fukuzawa, S.; Nakama, Y.; Hirayama, H.; Matsumoto, N.; Kosuge, M.; et al. Randomized controlled trial of TY-51924, a novel hydrophilic NHE inhibitor, in acute myocardial infarction. J. Cardiol. 2016, 67, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazmuri, R.J.; Radhakrishnan, J.; Ayoub, I.M. Sodium-hydrogen exchanger isoform-1 inhibition: A promising pharmacological intervention for resuscitation from cardiac arrest. Molecules 2019, 24, 1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhalla, N.S.; Temsah, R.M.; Netticacadan, T.; Sandhu, M.S. Calcium overload in ischemia/reperfusion injury. In Heart Physiology and Pathophysiology, 4th ed.; Sperelakis, N., Kurachi, Y., Terzic, A., Cohen, M., Eds.; Academic Press: San Diego, PA, USA, 2001; pp. 949–965. [Google Scholar]
- Bompotis, G.C.; Deftereos, S.; Angelidis, C.; Choidis, E.; Panagopoulou, V.; Kaoukis, A.; Vassilikos, V.P.; Cleman, M.W.; Giannopoulos, G. Altered calcium handling in reperfusion injury. Med. Chem. 2016, 12, 114–130. [Google Scholar] [CrossRef]
- Fauconnier, J.; Roberge, S.; Saint, N.; Lacampagne, A. Type 2 ryanodine receptor: A novel therapeutic target in myocardial ischemia/reperfusion. Pharmacol. Ther. 2013, 138, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Li, S. The Na+/Ca2+ exchanger in cardiac ischemia/reperfusion injury. Med. Sci. Monit. 2012, 18, RA161–RA165. [Google Scholar] [CrossRef] [Green Version]
- Saini, H.K.; Dhalla, N.S. Defective calcium handling in cardiomyocytes isolated from hearts subjected to ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2260–H2270. [Google Scholar] [CrossRef]
- He, F.; Wu, Q.; Xu, B.; Wang, X.; Wu, J.; Huang, L.; Cheng, J. Suppression of Stim1 reduced intracellular calcium concentration and attenuated hypoxia/reoxygenation induced apoptosis in H9C2 cells. Biosci. Rep. 2017, 37, BSR20171249. [Google Scholar] [CrossRef] [Green Version]
- Müller, B.A.; Dhalla, N.S. Mechanisms of the beneficial actions of ischemic preconditioning on subcellular remodeling in ischemic-reperfused heart. Curr. Cardiol. Rev. 2010, 6, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, H.K.; Machackova, J.; Dhalla, N.S. Role of reactive oxygen species in ischemic preconditioning of subcellular organelles in the heart. Antioxid. Redox Signal. 2004, 6, 393–404. [Google Scholar] [CrossRef]
- Fernandez Rico, C.; Konate, K.; Josse, E.; Nargeot, J.; Barrère-Lemaire, S.; Boisguérin, P. Therapeutic peptides to treat myocardial ischemia-reperfusion injury. Front. Cardiovasc. Med. 2022, 9, 792885. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Horvath, C.; Abdul-Ghani, S.; Varga, Z.V.; Suleiman, M.S.; Dhalla, N.S. Interplay of oxidative stress and necrosis-like cell death in cardiac ischemia/reperfusion injury: A focus on necroptosis. Biomedicines. 2022, 10, 127. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Zhang, L.; Jin, Z.; Kasumov, T.; Chen, Y.R. Mitochondrial redox regulation and myocardial ischemia-reperfusion injury. Am. J. Physiol. Cell Physiol. 2022, 322, C12–C23. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, R.; Wang, C. The role of mitochondrial quality control in cardiac ischemia/reperfusion injury. Oxid. Med. Cell Longev. 2021, 2021, 5543452. [Google Scholar] [CrossRef]
- Zucchi, R.; Ronca, F.; Ronca-Testoni, S. Modulation of sarcoplasmic reticulum function: A new strategy in cardioprotection? Pharmacol. Ther. 2001, 89, 47–65. [Google Scholar] [CrossRef]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef] [Green Version]
- Jovanović, A. Cardioprotective signalling: Past, present and future. Eur. J. Pharmacol. 2018, 833, 314–319. [Google Scholar] [CrossRef]
- Caricati-Neto, A.; Errante, P.R.; Menezes-Rodrigues, F.S. Recent advances in pharmacological and non-pharmacological strategies of cardioprotection. Int. J. Mol. Sci. 2019, 20, 4002. [Google Scholar] [CrossRef] [Green Version]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Obal, D.; Dai, S.; Keith, R.; Dimova, N.; Kingery, J.; Zheng, Y.T.; Zweier, J.; Velayutham, M.; Prabhu, S.D.; Li, Q.; et al. Cardiomyocyte-restricted overexpression of extracellular superoxide dismutase increases nitric oxide bioavailability and reduces infarct size after ischemia/reperfusion. Basic Res. Cardiol. 2012, 107, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G. Electron paramagnetic resonance oximetry and redoximetry. Methods Mol. Biol. 2010, 594, 85–105. [Google Scholar]
- Khan, M.; Brauner, M.E.; Plewa, M.C.; Kutala, V.K.; Angelos, M.; Kuppusamy, P. Effect of pulmonary-generated reactive oxygen species on left-ventricular dysfunction associated with cardio-pulmonary ischemia-reperfusion injury. Cell Biochem. Biophys. 2013, 67, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Saini-Chohan, H.K.; Dhalla, N.S. Attenuation of ischemia-reperfusion-induced alterations in intracellular Ca2+ in cardiomyocytes from hearts treated with N-acetylcysteine and N-mercaptopropionylglycine. Can. J. Physiol. Pharmacol. 2009, 87, 1110–1119. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The role of mitochondria in the mechanisms of cardiac ischemia-reperfusion injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.C.; Lien, C.F.; Lee, W.S.; Chang, H.R.; Hsu, Y.C.; Luo, Y.P.; Jeng, J.R.; Hsieh, J.C.; Yang, K.T. Intermittent hypoxia prevents myocardial mitochondrial Ca2+ overload and cell death during ischemia/reperfusion: The role of reactive oxygen species. Cells 2019, 8, 564. [Google Scholar] [CrossRef] [Green Version]
- Tappia, P.S.; Asemu, G.; Rodriguez-Leyva, D. Phospholipase C as a potential target for cardioprotection during oxidative stress. Can. J. Physiol. Pharmacol. 2010, 88, 249–263. [Google Scholar] [CrossRef]
- Matucci, R.; Bennardini, F.; Sciammarella, M.L.; Baccaro, C.; Stendardi, I.; Franconi, F.; Giotti, A. [3H]-nitrendipine binding in membranes obtained from hypoxic and reoxygenated heart. Biochem. Pharmacol. 1987, 36, 1059–1062. [Google Scholar] [CrossRef]
- Nayler, W.G.; Dillon, J.S.; Elz, J.S.; McKelvie, M. An effect of ischemia on myocardial dihydropyridine binding sites. Eur. J. Pharmacol. 1985, 115, 81–89. [Google Scholar] [CrossRef]
- Kaneko, M.; Lee, S.L.; Wolf, C.M.; Dhalla, N.S. Reduction of calcium channel antagonist binding sites by oxygen free radicals in rat heart. J. Mol. Cell. Cardiol. 1989, 21, 935–943. [Google Scholar] [CrossRef]
- Dixon, I.M.; Eyolfson, D.A.; Dhalla, N.S. Sarcolemmal Na+-Ca2+ exchange activity in hearts subjected to hypoxia reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 1987, 253, H1026–H1034. [Google Scholar] [CrossRef] [PubMed]
- Conway, S.J.; Koushik, S.V. Cardiac sodium–calcium exchanger: A double-edged sword. Cardiovasc. Res. 2001, 51, 194–197. [Google Scholar] [CrossRef] [Green Version]
- Javier Inserte, J.; Garcia-Dorado, D.; Ruiz-Meana, M.; Padilla, F.; Barrabés, J.A.; Pina, P.; Agulló, L.; Piper, H.M.; Soler-Soler, J. Effect of inhibition of Na+/Ca2+ exchanger at the time of myocardial reperfusion on hypercontracture and cell death. Cardiovasc. Res. 2002, 55, 739–748. [Google Scholar]
- Kaneko, M.; Beamish, R.E.; Dhalla, N.S. Depression of heart sarcolemmal Ca2+-pump activity by oxygen free radicals. Am. J. Physiol. Heart Circ. Physiol. 1989, 256, H368–H374. [Google Scholar] [CrossRef]
- Kaneko, M.; Elimban, V.; Dhalla, N.S. Mechanism for depression of heart sarcolemmal Ca2+ pump by oxygen free radicals. Am. J. Physiol. Heart Circ. Physiol. 1989, 257, H804–H811. [Google Scholar] [CrossRef]
- Hata, T.; Kaneko, M.; Beamish, R.E.; Dhalla, N.S. Influence of oxygen free radicals on heart sarcolemmal Na+-Ca2+ exchange. Coron. Artery Dis. 1991, 2, 397–407. [Google Scholar] [CrossRef]
- Pratt, R.D.; Brickman, C.R.; Cottrill, C.L.; Shapiro, J.I.; Liu, J. The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species. Int. J. Mol. Sci. 2018, 19, 2600. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.; Singal, P.K.; Dhalla, N.S. Alterations in heart sarcolemmal Ca2+-ATPase and Ca2+-binding activities due to oxygen free radicals. Basic Res. Cardiol. 1990, 85, 45–54. [Google Scholar] [CrossRef]
- Ostadal, P.; Elmoselhi, A.B.; Zdobnicka, I.; Lukas, A.; Elimban, V.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced changes in Na+-K+-ATPase isoform expression in rat heart. Antioxid. Redox Signal. 2004, 6, 914–923. [Google Scholar] [CrossRef]
- Elmoselhi, A.B.; Lukas, A.; Ostadal, P.; Dhalla, N.S. Preconditioning attenuates ischemia-reperfusion-induced remodeling of Na+-K+ATPase in hearts. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1055–H1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Nie, Y.; Chaudhry, M.; Bai, F.; Chuang, J.; Sodhi, K.; Shapiro, J.I. The redox-sensitive Na/K-ATPase signaling in uremic cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lilly, M.N.; Shapiro, J.I. Targeting Na/K-ATPase signaling: A new approach to control oxidative stress. Curr. Pharm. Des. 2018, 24, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Petrushanko, I.Y.; Hernansanz-Agustín, P.; Martínez-Ruiz, A. “Oxygen Sensing” by Na,K-ATPase: These miraculous thiols. Front. Physiol. 2016, 7, 314. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.; Panagia, V.; Paolillo, G.; Majumder, S.; Ou, C.; Dhalla, N.S. Inhibition of cardiac phosphatidylethanolamine N-methylation by oxygen free radicals. Biochim. Biophys. Acta 1990, 1021, 33–38. [Google Scholar] [CrossRef]
- Kato, K.; Shao, Q.; Elimban, V.; Lukas, A.; Dhalla, N.S. Mechanism of depression in cardiac sarcolemmal Na+-K+-ATPase by hypochlorous acid. Am. J. Physiol. Cell Physiol. 1998, 275, C826–C831. [Google Scholar] [CrossRef]
- Shao, Q.; Matsubara, T.; Bhatt, S.K.; Dhalla, N.S. Inhibition of cardiac sarcolemma Na+-K+ ATPase by oxyradical generating systems. Mol. Cell. Biochem. 1995, 147, 139–144. [Google Scholar] [CrossRef]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef] [Green Version]
- Santacruz-Toloza, L.; Ottolia, M.; Nicoll, D.A.; Philipson, K.D. Functional analysis of a disulfide bond in the cardiac Na+-Ca2+ exchanger. J. Biol. Chem. 2000, 275, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Chakraborti, S.; Das, S.; Kar, P.; Ghosh, B.; Samanta, K.; Kolley, S.; Ghosh, S.; Roy, S.; Chakraborti, T. Calcium signaling phenomena in heart diseases: A perspective. Mol. Cell. Biochem. 2007, 298, 1–40. [Google Scholar] [CrossRef]
- Villa-Abrille, M.C.; Sidor, A.; O’Rourke, B. Insulin effects on cardiac Na+/Ca2+ exchanger activity: Role of the cytoplasmic regulatory loop. J. Biol. Chem. 2008, 283, 16505–16513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, T.; Dhalla, N.S. Effect of oxygen free radicals on cardiac contractile activity and sarcolemmal Na+-Ca2+ exchange. J. Cardiovasc. Pharmacol. Ther. 1996, 1, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Dhalla, N.S. Relationship between mechanical dysfunction and depression of sarcolemmal Ca2+-pump activity in hearts perfused with oxygen free radicals. Mol. Cell. Biochem. 1996, 160–161, 179–185. [Google Scholar] [CrossRef]
- Aleksandrova, E.A. Calcium- transporting systems and calcium regulation in cardiomyocytes. Usp. Fiziol. Nauk. 2001, 32, 40–48. [Google Scholar]
- Matsubara, T.; Musat-Marcu, S.; Misra, H.P.; Dhalla, N.S. Protective effect of vanadate on oxyradical-induced changes in isolated perfused heart. Mol. Cell. Biochem. 1995, 153, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Musat, S.; Dhalla, N.S. Alteration in cardiac sarcolemmal ATP receptors by oxyradicals. Ann. N. Y. Acad. Sci. 1996, 793, 1–12. [Google Scholar] [CrossRef]
- Persad, S.; Takeda, S.; Panagia, V.; Dhalla, N.S. Beta-adrenoceptor-linked signal transduction in ischemic-reperfused heart and scavenging of oxyradicals. J. Mol. Cell. Cardiol. 1997, 29, 545–558. [Google Scholar] [CrossRef]
- Persad, S.; Takeda, S.; Dhalla, N.S. Alternations in beta-adrenoceptor mechanisms in hearts perfused with xanthine plus xanthine oxidase. J. Cardiovasc. Pharmacol. Ther. 1997, 2, 115–124. [Google Scholar] [CrossRef]
- Persad, S.; Panagia, V.; Dhalla, N.S. Role of H2O2 in changing beta-adrenoceptor and adenylyl cyclase in ischemia-reperfused hearts. Mol. Cell. Biochem. 1998, 186, 99–106. [Google Scholar] [CrossRef]
- Persad, S.; Dhalla, N.S. Modification of beta-adrenoceptors and adenylyl cyclase in hearts perfused with hypochlorous acid. Can. J. Physiol. Pharmacol. 1998, 76, 961–966. [Google Scholar] [CrossRef]
- Persad, S.; Elimban, V.; Kaila, J.; Dhalla, N.S. Biphasic alterations in cardiac beta-adrenoceptor signal transduction mechanism due to oxyradicals. J. Pharmacol. Exp. Ther. 1997, 282, 1623–1631. [Google Scholar] [PubMed]
- Persad, S.; Rupp, H.; Jindal, R.; Arneja, J.; Dhalla, N.S. Modification of cardiac beta-adrenoceptor mechanisms by H2O2. Am. J. Physiol. Heart Circ. Physiol. 1998, 274, H416–H423. [Google Scholar] [CrossRef] [PubMed]
- Persad, S.; Elimban, V.; Siddiqui, F.; Dhalla, N.S. Alterations in cardiac membrane beta-adrenoceptors and adenylyl cyclase due to hypochlorous acid. J. Mol. Cell. Cardiol. 1999, 31, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sato, N.; Kudej, R.K.; Uechi, M.; Asai, K.; Shen, Y.T.; Ishikawa, Y.; Vatner, S.F.; Vatner, D.E. β-adrenergic receptor signalling in stunned myocardium of conscious pigs. J. Mol. Cell. Cardiol. 1997, 29, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Haenen, G.R.; Veerman, M.; Bast, A. Reduction of beta-adrenoceptor function by oxidative stress in the heart. Free Radic. Biol. Med. 1990, 9, 279–288. [Google Scholar] [CrossRef]
- Kaneko, M.; Chapman, D.C.; Ganguly, P.K.; Beamish, R.E.; Dhalla, N.S. Modification of cardiac adrenergic receptors by oxygen free radicals. Am. J. Physiol. Heart Circ. Physiol. 1991, 260, H821–H826. [Google Scholar] [CrossRef] [PubMed]
- Asemu, G.; Tappia, P.S.; Dhalla, N.S. Identification of the changes in phospholipase C isozymes in ischemic-reperfused rat heart. Arch. Biochem. Biophys. 2003, 411, 174–182. [Google Scholar] [CrossRef]
- Asemu, G.; Dent, M.; Singal, T.; Dhalla, N.S.; Tappia, P.S. Differential changes in phospholipase D and phosphatidate phosphohydralase activities in ischemia-reperfusion of rat heart. Arch. Biochem. Biophys. 2005, 436, 136–144. [Google Scholar] [CrossRef]
- Munakata, M.; Stamm, C.; Friehs, I.; Zurakowski, D.; Cowan, D.B.; Cao-Danh, H.; McGowan, F.X., Jr.; del Nido, P.J. Protective effects of protein kinase C during myocardial ischemia require activation of phosphatidyl-inositol specific phospholipase C. Ann. Thorac. Surg. 2002, 73, 1236–1245. [Google Scholar] [CrossRef]
- Askenasy, N.; Vivi, A.; Tassini, M.; Navon, G.; Farkas, D.L. NMR spectroscopic characterization of sarcolemmal permeability during myocardial ischemia and reperfusion. J. Mol. Cell. Cardiol. 2001, 33, 1421–1433. [Google Scholar] [CrossRef]
- Kyoi, S.; Otani, H.; Sumida, T.; Okada, T.; Osako, M.; Imamura, H.; Kamihata, H.; Matsubara, H.; Iwasaka, T. Loss of intracellular dystrophin: A potential mechanism for myocardial reperfusion injury. Circ. J. 2003, 67, 725–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative stress as a mechanism for functional alterations in cardiac hypertrophy and heart failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.F. Emerging role for antioxidant therapy in protection against diabetic cardiac complications: Experimental and clinical evidence for utilization of classic and new antioxidants. Curr. Cardiol. Rev. 2008, 4, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddall, H.K.; Yellon, D.M.; Ong, S.B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PLoS ONE 2013, 8, e62400. [Google Scholar] [CrossRef]
- Morrey, C.; Brazin, J.; Seyedi, N.; Corti, F.; Silver, R.B.; Levi, R. Interaction between sensory C-fibers and cardiac mast cells in ischemia/reperfusion: Activation of a local renin-angiotensin system culminating in severe arrhythmic dysfunction. J. Pharmacol. Exp. Ther. 2010, 335, 76–84. [Google Scholar] [CrossRef]
- Goldhaber, J.I.; Qayyum, M.S. Oxygen free radicals and excitation-contraction coupling. Antioxid. Redox. Signal. 2000, 2, 55–64. [Google Scholar] [CrossRef]
- Basu, D.K.; Karmazyn, M. Injury to rat hearts produced by an exogenous free radical generating system. Study into the role of arachidonic acid and eicosanoids. J. Pharmacol. Exp. Ther. 1987, 242, 673–685. [Google Scholar]
- Eley, D.W.; Korecky, B.; Fliss, H. Dithiothreitol restores contractile function to oxidant-injured cardiac muscle. Am. J. Physiol. Heart Circ. Physiol. 1989, 257, H1321–H1325. [Google Scholar] [CrossRef]
- Manson, N.H.; Hess, M.L. Interaction of oxygen free radicals and cardiac sarcoplasmic reticulum: Proposed role in the pathogenesis of endotoxin shock. Circ. Shock. 1983, 10, 205–213. [Google Scholar]
- Rowe, G.T.; Manson, N.H.; Caplan, M.; Hess, M.L. Hydrogen peroxide and hydroxyl radical mediation of activated leukocyte depression of cardiac sarcoplasmic reticulum. Participation of the cyclooxygenase pathway. Circ. Res. 1983, 53, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Okabe, E.; Hess, M.L.; Oyama, M.; Ito, H. Characterization of free radical-mediated damage of canine cardiac sarcoplasmic reticulum. Arch. Biochem. Biophys. 1983, 225, 164–177. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The role of oxidative stress in cardiac disease: From physiological response to injury factor. Oxid. Med. Cell Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Shah, A.K.; Dhalla, N.S. Role of oxidative stress in the genesis of ventricular arrhythmias. Int. J. Mol. Sci. 2020, 21, 4200. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.L.; Webb, C.; Moffat, M.; McIver, D.; del Maestro, R. Activated neutrophils depress myocardial function in the perfused rabbit heart. Can. J. Cardiol. 1991, 7, 323–330. [Google Scholar] [PubMed]
- Ford, D.A. Lipid oxidation by hypochlorous acid: Chlorinated lipids in atherosclerosis and myocardial ischemia. Clin. Lipidol. 2010, 5, 835–852. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Xu, J.; Tao, L.; Wang, S.; Xiang, H.; Tang, Y. Synergetic protective effect of remote ischemic preconditioning and prolyl 4-hydroxylase inhibition in ischemic cardiac injury. Mol. Med. Rep. 2022, 25, 80. [Google Scholar] [CrossRef]
- Khaliulin, I.; Ascione, R.; Maslov, L.N.; Amal, H.; Suleiman, M.S. Preconditioning or postconditioning with 8-Br-cAMP-AM protects the heart against regional ischemia and reperfusion: A role for mitochondrial permeability transition. Cells 2021, 10, 1223. [Google Scholar] [CrossRef]
- Hentia, C.; Rizzato, A.; Camporesi, E.; Yang, Z.; Muntean, D.M.; Săndesc, D.; Bosco, G. An overview of protective strategies against ischemia/reperfusion injury: The role of hyperbaric oxygen preconditioning. Brain Behav. 2018, 8, e00959. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef]
- Nuñez, R.E.; Javadov, S.; Escobales, N. Angiotensin II-preconditioning is associated with increased PKCε/PKCδ ratio and prosurvival kinases in mitochondria. Clin. Exp. Pharmacol. Physiol. 2017, 44, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Guo, S.; Wu, H.K.; Lv, F.; Jin, L.; Zhang, M.; Xie, P.; Wang, Y.; Song, Y.; Wu, F.; et al. Cardiac ischemic preconditioning promotes MG53 secretion through H2O2-activated protein kinase C-δ signaling. Circulation 2020, 142, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Wang, L.; Zhang, J.; Pan, T.; Yu, Y.; Lu, J.; Zhou, P.; Yang, H.; Li, P. Activated PKB/GSK-3βsynergizes with PKC-δ signaling in attenuating myocardial ischemia/reperfusion injury via potentiation of NRF2 activity: Therapeutic efficacy of dihydrotanshinone-I. Acta Pharm. Sin B 2021, 11, 71–88. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, E.; Baldassari, F.; Bononi, A.; Wieckowski, M.R.; Pinton, P. Oxidative stress in cardiovascular diseases and obesity: Role of p66Shc and protein kinase C. Oxid. Med. Cell Longev. 2013, 2013, 564961. [Google Scholar] [CrossRef] [Green Version]
- Churchill, E.N.; Disatnik, M.H.; Mochly-Rosen, D. Time-dependent and ethanol-induced cardiac protection from ischemia mediated by mitochondrial translocation of varepsilon PKC and activation of aldehyde dehydrogenase 2. J. Mol. Cell. Cardiol. 2009, 46, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Apostolakis, E.; Baikoussis, N.G.; Papakonstantinou, N.A. The role of myocardial ischaemic preconditioning during beating heart surgery: Biological aspect and clinical outcome. Interact. Cardiovasc. Thorac. Surg. 2012, 14, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Asemu, G.; Dhalla, N.S.; Tappia, P.S. Inhibition of PLC improves postischemic recovery in isolated rat heart. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2598–H2605. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.J.; Saini, H.K.; Turan, B.; Liu, P.P.; Dhalla, N.S. Pentoxifylline attenuate cardiac dysfunction and reduces TNF-α level in ischemia-reperfused heart. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H832–H839. [Google Scholar] [CrossRef]
- Beresewicz, A.; Czarnowska, E.; Maczewski, M. Ischemic preconditioning and superoxide dismutase protect against endothelial dysfunction and endothelium glycocalyx disruption in the postischemic guinea-pig hearts. Mol. Cell. Biochem. 1998, 186, 87–97. [Google Scholar] [CrossRef]
- Lalu, M.M.; Csonka, C.; Giricz, Z.; Csont, T.; Schulz, R.; Ferdinandy, P. Preconditioning decreases ischemia/reperfusion-induced release and activation of matrix metalloproteinase-2. Biochem. Biophys. Res. Commun. 2002, 296, 937–941. [Google Scholar] [CrossRef]
- Cheung, P.Y.; Sawicki, G.; Wozniak, M.; Wang, W.; Radomski, M.W.; Schulz, R. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation 2000, 101, 1833–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, R.; Castillo, R.; Cereceda, M.; Asenjo, R.; Zamorano, J.; Araya, J. Non-hypoxic preconditioning of myocardium against postoperative atrial fibrillation: Mechanism based on enhancement of the antioxidant defense system. Med. Hypotheses 2007, 69, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.; Huot, J. Modulation of actin dynamics during stress and physiological stimulation by a signaling pathway involving p38 MAP kinase and heat-shock protein 27. Biochem. Cell Biol. 1995, 73, 703–707. [Google Scholar] [CrossRef]
- Das, D.K.; Maulik, N.; Sato, M.; Ray, P.S. Reactive oxygen species function as second messenger during ischemic preconditioning of heart. Mol. Cell. Biochem. 1999, 196, 59–67. [Google Scholar] [CrossRef]
- Singh, L.; Kulshrestha, R.; Singh, N.; Jaggi, A.S. Mechanisms involved in adenosine pharmacological preconditioning-induced cardioprotection. Korean J. Physiol. Pharmacol. 2018, 22, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Fryer, R.M.; Auchampach, J.A.; Gross, G.J. Therapeutic receptor targets of ischemic preconditioning. Cardiovasc. Res. 2002, 55, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Pachauri, P.; Garabadu, D.; Goyal, A.; Upadhyay, P.K. Angiotensin (1-7) facilitates cardioprotection of ischemic preconditioning on ischemia-reperfusion-challenged rat heart. Mol. Cell. Biochem. 2017, 430, 99–113. [Google Scholar] [CrossRef]
- Zhang, J.; Simpson, P.C.; Jensen, B.C. Cardiac α1A-adrenergic receptors: Emerging protective roles in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H725–H733. [Google Scholar] [CrossRef]
- Buchholz, B.; Annunzio, D.V.; Giani, J.F.; Siachoque, N.; Dominici, F.P.; Turyn, D.; Perez, V.; Donato, M.; Gelpi, R.J. Ischemic postconditioning reduces infarct size through the α1-adrenergic receptor pathway. J. Cardiovasc. Pharmacol. 2014, 63, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Headrick, J.P.; See Hoe, L.E.; Du Toit, E.F.; Peart, J.N. Opioid receptors and cardioprotection—‘Opioidergic conditioning’ of the heart. Br. J. Pharmacol. 2015, 172, 2026–2050. [Google Scholar] [CrossRef] [Green Version]
- Fraessdorf, J.; Hollmann, M.W.; Hanschmann, I.; Heinen, A.; Weber, N.C.; Preckel, B.; Huhn, R. Role of endogenous opioid system in ischemic-induced late preconditioning. PLoS ONE 2015, 10, e0134283. [Google Scholar]
- Anzawa, R.; Seki, S.; Nagoshi, T.; Taniguchi, I.; Feuvray, D.; Yoshimura, M. The role of Na+/H+ exchanger in Ca2+ overload and ischemic myocardial damage in hearts from type 2 diabetic db/db mice. Cardiovasc. Diabetol. 2012, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.H.; Long, C.; Liu, J.; Liu, B. Inhibition of the Na+/H+ exchanger protects the immature rabbit myocardium from ischemia and reperfusion injury. Pediatr. Cardiol. 2008, 29, 113–120. [Google Scholar] [CrossRef]
- Ostadal, P.; Elmoselhi, A.B.; Zdobnicka, I.; Lukas, A.; Chapman, D.; Dhalla, N.S. Ischemia-reperfusion alters gene expression of Na+-K+ ATPase isoforms in rat heart. Biochem. Biophys. Res. Commun. 2003, 306, 457–462. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Saini, H.K.; Tappia, P.S.; Sethi, R.; Mengi, S.A.; Gupta, S.K. Potential role and mechanisms of subcellular remodeling in cardiac dysfunction due to ischemic heart disease. J. Cardiovasc. Med. 2007, 8, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Zahler, R.; Brines, M.; Kashgarian, M.; Benz, E.J., Jr.; Gilmore-Hebert, M. The cardiac conduction system in the rat expresses the α2 and α3 isoforms of the Na+,K+-ATPase. Proc. Natl. Acad. Sci. USA 1992, 89, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Akera, T. O2 free radicals: Cause of ischemia-reperfusion injury to cardiac Na+-K+-ATPase. Am. J. Physiol. 1987, 252, H252–H257. [Google Scholar] [CrossRef]
- Aizman, O.; Aperia, A. Na,K-ATPase as a signal transducer. Ann. N. Y. Acad. Sci. 2003, 986, 489–496. [Google Scholar] [CrossRef]
- Shapiro, J.I.; Tian, J. Signaling through the Na/K-ATPase: Implications for cardiac fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H29–H30. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Cai, T. Na+-K+-ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 2003, 3, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Marck, P.V.; Pierre, S.V. Na/K-ATPase Signaling and Cardiac Pre/Postconditioning with Cardiotonic Steroids. Int. J. Mol. Sci. 2018, 19, 2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Staehr, C.; Zeng, F.; Bouzinova, E.V.; Matchkov, V.V. The Na,K-ATPase in vascular smooth muscle cells. Curr. Top. Membr. 2019, 83, 151–175. [Google Scholar]
- Pierre, S.V.; Yang, C.; Yuan, Z.; Seminerio, J.; Mouas, C.; Garlid, K.D.; Dos-Santos, P.; Xie, Z. Ouabain triggers preconditioning through activation of the Na+,K+-ATPase signaling cascade in rat hearts. Cardiovasc. Res. 2007, 73, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belliard, A.; Gulati, G.K.; Duan, Q.; Alves, R.; Brewer, S.; Madan, N.; Sottejeau, Y.; Wang, X.; Kalisz, J.; Pierre, S.V. Ischemia/reperfusion-induced alterations of enzymatic and signaling functions of the rat cardiac Na+/K+-ATPase: Protection by ouabain preconditioning. Physiol. Rep. 2016, 4, e12991. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Murakami, T.; Iwase, T.; Nagai, K.; Nawada, R.; Kouchi, I.; Akao, M.; Sasayama, S. KATP channels contribute to the cardioprotection of preconditioning independent of anaesthetics in rabbit hearts. J. Mol. Cell. Cardiol. 1997, 29, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imahashi, K.; Nishimura, T.; Yoshioka, J.; Kusuoka, H. Role of intracellular Na+ kinetics in preconditioned rat heart. Circ. Res. 2001, 88, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Slezak, J.; Tribulova, N.; Pristacova, J.; Uhrik, B.; Thomas, T.; Khaper, N.; Kaul, N.; Singal, P.K. Hydrogen peroxide changes in ischemic and reperfused heart. Cytochemistry and biochemical and X-ray microanalysis. Am. J. Pathol. 1995, 147, 772–781. [Google Scholar]
- Gross, G.J.; Auchampach, J.A. Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ. Res. 1992, 70, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Tokube, K.; Kiyosue, T.; Arita, M. Openings of cardiac KATP channel by oxygen free radicals produced by xanthine oxidase reaction. Am. J. Physiol. 1996, 271, H478–H489. [Google Scholar] [CrossRef]
- Gross, G.J.; Fryer, R.M. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ. Res. 1999, 84, 973–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovic, A.; Jovanovic, S.; Lorenz, E.; Terzic, A. Recombinant cardiac ATP-sensitive K+ channel subunits confer resistance to chemical hypoxia-reoxygenation injury. Circulation 1998, 98, 1548–1555. [Google Scholar] [CrossRef] [Green Version]
- Marinovic, J.; Bosnjak, Z.J.; Stadnicka, A. Distinct roles for sarcolemmal and mitochondrial adenosine triphosphate-sensitive potassium channels in isoflurane-induced protection against oxidative stress. Anesthesiology 2006, 105, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Gumina, R.J.; Pucar, D.; Bast, P.; Hodgson, D.M.; Kurtz, C.E.; Dzeja, P.P.; Miki, T.; Seino, S.; Terzic, A. Knockout of Kir6.2 negates ischemic preconditioning-induced protection of myocardial energetics. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2106–H2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinovic, J.; Ljubkovic, M.; Stadnicka, A.; Bosnjak, Z.J.; Bienengraeber, M. Role of sarcolemmal ATP-sensitive potassium channel in oxidative stress-induced apoptosis: Mitochondrial connection. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1317–H1325. [Google Scholar] [CrossRef]
- Nakamura, T.; Hayashi, H.; Satoh, H.; Katoh, H.; Kaneko, M.; Terada, H. A single cell model of myocardial reperfusion injury: Changes in intracellular Na+ and Ca2+ concentrations in guinea pig ventricular myocytes. Mol. Cell. Biochem. 1999, 194, 147–157. [Google Scholar] [CrossRef]
- Nayler, W.G. The role of calcium in the ischemic myocardium. Am. J. Pathol. 1981, 102, 262–270. [Google Scholar]
- Nayler, W.G.; Panagiotopoulos, S.; Elz, J.S.; Daly, M.J. Calcium-mediated damage during post-ischaemic reperfusion. J. Mol. Cell. Cardiol. 1988, 20, 41–54. [Google Scholar] [CrossRef]
- Temsah, R.M.; Netticadan, T.; Chapman, D.; Takeda, S.; Mochizuki, S.; Dhalla, N.S. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am. J. Physiol. 1999, 277, H584–H594. [Google Scholar] [CrossRef]
- Osada, M.; Netticadan, T.; Tamura, K.; Dhalla, N.S. Modification of ischemia-reperfusion-induced changes in cardiac sarcoplasmic reticulum by preconditioning. Am. J. Physiol. 1998, 274, H2025–H2034. [Google Scholar] [CrossRef]
- Temsah, R.M.; Kawabata, K.; Chapman, D.; Dhalla, N.S. Preconditioning prevents alterations in cardiac SR gene expression due to ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1461–H1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuster, G.M.; Lancel, S.; Zhang, J.; Communal, C.; Trucillo, M.P.; Lim, C.C.; Pfister, O.; Weinberg, E.O.; Cohen, R.A.; Liao, R.; et al. Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic. Biol. Med. 2010, 48, 1182–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, P.; Babusikova, E.; Lehotsky, J.; Dobrota, D. Free radical-induced protein modification and inhibition of Ca2+-ATPase of cardiac sarcoplasmic reticulum. Mol. Cell. Biochem. 2003, 248, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, R.; Yu, G.; Galbani, P.; Mariani, M.; Ronca, G.; Ronca-Testoni, S. Sulfhydryl redox state affects susceptibility to ischemia and sarcoplasmic reticulum Ca2+ release in rat heart. Implications for ischemic preconditioning. Circ. Res. 1998, 83, 908–915. [Google Scholar] [CrossRef] [Green Version]
- Lotz, C.; Herrmann, J.; Notz, Q.; Meybohm, P.; Kehl, F. Mitochondria and pharmacologic cardiac conditioning-at the heart of ischemic injury. Int. J. Mol. Sci. 2021, 22, 3224. [Google Scholar] [CrossRef]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria "THE" target of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef] [Green Version]
- Vishwakarma, V.K.; Upadhyay, P.K.; Chaurasiya, H.S.; Srivasatav, R.K.; Ansari, T.M.; Srivastava, V. Mechanistic pathways of ATP sensitive potassium channels referring to cardio-protective effects and cellular functions. Drug Res. 2019, 69, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Matejíková, J.; Kucharská, J.; Pintérová, M.; Pancza, D.; Ravingerová, T. Protection against ischemia-induced ventricular arrhythmias and myocardial dysfunction conferred by preconditioning in the rat heart: Involvement of mitochondrial K(ATP) channels and reactive oxygen species. Physiol. Res. 2009, 58, 9–19. [Google Scholar] [CrossRef]
- Forbes, R.A.; Steenbergen, C.; Murphy, E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ. Res. 2001, 88, 802–809. [Google Scholar] [CrossRef] [Green Version]
- Pagliaro, P.; Mancardi, D.; Rastaldo, R.; Penna, C.; Gattullo, D.; Miranda, K.M.; Feelisch, M.; Wink, D.A.; Kass, D.A.; Paolocci, N. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic. Biol. Med. 2003, 34, 33–43. [Google Scholar] [CrossRef]
- Mancardi, D.; Pagliaro, P.; Ridnour, L.A.; Tocchetti, C.G.; Miranda, K.; Juhaszova, M.; Sollott, S.J.; Wink, D.A.; Paolocci, N. HNO protects the myocardium against reperfusion injury, inhibiting the mPTP opening via PKCε activation. Antioxidants 2022, 11, 382. [Google Scholar] [CrossRef] [PubMed]
- Vanden Hoek, T.L.; Li, C.; Shao, Z.; Schumacker, P.T.; Becker, L.B. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol. 1997, 29, 2571–2583. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.M.; Eells, J.T.; Hsu, A.K.; Henry, M.M.; Gross, G.J. Ischemic preconditioning in rats: Role of mitochondrial K(ATP) channel in preservation of mitochondrial function. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H305–H3012. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. What makes the mitochondria a killer? Can we condition them to be less destructive? Biochim. Biophys. Acta 2011, 1813, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Perrelli, M.G.; Pagliaro, P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: Therapeutic implications. Antioxid. Redox Signal. 2013, 18, 556–599. [Google Scholar] [CrossRef]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [Green Version]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Fontaine, E.; Bernardi, P. Progress on the mitochondrial permeability transition pore: Regulation by complex I and ubiquinone analogs. J. Bioenerg. Biomembr. 1999, 31, 335–345. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Brookes, P.S. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: Implications for ischemic preconditioning. Biochim. Biophys. Acta 2008, 1777, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Nadtochiy, S.M.; Baker, P.R.; Freeman, B.A.; Brookes, P.S. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: Implications for cardioprotection. Cardiovasc. Res. 2009, 82, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batthyany, C.; Schopfer, F.J.; Baker, P.R.; Durán, R.; Baker, L.M.; Huang, Y.; Cerveñansky, C.; Branchaud, B.P.; Freeman, B.A. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J. Biol. Chem. 2006, 281, 20450–20463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabe, K.; Nasa, Y.; Sato, M.; Iijima, R.; Takeo, S. Preconditioning preserves mitochondrial function and glycolytic flux during an early period of reperfusion in perfused rat hearts. Cardiovasc. Res. 1997, 33, 677–685. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Eerbeek, O.; Meijer, A.J. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H496–H499. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Shukair, S.; Naik, T.J.; Moazed, F.; Ardehali, H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol. Cell Biol. 2008, 28, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Osada, M.; Netticadan, T.; Kawabata, K.; Tamura, K.; Dhalla, N.S. Ischemic preconditioning prevents I/R-induced alterations in SR calcium-calmodulin protein kinase II. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1791–H1798. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xu, Y.J.; Saini, H.K.; Turan, B.; Liu, P.P.; Dhalla, N.S. TNF-α as a potential mediator of cardiac dysfunction due to intracellular Ca2+-overload. Biochem. Biophys. Res. Commun. 2005, 327, 57–63. [Google Scholar] [CrossRef]
- Qian, W.; Zhao, C.; Li, D.; Dai, R. Mechanism of interleukin-1 receptor antagonist protection against myocardial ischaemia/reperfusion-induced injury. Arch. Cardiovasc. Dis. 2018, 111, 545–554. [Google Scholar] [CrossRef]
- Wang, H.Y.; Liu, X.Y.; Han, G.; Wang, Z.Y.; Li, X.X.; Jiang, Z.M.; Jiang, C.M. LPS induces cardiomyocyte injury through calcium-sensing receptor. Mol. Cell. Biochem. 2013, 379, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Smart, N.; Mojet, M.H.; Latchman, D.S.; Marber, M.S.; Duchen, M.R.; Heads, R.J. IL-6 induces PI 3-kinase and nitric oxide-dependent protection and preserves mitochondrial function in cardiomyocytes. Cardiovasc. Res. 2006, 69, 164–177. [Google Scholar] [CrossRef]
- Meldrum, D.R. Tumor necrosis factor in the heart. Am. J. Physiol. 1998, 274, R577–R595. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Zarębiński, M.; Pawluczuk, P.; Szukiewicz, D. Decreased effectiveness of ischemic heart preconditioning in the state of chronic inflammation. Med. Hypotheses 2015, 85, 675–679. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Yin, B.L. Pro-inflammatory cytokines may induce late preconditioning in unstable angina patients. Med. Hypotheses 2006, 67, 1121–1124. [Google Scholar] [CrossRef]
- Zubakov, D.; Hoheisel, J.D.; Kluxen, F.W.; Brändle, M.; Ehring, T.; Hentsch, B.; Frohme, M. Late ischemic preconditioning of the myocardium alters the expression of genes involved in inflammatory response. FEBS. Lett. 2003, 547, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Lecour, S.; Smith, R.M.; Woodward, B.; Opie, L.H.; Rochette, L.; Sack, M.N. Identification of a novel role for sphingolipid signaling in TNFα and ischemic preconditioning mediated cardioprotection. J. Mol. Cell. Cardiol. 2002, 34, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldrum, D.R.; Dinarello, C.A.; Shames, B.D.; Cleveland, J.C., Jr.; Cain, B.S.; Banerjee, A.; Meng, X.; Harken, A.H. Ischemic preconditioning decreases postischemic myocardial tumor necrosis factor-α production. Potential ultimate effector mechanism of preconditioning. Circulation 1998, 98, II214–II218. [Google Scholar] [PubMed]
- Xiong, J.; Wang, Q.; Xue, F.S.; Yuan, Y.J.; Li, S.; Liu, J.H.; Liao, X.; Zhang, Y.M. Comparison of cardioprotective and anti-inflammatory effects of ischemia pre- and postconditioning in rats with myocardial ischemia-reperfusion injury. Inflamm. Res. 2011, 60, 547–554. [Google Scholar] [CrossRef]
- Maulik, N.; Engelman, R.M.; Wei, Z.; Lu, D.; Rousou, J.A.; Das, D.K. Interleukin-1 α preconditioning reduces myocardial ischemia reperfusion injury. Circulation 1993, 88, II387–II394. [Google Scholar]
- McGinnis, G.R.; Ballmann, C.; Peters, B.; Nanayakkara, G.; Roberts, M.; Amin, R.; Quindry, J.C. Interleukin-6 mediates exercise preconditioning against myocardial ischemia reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1423–H1433. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Qiao, Z.; Xu, B. Effects of ischemic preconditioning on myocardium Caspase-3, SOCS-1, SOCS-3, TNF-α and IL-6 mRNA expression levels in myocardium IR rats. Mol. Biol. Rep. 2013, 40, 5741–5748. [Google Scholar] [CrossRef]
- Hiasa, G.; Hamada, M.; Ikeda, S.; Hiwada, K. Ischemic preconditioning and lipopolysaccharide attenuate nuclear factor-κB activation and gene expression of inflammatory cytokines in the ischemia-reperfused rat heart. Jpn. Circ. J. 2001, 65, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Zuurbier, C.J.; Jong, W.M.; Eerbeek, O.; Koeman, A.; Pulskens, W.P.; Butter, L.M.; Leemans, J.C.; Hollmann, M.W. Deletion of the innate immune NLRP3 receptor abolishes cardiac ischemic preconditioning and is associated with decreased Il-6/STAT3 signaling. PLoS ONE 2012, 7, e40643. [Google Scholar]
- Dawn, B.; Xuan, Y.T.; Guo, Y.; Rezazadeh, A.; Stein, A.B.; Hunt, G.; Wu, W.J.; Tan, W.; Bolli, R. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc. Res. 2004, 64, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, A.L.; Hryshko, L.V.; Dhalla, N.S. Extracellular and intracellular proteases in cardiac dysfunction due to ischemia-reperfusion injury. Int. J. Cardiol. 2013, 164, 39–47. [Google Scholar] [CrossRef]
- Batista-Almeida, D.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Girao, H. The role of proteostasis in the regulation of cardiac intercellular communication. Adv. Exp. Med. Biol. 2020, 1233, 279–302. [Google Scholar]
- Singh, R.B.; Hryshko, L.; Freed, D.; Dhalla, N.S. Activation of proteolytic enzymes and depression of the sarcolemmal Na+/K+-ATPase in ischemia-reperfused heart may be mediated through oxidative stress. Can. J. Physiol. Pharmacol. 2012, 90, 249–260. [Google Scholar] [CrossRef]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Bárcena, C.; López-Otín, C.; Yehia, G.; et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Mata, A.; Cadenas, S. The antioxidant transcription factor Nrf2 in cardiac ischemia-reperfusion injury. Int. J. Mol. Sci. 2021, 22, 11939. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, J.; Strom, J.; Lee, S.; Chen, Q.M. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta 2014, 1842, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Ucar, B.I.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological protection against ischemia-reperfusion injury by regulating the Nrf2-Keap1-ARE signaling pathway. Antioxidants 2021, 10, 823. [Google Scholar] [CrossRef]
- Cheng, L.; Jin, Z.; Zhao, R.; Ren, K.; Deng, C.; Yu, S. Resveratrol attenuates inflammation and oxidative stress induced by myocardial ischemia-reperfusion injury: Role of Nrf2/ARE pathway. Int. J. Clin. Exp. Med. 2015, 8, 10420–10428. [Google Scholar] [PubMed]
- Purdom-Dickinson, S.E.; Lin, Y.; Dedek, M.; Morrissy, S.; Johnson, J.; Chen, Q.M. Induction of antioxidant and detoxification response by oxidants in cardiomyocytes: Evidence from gene expression profiling and activation of Nrf2 transcription factor. J. Mol. Cell. Cardiol. 2007, 42, 159–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, V.O.; Menshchikova, E.B.; Zenkov, N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry. 2011, 76, 407–422. [Google Scholar] [CrossRef]
- Maldonado, E.; Rojas, D.A.; Urbina, F.; Solari, A. The use of antioxidants as potential co-adjuvants to treat chronic chagas disease. Antioxidants 2021, 10, 1022. [Google Scholar] [CrossRef]
- Wang, J.; De-Qiong, X.; Hong, D.Q.; Zhang, Q.Q.; Zhang, J. Attenuation of myocardial ischemia reperfusion injury by Geniposide preconditioning in diabetic rats. Curr. Res. Transl. Med. 2019, 67, 35–40. [Google Scholar] [CrossRef]
- Yu, L.M.; Dong, X.; Xue, X.D.; Zhang, J.; Li, Z.; Wu, H.J.; Yang, Z.L.; Yang, Y.; Wang, H.S. Protection of the myocardium against ischemia/reperfusion injury by punicalagin through an SIRT1-NRF-2-HO-1-dependent mechanism. Chem. Biol. Interact. 2019, 306, 152–162. [Google Scholar] [CrossRef]
- Radjendirane, V.; Joseph, P.; Lee, Y.H.; Kimura, S.; Klein-Szanto, A.J.; Gonzalez, F.J.; Jaiswal, A.K. Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity. J. Biol. Chem. 1998, 273, 7382–7389. [Google Scholar] [CrossRef] [Green Version]
- Ku, B.M.; Joo, Y.; Mun, J.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Kim, H.J. Heme oxygenase protects hippocampal neurons from ethanol-induced neurotoxicity. Neurosci. Lett. 2006, 405, 168–171. [Google Scholar] [CrossRef]
- Refaie, M.M.M.; Shehata, S.; Ibrahim, R.A.; Bayoumi, A.M.A.; Abdel-Gaber, S.A. Dose-dependent cardioprotective effect of hemin in doxorubicin-induced cardiotoxicity via Nrf-2/HO-1 and TLR-5/NF-κB/TNF-α signaling pathways. Cardiovasc. Toxicol. 2021, 21, 1033–1044. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, Y.; He, H.; Wei, W. Liraglutide restores late cardioprotective effects of remote preconditioning in diabetic rats via activation of hydrogen sulfide and nuclear factor erythroid 2-related factor 2 signaling pathway. Acta Cir. Bras. 2021, 36, e360207. [Google Scholar] [CrossRef]
- Huang, X.S.; Chen, H.P.; Yu, H.H.; Yan, Y.F.; Liao, Z.P.; Huang, Q.R. Nrf2-dependent upregulation of antioxidative enzymes: A novel pathway for hypoxic preconditioning-mediated delayed cardioprotection. Mol. Cell. Biochem. 2014, 385, 33–41. [Google Scholar] [CrossRef]
- Yan, Y.F.; Chen, H.P.; Huang, X.S.; Qiu, L.Y.; Liao, Z.P.; Huang, Q.R. DJ-1 mediates the delayed cardioprotection of hypoxic preconditioning through activation of Nrf2 and subsequent upregulation of antioxidative enzymes. J. Cardiovasc. Pharmacol. 2015, 66, 148–158. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, Y.; Lei, H.; Cai, Y.; Shen, J.; Zhu, P.; He, Q.; Zhao, M. The Nrf-2/HO-1 Signaling Axis: A Ray of Hope in Cardiovascular Diseases. Cardiol. Res. Pract. 2020, 2020, 5695723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Shi, X.; Cheng, L.; Han, J.; Mu, J. Hydrogen sulfide restores cardioprotective effects of remote ischemic preconditioning in aged rats via HIF-1α/Nrf2 signaling pathway. Korean J. Physiol. Pharmacol. 2021, 25, 239–249. [Google Scholar] [CrossRef]
- Cominacini, L.; Mozzini, C.; Garbin, U.; Pasini, A.; Stranieri, C.; Solani, E.; Vallerio, P.; Tinelli, I.A.; Fratta Pasini, A. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free Radic. Biol. Med. 2015, 88, 233–242. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, Z.; Yao, J.; Zhao, G.; Fa, X.; Niu, J. Participation of protein kinase C in the activation of Nrf2 signaling by ischemic preconditioning in the isolated rabbit heart. Mol. Cell. Biochem. 2013, 372, 169–179. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Control | IP | I/R | IP + I/R | |
|---|---|---|---|---|
| Na+-K+-ATPase activity (µmol Pi/mg/h) | 15.0 ± 0.6 | 14.8 ± 0.8 | 11.3 ± 0.7 * | 15.4 ± 0.7 # |
| Protein content (arbitrary units) | ||||
| Na+-K+-ATPase α1 | 15.7 ± 1.3 | 15.0 ± 2.1 | 11.2 ± 1.4 * | 13.2 ± 1.8 |
| Na+-K+-ATPase α2 | 7.3 ± 1.8 | 7.1 ± 1.3 | 2.1 ± 1.5 * | 6.2 ± 1.1 # |
| Na+-K+-ATPase α3 | 2.3 ± 0.2 | 2.2 ± 0.5 | 0.8 ± 0.4 * | 1.4 ± 0.5 # |
| mRNA expression levels (% of control arbitrary units) | ||||
| Na+-K+-ATPase α1 | 100 | 140 ± 11 * | 79 ± 3 * | 95 ± 2 |
| Na+-K+-ATPase α2 | 100 | 90 ± 5 | 51 ± 4 * | 78 ± 4 # |
| Na+-K+-ATPase α3 | 100 | 100 ± 20 | 32 ± 5 * | 61 ± 12 # |
| SR Ca2+-Release (nmol/mg/15 s) | Preischemia | Postischemia | Reperfusion |
|---|---|---|---|
| Control | 17.5 ± 5.1 | 2.0 ± 0.5 # | 2.5 ± 0.5 # |
| Precondition | 6.7 ± 5.2 * | 7.3 ± 2.2 * | 11.2 ± 4.5 * |
| SR Ca2+-uptake (nmol/mg/min) | |||
| Control | 70.8 ± 8.1 | 8.0 ± 1.1 # | 9.5 ± 2.0 # |
| Precondition | 38.6 ± 6.4 * | 28.3 ± 3.7 * | 24.5 ± 1.5 * |
| SR Ca2+-stimulated ATPase activity (nmol Pi/mg/min) | |||
| Control | 199.8 ± 28.4 | 102.0 ± 20.4 @ | 65.5 ± 13.4 @ |
| Precondition | 201.4 ± 16.9 | 203.5 ± 26.4 ! | 146.1 ± 19.6 ! |
| A: mRNA Expression Levels (% of Control Arbitrary Units) | IP | I/R | IP + I/R |
|---|---|---|---|
| RyR | 68.2 ± 4.4 * | 35.7 ± 4.2 * | 72.1 ± 4.7 *,# |
| SERCA | 76.9 ± 4.9 * | 70.8 ± 2.4 * | 90.3 ± 3.4 *,# |
| PLB | 74.1 ± 3.2 * | 40.6 ± 3.6 * | 67.5 ± 4.8 *,# |
| Calsequestrin | 100 ± 3.5 | 50.8 ± 4.5 * | 71.8 ± 6.4 *,# |
| B: Relative protein content (% of control arbitrary units) | |||
| RyR | 73.4 ± 8.2 * | 51.7 ± 9.1 # | 923 ± 8.6 * |
| SERCA | 101.1 ± 18.1 | 45.4 ± 2.2 # | 81.2 ± 17.1 * |
| PLB | 118.1 ± 19.2 | 100.3 ± 18.3 | 105.5 ± 19.4 |
| Calsequestrin | n.d. | n.d. | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tappia, P.S.; Shah, A.K.; Ramjiawan, B.; Dhalla, N.S. Modification of Ischemia/Reperfusion-Induced Alterations in Subcellular Organelles by Ischemic Preconditioning. Int. J. Mol. Sci. 2022, 23, 3425. https://doi.org/10.3390/ijms23073425
Tappia PS, Shah AK, Ramjiawan B, Dhalla NS. Modification of Ischemia/Reperfusion-Induced Alterations in Subcellular Organelles by Ischemic Preconditioning. International Journal of Molecular Sciences. 2022; 23(7):3425. https://doi.org/10.3390/ijms23073425
Chicago/Turabian StyleTappia, Paramjit S., Anureet K. Shah, Bram Ramjiawan, and Naranjan S. Dhalla. 2022. "Modification of Ischemia/Reperfusion-Induced Alterations in Subcellular Organelles by Ischemic Preconditioning" International Journal of Molecular Sciences 23, no. 7: 3425. https://doi.org/10.3390/ijms23073425