
Blood–Brain Barrier Disruption and Its Involvement in Neurodevelopmental and Neurodegenerative Disorders

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Development of the Blood–Brain Barrier (BBB)
3. Components of the Blood–Brain Barrier (BBB)
3.1. Brain Microvascular Endothelial Cells (BMVECs)
BMVECs in Brain Vascular Contraction
3.2. Astrocytes
3.3. Pericytes
3.4. Basement Membrane (BM)
3.5. Transport across the BBB
4. Mechanisms Altering Blood–Brain Barrier (BBB) Function
4.1. Genetic Factors
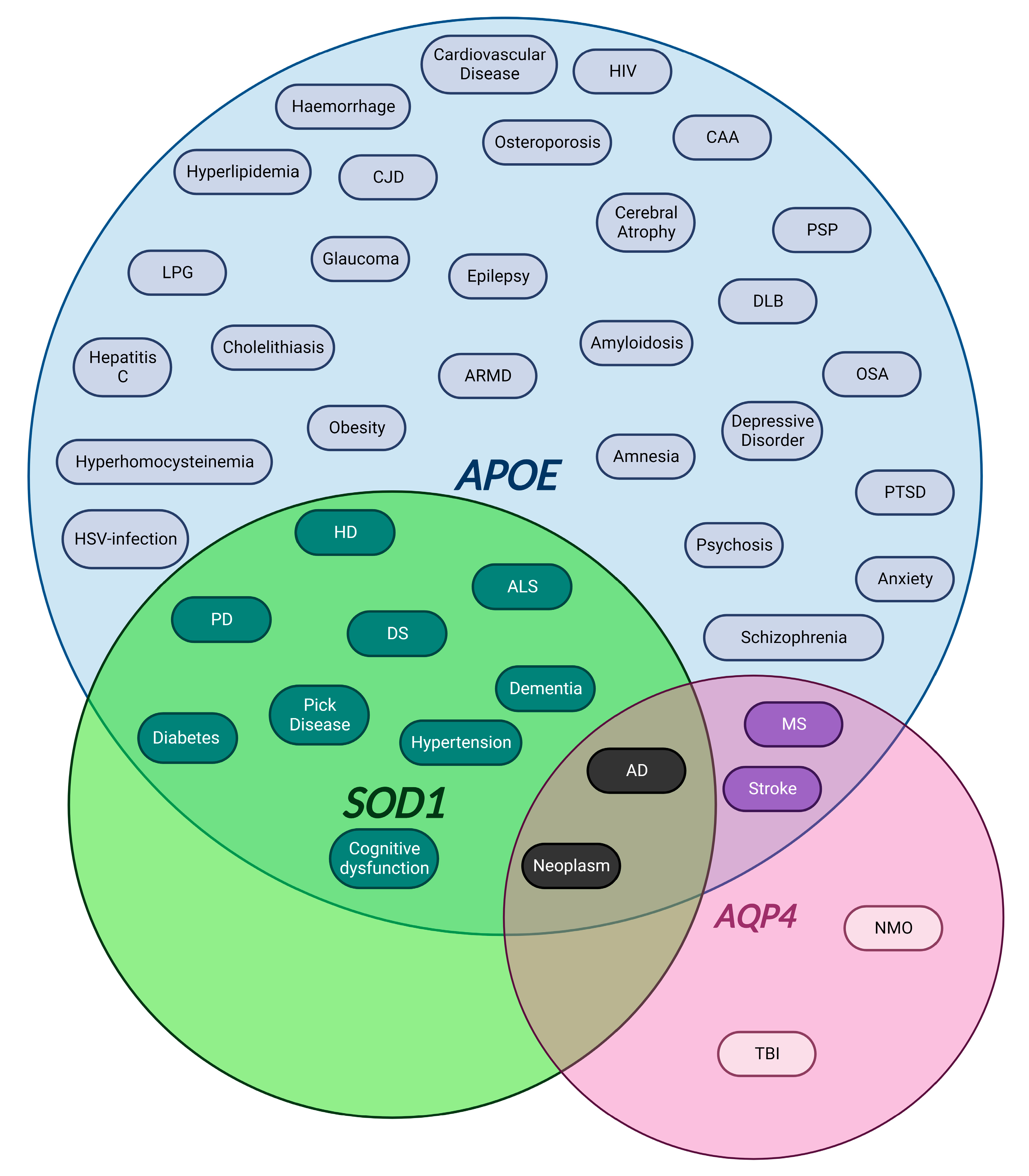
4.1.1. APOE
4.1.2. SOD1
4.1.3. AQP4
4.2. Oxidative Stress
4.2.1. Antioxidant Defences
Enzymes
Non-Enzymatic Anti-Oxidants
4.3. Neuroinflammation
4.4. Injures to the BBB
4.5. Gut–Brain Axis: Microbiota
5. Potential Therapies
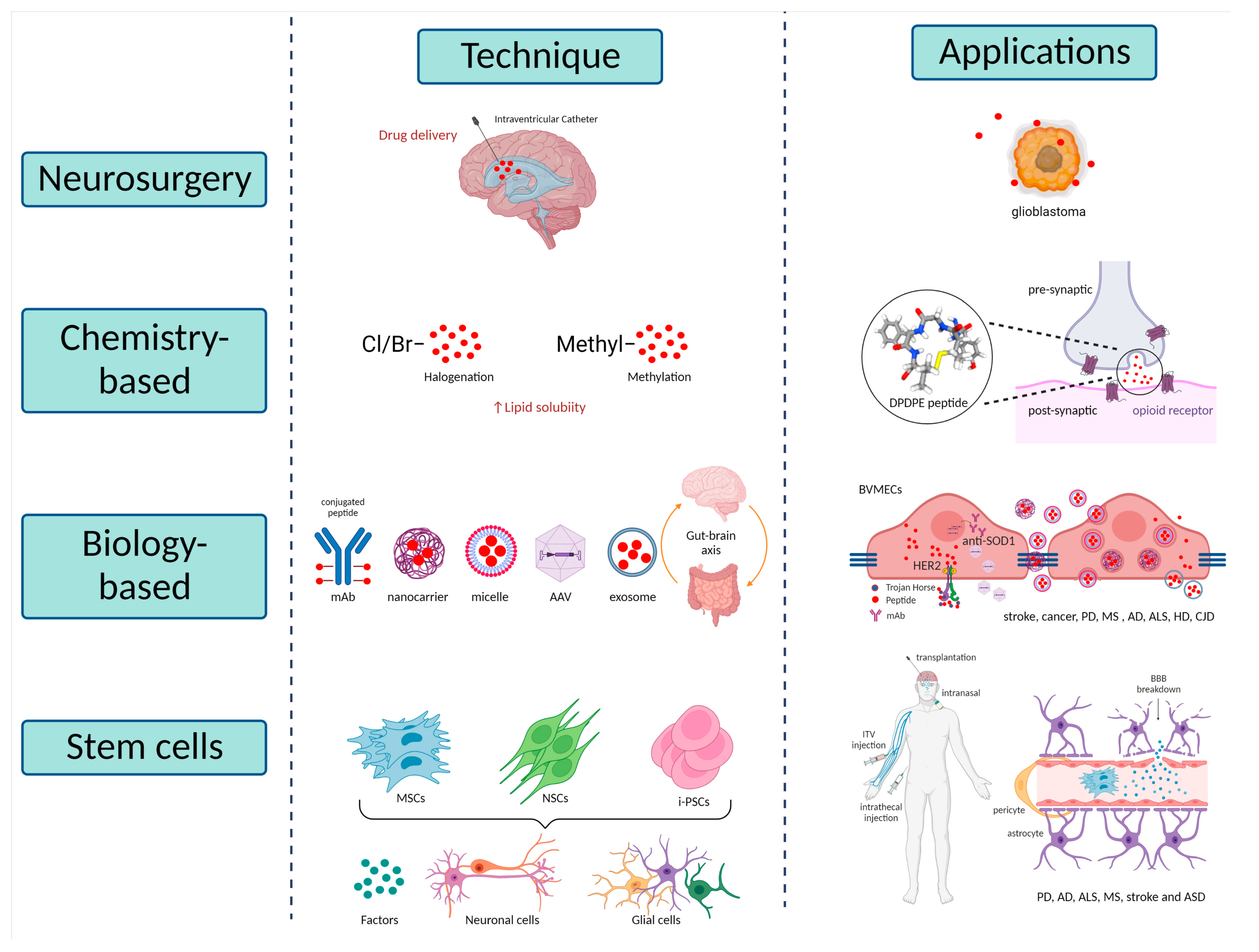
5.1. Neurosurgical-Based Techniques
5.2. Chemistry-Based Techniques
5.3. Biology-Based Techniques
5.3.1. Trojan Horse Technology
5.3.2. Nanotechnology Approaches
5.3.3. Exosomes Therapies
5.3.4. Microbiome Therapies
5.4. Stem Cell-Based Techniques
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Sharif, Y.; Jumah, F.; Coplan, L.; Krosser, A.; Sharif, K.; Tubbs, R.S. Blood Brain Barrier: A Review of Its Anatomy and Physiology in Health and Disease. Clin. Anat. 2018, 31, 812–823. [Google Scholar] [CrossRef]
- Coomber, B.L.; Stewart, P.A. Morphometric Analysis of CNS Microvascular Endothelium. Microvasc. Res. 1985, 30, 99–115. [Google Scholar] [CrossRef]
- Gross, P.M.; Sposito, N.M.; Pettersen, S.E.; Fenstermacher, J.D. Differences in Function and Structure of the Capillary Endothelium in Gray Matter, White Matter and a Circumventricular Organ of Rat Brain. Blood Vessel. 1986, 23, 261–270. [Google Scholar] [CrossRef]
- Zhao, R.; Pollack, G.M. Regional Differences in Capillary Density, Perfusion Rate, and P-Glycoprotein Activity: A Quantitative Analysis of Regional Drug Exposure in the Brain. Biochem. Pharmacol. 2009, 78, 1052–1059. [Google Scholar] [CrossRef]
- Wilhelm, I.; Nyúl-Tóth, Á.; Suciu, M.; Hermenean, A.; Krizbai, I.A. Heterogeneity of the Blood-Brain Barrier. Tissue Barriers 2016, 4, e1143544. [Google Scholar] [CrossRef]
- Wang, Q.-P.; Guan, J.-L.; Pan, W.; Kastin, A.J.; Shioda, S. A Diffusion Barrier Between the Area Postrema and Nucleus Tractus Solitarius. Neurochem. Res. 2008, 33, 2035–2043. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [Green Version]
- Delaney, C.; Campbell, M. The Blood Brain Barrier: Insights from Development and Ageing. Tissue Barriers 2017, 5, e1373897. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Marín-Padilla, M. The Human Brain Intracerebral Microvascular System: Development and Structure. Front. Neuroanat. 2012, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raab, S.; Beck, H.; Gaumann, A.; Yüce, A.; Gerber, H.-P.; Plate, K.; Hammes, H.-P.; Ferrara, N.; Breier, G. Impaired Brain Angiogenesis and Neuronal Apoptosis Induced by Conditional Homozygous Inactivation of Vascular Endothelial Growth Factor. Thromb. Haemost. 2004, 91, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/β-Catenin Signaling Is Required for CNS, but Not Non-CNS, Angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, C.Y.; Nusse, R. The Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [Green Version]
- Tran, K.A.; Zhang, X.; Predescu, D.; Huang, X.; Machado, R.F.; Göthert, J.R.; Malik, A.B.; Valyi-Nagy, T.; Zhao, Y.-Y. Endothelial β-Catenin Signaling Is Required for Maintaining Adult Blood-Brain Barrier Integrity and Central Nervous System Homeostasis. Circulation 2016, 133, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/Beta-Catenin Signaling Controls Development of the Blood-Brain Barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, M.; Francis, D.; Gisbert, D.; Gomes, F.C.A.; Stipursky, J. Radial Glia Cells Control Angiogenesis in the Developing Cerebral Cortex Through TGF-Β1 Signaling. Mol. Neurobiol. 2018, 55, 3660–3675. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes Are Required for Blood-Brain Barrier Integrity during Embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Hupe, M.; Li, M.X.; Kneitz, S.; Davydova, D.; Yokota, C.; Kele, J.; Hot, B.; Stenman, J.M.; Gessler, M. Gene Expression Profiles of Brain Endothelial Cells during Embryonic Development at Bulk and Single-Cell Levels. Sci. Signal. 2017, 10, eaag2476. [Google Scholar] [CrossRef]
- Corada, M.; Orsenigo, F.; Bhat, G.P.; Conze, L.L.; Breviario, F.; Cunha, S.I.; Claesson-Welsh, L.; Beznoussenko, G.V.; Mironov, A.A.; Bacigaluppi, M.; et al. Fine-Tuning of Sox17 and Canonical Wnt Coordinates the Permeability Properties of the Blood-Brain Barrier. Circ. Res. 2019, 124, 511–525. [Google Scholar] [CrossRef]
- Engelhardt, B.; Risau, W. Chapter 2: Development of the Blood-Brain Barrier. In New Concepts of a Blood–Brain Barrier; Springer: Boston, MA, USA, 1995. [Google Scholar] [CrossRef]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-Beta in Recruitment of Vascular Smooth Muscle Cells and Pericytes during Embryonic Blood Vessel Formation in the Mouse. Development 1999, 126, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.; Johansson, B.R.; Levéen, P.; Betsholtz, C. Pericyte Loss and Microaneurysm Formation in PDGF-B-Deficient Mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central Nervous System Pericytes in Health and Disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, Maintenance and Disruption of the Blood-Brain Barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of Pericytes Leads to Endothelial Hyperplasia and Abnormal Vascular Morphogenesis. J. Cell Biol. 2001, 153, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.-J.; Kim, K.-W. SSeCKS Regulates Angiogenesis and Tight Junction Formation in Blood-Brain Barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a Natural Antagonist for Tie2 That Disrupts in Vivo Angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- Nagase, T.; Nagase, M.; Machida, M.; Fujita, T. Hedgehog Signalling in Vascular Development. Angiogenesis 2008, 11, 71–77. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β Induces Blood-Brain Barrier Disruption by Downregulating Sonic Hedgehog in Astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [Green Version]
- Saunders, N.R.; Daneman, R.; Dziegielewska, K.M.; Liddelow, S.A. Transporters of the Blood-Brain and Blood-CSF Interfaces in Development and in the Adult. Mol. Aspects Med. 2013, 34, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Hagan, N.; Ben-Zvi, A. The Molecular, Cellular, and Morphological Components of Blood-Brain Barrier Development during Embryogenesis. Semin. Cell Dev. Biol. 2015, 38, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Mizee, M.R.; Wooldrik, D.; Lakeman, K.A.M.; van het Hof, B.; Drexhage, J.A.R.; Geerts, D.; Bugiani, M.; Aronica, E.; Mebius, R.E.; Prat, A.; et al. Retinoic Acid Induces Blood-Brain Barrier Development. J. Neurosci. 2013, 33, 1660–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, C.; Stepniak, B.; Schneider, A.; Papiol, S.; Tantra, M.; Begemann, M.; Sirén, A.-L.; Pardo, L.A.; Sperling, S.; Mohd Jofrry, S.; et al. Neuropsychiatric Disease Relevance of Circulating Anti-NMDA Receptor Autoantibodies Depends on Blood-Brain Barrier Integrity. Mol. Psychiatry 2014, 19, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [Green Version]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit—Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Villegas, J.C.; Broadwell, R.D. Transcytosis of Protein through the Mammalian Cerebral Epithelium and Endothelium. II. Adsorptive Transcytosis of WGA-HRP and the Blood-Brain and Brain-Blood Barriers. J. Neurocytol. 1993, 22, 67–80. [Google Scholar] [CrossRef]
- Henninger, D.D.; Panés, J.; Eppihimer, M.; Russell, J.; Gerritsen, M.; Anderson, D.C.; Granger, D.N. Cytokine-Induced VCAM-1 and ICAM-1 Expression in Different Organs of the Mouse. J. Immunol. 1997, 158, 1825–1832. [Google Scholar]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the Blood-Brain Barrier: Molecular Anatomy and Possible Investigation Approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-Resistance Gene (P-Glycoprotein) Is Expressed by Endothelial Cells at Blood-Brain Barrier Sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittapalli, R.K.; Manda, V.K.; Adkins, C.E.; Geldenhuys, W.J.; Lockman, P.R. Exploiting Nutrient Transporters at the Blood-Brain Barrier to Improve Brain Distribution of Small Molecules. Ther. Deliv. 2010, 1, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The Large Apparent Work Capability of the Blood-Brain Barrier: A Study of the Mitochondrial Content of Capillary Endothelial Cells in Brain and Other Tissues of the Rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Hladky, S.B.; Barrand, M.A. Fluid and Ion Transfer across the Blood-Brain and Blood-Cerebrospinal Fluid Barriers; a Comparative Account of Mechanisms and Roles. Fluids Barriers CNS 2016, 13, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J. Dynamics of CNS Barriers: Evolution, Differentiation, and Modulation. Cell. Mol. Neurobiol. 2005, 25, 5–23. [Google Scholar] [CrossRef]
- Pardridge, W.M. Molecular Biology of the Blood-Brain Barrier. Mol. Biotechnol. 2005, 30, 57–70. [Google Scholar] [CrossRef]
- Yakubu, M.A.; Nsaif, R.H.; Oyekan, A.O. Peroxisome Proliferator-Activated Receptor Alpha Activation-Mediated Regulation of Endothelin-1 Production via Nitric Oxide and Protein Kinase C Signaling Pathways in Piglet Cerebral Microvascular Endothelial Cell Culture. J. Pharmacol. Exp. Ther. 2007, 320, 774–781. [Google Scholar] [CrossRef] [Green Version]
- Kostov, K.; Blazhev, A.; Atanasova, M.; Dimitrova, A. Serum Concentrations of Endothelin-1 and Matrix Metalloproteinases-2, -9 in Pre-Hypertensive and Hypertensive Patients with Type 2 Diabetes. Int. J. Mol. Sci. 2016, 17, 1182. [Google Scholar] [CrossRef]
- Maguire, J.J.; Davenport, A.P. Endothelin Receptors and Their Antagonists. Semin. Nephrol. 2015, 35, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Kelland, N.F.; Kuc, R.E.; McLean, D.L.; Azfer, A.; Bagnall, A.J.; Gray, G.A.; Gulliver-Sloan, F.H.; Maguire, J.J.; Davenport, A.P.; Kotelevtsev, Y.V.; et al. Endothelial Cell-Specific ETB Receptor Knockout: Autoradiographic and Histological Characterisation and Crucial Role in the Clearance of Endothelin-1. Can. J. Physiol. Pharmacol. 2010, 88, 644–651. [Google Scholar] [CrossRef]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Salvetti, A. Vascular Effects of Endothelin-1 in Essential Hypertension: Relationship with Cyclooxygenase-Derived Endothelium-Dependent Contracting Factors and Nitric Oxide. J. Cardiovasc. Pharmacol. 2000, 35, S37–S40. [Google Scholar] [CrossRef] [PubMed]
- Mitsumori, T.; Furuyashiki, T.; Momiyama, T.; Nishi, A.; Shuto, T.; Hayakawa, T.; Ushikubi, F.; Kitaoka, S.; Aoki, T.; Inoue, H.; et al. Thromboxane Receptor Activation Enhances Striatal Dopamine Release, Leading to Suppression of GABAergic Transmission and Enhanced Sugar Intake. Eur. J. Neurosci. 2011, 34, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Shirasaki, Y.; Fukunaga, K. Microsphere Embolism-Induced Endothelial Nitric Oxide Synthase Expression Mediates Disruption of the Blood-Brain Barrier in Rat Brain. J. Neurochem. 2006, 99, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; d’Uscio, L.V.; Katusic, Z.S. Supplementation of Nitric Oxide Attenuates AβPP and BACE1 Protein in Cerebral Microcirculation of ENOS-Deficient Mice. J. Alzheimers. Dis. 2013, 33, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushner, P.D.; Stephenson, D.T.; Wright, S. Reactive Astrogliosis Is Widespread in the Subcortical White Matter of Amyotrophic Lateral Sclerosis Brain. J. Neuropathol. Exp. Neurol. 1991, 50, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte Dysfunction in Alzheimer Disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Krencik, R.; Ullian, E.M.; Tsai, H.; Deneen, B.; Richardson, W.D.; Barres, B.A.; Rowitch, D.H. Astrocytes and Disease: A Neurodevelopmental Perspective. Genes Dev. 2012, 26, 891–907. [Google Scholar] [CrossRef] [Green Version]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Derivation of Blood-Brain Barrier Endothelial Cells from Human Pluripotent Stem Cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Roessmann, U.; Gambetti, P. Astrocytes in the Developing Human Brain. An Immunohistochemical Study. Acta Neuropathol. 1986, 70, 308–313. [Google Scholar] [CrossRef]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a Source for Extracellular Matrix Molecules and Cytokines. Front. Pharmacol. 2012, 3, 120. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial Influence on the Blood Brain Barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xu, Z.; Xie, Z.; Rallo, M.; Duffy, A.; Matise, M.P. Inactivation of Hedgehog Signal Transduction in Adult Astrocytes Results in Region-Specific Blood–Brain Barrier Defects. Proc. Natl. Acad. Sci. USA 2021, 118, e2017779118. [Google Scholar] [CrossRef] [PubMed]
- András, I.E.; Deli, M.A.; Veszelka, S.; Hayashi, K.; Hennig, B.; Toborek, M. The NMDA and AMPA/KA Receptors Are Involved in Glutamate-Induced Alterations of Occludin Expression and Phosphorylation in Brain Endothelial Cells. J. Cereb. Blood Flow Metab. 2007, 27, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Sharp, C.D.; Hines, I.; Houghton, J.; Warren, A.; Jackson, T.H., 4th; Jawahar, A.; Nanda, A.; Elrod, J.W.; Long, A.; Chi, A.; et al. Glutamate Causes a Loss in Human Cerebral Endothelial Barrier Integrity through Activation of NMDA Receptor. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2592–H2598. [Google Scholar] [CrossRef]
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-Mediated Blood-Brain Barrier Opening: Implications for Neuroprotection and Drug Delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and Neuronal Control of Brain Blood Flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [Green Version]
- O’Kane, R.L.; Martínez-López, I.; DeJoseph, M.R.; Viña, J.R.; Hawkins, R.A. Na+-Dependent Glutamate Transporters (EAAT1, EAAT2, and EAAT3) of the Blood-Brain Barrier. A Mechanism for Glutamate Removal. J. Biol. Chem. 1999, 274, 31891–31895. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.; Plum, F. Biochemistry and Physiology of Brain Ammonia. Physiol. Rev. 1987, 67, 440–519. [Google Scholar] [CrossRef]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and Molecular Basis for the Regulation of Inflammation by TGF-Beta. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef]
- Tran, N.D.; Correale, J.; Schreiber, S.S.; Fisher, M. Transforming Growth Factor-Beta Mediates Astrocyte-Specific Regulation of Brain Endothelial Anticoagulant Factors. Stroke 1999, 30, 1671–1678. [Google Scholar] [CrossRef]
- Verkman, A.S.; Binder, D.K.; Bloch, O.; Auguste, K.; Papadopoulos, M.C. Three Distinct Roles of Aquaporin-4 in Brain Function Revealed by Knockout Mice. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandebroek, A.; Yasui, M. Regulation of AQP4 in the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Fujihara, K.; Nakashima, I.; Misu, T.; Miyazawa, I.; Nakamura, M.; Watanabe, S.; Shiga, Y.; Kanaoka, C.; Fujimori, J.; et al. Anti-Aquaporin-4 Antibody Is Involved in the Pathogenesis of NMO: A Study on Antibody Titre. Brain 2007, 130, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Bell, R.D.; Wang, J.; Zlokovic, B.V. Blood-Spinal Cord Barrier Pericyte Reductions Contribute to Increased Capillary Permeability. J. Cereb. Blood Flow Metab. 2012, 32, 1841–1852. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood-Spinal Cord Barrier Breakdown and Pericyte Reductions in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Berthiaume, A.-A.; Grant, R.I.; McDowell, K.P.; Underly, R.G.; Hartmann, D.A.; Levy, M.; Bhat, N.R.; Shih, A.Y. Dynamic Remodeling of Pericytes In Vivo Maintains Capillary Coverage in the Adult Mouse Brain. Cell Rep. 2018, 22, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary Pericytes Regulate Cerebral Blood Flow in Health and Disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood-Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, R.K. Transendothelial Transport and Its Role in Therapeutics. Int. Sch. Res. Not. 2014, 2014, 309404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, L.E.; Soriano, P. PDGFRβ Signaling Regulates Mural Cell Plasticity and Inhibits Fat Development. Dev. Cell 2011, 20, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeten, K.M.; Akassoglou, K. Extracellular Matrix and Matrix Receptors in Blood–Brain Barrier Formation and Stroke. Dev. Neurobiol. 2011, 71, 1018–1039. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.; Sorokin, L. The Blood-Brain and the Blood-Cerebrospinal Fluid Barriers: Function and Dysfunction. Semin. Immunopathol. 2009, 31, 497–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sado, Y.; Kagawa, M.; Kishiro, Y.; Sugihara, K.; Naito, I.; Seyer, J.M.; Sugimoto, M.; Oohashi, T.; Ninomiya, Y. Establishment by the Rat Lymph Node Method of Epitope-Defined Monoclonal Antibodies Recognizing the Six Different α Chains of Human Type IV Collagen. Histochem. Cell Biol. 1995, 104, 267–275. [Google Scholar] [CrossRef]
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV Is Essential for Basement Membrane Stability but Dispensable for Initiation of Its Assembly during Early Development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Nirwane, A.; Yao, Y. Basement Membrane and Blood-Brain Barrier. Stroke Vasc. Neurol. 2018, 4, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Milner, R. Fibronectin Promotes Brain Capillary Endothelial Cell Survival and Proliferation through Alpha5beta1 and Alphavbeta3 Integrins via MAP Kinase Signalling. J. Neurochem. 2006, 96, 148–159. [Google Scholar] [CrossRef]
- George, E.L.; Georges-Labouesse, E.N.; Patel-King, R.S.; Rayburn, H.; Hynes, R.O. Defects in Mesoderm, Neural Tube and Vascular Development in Mouse Embryos Lacking Fibronectin. Development 1993, 119, 1079–1091. [Google Scholar] [CrossRef]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and Function of Laminins in the Embryonic and Mature Vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Chen, Z.-L.; Norris, E.H.; Strickland, S. Astrocytic Laminin Regulates Pericyte Differentiation and Maintains Blood Brain Barrier Integrity. Nat. Commun. 2014, 5, 3413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes, M.J.; McClenahan, F.K.; Leiton, C.V.; Aranmolate, A.; Shan, X.; Colognato, H. The Extracellular Matrix Protein Laminin A2 Regulates the Maturation and Function of the Blood-Brain Barrier. J. Neurosci. 2014, 34, 15260–15280. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y. Laminin: Loss-of-Function Studies. Cell. Mol. Life Sci. 2017, 74, 1095–1115. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, F.; Cole, S.; Green, C.; van de Waterbeemd, H. Lipophilicity and Other Parameters Affecting Brain Penetration. Curr. Med. Chem. Cent. Nerv. Syst. Agents 2002, 2, 229–240. [Google Scholar] [CrossRef]
- Levin, V.A. Relationship of Octanol/Water Partition Coefficient and Molecular Weight to Rat Brain Capillary Permeability. J. Med. Chem. 1980, 23, 682–684. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.-F. Efflux Transporters in Blood-Brain Interfaces of the Developing Brain. Front. Neurosci. 2015, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Vanlandewijck, M.; Raschperger, E.; Andaloussi Mäe, M.; Jung, B.; Lebouvier, T.; Ando, K.; Hofmann, J.; Keller, A.; Betsholtz, C. Analysis of the Brain Mural Cell Transcriptome. Sci. Rep. 2016, 6, 35108. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Dziegielewska, K.M.; Ek, C.J.; Habgood, M.D.; Bauer, H.; Bauer, H.-C.; Lindsay, H.; Wakefield, M.J.; Strazielle, N.; Kratzer, I.; et al. Correction: Mechanisms That Determine the Internal Environment of the Developing Brain: A Transcriptomic, Functional and Ultrastructural Approach. PLoS ONE 2016, 11, e0147680. [Google Scholar] [CrossRef]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE Axis in Neuroinflammation, Neurodegeneration and Its Emerging Role in the Pathogenesis of Amyotrophic Lateral Sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef]
- Bakker, E.N.T.P.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.J.; Weller, R.O.; Carare, R.O. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederberg, H.H.C.; Uhd, N.C.; Brodin, B. Glutamate Efflux at the Blood-Brain Barrier: Cellular Mechanisms and Potential Clinical Relevance. Arch. Med. Res. 2014, 45, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bodles-Brakhop, A.M.; Barger, S.W. A Role for P-Glycoprotein in Clearance of Alzheimer Amyloid β -Peptide from the Brain. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The Glutamate Hypothesis in ALS: Pathophysiology and Drug Development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef]
- Cowan, C.M.; Raymond, L.A. Selective Neuronal Degeneration in Huntington’s Disease. Curr. Top. Dev. Biol. 2006, 75, 25–71. [Google Scholar] [CrossRef]
- Uno, Y.; Coyle, J.T. Glutamate Hypothesis in Schizophrenia. Psychiatry Clin. Neurosci. 2019, 73, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Yan, S.D.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/Amyloid β;-Peptide Interaction Mediates Differential Brain Efflux of A β; Isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Harel, T.; Quek, D.Q.Y.; Wong, B.H.; Cazenave-Gassiot, A.; Wenk, M.R.; Fan, H.; Berger, I.; Shmueli, D.; Shaag, A.; Silver, D.L.; et al. Homozygous Mutation in MFSD2A, Encoding a Lysolipid Transporter for Docosahexanoic Acid, Is Associated with Microcephaly and Hypomyelination. Neurogenetics 2018, 19, 227–235. [Google Scholar] [CrossRef]
- Qosa, H.; Miller, D.S.; Pasinelli, P.; Trotti, D. Regulation of ABC Efflux Transporters at Blood-Brain Barrier in Health and Neurological Disorders. Brain Res. 2015, 1628, 298–316. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- NCBI APOE Apolipoprotein E [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/348 (accessed on 15 June 2020).
- Boyles, J.K.; Pitas, R.E.; Wilson, E.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E Associated with Astrocytic Glia of the Central Nervous System and with Nonmyelinating Glia of the Peripheral Nervous System. J. Clin. Investig. 1985, 76, 1501–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, L.M.; Thomas, R.; Marottoli, F.M.; Koster, K.P.; Kanekiyo, T.; Morris, A.W.J.; Bu, G. The Role of APOE in Cerebrovascular Dysfunction. Acta Neuropathol. 2016, 131, 709–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Mucke, L. Alzheimer Mechanisms and Therapeutic Strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E Controls Cerebrovascular Integrity via Cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Halliday, M.R.; Rege, S.V.; Ma, Q.; Zhao, Z.; Miller, C.A.; Winkler, E.A.; Zlokovic, B.V. Accelerated Pericyte Degeneration and Blood-Brain Barrier Breakdown in Apolipoprotein E4 Carriers with Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2016, 36, 216–227. [Google Scholar] [CrossRef]
- Apostolova, L.G.; Mosconi, L.; Thompson, P.M.; Green, A.E.; Hwang, K.S.; Ramirez, A.; Mistur, R.; Tsui, W.H.; de Leon, M.J. Subregional Hippocampal Atrophy Predicts Alzheimer’s Dementia in the Cognitively Normal. Neurobiol. Aging 2010, 31, 1077–1088. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Brundel, M.; Heringa, S.M.; de Bresser, J.; Koek, H.L.; Zwanenburg, J.J.M.; Jaap Kappelle, L.; Luijten, P.R.; Biessels, G.J. High Prevalence of Cerebral Microbleeds at 7Tesla MRI in Patients with Early Alzheimer’s Disease. J. Alzheimers. Dis. 2012, 31, 259–263. [Google Scholar] [CrossRef]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular Injury and Blood-Brain Barrier Leakage in Alzheimer’s Disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, Z. ApoE and Neurodegenerative Diseases in Aging. Adv. Exp. Med. Biol. 2018, 1086, 77–92. [Google Scholar] [CrossRef]
- Yonashiro, R.; Sugiura, A.; Miyachi, M.; Fukuda, T.; Matsushita, N.; Inatome, R.; Ogata, Y.; Suzuki, T.; Dohmae, N.; Yanagi, S. Mitochondrial Ubiquitin Ligase MITOL Ubiquitinates Mutant SOD1 and Attenuates Mutant SOD1-Induced Reactive Oxygen Species Generation. Mol. Biol. Cell 2009, 20, 4524–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative Stress in ALS: Key Role in Motor Neuron Injury and Therapeutic Target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Heath, P.R.; Kirby, J.; Wharton, S.B.; Cookson, M.R.; Menzies, F.M.; Banks, R.E.; Shaw, P.J. Analysis of the Cytosolic Proteome in a Cell Culture Model of Familial Amyotrophic Lateral Sclerosis Reveals Alterations to the Proteasome, Antioxidant Defenses, and Nitric Oxide Synthetic Pathways. J. Biol. Chem. 2003, 278, 6371–6383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbuzova-Davis, S.; Rodrigues, M.C.O.; Hernandez-Ontiveros, D.G.; Louis, M.K.; Willing, A.E.; Borlongan, C.V.; Sanberg, P.R. Amyotrophic Lateral Sclerosis: A Neurovascular Disease. Brain Res. 2011, 1398, 113–125. [Google Scholar] [CrossRef]
- Cowley, P.M.; Nair, D.R.; DeRuisseau, L.R.; Keslacy, S.; Atalay, M.; DeRuisseau, K.C. Oxidant Production and SOD1 Protein Expression in Single Skeletal Myofibers from Down Syndrome Mice. Redox Biol. 2017, 13, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Muchová, J.; Žitňanová, I.; Ďuračková, Z. Oxidative Stress and Down Syndrome. Do Antioxidants Play a Role in Therapy? Physiol. Res. 2014, 63, 535–542. [Google Scholar] [CrossRef]
- Ram, G.; Chinen, J. Infections and Immunodeficiency in Down Syndrome. Clin. Exp. Immunol. 2011, 164, 9–16. [Google Scholar] [CrossRef]
- Amiry-Moghaddam, M.; Otsuka, T.; Hurn, P.D.; Traystman, R.J.; Haug, F.-M.; Froehner, S.C.; Adams, M.E.; Neely, J.D.; Agre, P.; Ottersen, O.P.; et al. An Alpha-Syntrophin-Dependent Pool of AQP4 in Astroglial End-Feet Confers Bidirectional Water Flow between Blood and Brain. Proc. Natl. Acad. Sci. USA 2003, 100, 2106–2111. [Google Scholar] [CrossRef] [Green Version]
- Nagelhus, E.A.; Mathiisen, T.M.; Ottersen, O.P. Aquaporin-4 in the Central Nervous System: Cellular and Subcellular Distribution and Coexpression with KIR4.1. Neuroscience 2004, 129, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Verkman, A.S. Impaired Hearing in Mice Lacking Aquaporin-4 Water Channels. J. Biol. Chem. 2001, 276, 31233–31237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skucas, V.A.; Mathews, I.B.; Yang, J.; Cheng, Q.; Treister, A.; Duffy, A.M.; Verkman, A.S.; Hempstead, B.L.; Wood, M.A.; Binder, D.K.; et al. Impairment of Select Forms of Spatial Memory and Neurotrophin-Dependent Synaptic Plasticity by Deletion of Glial Aquaporin-4. J. Neurosci. 2011, 31, 6392–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadoun, S.; Papadopoulos, M.C.; Watanabe, H.; Yan, D.; Manley, G.T.; Verkman, A.S. Involvement of Aquaporin-4 in Astroglial Cell Migration and Glial Scar Formation. J. Cell Sci. 2005, 118, 5691–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The Spectrum of Neuromyelitis Optica. Lancet. Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Lee, S. Expression of Astrocytic Markers Aquaporin 4 and Connexin 43 Is Altered in Brains of Subjects with Autism. Synapse 2008, 62, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, J.A.; Szu, J.I.; Binder, D.K. The Role of Aquaporin-4 in Synaptic Plasticity, Memory and Disease. Brain Res. Bull. 2018, 136, 118–129. [Google Scholar] [CrossRef]
- Smith, A.J.; Duan, T.; Verkman, A.S. Aquaporin-4 Reduces Neuropathology in a Mouse Model of Alzheimer’s Disease by Remodeling Peri-Plaque Astrocyte Structure. Acta Neuropathol. Commun. 2019, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Opdal, S.H.; Vege, Å.; Stray-Pedersen, A.; Rognum, T.O. The Gene Encoding the Inwardly Rectifying Potassium Channel Kir4.1 May Be Involved in Sudden Infant Death Syndrome. Acta Paediatr. 2017, 106, 1474–1480. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, M.; Park, W.; Lim, J.S.; Lee, E.; Cha, H.; Ahn, J.S.; Kim, J.H.; Hong, S.H.; Park, J.E.; et al. Upregulation of AQP4 Improves Blood-Brain Barrier Integrity and Perihematomal Edema Following Intracerebral Hemorrhage. Neurotherapeutics 2021, 18, 2692–2706. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Churg, A. Interactions of Exogenous or Evoked Agents and Particles: The Role of Reactive Oxygen Species. Free Radic. Biol. Med. 2003, 34, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-Induced Oxidative Stress in Brain Endothelial Cells Causes Blood-Brain Barrier Dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haorah, J.; Schall, K.; Ramirez, S.H.; Persidsky, Y. Activation of Protein Tyrosine Kinases and Matrix Metalloproteinases Causes Blood-Brain Barrier Injury: Novel Mechanism for Neurodegeneration Associated with Alcohol Abuse. Glia 2008, 56, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ighodaro, O.M.; Akinloye, O.A. First Line Defence Antioxidants-Superoxide Dismutase (SOD), Catalase (CAT) and Glutathione Peroxidase (GPX): Their Fundamental Role in the Entire Antioxidant Defence Grid. Alexandria J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-Related Factor 2 (Nrf2), a NF-E2-like Basic Leucine Zipper Transcriptional Activator That Binds to the Tandem NF-E2/AP1 Repeat of the Beta-Globin Locus Control Region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Sun, J.; Zhang, W.; Yu, J.; Zhuang, C. Transcription Factor NRF2 as a Promising Therapeutic Target for Alzheimer’s Disease. Free Radic. Biol. Med. 2020, 159, 87–102. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.-Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in Mitochondrial Superoxide Dismutase Modulates Alzheimer’s Disease-like Pathology and Accelerates the Onset of Behavioral Changes in Human Amyloid Precursor Protein Transgenic Mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, K.; Gray, E.; Mallam, E.; Scolding, N.; Wilkins, A. Inflammatory Cytokine Induced Regulation of Superoxide Dismutase 3 Expression by Human Mesenchymal Stem Cells. Stem cell Rev. reports 2010, 6, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Chinnery, R.M.; Thagesen, H.; Borthwick, G.M.; Ince, P.G. Immunocytochemical Study of the Distribution of the Free Radical Scavenging Enzymes Cu/Zn Superoxide Dismutase (SOD1); MN Superoxide Dismutase (MN SOD) and Catalase in the Normal Human Spinal Cord and in Motor Neuron Disease. J. Neurol. Sci. 1997, 147, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Miyamoto, T.; Yamaji, K.; Masuho, Y.; Hayashi, M.; Hayashi, H.; Onozaki, K. A Human Erythrocyte-Derived Growth-Promoting Factor with a Wide Target Cell Spectrum: Identification as Catalase. Cancer Res. 1995, 55, 1586–1589. [Google Scholar] [PubMed]
- Flagg, E.W.; Coates, R.J.; Jones, D.P.; Eley, J.W.; Gunter, E.W.; Jackson, B.; Greenberg, R.S. Plasma Total Glutathione in Humans and Its Association with Demographic and Health-Related Factors. Br. J. Nutr. 1993, 70, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Glutathione! Integr. Med. Encinitas 2014, 13, 8–12. [Google Scholar]
- Dringen, R. Metabolism and Functions of Glutathione in Brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Huang, S.-F.; Othman, A.; Koshkin, A.; Fischer, S.; Fischer, D.; Zamboni, N.; Ono, K.; Sawa, T.; Ogunshola, O.O. Astrocyte Glutathione Maintains Endothelial Barrier Stability. Redox Biol. 2020, 34, 101576. [Google Scholar] [CrossRef]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C Crosses the Blood-Brain Barrier in the Oxidized Form through the Glucose Transporters. J. Clin. Investig. 1997, 100, 2842–2848. [Google Scholar] [CrossRef] [Green Version]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef] [Green Version]
- Telang, S.; Clem, A.L.; Eaton, J.W.; Chesney, J. Depletion of Ascorbic Acid Restricts Angiogenesis and Retards Tumor Growth in a Mouse Model. Neoplasia 2007, 9, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Chen, J.-Y.; Wu, M.-H.; Hu, M.-L. Therapeutic Treatment with Vitamin C Reduces Focal Cerebral Ischemia-Induced Brain Infarction in Rats by Attenuating Disruptions of Blood Brain Barrier and Cerebral Neuronal Apoptosis. Free Radic. Biol. Med. 2020, 155, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.O.; Starkey, S.R.; Stipetic, K.; Divers, T.J.; Summers, B.A.; de Lahunta, A. The Role of Dietary Antioxidant Insufficiency on the Permeability of the Blood-Brain Barrier. J. Neuropathol. Exp. Neurol. 2008, 67, 1187–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohil, K.; Vasu, V.T.; Cross, C.E. Dietary Alpha-Tocopherol and Neuromuscular Health: Search for Optimal Dose and Molecular Mechanisms Continues! Mol. Nutr. Food Res. 2010, 54, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.P.R. Vitamin E and Neurological Function. Mol. Nutr. Food Res. 2010, 54, 710–718. [Google Scholar] [CrossRef] [PubMed]
- D’Antona, S.; Caramenti, M.; Porro, D.; Castiglioni, I.; Cava, C. Amyotrophic Lateral Sclerosis: A Diet Review. Foods 2021, 10, 3128. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Khan, F.; Young, C.A.; Galea, M. Symptomatic Treatments for Amyotrophic Lateral Sclerosis/Motor Neuron Disease. Cochrane database Syst. Rev. 2017, 1, CD011776. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Simi, A.; Tsakiri, N.; Wang, P.; Rothwell, N.J. Interleukin-1 and Inflammatory Neurodegeneration. Biochem. Soc. Trans. 2007, 35, 1122–1126. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, G.A. Matrix Metalloproteinases and Their Multiple Roles in Neurodegenerative Diseases. Lancet. Neurol. 2009, 8, 205–216. [Google Scholar] [CrossRef]
- Strbian, D.; Karjalainen-Lindsberg, M.-L.; Tatlisumak, T.; Lindsberg, P.J. Cerebral Mast Cells Regulate Early Ischemic Brain Swelling and Neutrophil Accumulation. J. Cereb. Blood Flow Metab. 2006, 26, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, G.; Saad, K.; Chirumbolo, S.; Kern, J.K.; Geier, D.A.; Geier, M.R.; Urbina, M.A. Immune Dysfunction and Neuroinflammation in Autism Spectrum Disorder. Acta Neurobiol. Exp. Wars 2016, 76, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. How Neuroinflammation Contributes to Neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Inflammatory Neuroprotection Following Traumatic Brain Injury. Science 2016, 353, 783–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast Cells and Neutrophils Mediate Peripheral Motor Pathway Degeneration in ALS. JCI insight 2018, 3, e123249. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int. J. Mol. Sci. 2018, 19, 2693. [Google Scholar] [CrossRef] [Green Version]
- Zenaro, E.; Pietronigro, E.; Bianca, V.D.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils Promote Alzheimer’s Disease–like Pathology and Cognitive Decline via LFA-1 Integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Beaman, C.; Kozii, K.; Hilal, S.; Liu, M.; Spagnolo-Allende, A.J.; Polanco-Serra, G.; Chen, C.; Cheng, C.-Y.; Zambrano, D.; Arikan, B.; et al. Cerebral Microbleeds, Cerebral Amyloid Angiopathy, and Their Relationships to Quantitative Markers of Neurodegeneration. Neurology 2022, 98, e1605. [Google Scholar] [CrossRef]
- Cash, A.; Theus, M.H. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef]
- Price, L.; Wilson, C.; Grant, G. Chapter 4 Blood-Brain Barrier Pathophysiology Following Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2016. [Google Scholar]
- Ferrari, D.C.; Nesic, O.; Perez-Polo, J.R. Perspectives on Neonatal Hypoxia/Ischemia-Induced Edema Formation. Neurochem. Res. 2010, 35, 1957–1965. [Google Scholar] [CrossRef]
- Hamrick, S.E.G.; Ferriero, D.M. The Injury Response in the Term Newborn Brain: Can We Neuroprotect? Curr. Opin. Neurol. 2003, 16, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Disdier, C.; Stonestreet, B.S. Hypoxic-Ischemic-Related Cerebrovascular Changes and Potential Therapeutic Strategies in the Neonatal Brain. J. Neurosci. Res. 2020, 98, 1468–1484. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Melendez, M.; Yawno, T.; Allison, B.J.; Jenkin, G.; Wallace, E.M.; Miller, S.L. Cerebrovascular Adaptations to Chronic Hypoxia in the Growth Restricted Lamb. Int. J. Dev. Neurosci. 2015, 45, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Baschat, A.A. Neurodevelopment Following Fetal Growth Restriction and Its Relationship with Antepartum Parameters of Placental Dysfunction. Ultrasound Obstet. Gynecol. 2011, 37, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, K.; Devaskar, S.U. Intrauterine Growth Restriction: Postnatal Monitoring and Outcomes. Pediatr. Clin. N. Am. 2019, 66, 403–423. [Google Scholar] [CrossRef]
- Chandra, S.; Alam, M.T.; Dey, J.; Sasidharan, B.C.P.; Ray, U.; Srivastava, A.K.; Gandhi, S.; Tripathi, P.P. Healthy Gut, Healthy Brain: The Gut Microbiome in Neurodegenerative Disorders. Curr. Top. Med. Chem. 2020, 20, 1142–1153. [Google Scholar] [CrossRef]
- Zhu, X.; Li, B.; Lou, P.; Dai, T.; Chen, Y.; Zhuge, A.; Yuan, Y.; Li, L. The Relationship Between the Gut Microbiome and Neurodegenerative Diseases. Neurosci. Bull. 2021, 37, 1510–1522. [Google Scholar] [CrossRef]
- Mayer, E.A.; Knight, R.; Mazmanian, S.K.; Cryan, J.F.; Tillisch, K. Gut Microbes and the Brain: Paradigm Shift in Neuroscience. J. Neurosci. 2014, 34, 15490–15496. [Google Scholar] [CrossRef] [Green Version]
- Bohórquez, D.V.; Shahid, R.A.; Erdmann, A.; Kreger, A.M.; Wang, Y.; Calakos, N.; Wang, F.; Liddle, R.A. Neuroepithelial Circuit Formed by Innervation of Sensory Enteroendocrine Cells. J. Clin. Investig. 2015, 125, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Breen, D.P.; Halliday, G.M.; Lang, A.E. Gut–Brain Axis and the Spread of α-Synuclein Pathology: Vagal Highway or Dead End? Mov. Disord. 2019, 34, 307–316. [Google Scholar] [CrossRef]
- Pulikkan, J.; Mazumder, A.; Grace, T. Role of the Gut Microbiome in Autism Spectrum Disorders. Adv. Exp. Med. Biol. 2019, 1118, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The Gut Microbiota Influences Blood-Brain Barrier Permeability in Mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, E.Y.; McBride, S.W.; Chow, J.; Mazmanian, S.K.; Patterson, P.H. Modeling an Autism Risk Factor in Mice Leads to Permanent Immune Dysregulation. Proc. Natl. Acad. Sci. USA 2012, 109, 12776–12781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureda, A.; Daglia, M.; Castilla, A.S.; Sanadgol, N.; Nabavi, S.F.; Khan, H.; Belwal, T.; Jeandet, P.; Marchese, A.; Pistollato, F.; et al. Oral Microbiota and Alzheimer’s Disease: Do All Roads Lead to Rome? Pharmacol. Res. 2020, 151, 104582. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I. Update on Bacteraemia Related to Dental Procedures. Transfus. Apher. Sci. 2008, 39, 173–178. [Google Scholar] [CrossRef]
- Shoemark, D.K.; Allen, S.J. The Microbiome and Disease: Reviewing the Links between the Oral Microbiome, Aging, and Alzheimer’s Disease. J. Alzheimers. Dis. 2015, 43, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Capsoni, S.; Carucci, N.M.; Cattaneo, A. Pathogen Free Conditions Slow the Onset of Neurodegeneration in a Mouse Model of Nerve Growth Factor Deprivation. J. Alzheimer’s Dis. 2012, 31, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ghersi-Egea, J.F.; Leninger-Muller, B.; Suleman, G.; Siest, G.; Minn, A. Localization of Drug-Metabolizing Enzyme Activities to Blood-Brain Interfaces and Circumventricular Organs. J. Neurochem. 1994, 62, 1089–1096. [Google Scholar] [CrossRef]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef]
- Cohen-Pfeffer, J.L.; Gururangan, S.; Lester, T.; Lim, D.A.; Shaywitz, A.J.; Westphal, M.; Slavc, I. Intracerebroventricular Delivery as a Safe, Long-Term Route of Drug Administration. Pediatr. Neurol. 2017, 67, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Gu, X.; Gao, X. 13. Nanotherapeutic Strategies for the Treatment of Neurodegenerative Diseases. In Brain Targeted Drug Delivery Systems: A Focus on Nanotechnology and Nanoparticulates; Gao, H., Gao, X., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 321–356. ISBN 978-0-12-814001-7. [Google Scholar]
- Gentry, C.L.; Egleton, R.D.; Gillespie, T.; Abbruscato, T.J.; Bechowski, H.B.; Hruby, V.J.; Davis, T.P. The Effect of Halogenation on Blood-Brain Barrier Permeability of a Novel Peptide Drug. Peptides 1999, 20, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Witt, K.A.; Slate, C.A.; Egleton, R.D.; Huber, J.D.; Yamamura, H.I.; Hruby, V.J.; Davis, T.P. Assessment of Stereoselectivity of Trimethylphenylalanine Analogues of δ-Opioid [D-Pen2,D-Pen5]-Enkephalin. J. Neurochem. 2000, 75, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J. Peptides and the Blood-Brain Barrier: Lipophilicity as a Predictor of Permeability. Brain Res. Bull. 1985, 15, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Molecular Trojan Horses for Blood-Brain Barrier Drug Delivery. Curr. Opin. Pharmacol. 2006, 6, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Pencea, V.; Bingaman, K.D.; Wiegand, S.J.; Luskin, M.B. Infusion of Brain-Derived Neurotrophic Factor into the Lateral Ventricle of the Adult Rat Leads to New Neurons in the Parenchyma of the Striatum, Septum, Thalamus, and Hypothalamus. J. Neurosci. 2001, 21, 6706–6717. [Google Scholar] [CrossRef] [Green Version]
- Ventriglia, M.; Bocchio Chiavetto, L.; Benussi, L.; Binetti, G.; Zanetti, O.; Riva, M.A.; Gennarelli, M. Association between the BDNF 196 A/G Polymorphism and Sporadic Alzheimer’s Disease. Mol. Psychiatry 2002, 7, 136–137. [Google Scholar] [CrossRef] [Green Version]
- Neves-Pereira, M.; Mundo, E.; Muglia, P.; King, N.; Macciardi, F.; Kennedy, J.L. The Brain-Derived Neurotrophic Factor Gene Confers Susceptibility to Bipolar Disorder: Evidence from a Family-Based Association Study. Am. J. Hum. Genet. 2002, 71, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Biologic TNFα-Inhibitors That Cross the Human Blood-Brain Barrier. Bioeng. Bugs 2010, 1, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Delivery of Biologics Across the Blood–Brain Barrier with Molecular Trojan Horse Technology. BioDrugs 2017, 31, 503–519. [Google Scholar] [CrossRef]
- Naqvi, S.; Panghal, A.; Flora, S.J.S. Nanotechnology: A Promising Approach for Delivery of Neuroprotective Drugs. Front. Neurosci. 2020, 14, 494. [Google Scholar] [CrossRef]
- Patel, P.; Kriz, J.; Gravel, M.; Soucy, G.; Bareil, C.; Gravel, C.; Julien, J.-P. Adeno-Associated Virus–Mediated Delivery of a Recombinant Single-Chain Antibody Against Misfolded Superoxide Dismutase for Treatment of Amyotrophic Lateral Sclerosis. Mol. Ther. 2014, 22, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.M.; Fernando, S.M.; Grad, L.I.; Hill, A.F.; Turner, B.J.; Yerbury, J.J.; Cashman, N.R. Disease Mechanisms in ALS: Misfolded SOD1 Transferred Through Exosome-Dependent and Exosome-Independent Pathways. Cell. Mol. Neurobiol. 2016, 36, 377–381. [Google Scholar] [CrossRef]
- Jeon, I.; Cicchetti, F.; Cisbani, G.; Lee, S.; Li, E.; Bae, J.; Lee, N.; Li, L.; Im, W.; Kim, M.; et al. Human-to-Mouse Prion-like Propagation of Mutant Huntingtin Protein. Acta Neuropathol. 2016, 132, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saá, P.; Yakovleva, O.; de Castro, J.; Vasilyeva, I.; De Paoli, S.H.; Simak, J.; Cervenakova, L. First Demonstration of Transmissible Spongiform Encephalopathy-Associated Prion Protein (PrPTSE) in Extracellular Vesicles from Plasma of Mice Infected with Mouse-Adapted Variant Creutzfeldt-Jakob Disease by in Vitro Amplification. J. Biol. Chem. 2014, 289, 29247–29260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokubo, H.; Saido, T.C.; Iwata, N.; Helms, J.B.; Shinohara, R.; Yamaguchi, H. Part of Membrane-Bound Abeta Exists in Rafts within Senile Plaques in Tg2576 Mouse Brain. Neurobiol. Aging 2005, 26, 409–418. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Klyubin, I.; Kim, Y.; Jung, J.H.; Mably, A.J.; O’Dowd, S.T.; Lynch, T.; Kanmert, D.; Lemere, C.A.; Finan, G.M.; et al. Exosomes Neutralize Synaptic-Plasticity-Disrupting Activity of Aβ Assemblies in Vivo. Mol. Brain 2013, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Vyas, Y.; Montgomery, J.M.; Cheyne, J.E. Hippocampal Deficits in Amyloid-β-Related Rodent Models of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 266. [Google Scholar] [CrossRef]
- Shpyleva, S.; Ivanovsky, S.; de Conti, A.; Melnyk, S.; Tryndyak, V.; Beland, F.A.; James, S.J.; Pogribny, I.P. Cerebellar Oxidative DNA Damage and Altered DNA Methylation in the BTBR T+tf/J Mouse Model of Autism and Similarities with Human Post Mortem Cerebellum. PLoS ONE 2014, 9, e113712. [Google Scholar] [CrossRef] [Green Version]
- Perets, N.; Betzer, O.; Shapira, R.; Brenstein, S.; Angel, A.; Sadan, T.; Ashery, U.; Popovtzer, R.; Offen, D. Golden Exosomes Selectively Target Brain Pathologies in Neurodegenerative and Neurodevelopmental Disorders. Nano Lett. 2019, 19, 3422–3431. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.F.; Alonso-Juarez, M. The Link between Gut Dysbiosis and Neuroinflammation in Parkinson’s Disease. Neuroscience 2020, 432, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Santangelo, R. Alzheimer’s Disease and Gut Microbiota Modifications: The Long Way between Preclinical Studies and Clinical Evidence. Pharmacol. Res. 2018, 129, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Tillisch, K.; Labus, J.; Kilpatrick, L.; Jiang, Z.; Stains, J.; Ebrat, B.; Guyonnet, D.; Legrain-Raspaud, S.; Trotin, B.; Naliboff, B.; et al. Consumption of Fermented Milk Product with Probiotic Modulates Brain Activity. Gastroenterology 2013, 144, 1394–1401, 1401.e1-4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messaoudi, M.; Lalonde, R.; Violle, N.; Javelot, H.; Desor, D.; Nejdi, A.; Bisson, J.-F.; Rougeot, C.; Pichelin, M.; Cazaubiel, M.; et al. Assessment of Psychotropic-like Properties of a Probiotic Formulation (Lactobacillus Helveticus R0052 and Bifidobacterium Longum R0175) in Rats and Human Subjects. Br. J. Nutr. 2011, 105, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messaoudi, M.; Violle, N.; Bisson, J.-F.; Desor, D.; Javelot, H.; Rougeot, C. Beneficial Psychological Effects of a Probiotic Formulation (Lactobacillus Helveticus R0052 and Bifidobacterium Longum R0175) in Healthy Human Volunteers. Gut Microbes 2011, 2, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Sasmita, A.O. Modification of the Gut Microbiome to Combat Neurodegeneration. Rev. Neurosci. 2019, 30, 795–805. [Google Scholar] [CrossRef]
- Goyal, D.; Ali, S.A.; Singh, R.K. Emerging Role of Gut Microbiota in Modulation of Neuroinflammation and Neurodegeneration with Emphasis on Alzheimer’s Disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 106, 110112. [Google Scholar] [CrossRef]
- Kouchaki, E.; Tamtaji, O.R.; Salami, M.; Bahmani, F.; Daneshvar Kakhaki, R.; Akbari, E.; Tajabadi-Ebrahimi, M.; Jafari, P.; Asemi, Z. Clinical and Metabolic Response to Probiotic Supplementation in Patients with Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Trial. Clin. Nutr. 2017, 36, 1245–1249. [Google Scholar] [CrossRef]
- Cho, Y.; Son, H.J.; Kim, E.-M.; Choi, J.H.; Kim, S.T.; Ji, I.J.; Choi, D.H.; Joh, T.H.; Kim, Y.S.; Hwang, O. Doxycycline Is Neuroprotective against Nigral Dopaminergic Degeneration by a Dual Mechanism Involving MMP-3. Neurotox. Res. 2009, 16, 361–371. [Google Scholar] [CrossRef]
- Lazzarini, M.; Martin, S.; Mitkovski, M.; Vozari, R.R.; Stühmer, W.; Bel, E. Del Doxycycline Restrains Glia and Confers Neuroprotection in a 6-OHDA Parkinson Model. Glia 2013, 61, 1084–1100. [Google Scholar] [CrossRef]
- McElhanon, B.O.; McCracken, C.; Karpen, S.; Sharp, W.G. Gastrointestinal Symptoms in Autism Spectrum Disorder: A Meta-Analysis. Pediatrics 2014, 133, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labusca, L.; Herea, D.D.; Mashayekhi, K. Stem Cells as Delivery Vehicles for Regenerative Medicine-Challenges and Perspectives. World J. Stem Cells 2018, 10, 43–56. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Sonninen, T.-M.; Peltonen, S.; Koistinaho, J.; Lehtonen, Š. Blood-Brain Barrier and Neurodegenerative Diseases—Modeling with IPSC-Derived Brain Cells. Int. J. Mol. Sci. 2021, 22, 7710. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Kibayashi, T.; Katayama, T.; Yamashita, Y.; Suzuki, S.; Kawamata, J.; Honmou, O.; Minami, M.; Shimohama, S. Mesenchymal Stem Cells Transmigrate across Brain Microvascular Endothelial Cell Monolayers through Transiently Formed Inter-Endothelial Gaps. Neurosci. Lett. 2011, 502, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Volkman, R.; Offen, D. Concise Review: Mesenchymal Stem Cells in Neurodegenerative Diseases. Stem Cells 2017, 35, 1867–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perets, N.; Segal-Gavish, H.; Gothelf, Y.; Barzilay, R.; Barhum, Y.; Abramov, N.; Hertz, S.; Morozov, D.; London, M.; Offen, D. Long Term Beneficial Effect of Neurotrophic Factors-Secreting Mesenchymal Stem Cells Transplantation in the BTBR Mouse Model of Autism. Behav. Brain Res. 2017, 331, 254–260. [Google Scholar] [CrossRef]
- Behnan, J.; Stangeland, B.; Langella, T.; Finocchiaro, G.; Tringali, G.; Meling, T.R.; Murrell, W. Identification and Characterization of a New Source of Adult Human Neural Progenitors. Cell Death Dis. 2017, 8, e2991. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Weick, J.P.; Liu, H.; Krencik, R.; Zhang, X.; Ma, L.; Zhou, G.; Ayala, M.; Zhang, S.-C. Medial Ganglionic Eminence–like Cells Derived from Human Embryonic Stem Cells Correct Learning and Memory Deficits. Nat. Biotechnol. 2013, 31, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Lien, C.-C.; Hou, W.-H.; Chiang, P.-M.; Tsai, K.-J. Gain of BDNF Function in Engrafted Neural Stem Cells Promotes the Therapeutic Potential for Alzheimer’s Disease. Sci. Rep. 2016, 6, 27358. [Google Scholar] [CrossRef] [Green Version]
- Blurton-Jones, M.; Spencer, B.; Michael, S.; Castello, N.A.; Agazaryan, A.A.; Davis, J.L.; Müller, F.-J.; Loring, J.F.; Masliah, E.; LaFerla, F.M. Neural Stem Cells Genetically-Modified to Express Neprilysin Reduce Pathology in Alzheimer Transgenic Models. Stem Cell Res. Ther. 2014, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doss, M.X.; Sachinidis, A. Current Challenges of IPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picanço-Castro, V.; Moreira, L.F.; Kashima, S.; Covas, D.T. Can Pluripotent Stem Cells Be Used in Cell-Based Therapy? Cell. Reprogram. 2014, 16, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.-Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized IPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- Madrid, M.; Sumen, C.; Aivio, S.; Saklayen, N. Autologous Induced Pluripotent Stem Cell–Based Cell Therapies: Promise, Progress, and Challenges. Curr. Protoc. 2021, 1, e88. [Google Scholar] [CrossRef]
- Kondziolka, D. Reaching the Far Corners of Neurosurgery. Neurosurgery 2022, 91, 525–526. [Google Scholar] [CrossRef]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S. Pulmonary Passage Is a Major Obstacle for Intravenous Stem Cell Delivery: The Pulmonary First-Pass Effect. Stem Cells Dev. 2008, 18, 683–692. [Google Scholar] [CrossRef]
- Zhang, Y.-T.; He, K.-J.; Zhang, J.-B.; Ma, Q.-H.; Wang, F.; Liu, C.-F. Advances in Intranasal Application of Stem Cells in the Treatment of Central Nervous System Diseases. Stem Cell Res. Ther. 2021, 12, 210. [Google Scholar] [CrossRef]
- Berry, J.D.; Cudkowicz, M.E.; Windebank, A.J.; Staff, N.P.; Owegi, M.; Nicholson, K.; McKenna-Yasek, D.; Levy, Y.S.; Abramov, N.; Kaspi, H.; et al. NurOwn, Phase 2, Randomized, Clinical Trial in Patients with ALS: Safety, Clinical, and Biomarker Results. Neurology 2019, 93, e2294–e2305. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aragón-González, A.; Shaw, P.J.; Ferraiuolo, L. Blood–Brain Barrier Disruption and Its Involvement in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2022, 23, 15271. https://doi.org/10.3390/ijms232315271
Aragón-González A, Shaw PJ, Ferraiuolo L. Blood–Brain Barrier Disruption and Its Involvement in Neurodevelopmental and Neurodegenerative Disorders. International Journal of Molecular Sciences. 2022; 23(23):15271. https://doi.org/10.3390/ijms232315271
Chicago/Turabian StyleAragón-González, Ana, Pamela J. Shaw, and Laura Ferraiuolo. 2022. "Blood–Brain Barrier Disruption and Its Involvement in Neurodevelopmental and Neurodegenerative Disorders" International Journal of Molecular Sciences 23, no. 23: 15271. https://doi.org/10.3390/ijms232315271