
Milk Exosomal microRNAs: Postnatal Promoters of β Cell Proliferation but Potential Inducers of β Cell De-Differentiation in Adult Life


Abstract
:1. Introduction
2. Exosome Crosstalk with Pancreatic β Cells
3. Postnatal MEX miR Signaling Promotes Pancreatic β Cell Proliferation
3.1. The Role of MEX miRs in AKT-mTORC1-Mediated β Cell Proliferation
3.2. The Role of Long Non-Coding RNA H19 for β Cell Proliferation
3.3. H19 Acts as a Sponge of let-7 miRs, miR-29a, and miR-29b
3.4. LIN28 and let-7
3.5. MEX miR-Mediated Regulation of p53 and miR-29b
3.6. MYC, MAX, and H19 Expression
3.7. FTO α-Ketoglutarate-Dependent Dioxygenase
4. MEX miR Signaling and Functional Immaturity of β Cells
4.1. Pyruvate Kinase
4.2. AMP-Activated Protein Kinase
4.3. MiR-375
4.4. PPARGC1A and CPT1A
4.5. Estrogen-Related Receptor-γ
4.6. DNA Methyltransferase 3A
4.7. β Cell Glycolysis: Impact of p53, TIGAR, SCO2, and HIF-1α
4.8. MAFB
4.9. ABCA1
4.10. TGF-β and FoxO1 Signaling
4.11. NEUROD1 and NKX2.2
5. Weaning: The Metabolic Switch Induced by Fading of MEX miR Signaling
6. Perinatal Factors Disturbing MEX miR-Regulated β Cell Homeostasis
6.1. Preterm Birth
6.2. Maternal Stress during Pregnancy
6.3. Maternal Obesity
6.4. Gestational Diabetes Mellitus
6.5. Maternal Diet
6.6. Caesarean Delivery
6.7. Changes of miR Levels during Breastfeeding
6.8. Duration of Breastfeeding
6.9. Human Donor Milk
6.10. MiR-Deficient Artificial Formula Feeding
7. Bovine MEX miR Signaling during Adult Human Life
7.1. Type 1 Diabetes Mellitus
7.2. Type 2 Diabetes Mellitus
7.3. Metformin
8. Discussion
9. Conclusions
10. Limitations
11. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter, subfamily A, member1 |
| AMPK | AMP-activated protein kinase |
| AQP7 | aquaporin 7 |
| ATG5 | autophagy-related 5 |
| ATM | adipose tissue macrophage |
| BAT | brown adipose tissue |
| BCAA | branched-chain amino acid |
| BECN1 | beclin 1 |
| BIRC5 | baculoviral IAP repeat-containing protein 5 |
| bmMDE | bone marrow mesenchymal stem cell-derived exosome |
| CDK4 | cyclin-dependent kinase 4 |
| CDKN1A | cyclin-dependent kinase inhibitor 1A |
| CDKN1B | cyclin-dependent kinase inhibitor 1B |
| CDKN2A | cyclin-dependent kinase inhibitor 2A |
| CHREBP | carbohydrate response element-binding protein |
| CPT1A | carnitine palmitoyltransferase 1, liver |
| DNMT1 | DNA methyltransferase 1 |
| DNMT3A | DNA methyltransferase 3A |
| DNMT3B | DNA methyltransferase 3B |
| E2F1 | E2F transcription factor 1 |
| ELF3 | E74-like factor 3 |
| ER | endoplasmic reticulum |
| ERRγ | estrogen-related receptor γ |
| ESRRG | estrogen-related receptor γ |
| EV | extracellular vesicle |
| FFAR4 | free fatty acid receptor 4 |
| FIH-1 | factor inhibiting HIF-1 |
| FoxO1A | forkhead box O1A |
| FoxO3A | forkhead box O3A |
| FoxP3 | forkhead box P3 |
| FTO | α-ketoglutarate-dependent dioxigenase |
| GDM | gestational diabetes mellitus |
| GH | growth hormone |
| GHR | growth hormone receptor |
| GLUT1 | glucose transporter 1 |
| GSIS | glucose-stimulated insulin secretion |
| GSK3β | glycogen synthase kinase 3β |
| H19 | long non-coding RNA H19 |
| HIF-1α | hypoxia-inducible factor 1α |
| HIF1AN | hypoxia-inducible factor 1α inhibitor |
| HK1 | hexokinase 1 |
| HoP | Holder pasteurization |
| HPP | high pressure processing |
| IGF-1 | insulin-like growth factor 1 |
| IGF1R | insulin-like growth factor 1 receptor |
| IGF2BP2 | insulin-like growth factor 2 mRNA-binding protein 2 (IMP2) |
| INS | insulin |
| INSR | insulin receptor |
| IRS2 | insulin receptor substrate 2 |
| JAK2 | anus kinase 2 |
| KATP | ATP-sensitive potassium channel |
| LAMP2A | lysosome-associated membrane protein type 2A |
| LDHA | lactate dehydrogenase A |
| LIN28A | LIN28 homolog A |
| LIN28B | LIN28 homolog B |
| LKB1 | serine/threonine protein kinase 11 |
| MAFA | MAF bZIP transcription factor A |
| MAFB | MAF bZIP transcription factor B |
| MAPK | mitogen-activated protein kinase kinase 3 |
| MAX | MAX protein |
| m6A | N6-methyladenosine |
| MCT1 | monocarboxylate transporter 1 |
| MDM2 | MDM2 protooncogene |
| MDM4 | MDM4 regulator of p53 |
| MECP2 | methyl CpG binding protein 2 |
| METTL3 | methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit |
| METTL14 | methyltransferase 14, N6-adenosine-methyltransferase complex catalytic subunit |
| MEX | milk exosome |
| MGST1 | glutathione S-transferase, microsomal, 1 |
| miR | micro-ribonucleic acid |
| MSC | mesenchymal stem cell |
| MYC | MYC protooncogen, bHLH transcription factor |
| mTORC1 | mechanistic target of rapamycin complex 1 |
| NANOG | NANOG homeobox |
| NCOA1 | nuclear receptor coactivator 1 |
| NEUROD1 | neurogenic differentiation 1 |
| NKX2.2 | NK2 homeobox 2 |
| NKX6.1 | NK6 homeobox 1 |
| NRIP1 | nuclear receptor-interacting protein 1 |
| OCT4 | POU domain, class 5, transcription factor 1 |
| OXPHOS | oxidative phosphorylation |
| PAX4 | paired box gene 4 |
| PAX6 | paired box gene 6 |
| PBMC | peripheral blood mononuclear cell |
| PDGFRA | platelet-derived growth factor receptor α |
| PDH | pyruvate dehydrogenase |
| PDX-1 | pancreas/duodenum homeobox protein 1 |
| PGC-1α | peroxisome proliferator-activated receptor-γ coactivator-1α |
| PI3K | phosphatidylinositol-3 kinase |
| PIK3IP1 | phosphatidylinositol 3-kinase-interacting protein 1 |
| PPAR-γ | peroxisome proliferator-activated receptor-γ |
| PRAS40 | proline-rich AKT substrate 40-KD |
| PRKAA1 | protein kinase, AMP-activated, catalytic, α1 |
| PRKAG2 | protein kinase, AMP-activated, noncatalytic, γ-2; |
| PRL | prolactin |
| PRLR | prolactin receptor |
| PTEN | phosphatase and tensin homolog |
| PUFA | polyunsaturated fatty acid |
| RIP140 | receptor interacting protein 140 |
| RAF1 | RAF1 protooncogene, serine/threonine kinase |
| SCO2 | SCO cytochrome c oxidase assembly protein 2 |
| SESN1 | sestrin 1 |
| SESN2 | sestrin 2 |
| SLC2A1 | solute carrier family 2, member 1 |
| SLC2A2 | solute carrier family 2, member 2 |
| SLC16A1 | solute carrier family 16 (monocarboxylic acid transporter), member 1 |
| SMAD2 | SMAD family member 2 |
| SMAD3 | SMAD family member 3 |
| SNCA | synuclein α |
| STZ | streptozotocin |
| TIGAR | TP53-induced glycolysis and apoptosis regulator |
| T1DM | type 1 diabetes mellitus |
| T2DM | type 2 diabetes mellitus |
| TGF-β | transforming growth factor-β |
| TGFBR2 | transforming growth factor-β receptor type 2 |
| TNFRSF10B | tumor necrosis factor receptor superfamily, member 10B; |
| TP53 | tumor protein p53 |
| TSC2 | tuberin |
| UHT | ultraheat treatment |
| USP4 | ubiquitin-specific protease 4 |
| UTR | untranslated region |
| VHL | von Hippel-Lindau tumor suppressor |
References
- He, X.; Kuang, G.; Wu, Y.; Ou, C. Emerging roles of exosomal miRNAs in diabetes mellitus. Clin. Transl. Med. 2021, 11, e468. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, U.; Yzydorczyk, C.; Siddeek, B.; Benahmed, M. Epigenetics and neonatal nutrition. Early Hum. Dev. 2014, 90 (Suppl. S2), S23–S24. [Google Scholar] [CrossRef]
- Marousez, L.; Lesage, J.; Eberlé, D. Epigenetics: Linking early postnatal nutrition to obesity programming? Nutrients 2019, 11, 2966. [Google Scholar] [CrossRef]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef]
- Liu, J.S.E.; Hebrok, M. All mixed up: Defining roles for beta-cell subtypes in mature islets. Genes Dev. 2017, 31, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.M.; Gannon, M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J. Mol. Endocrinol. 2007, 38, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Jacovetti, C.; Regazzi, R. Mechanisms underlying the expansion and functional maturation of β-cells in newborns: Impact of the nutritional environment. Int. J. Mol. Sci. 2022, 23, 2096. [Google Scholar] [CrossRef] [PubMed]
- Rohli, K.E.; Boyer, C.K.; Blom, S.E.; Stephens, S.B. Nutrient regulation of pancreatic islet β-cell secretory capacity and insulin production. Biomolecules 2022, 12, 335. [Google Scholar] [CrossRef]
- Plaisance, V.; Waeber, G.; Regazzi, R.; Abderrahmani, A. Role of microRNAs in islet beta-cell compensation and failure during diabetes. J. Diabetes Res. 2014, 2014, 618652. [Google Scholar] [CrossRef]
- Guay, C.; Regazzi, R. MicroRNAs and the functional β cell mass: For better or worse. Diabetes Metab. 2015, 41, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Abderrahmani, A.; Jacovetti, C.; Regazzi, R. Lessons from neonatal β-cell epigenomic for diabetes prevention and treatment. Trends Endocrinol. Metab. 2022, 33, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Jacovetti, C.; Matkovich, S.J.; Rodriguez-Trejo, A.; Guay, C.; Regazzi, R. Postnatal β-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nat. Commun. 2015, 6, 8084. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Schmitz, G. MicroRNAs: Milk’s epigenetic regulators. Best. Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Zempleni, J.; Aguilar-Lozano, A.; Sadri, M.; Sukreet, S.; Manca, S.; Wu, D.; Zhou, F.; Mutai, E. Biological activities of extracellular vesicles and their cargos from bovine and human milk in humans and implications for infants. J. Nutr. 2017, 147, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Golan-Gerstl, R.; Elbaum Shiff, Y.; Moshayoff, V.; Schecter, D.; Leshkowitz, D.; Reif, S. Characterization and biological function of milk-derived miRNAs. Mol. Nutr. Food Res. 2017, 61, 1700009. [Google Scholar] [CrossRef] [PubMed]
- Benmoussa, A.; Provost, P. Milk microRNAs in health and disease. Compr. Rev. Food Sci. Food Saf. 2019, 18, 703–722. [Google Scholar] [CrossRef]
- Zempleni, J.; Sukreet, S.; Zhou, F.; Wu, D.; Mutai, E. Milk-derived exosomes and metabolic regulation. Annu. Rev. Anim. Biosci. 2019, 7, 245–262. [Google Scholar] [CrossRef]
- Lönnerdal, B. Human milk microRNAs/exosomes: Composition and biological effects. Nestle Nutr. Inst. Workshop Ser. 2019, 90, 83–92. [Google Scholar]
- Melnik, B.C.; Stremmel, W.; Weiskirchen, R.; John, S.M.; Schmitz, G. Exosome-derived microRNAs of human milk and their effects on infant health and development. Biomolecules 2021, 11, 851. [Google Scholar] [CrossRef]
- Jiang, X.; You, L.; Zhang, Z.; Cui, X.; Zhong, H.; Sun, X.; Ji, C.; Chi, X. Biological properties of milk-derived extracellular vesicles and their physiological functions in infant. Front. Cell Dev. Biol. 2021, 9, 693534. [Google Scholar] [CrossRef]
- van Herwijnen, M.J.C.; Driedonks, T.A.P.; Snoek, B.L.; Kroon, A.M.T.; Kleinjan, M.; Jorritsma, R.; Pieterse, C.M.J.; Hoen, E.N.M.N.; Wauben, M.H.M. Abundantly present miRNAs in milk-derived extracellular vesicles are conserved between mammals. Front. Nutr. 2018, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Benmoussa, A.; Lee, C.H.; Laffont, B.; Savard, P.; Laugier, J.; Boilard, E.; Gilbert, C.; Fliss, I.; Provost, P. Commercial dairy cow milk microRNAs resist digestion under simulated gastrointestinal tract conditions. J. Nutr. 2016, 146, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Du, X.; Li, J.; Lönnerdal, B. Human milk exosomes and their microRNAs survive digestion in vitro and are taken up by human intestinal cells. Mol. Nutr. Food Res. 2017, 61, 1700082. [Google Scholar] [CrossRef] [PubMed]
- Rani, P.; Vashisht, M.; Golla, N.; Vashisht, M.; Shandilya, S. Milk miRNAs encapsulated in exosomes are stable to human digestion and permeable to intestinal barrier in vitro. J. Funct. Foods 2017, 34, 431–439. [Google Scholar] [CrossRef]
- Wolf, T.; Baier, S.R.; Zempleni, J. The intestinal transport of bovine milk exosomes is mediated by endocytosis in human colon carcinoma Caco-2 cells and rat small intestinal IEC-6 Cells. J. Nutr. 2015, 145, 2201–2206. [Google Scholar] [CrossRef]
- Kusuma, R.J.; Manca, S.; Friemel, T.; Sukreet, S.; Nguyen, C.; Zempleni, J. Human vascular endothelial cells transport foreign exosomes from cow’s milk by endocytosis. Am. J. Physiol. Cell Physiol. 2016, 310, C800–C807. [Google Scholar] [CrossRef]
- Wang, L.; Sadri, M.; Giraud, D.; Zempleni, J. RNase H2-dependent polymerase chain reaction and elimination of confounders in sample collection, storage, and analysis strengthen evidence that microRNAs in bovine milk are bioavailable in humans. J. Nutr. 2018, 148, 153–159. [Google Scholar] [CrossRef]
- López de Las Hazas, M.C.; Del Pozo-Acebo, L.; Hansen, M.S.; Gil-Zamorano, J.; Mantilla-Escalante, D.C.; Gómez-Coronado, D.; Marín, F.; Garcia-Ruiz, A.; Rasmussen, J.T.; Dávalos, A. Dietary bovine milk miRNAs transported in extracellular vesicles are partially stable during GI digestion, are bioavailable and reach target tissues but need a minimum dose to impact on gene expression. Eur. J. Nutr. 2022, 61, 1043–1056. [Google Scholar] [CrossRef]
- Lin, D.; Chen, T.; Xie, M.; Li, M.; Zeng, B.; Sun, R.; Zhu, Y.; Ye, D.; Wu, J.; Sun, J.; et al. Oral administration of bovine and porcine milk exosome alter miRNAs profiles in piglet serum. Sci. Rep. 2020, 10, 6983. [Google Scholar] [CrossRef]
- Manca, S.; Upadhyaya, B.; Mutai, E.; Desaulniers, A.T.; Cederberg, R.A.; White, B.R.; Zempleni, J. Milk exosomes are bioavailable and distinct microRNA cargos have unique tissue distribution patterns. Sci. Rep. 2018, 8, 11321. [Google Scholar] [CrossRef]
- Aguilar-Lozano, A.; Baier, S.; Grove, R.; Shu, J.; Giraud, D.; Leiferman, A.; Mercer, K.E.; Cui, J.; Badger, T.M.; Adamec, J.; et al. Concentrations of purine metabolites are elevated in fluids from adults and infants and in livers from mice fed diets depleted of bovine milk exosomes and their RNA cargos. J. Nutr. 2018, 148, 1886–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betker, J.L.; Angle, B.M.; Graner, M.W.; Anchordoquy, T.J. The potential of exosomes from cow milk for oral delivery. J. Pharm. Sci. 2019, 108, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Sadri, M.; Shu, J.; Kachman, S.D.; Cui, J.; Zempleni, J. Milk exosomes and miRNA cross the placenta and promote embryo survival in mice. Reproduction 2020, 160, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Sanwlani, R.; Fonseka, P.; Mathivanan, S. Are dietary extracellular vesicles bioavailable and functional in consuming organisms? Subcell. Biochem. 2021, 97, 509–521. [Google Scholar] [PubMed]
- Samuel, M.; Fonseka, P.; Sanwlani, R.; Gangoda, L.; Chee, S.H.; Keerthikumar, S.; Spurling, A.; Chitti, S.V.; Zanker, D.; Ang, C.S.; et al. Oral administration of bovine milk-derived extracellular vesicles induces senescence in the primary tumor but accelerates cancer metastasis. Nat. Commun. 2021, 12, 3950. [Google Scholar] [CrossRef]
- Melnik, B.C. Milk exosomal miRNAs: Potential drivers of AMPK-to-mTORC1 switching in β-cell de-differentiation of type 2 diabetes mellitus. Nutr. Metab. 2019, 16, 85. [Google Scholar] [CrossRef]
- Zhang, A.; Li, D.; Liu, Y.; Li, J.; Zhang, Y.; Zhang, C.Y. Islet β cell: An endocrine cell secreting miRNAs. Biochem. Biophys. Res. Commun. 2018, 495, 1648–1654. [Google Scholar] [CrossRef]
- Guay, C.; Menoud, V.; Rome, S.; Regazzi, R. Horizontal transfer of exosomal microRNAs transduce apoptotic signals between pancreatic beta-cells. Cell Commun. Signal. 2015, 13, 17. [Google Scholar] [CrossRef]
- Qian, B.; Yang, Y.; Tang, N.; Wang, J.; Sun, P.; Yang, N.; Chen, F.; Wu, T.; Sun, T.; Li, Y.; et al. M1 macrophage-derived exosomes impair beta cell insulin secretion via miR-212-5p by targeting SIRT2 and inhibiting Akt/GSK-3β/β-catenin pathway in mice. Diabetologia 2021, 64, 2037–2051. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Kumar, A.P.; Aref, A.R.; Zarrabi, A.; Mostafavi, E. Exosomes as promising nanostructures in diabetes mellitus: From insulin sensitivity to ameliorating diabetic complications. Int. J. Nanomed. 2022, 17, 1229–1253. [Google Scholar] [CrossRef]
- Guay, C.; Kruit, J.K.; Rome, S.; Menoud, V.; Mulder, N.L.; Jurdzinski, A.; Mancarella, F.; Sebastiani, G.; Donda, A.; Gonzalez, B.J.; et al. Lymphocyte-derived exosomal microRNAs promote pancreatic β cell death and may contribute to type 1 diabetes development. Cell Metab. 2019, 29, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesmundo, I.; Pardini, B.; Gargantini, E.; Gamba, G.; Birolo, G.; Fanciulli, A.; Banfi, D.; Congiusta, N.; Favaro, E.; Deregibus, M.C.; et al. Adipocyte-derived extracellular vesicles regulate survival and function of pancreatic β cells. JCI Insight 2021, 6, e141962. [Google Scholar] [CrossRef] [PubMed]
- Jalabert, A.; Vial, G.; Guay, C.; Wiklander, O.P.; Nordin, J.Z.; Aswad, H.; Forterre, A.; Meugnier, E.; Pesenti, S.; Regazzi, R.; et al. Exosome-like vesicles released from lipid-induced insulin-resistant muscles modulate gene expression and proliferation of beta recipient cells in mice. Diabetologia 2016, 59, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- James-Allan, L.B.; Devaskar, S.U. Extracellular vesicles and their role in gestational diabetes mellitus. Placenta 2021, 113, 15–22. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, T.; Sun, D.; Cheng, G.; Ren, H.; Hong, H.; Chen, L.; Jiao, X.; Du, Y.; Zou, Y.; et al. Diagnostic value of dysregulated microribonucleic acids in the placenta and circulating exosomes in gestational diabetes mellitus. J. Diabetes Investig. 2021, 12, 1490–1500. [Google Scholar] [CrossRef]
- Gao, H.; Luo, Z.; Jin, Z.; Ji, Y.; Ying, W. Adipose tissue macrophages modulate obesity-associated β cell adaptations through secreted miRNA-containing extracellular vesicles. Cells 2021, 10, 2451. [Google Scholar] [CrossRef]
- He, Q.; Song, J.; Cui, C.; Wang, J.; Hu, H.; Guo, X.; Yang, M.; Wang, L.; Yan, F.; Liang, K.; et al. Mesenchymal stem cell-derived exosomal miR-146a reverses diabetic β-cell dedifferentiation. Stem Cell Res. Ther. 2021, 12, 449. [Google Scholar] [CrossRef]
- Sun, Y.; Shi, H.; Yin, S.; Ji, C.; Zhang, X.; Zhang, B.; Wu, P.; Shi, Y.; Mao, F.; Yan, Y.; et al. Human mesenchymal stem cell derived exosomes alleviate type 2 diabetes mellitus by reversing peripheral insulin resistance and relieving β-cell destruction. ACS Nano 2018, 12, 7613–7628. [Google Scholar] [CrossRef]
- Chang, W.; Li, M.; Song, L.; Miao, S.; Yu, W.; Wang, J. Noncoding RNAs from tissue-derived small extracellular vesicles: Roles in diabetes and diabetic complications. Mol. Metab. 2022, 58, 101453. [Google Scholar] [CrossRef]
- Saeedi Borujeni, M.J.; Esfandiary, E.; Taheripak, G.; Codoñer-Franch, P.; Alonso-Iglesias, E.; Mirzaei, H. Molecular aspects of diabetes mellitus: Resistin, microRNA, and exosome. J. Cell Biochem. 2018, 119, 1257–1272. [Google Scholar] [CrossRef]
- Castaño, C.; Novials, A.; Párrizas, M. Exosomes and diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3107. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Wang, J. Exosomes and their noncoding RNA cargo are emerging as new modulators for diabetes mellitus. Cells 2019, 8, 853. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.; Luo, S.; Xiao, Y.; Xia, Y.; Li, X.; Huang, G.; Xie, Z.; Zhou, Z. Emerging roles of exosomes in T1DM. Front. Immunol. 2020, 11, 593348. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Song, X.; Yu, H.; Sun, J.; Zhao, Y. Therapeutic potentials of extracellular vesicles for the treatment of diabetes and diabetic complications. Int. J. Mol. Sci. 2020, 21, 5163. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Evans, M.K. Extracellular vesicles as signaling mediators in type 2 diabetes mellitus. Am. J. Physiol. Cell Physiol. 2020, 318, C1189–C1199. [Google Scholar] [CrossRef]
- Sharma, R.; Kumari, M.; Mishra, S.; Chaudhary, D.K.; Kumar, A.; Avni, B.; Tiwari, S. Exosomes secreted by umbilical cord blood-derived mesenchymal stem cell attenuate diabetes in mice. J. Diabetes Res. 2021, 2021, 9534574. [Google Scholar] [CrossRef]
- Chidester, S.; Livinski, A.A.; Fish, A.F.; Joseph, P.V. The role of extracellular vesicles in β-cell function and viability: A scoping review. Front. Endocrinol. 2020, 11, 375. [Google Scholar] [CrossRef]
- Wang, P.; Fiaschi-Taesch, N.M.; Vasavada, R.C.; Scott, D.K.; García-Ocaña, A.; Stewart, A.F. Diabetes mellitus—Advances and challenges in human β-cell proliferation. Nat. Rev. Endocrinol. 2015, 11, 201–212. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Bonner-Weir, S. Pancreatic β cell regeneration as a possible therapy for diabetes. Cell Metab. 2018, 27, 57–67. [Google Scholar] [CrossRef]
- Butler, P.C.; Meier, J.J.; Butler, A.E.; Bhushan, A. The replication of beta cells in normal physiology, in disease and for therapy. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 758–768. [Google Scholar] [CrossRef]
- Kushner, J.A. The role of aging upon β cell turnover. J. Clin. Investig. 2013, 123, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Fatrai, S.; Elghazi, L.; Balcazar, N.; Cras-Méneur, C.; Krits, I.; Kiyokawa, H.; Bernal-Mizrachi, E. Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes 2006, 55, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Elghazi, L.; Balcazar, N.; Bernal-Mizrachi, E. Emerging role of protein kinase B/Akt signaling in pancreatic beta-cell mass and function. Int. J. Biochem. Cell Biol. 2006, 38, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Elghazi, L.; Rachdi, L.; Weiss, A.J.; Cras-Méneur, C.; Bernal-Mizrachi, E. Regulation of beta-cell mass and function by the Akt/protein kinase B signalling pathway. Diabetes Obes. Metab. 2007, 9 (Suppl. S2), 147–157. [Google Scholar] [CrossRef] [PubMed]
- Balcazar, N.; Sathyamurthy, A.; Elghazi, L.; Gould, A.; Weiss, A.; Shiojima, I.; Walsh, K.; Bernal-Mizrachi, E. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J. Biol. Chem. 2009, 284, 7832–7842. [Google Scholar] [CrossRef]
- Balcazar Morales, N.; Aguilar de Plata, C. Role of AKT/mTORC1 pathway in pancreatic β-cell proliferation. Colomb. Med. (Cali.) 2012, 43, 235–243. [Google Scholar] [CrossRef]
- Blandino-Rosano, M.; Chen, A.Y.; Scheys, J.O.; Alejandro, E.U.; Gould, A.P.; Taranukha, T.; Elghazi, L.; Cras-Méneur, C.; Bernal-Mizrachi, E. mTORC1 signaling and regulation of pancreatic β-cell mass. Cell Cycle 2012, 11, 1892–1902. [Google Scholar] [CrossRef]
- Helman, A.; Cangelosi, A.L.; Davis, J.C.; Pham, Q.; Rothman, A.; Faust, A.L.; Straubhaar, J.R.; Sabatini, D.M.; Melton, D.A. A nutrient-sensing transition at birth triggers glucose-responsive insulin secretion. Cell Metab. 2020, 31, 1004–1016. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Nie, A.; Ni, Q.; Li, F.; Ning, G.; Li, X.; Gu, Y.; Wang, Q. mTORC1 pathway mediates beta cell compensatory proliferation in 60 % partial-pancreatectomy mice. Endocrine 2016, 53, 117–128. [Google Scholar] [CrossRef]
- Melnik, B.C.; John, S.M.; Schmitz, G. Milk is not just food but most likely a genetic transfection system activating mTORC1 signaling for postnatal growth. Nutr. J. 2013, 12, 103. [Google Scholar] [CrossRef]
- Howell, J.J.; Ricoult, S.J.; Ben-Sahra, I.; Manning, B.D. A growing role for mTOR in promoting anabolic metabolism. Biochem. Soc. Trans. 2013, 41, 906–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Fingar, D.C. Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 2014, 36, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Ozkan, H.; Tuzun, F.; Taheri, S.; Korhan, P.; Akokay, P.; Yılmaz, O.; Duman, N.; Özer, E.; Tufan, E.; Kumral, A.; et al. Epigenetic programming through breast milk and its impact on milk-siblings mating. Front. Genet. 2020, 11, 569232. [Google Scholar] [CrossRef]
- Chen, T.; Xie, M.Y.; Sun, J.J.; Ye, R.S.; Cheng, X.; Sun, R.P.; Wei, L.M.; Li, M.; Lin, D.L.; Jiang, Q.Y.; et al. Porcine milk-derived exosomes promote proliferation of intestinal epithelial cells. Sci. Rep. 2016, 6, 33862. [Google Scholar] [CrossRef]
- Liu, Z.; Xie, Y.; Guo, J.; Su, X.; Zhao, C.; Zhang, C.; Qin, Q.; Dai, D.; Tuo, Y.; Li, Z.; et al. Comparison of porcine milk microRNA expression in milk exosomes versus whole swine milk and prediction of target genes. Arch. Anim. Breed. 2022, 65, 37–46. [Google Scholar] [CrossRef]
- Nielsen, J.H.; Svensson, C.; Galsgaard, E.D.; Møldrup, A.; Billestrup, N. Beta cell proliferation and growth factors. J. Mol. Med. 1999, 77, 62–66. [Google Scholar] [CrossRef]
- Huang, Y.; Chang, Y. Regulation of pancreatic islet beta-cell mass by growth factor and hormone signaling. Prog. Mol. Biol. Transl. Sci. 2014, 121, 321–349. [Google Scholar] [PubMed]
- Ma, F.; Wei, Z.; Shi, C.; Gan, Y.; Lu, J.; Frank, S.J.; Balducci, J.; Huang, Y. Signaling cross talk between growth hormone (GH) and insulin-like growth factor-I (IGF-I) in pancreatic islet β-cells. Mol. Endocrinol. 2011, 25, 2119–2133. [Google Scholar] [CrossRef] [PubMed]
- Carver, K.C.; Schuler, L.A. Prolactin does not require insulin-like growth factor intermediates but synergizes with insulin-like growth factor I in human breast cancer cells. Mol. Cancer Res. 2008, 6, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Carver, K.C.; Piazza, T.M.; Schuler, L.A. Prolactin enhances insulin-like growth factor I receptor phosphorylation by decreasing its association with the tyrosine phosphatase SHP-2 in MCF-7 breast cancer cells. J. Biol. Chem. 2010, 285, 8003–8012. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kaburagi, Y.; Ueki, K.; Tsuji, Y.; Stark, G.R.; Kerr, I.M.; Tsushima, T.; Akanuma, Y.; Komuro, I.; Tobe, K.; et al. Growth hormone and prolactin stimulate tyrosine phosphorylation of insulin receptor substrate-1, -2, and -3, their association with p85 phosphatidylinositol 3-kinase (PI3-kinase), and concomitantly PI3-kinase activation via JAK2 kinase. J. Biol. Chem. 1998, 273, 15719–15726. [Google Scholar] [CrossRef]
- Amaral, M.E.; Ueno, M.; Carvalheira, J.B.; Carneiro, E.M.; Velloso, L.A.; Saad, M.J.; Boschero, A.C. Prolactin-signal transduction in neonatal rat pancreatic islets and interaction with the insulin-signaling pathway. Horm. Metab. Res. 2003, 35, 282–289. [Google Scholar]
- Amaral, M.E.; Cunha, D.A.; Anhê, G.F.; Ueno, M.; Carneiro, E.M.; Velloso, L.A.; Bordin, S.; Boschero, A.C. Participation of prolactin receptors and phosphatidylinositol 3-kinase and MAP kinase pathways in the increase in pancreatic islet mass and sensitivity to glucose during pregnancy. J. Endocrinol. 2004, 183, 469–476. [Google Scholar] [CrossRef]
- Hügl, S.R.; White, M.F.; Rhodes, C.J. Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J. Biol. Chem. 1998, 273, 17771–17779. [Google Scholar] [CrossRef]
- Tuttle, R.L.; Gill, N.S.; Pugh, W.; Lee, J.P.; Koeberlein, B.; Furth, E.E.; Polonsky, K.S.; Naji, A.; Birnbaum, M.J. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat. Med. 2001, 7, 1133–1137. [Google Scholar] [CrossRef]
- Wrede, C.E.; Dickson, L.M.; Lingohr, M.K.; Briaud, I.; Rhodes, C.J. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J. Biol. Chem. 2002, 277, 49676–49684. [Google Scholar] [CrossRef]
- Withers, D.J.; Burks, D.J.; Towery, H.H.; Altamuro, S.L.; Flint, C.L.; White, M.F. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat. Genet. 1999, 23, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Beith, J.L.; Alejandro, E.U.; Johnson, J.D. Insulin stimulates primary beta-cell proliferation via Raf-1 kinase. Endocrinology 2008, 149, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Parra, C.; Jacovetti, C.; Dumortier, O.; Lee, K.; Peyot, M.L.; Guay, C.; Prentki, M.; Laybutt, D.R.; Van Obberghen, E.; Regazzi, R. Contribution of the long noncoding RNA H19 to β-cell mass expansion in neonatal and adult rodents. Diabetes 2018, 67, 2254–2267. [Google Scholar] [CrossRef] [PubMed]
- Blandino-Rosano, M.; Alejandro, E.U.; Sathyamurthy, A.; Scheys, J.O.; Gregg, B.; Chen, A.Y.; Rachdi, L.; Weiss, A.; Barker, D.J.; Gould, A.P.; et al. Enhanced beta cell proliferation in mice overexpressing a constitutively active form of Akt and one allele of p21Cip. Diabetologia 2012, 55, 1380–1389. [Google Scholar] [CrossRef]
- Brown, V.D.; Phillips, R.A.; Gallie, B.L. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol. Cell Biol. 1999, 19, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, M.; Ma, Y.; Nagasawa, K.; Toyoshima, F. A G-quadruplex structure at the 5′ end of the H19 coding region regulates H19 transcription. Sci. Rep. 2017, 8, 45815. [Google Scholar] [CrossRef]
- Faja, L.; Annicotte, J.S.; Miard, S.; Sarruf, D.; Watanabe, M.; Auwerx, J. Impaired pancreatic growth, beta cell mass, and beta cell function in E2F1(-/-) mice. J. Clin. Investig. 2004, 113, 1288–1295. [Google Scholar] [CrossRef]
- Grouwels, G.; Cai, Y.; Hoebeke, I.; Leuckx, G.; Heremans, Y.; Ziebold, U.; Stangé, G.; Chintinne, M.; Ling, Z.; Pipeleers, D.; et al. Ectopic expression of E2F1 stimulates beta-cell proliferation and function. Diabetes 2010, 59, 1435–1444. [Google Scholar] [CrossRef]
- Withers, D.J.; Gutierrez, J.S.; Towery, H.; Burks, D.J.; Ren, J.M.; Previs, S.; Zhang, Y.; Bernal, D.; Pons, S.; Shulman, G.I.; et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998, 391, 900–904. [Google Scholar] [CrossRef]
- Puri, S.; Roy, N.; Russ, H.A.; Leonhardt, L.; French, E.K.; Roy, R.; Bengtsson, H.; Scott, D.K.; Stewart, A.F.; Hebrok, M. Replication confers β cell immaturity. Nat. Commun. 2018, 9, 485. [Google Scholar] [CrossRef]
- Rosselot, C.; Baumel-Alterzon, S.; Li, Y.; Brill, G.; Lambertini, L.; Katz, L.S.; Lu, G.; Garcia-Ocaña, A.; Scott, D.K. The many lives of Myc in the pancreatic β-cell. J. Biol. Chem. 2021, 296, 100122. [Google Scholar] [CrossRef] [PubMed]
- Rosselot, C.; Kumar, A.; Lakshmipathi, J.; Zhang, P.; Lu, G.; Katz, L.S.; Prochownik, E.V.; Stewart, A.F.; Lambertini, L.; Scott, D.K.; et al. Myc is required for adaptive β-cell replication in young mice but is not sufficient in one-year-old mice fed with a high-fat diet. Diabetes 2019, 68, 1934–1949. [Google Scholar] [CrossRef] [PubMed]
- Barsyte-Lovejoy, D.; Lau, S.K.; Boutros, P.C.; Khosravi, F.; Jurisica, I.; Andrulis, I.L.; Tsao, M.S.; Penn, L.Z. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006, 66, 5330–5337. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Mo, J.; Luo, M.; Yu, Q.; Zhou, S.; Li, T.; Zhang, Y.; Luo, W. c-Myc-activated long non-coding RNA H19 downregulates miR-107 and promotes cell cycle progression of non-small cell lung cancer. Int. J. Clin Exp. Pathol. 2015, 8, 12400–12409. [Google Scholar]
- Zhang, E.; Li, W.; Yin, D.; De, W.; Zhu, L.; Sun, S.; Han, L. c-Myc-regulated long non-coding RNA H19 indicates a poor prognosis and affects cell proliferation in non-small-cell lung cancer. Tumour Biol. 2016, 37, 4007–4015. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Yang, F. The role of long non-coding RNA H19 in breast cancer. Oncol. Lett. 2020, 19, 7–16. [Google Scholar] [CrossRef]
- Yang, J.J.; She, Q.; Yang, Y.; Tao, H.; Li, J. DNMT1 controls LncRNA H19/ERK signal pathway in hepatic stellate cell activation and fibrosis. Toxicol. Lett. 2018, 295, 325–334. [Google Scholar] [CrossRef]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef]
- Shu, C.; Yan, D.; Chen, C.; Mo, Y.; Wu, L.; Gu, J.; Shah, N.K.; He, J.; Dong, S. Metformin exhibits its therapeutic effect in the treatment of pre-eclampsia via modulating the Met/H19/miR-148a-5p/P28 and Met/H19/miR-216-3p/EBI3 signaling pathways. Int. Immunopharmacol. 2019, 74, 105693. [Google Scholar] [CrossRef]
- Chen, J.; Qin, C.; Zhou, Y.; Chen, Y.; Mao, M.; Yang, J. Metformin may induce ferroptosis by inhibiting autophagy via lncRNA H19 in breast cancer. FEBS Open Bio. 2022, 12, 146–153. [Google Scholar] [CrossRef]
- Zeng, J.; Zhu, L.; Liu, J.; Zhu, T.; Xie, Z.; Sun, X.; Zhang, H. Metformin protects against oxidative stress injury induced by ischemia/reperfusion via regulation of the lncRNA-H19/miR-148a-3p/Rock2 axis. Oxid. Med. Cell. Longev. 2019, 2019, 8768327. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xiang, P.; Liu, L.; Sun, J.; Ye, S. Metformin inhibits extracellular matrix accumulation, inflammation and proliferation of mesangial cells in diabetic nephropathy by regulating H19/miR-143-3p/TGF-β1 axis. J. Pharm. Pharmacol. 2020, 72, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhou, J.; Gao, Y.; Ghazal, S.; Lu, L.; Bellone, S.; Yang, Y.; Liu, N.; Zhao, X.; Santin, A.D.; et al. Regulation of tumor cell migration and invasion by the H19/let-7 axis is antagonized by metformin-induced DNA methylation. Oncogene 2015, 34, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Aminimoghaddam, S.; Fooladi, B.; Noori, M.; Nickhah Klashami, Z.; Kakavand Hamidi, A.; Amoli, M.M. The effect of metformin on expression of long non-coding RNA H19 in endometrial cancer. Med. J. Islam. Repub. Iran 2021, 22, 155. [Google Scholar] [CrossRef]
- Kallen, A.N.; Zhou, X.B.; Xu, J.; Qiao, C.; Ma, J.; Yan, L.; Lu, L.; Liu, C.; Yi, J.S.; Zhang, H.; et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol. Cell 2013, 52, 101–112. [Google Scholar] [CrossRef]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef]
- Frost, R.J.; Olson, E.N. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc. Natl. Acad. Sci. USA 2011, 108, 21075–21080. [Google Scholar] [CrossRef]
- Jacovetti, C.; Rodriguez-Trejo, A.; Guay, C.; Sobel, J.; Gattesco, S.; Petrenko, V.; Saini, C.; Dibner, C.; Regazzi, R. MicroRNAs modulate core-clock gene expression in pancreatic islets during early postnatal life in rats. Diabetologia 2017, 60, 2011–2020. [Google Scholar] [CrossRef]
- He, H.; Wang, N.; Yi, X.; Tang, C.; Wang, D. Long non-coding RNA H19 regulates E2F1 expression by competitively sponging endogenous miR-29a-3p in clear cell renal cell carcinoma. Cell Biosci. 2017, 7, 65. [Google Scholar] [CrossRef]
- Yan, L.; Liu, G.; Wu, X. The umbilical cord mesenchymal stem cell-derived exosomal lncRNA H19 improves osteochondral activity through miR-29b-3p/FoxO3 axis. Clin. Transl. Med. 2021, 11, e255. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, Y.; Liu, W.; Huang, Y.; Shen, X.; Jing, R.; Pu, J.; Wang, X.; Ju, S.; Cong, H.; et al. LncRNA H19 overexpression induces bortezomib resistance in multiple myeloma by targeting MCL-1 via miR-29b-3p. Cell Death Dis. 2019, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhou, Y.; Chen, J.; Wang, T.; Li, Z.; Fu, Y.; Zhai, A.; Bi, C. Long noncoding RNA H19 acts as a miR-29b sponge to promote wound healing in diabetic foot ulcer. FASEB J. 2021, 35, e20526. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, C.; Meng, F.; Xu, P. Long non-coding RNA H19 inhibition ameliorates oxygen-glucose deprivation-induced cell apoptosis and inflammatory cytokine expression by regulating the microRNA-29b/SIRT1/PGC-1α axis. Mol. Med. Rep. 2021, 23, 131. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Tang, C.; Huang, B.; Gu, L.; Zhou, J.; Mo, Z.; Liu, C.; Liu, Y. LncRNA H19 drives proliferation of cardiac fibroblasts and collagen production via suppression of the miR-29a-3p/miR-29b-3p-VEGFA/TGF-β axis. Mol. Cells 2022, 45, 122–133. [Google Scholar] [CrossRef]
- Sung, Y.; Jeong, J.; Kang, R.J.; Choi, M.; Park, S.; Kwon, W.; Lee, J.; Jang, S.; Park, S.J.; Kim, S.H.; et al. Lin28a expression protects against streptozotocin-induced β-cell destruction and prevents diabetes in mice. Cell Biochem. Funct. 2019, 37, 139–147. [Google Scholar] [CrossRef]
- Zhou, X.; Nair, G.G.; Russ, H.A.; Belair, C.D.; Li, M.L.; Shveygert, M.; Hebrok, M.; Blelloch, R. LIN28B impairs the transition of hESC-derived β cells from the juvenile to adult state. Stem Cell Rep. 2020, 14, 9–20. [Google Scholar] [CrossRef]
- He, J.; Fan, L.; Shan, A.; Wang, W.; Ning, G.; Cao, Y.; Jiang, X. Let7b-5p inhibits insulin secretion and decreases pancreatic β-cell mass in mice. Mol. Cell. Endocrinol. 2022, 540, 111506. [Google Scholar]
- Munch, E.M.; Harris, R.A.; Mohammad, M.; Benham, A.L.; Pejerrey, S.M.; Showalter, L.; Hu, M.; Shope, C.D.; Maningat, P.D.; Gunaratne, P.H.; et al. Transcriptome profiling of microRNA by Next-Gen deep sequencing reveals known and novel miRNA species in the lipid fraction of human breast milk. PLoS ONE 2013, 8, e50564. [Google Scholar]
- Zhou, Q.; Li, M.; Wang, X.; Li, Q.; Wang, T.; Zhu, Q.; Zhou, X.; Wang, X.; Gao, X.; Li, X. Immune-related microRNAs are abundant in breast milk exosomes. Int. J. Biol. Sci. 2012, 8, 118–123. [Google Scholar] [CrossRef]
- Rubio, M.; Bustamante, M.; Hernandez-Ferrer, C.; Fernandez-Orth, D.; Pantano, L.; Sarria, Y.; Piqué-Borras, M.; Vellve, K.; Agramunt, S.; Carreras, R.; et al. Circulating miRNAs, isomiRs and small RNA clusters in human plasma and breast milk. PLoS ONE 2018, 13, e0193527. [Google Scholar] [CrossRef]
- Xie, M.Y.; Hou, L.J.; Sun, J.J.; Zeng, B.; Xi, Q.Y.; Luo, J.Y.; Chen, T.; Zhang, Y.L. Porcine milk exosome miRNAs attenuate LPS-induced apoptosis through inhibiting TLR4/NF-κB and p53 pathways in intestinal epithelial cells. J. Agric. Food Chem. 2019, 67, 9477–9491. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.M.; Zhang, K.; Zhang, J.H. Human breast milk-derived exosomal miR-148a-3p protects against necrotizing enterocolitis by regulating p53 and Sirtuin 1. Inflammation 2022, 45, 1254–1268. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Van Long, F.N.; Diaz, J.J. 40 years of research put p53 in translation. Cancers 2018, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Hoh, J.; Jin, S.; Parrado, T.; Edington, J.; Levine, A.J.; Ott, J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc. Natl. Acad. Sci. USA 2002, 99, 8467–8472. [Google Scholar] [CrossRef] [Green Version]
- el-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar]
- Pappas, K.; Xu, J.; Zairis, S.; Resnick-Silverman, L.; Abate, F.; Steinbach, N.; Ozturk, S.; Saal, L.H.; Su, T.; Cheung, P.; et al. p53 maintains baseline expression of multiple tumor suppressor genes. Mol. Cancer Res. 2017, 15, 1051–1062. [Google Scholar] [CrossRef]
- Werner, H.; Karnieli, E.; Rauscher, F.J.; LeRoith, D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 1996, 93, 8318–8323. [Google Scholar] [CrossRef]
- Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, a001057. [Google Scholar] [CrossRef]
- Krstic, J.; Reinisch, I.; Schupp, M.; Schulz, T.J.; Prokesch, A. p53 functions in adipose tissue metabolism and homeostasis. Int. J. Mol. Sci. 2018, 19, 2622. [Google Scholar] [CrossRef]
- Agarwal, S.; Bell, C.M.; Taylor, S.M.; Moran, R.G. p53 deletion or hotspot mutations enhance mTORC1 activity by altering lysosomal dynamics of TSC2 and Rheb. Mol. Cancer Res. 2016, 14, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Co, N.N.; Iglesias, D.; Celestino, J.; Kwan, S.Y.; Mok, S.C.; Schmandt, R.; Lu, K.H. Loss of LKB1 in high-grade endometrial carcinoma: LKB1 is a novel transcriptional target of p53. Cancer 2014, 120, 3457–3468. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Lima, S.; Takabe, K.; Newton, J.; Saurabh, K.; Young, M.M.; Leopoldino, A.M.; Hait, N.C.; Roberts, J.L.; Wang, H.G.; Dent, P.; et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018, 14, 942–957. [Google Scholar] [CrossRef]
- Kuribayashi, K.; Krigsfeld, G.; Wang, W.; Xu, J.; Mayes, P.A.; Dicker, D.T.; Wu, G.S.; El-Deiry, W.S. TNFSF10 (TRAIL), a p53 target gene that mediates p53-dependent cell death. Cancer Biol. Ther. 2008, 7, 2034–2038. [Google Scholar] [CrossRef] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Laptenko, O.; Prives, C. p53: Master of life, death, and the epigenome. Genes Dev. 2017, 31, 955–956. [Google Scholar] [CrossRef]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 2002, 277, 3247–3257. [Google Scholar] [CrossRef]
- Le, M.T.; Shyh-Chang, N.; Khaw, S.L.; Chin, L.; The, C.; Tay, J.; O’Day, E.; Korzh, V.; Yang, H.; Lal, A.; et al. Conserved regulation of p53 network dosage by microRNA-125b occurs through evolving miRNA-target gene pairs. PLoS Genet. 2011, 7, e1002242. [Google Scholar] [CrossRef]
- Quan, S.; Nan, X.; Wang, K.; Jiang, L.; Yao, J.; Xiong, B. Characterization of sheep milk extracellular vesicle-miRNA by sequencing and comparison with cow milk. Animals 2020, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Xi, Q.Y.; Ye, R.S.; Cheng, X.; Qi, Q.E.; Wang, S.B.; Shu, G.; Wang, L.N.; Zhu, X.T.; Jiang, Q.Y.; et al. Exploration of microRNAs in porcine milk exosomes. BMC Genom. 2014, 15, 100. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Kooka, A.; Kowatari, K.; Yoshizawa, M.; Chiba, N.; Takaguri, A.; Fukushi, Y.; Hongo, F.; Sato, H.; Wada, S. Expression profiles of hsa-miR-148a-3p and hsa-miR-125b-5p in human breast milk and infant formulae. Int. Breastfeed. J. 2022, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Baddela, V.S.; Nayan, V.; Rani, P.; Onteru, S.K.; Singh, D. Physicochemical biomolecular insights into Buffalo milk-derived nanovesicles. Appl. Biochem. Biotechnol. 2016, 178, 544–557. [Google Scholar] [CrossRef]
- Kumar, M.; Lu, Z.; Takwi, A.A.; Chen, W.; Callander, N.S.; Ramos, K.S.; Young, K.H.; Li, Y. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene 2011, 30, 843–853. [Google Scholar] [CrossRef]
- Gu, Y.; Li, M.; Wang, T.; Liang, Y.; Zhong, Z.; Wang, X.; Zhou, Q.; Chen, L.; Lang, Q.; He, Z.; et al. Lactation-related microRNA expression profiles of porcine breast milk exosomes. PLoS ONE 2012, 7, e43691. [Google Scholar] [CrossRef]
- Tingö, L.; Ahlberg, E.; Johansson, L.; Pedersen, S.A.; Chawla, K.; Sætrom, P.; Cione, E.; Simpson, M.R. Non-coding RNAs in human breast milk: A systematic review. Front. Immunol. 2021, 12, 725323. [Google Scholar] [CrossRef]
- Chen, X.; Gao, C.; Li, H.; Huang, L.; Sun, Q.; Dong, Y.; Tian, C.; Gao, S.; Dong, H.; Guan, D.; et al. Identification and characterization of microRNAs in raw milk during different periods of lactation, commercial fluid, and powdered milk products. Cell Res. 2010, 20, 1128–1137. [Google Scholar] [CrossRef]
- Melnik, B.C. Milk—A nutrient system of mammalian evolution promoting mTORC1-dependent translation. Int. J. Mol. Sci. 2015, 16, 17048–17087. [Google Scholar] [CrossRef]
- Reif, S.; Elbaum Shiff, Y.; Golan-Gerstl, R. Milk-derived exosomes (MDEs) have a different biological effect on normal fetal colon epithelial cells compared to colon tumor cells in a miRNA-dependent manner. J. Transl. Med. 2019, 17, 325. [Google Scholar] [CrossRef]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Mecocci, S.; Pietrucci, D.; Milanesi, M.; Pascucci, L.; Filippi, S.; Rosato, V.; Chillemi, G.; Capomaccio, S.; Cappelli, K. Transcriptomic characterization of cow, donkey and goat milk extracellular vesicles reveals their anti-inflammatory and immunomodulatory potential. Int. J. Mol. Sci. 2021, 22, 12759. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Mejía-Hernández, J.O.; Vijayakumaran, R.; Keam, S.P.; Haupt, Y. The long and the short of it: The MDM4 tail so far. J. Mol. Cell Biol. 2019, 11, 231–244. [Google Scholar] [CrossRef]
- Ceballos, E.; Delgado, M.D.; Gutierrez, P.; Richard, C.; Müller, D.; Eilers, M.; Ehinger, M.; Gullberg, U.; León, J. c-Myc antagonizes the effect of p53 on apoptosis and p21WAF1 transactivation in K562 leukemia cells. Oncogene 2000, 19, 2194–2204. [Google Scholar] [CrossRef]
- Jiang, Y.; Nishimura, W.; Devor-Henneman, D.; Kusewitt, D.; Wang, H.; Holloway, M.P.; Dohi, T.; Sabo, E.; Robinson, M.L.; Altieri, D.C.; et al. Postnatal expansion of the pancreatic beta-cell mass is dependent on survivin. Diabetes 2008, 57, 2718–2727. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wang, L.; Schroer, S.; Choi, D.; Chen, P.; Okada, H.; Woo, M. Perinatal survivin is essential for the establishment of pancreatic beta cell mass in mice. Diabetologia 2009, 52, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Pullen, T.J.; da Silva Xavier, G.; Kelsey, G.; Rutter, G.A. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol. Cell Biol. 2011, 31, 3182–3194. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, A.P.; Ramsay, A.J.; de la Rosa, J.; Varela, I.; Mariño, G.; Cadiñanos, J.; Lu, J.; Freije, J.M.; López-Otín, C. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 2011, 30, 2219–2232. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, J.H.; Ha, M.; Nam, J.W.; Kim, V.N. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat. Struct. Mol. Biol. 2009, 16, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.L.; Kurita, S.; Cazanave, S.C.; Bronk, S.F.; Werneburg, N.W.; Fernandez-Zapico, M.E. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J. Cell Biochem. 2010, 110, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, A.J.; Liu, Y.; Fang, Y.; Ding, X.; Liang, M. The miR-29 family: Genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol. Genom. 2012, 44, 237–244. [Google Scholar] [CrossRef]
- Melnik, B.C. Milk disrupts p53 and DNMT1, the guardians of the genome: Implications for acne vulgaris and prostate cancer. Nutr. Metab. 2017, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Rafizadeh, S.; Gorrepati, K.D.; Lupse, B.; Oberholzer, J.; Maedler, K.; Ardestani, A. Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia 2017, 60, 668–678. [Google Scholar] [CrossRef]
- de Candia, P.; Spinetti, G.; Specchia, C.; Sangalli, E.; La Sala, L.; Uccellatore, A.; Lupini, S.; Genovese, S.; Matarese, G.; Ceriello, A. A unique plasma microRNA profile defines type 2 diabetes progression. PLoS ONE 2017, 12, e0188980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati, B.; Land, H. Myc-Max-Mad: A transcription factor network controlling cell cycle progression, differentiation and death. Curr. Opin. Genet. Dev. 1994, 4, 102–108. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Mao, D.Y.; Watson, J.D.; Yan, P.S.; Barsyte-Lovejoy, D.; Khosravi, F.; Wong, W.W.; Farnham, P.J.; Huang, T.H.; Penn, L.Z. Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 2003, 13, 882–886. [Google Scholar] [CrossRef]
- TargetScanHuman 8.0. Human MAX ENST00000284165.6. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000284165.6&taxid=9606&members=.&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Prendergast, G.C.; Ziff, E.B. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 1991, 251, 186–189. [Google Scholar] [CrossRef]
- Kahn, S.; Liao, Y.; Du, X.; Xu, W.; Li, J.; Lönnerdal, B. Exosomal microRNAs in milk from mothers delivering preterm infants survive in vitro digestion and are taken up by human intestinal cells. Mol. Nutr. Food Res. 2018, 62, e1701050. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman 8.0. Human MECP2 ENST00000303391.6. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000303391.6&taxid=9606&members=.&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, S.J.; Tellam, R.L.; Morrison, J.L.; Muhlhausler, B.S.; Molloy, P.L. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin. Epigenet. 2015, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Aljofan, M.; Riethmacher, D. Anticancer activity of metformin: A systematic review of the literature. Future Sci. OA 2019, 5, FSO410. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Luo, J.; Yu, T.; Zhou, L.; Lv, H.; Shang, P. Anticancer mechanisms of metformin: A review of the current evidence. Life Sci. 2020, 254, 117717. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Schmitz, G. Metformin: An inhibitor of mTORC1 signaling. J. Endocrinol. Diabetes Obes. 2014, 2, 1029. [Google Scholar]
- De Jesus, D.F.; Zhang, Z.; Kahraman, S.; Brown, N.K.; Chen, M.; Hu, J.; Gupta, M.K.; He, C.; Kulkarni, R.N. m6A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat. Metab. 2019, 1, 765–774. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, J.; Lin, Z.; Zhang, W.; Wang, S.; Wang, W.; Wang, Q.; Ning, G. m6A mRNA methylation controls functional maturation in neonatal murine β-cells. Diabetes 2020, 69, 1708–1722. [Google Scholar] [CrossRef]
- Bornaque, F.; Delannoy, C.P.; Courty, E.; Rabhi, N.; Carney, C.; Rolland, L.; Moreno, M.; Gromada, X.; Bourouh, C.; Petit, P.; et al. Glucose regulates m6A methylation of RNA in pancreatic Islets. Cells 2022, 11, 291. [Google Scholar] [CrossRef]
- Lei, L.; Bai, Y.H.; Jiang, H.Y.; He, T.; Li, M.; Wang, J.P. A bioinformatics analysis of the contribution of m6A methylation to the occurrence of diabetes mellitus. Endocr. Connect. 2021, 10, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Beattie, G.M.; Itkin-Ansari, P.; Cirulli, V.; Leibowitz, G.; Lopez, A.D.; Bossie, S.; Mally, M.I.; Levine, F.; Hayek, A. Sustained proliferation of PDX-1+ cells derived from human islets. Diabetes 1999, 48, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Feanny, M.A.; Fagan, S.P.; Ballian, N.; Liu, S.H.; Li, Z.; Wang, X.; Fisher, W.; Brunicardi, F.C.; Belaguli, N.S. PDX-1 expression is associated with islet proliferation in vitro and in vivo. J. Surg. Res. 2008, 144, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Hayes, H.L.; Zhang, L.; Becker, T.C.; Haldeman, J.M.; Stephens, S.B.; Arlotto, M.; Moss, L.G.; Newgard, C.B.; Hohmeier, H.E. A Pdx-1-regulated soluble factor activates rat and human islet cell proliferation. Mol. Cell Biol. 2016, 36, 2918–2930. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887, Erratum in: Nat. Chem. Biol. 2012, 8, 1008. [Google Scholar] [CrossRef]
- Taneera, J.; Prasad, R.B.; Dhaiban, S.; Mohammed, A.K.; Haataja, L.; Arvan, P.; Hamad, M.; Groop, L.; Wollheim, C.B. Silencing of the FTO gene inhibits insulin secretion: An in vitro study using GRINCH cells. Mol. Cell. Endocrinol. 2018, 472, 10–17. [Google Scholar] [CrossRef]
- Russell, M.A.; Morgan, N.G. Conditional expression of the FTO gene product in rat INS-1 cells reveals its rapid turnover and a role in the profile of glucose-induced insulin secretion. Clin. Sci. 2011, 120, 403–413. [Google Scholar] [CrossRef]
- Sun, L.; Gao, M.; Qian, Q.; Guo, Z.; Zhu, P.; Wang, X.; Wang, H. Triclosan-induced abnormal expression of miR-30b regulates fto-mediated m6A methylation level to cause lipid metabolism disorder in zebrafish. Sci. Total Environ. 2021, 770, 145285. [Google Scholar] [CrossRef]
- Izumi, H.; Tsuda, M.; Sato, Y.; Kosaka, N.; Ochiya, T.; Iwamoto, H.; Namba, K.; Takeda, Y. Bovine milk exosomes contain microRNA and mRNA and are taken up by human macrophages. J. Dairy Sci. 2015, 98, 2920–2933. [Google Scholar] [CrossRef]
- TargetScanHuman 8.0. Human FTO ENST00000471389.1. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000471389.1&taxid=9606&showcnc=&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef]
- Regué, L.; Zhao, L.; Ji, F.; Wang, H.; Avruch, J.; Dai, N. RNA m6A reader IMP2/IGF2BP2 promotes pancreatic β-cell proliferation and insulin secretion by enhancing PDX1 expression. Mol. Metab. 2021, 48, 101209. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman 8.0. Human IGF2BP2 ENST00000382199.2. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000382199.2&taxid=9606&showcnc=.0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Dai, N.; Rapley, J.; Angel, M.; Yanik, M.F.; Blower, M.D.; Avruch, J. mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 2011, 25, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Ji, F.; Wright, J.; Minichiello, L.; Sadreyev, R.; Avruch, J. IGF2 mRNA binding protein-2 is a tumor promoter that drives cancer proliferation through its client mRNAs IGF2 and HMGA1. Elife 2017, 6, e27155. [Google Scholar] [CrossRef] [PubMed]
- Dai, N. The diverse functions of IMP2/IGF2BP2 in metabolism. Trends Endocrinol. Metab. 2020, 31, 670–679. [Google Scholar] [CrossRef]
- Kaung, H.L. Growth dynamics of pancreatic islet cell populations during fetal and neonatal development of the rat. Dev. Dyn. 1994, 200, 163–175. [Google Scholar] [CrossRef]
- Schuit, F. Epigenetic programming of glucose-regulated insulin release. J. Clin. Investig. 2015, 125, 2565–2568. [Google Scholar] [CrossRef]
- Lewandowski, S.L.; Cardone, R.L.; Foster, H.R.; Ho, T.; Potapenko, E.; Poudel, C.; VanDeusen, H.R.; Sdao, S.M.; Alves, T.C.; Zhao, X.; et al. Pyruvate kinase controls signal strength in the insulin secretory pathway. Cell Metab. 2020, 32, 736–750. [Google Scholar] [CrossRef]
- Collier, J.J.; Doan, T.T.; Daniels, M.C.; Schurr, J.R.; Kolls, J.K.; Scott, D.K. c-Myc is required for the glucose-mediated induction of metabolic enzyme genes. J. Biol. Chem. 2003, 278, 6588–6595. [Google Scholar] [CrossRef]
- Collier, J.J.; Zhang, P.; Pedersen, K.B.; Burke, S.J.; Haycock, J.W.; Scott, D.K. c-Myc and ChREBP regulate glucose-mediated expression of the L-type pyruvate kinase gene in INS-1-derived 832/13 cells. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E48–E56. [Google Scholar] [CrossRef]
- Lien, Y.C.; Won, K.J.; Simmons, R.A. Transcriptomic and quantitative proteomic profiling reveals signaling pathways critical for pancreatic islet maturation. Endocrinology 2020, 161, bqaa187. [Google Scholar] [CrossRef]
- Kone, M.; Pullen, T.J.; Sun, G.; Ibberson, M.; Martinez-Sanchez, A.; Sayers, S.; Nguyen-Tu, M.S.; Kantor, C.; Swisa, A.; Dor, Y.; et al. LKB1 and AMPK differentially regulate pancreatic β-cell identity. FASEB J. 2014, 28, 4972–4985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing glucose as well as cellular energy status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Piao, H.L.; Hardie, D.G.; Lin, S.C. Aldolase is a sensor for both low and high glucose, linking to AMPK and mTORC1. Cell Res. 2021, 31, 478–481. [Google Scholar]
- Li, M.; Zhang, C.S.; Zong, Y.; Feng, J.W.; Ma, T.; Hu, M.; Lin, Z.; Li, X.; Xie, C.; Wu, Y.; et al. Transient receptor potential V channels are essential for glucose sensing by aldolase and AMPK. Cell Metab. 2019, 30, 508–524.e12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Guo, H.; Zhang, C.S.; Lin, S.Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Zong, Y.; Lin, S.C. Determining AMPK activation via the lysosomal v-ATPase-Ragulator-AXIN/LKB1 axis. Methods Mol. Biol. 2018, 1732, 393–411. [Google Scholar]
- Beall, C.; Watterson, K.R.; McCrimmon, R.J.; Ashford, M.L. AMPK modulates glucose-sensing in insulin-secreting cells by altered phosphotransfer to KATP channels. J. Bioenerg. Biomembr. 2013, 45, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Beall, C.; Piipari, K.; Al-Qassab, H.; Smith, M.A.; Parker, N.; Carling, D.; Viollet, B.; Withers, D.J.; Ashford, M.L. Loss of AMP-activated protein kinase alpha2 subunit in mouse beta-cells impairs glucose-stimulated insulin secretion and inhibits their sensitivity to hypoglycaemia. Biochem. J. 2010, 429, 323–333. [Google Scholar] [CrossRef]
- Chang, T.J.; Chen, W.P.; Yang, C.; Lu, P.H.; Liang, Y.C.; Su, M.J.; Lee, S.C.; Chuang, L.M. Serine-385 phosphorylationm of inwardly rectifying K+ channel subunit (Kir6.2) by AMP-dependent protein kinase plays a key role in rosiglitazone-induced closure of the K(ATP) channel and insulin secretion in rats. Diabetologia 2009, 52, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, J.L.; Hellberg, K.; Luo, E.C.; Van Nostrand, E.L.; Dayn, A.; Yu, J.; Shokhirev, M.N.; Dayn, Y.; Yeo, G.W.; Shaw, R.J. AMPK regulation of Raptor and TSC2 mediate metformin effects on transcriptional control of anabolism and inflammation. Genes Dev. 2020, 34, 1330–1344. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Dunlop, E.A. The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 2017, 474, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, R.; Tran, S.; Shah, A.N.; Sun, G.; Valdearcos, M.; Marchetti, P.; Masini, M.; Swisa, A.; Giacometti, S.; Bernal-Mizrachi, E.; et al. mTORC1 to AMPK switching underlies β-cell metabolic plasticity during maturation and diabetes. J. Clin. Investig. 2019, 129, 4124–4137. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman Release 8.0. Human PRKAA1. Available online: http://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000397128.2&taxid=9606&members=.&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- TargetScanHuman Release 8.0. Human PRKAG2. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000287878.4&taxid=9606&showcnc=0.&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zou, M.H. AMPK, mitochondrial function, and cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef]
- Reif, S.; Elbaum-Shiff, Y.; Koroukhov, N.; Shilo, I.; Musseri, M.; Golan-Gerstl, R. Cow and human milk-derived exosomes ameliorate colitis in DSS murine model. Nutrients 2020, 12, 2589. [Google Scholar] [CrossRef]
- Yun, B.; Kim, Y.; Park, D.J.; Oh, S. Comparative analysis of dietary exosome-derived microRNAs from human, bovine and caprine colostrum and mature milk. J. Anim. Sci. Technol. 2021, 63, 593–602. [Google Scholar] [CrossRef]
- El Ouaamari, A.; Baroukh, N.; Martens, G.A.; Lebrun, P.; Pipeleers, D.; van Obberghen, E. miR-375 targets 3′-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes 2008, 57, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, O.; Fabris, G.; Pisani, D.F.; Casamento, V.; Gautier, N.; Hinault, C.; Lebrun, P.; Duranton, C.; Tauc, M.; Dalle, S.; et al. microRNA-375 regulates glucose metabolism-related signaling for insulin secretion. J. Endocrinol. 2020, 244, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman Release 8.0. Human PPARGC1A ENST00000264867. Available online: http://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000264867.2&taxid=9606&members=&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Tan, L.; Chen, Z.; Teng, M.; Chen, B.; Xu, H. MicroRNA profiling reveals an abundant ssc-miRNA-148a-3p that promotes proliferation and differentiation during intramuscular adipogenesis in Chinese Guizhou Congjiang pigs breeds by targeting PPARGC1A. Res. Sq. 2022. Available online: https://assets.researchsquare.com/files/rs-818015/v1/8127f26d-0c88-4ec9-8da4-7ed1ec056a19.pdf?c=1631888228 (accessed on 2 June 2022).
- Kaufman, B.A.; Li, C.; Soleimanpour, S.A. Mitochondrial regulation of β-cell function: Maintaining the momentum for insulin release. Mol. Aspects Med. 2015, 42, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Del Guerra, S.; Lupi, R.; Rönn, T.; Granhall, C.; Luthman, H.; Masiello, P.; Marchetti, P.; Groop, L.; Del Prato, S. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008, 51, 615–622. [Google Scholar] [CrossRef]
- Wagschal, A.; Najafi-Shoushtari, S.H.; Wang, L.; Goedeke, L.; Sinha, S.; de Lemos, A.S.; Black, J.C.; Ramírez, C.M.; Li, Y.; Tewhey, R.; et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 2015, 21, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Aspects Med. 2004, 25, 495–520. [Google Scholar] [CrossRef]
- Xie, T.; So, W.Y.; Li, X.Y.; Leung, P.S. Fibroblast growth factor 21 protects against lipotoxicity-induced pancreatic β-cell dysfunction via regulation of AMPK signaling and lipid metabolism. Clin. Sci. 2019, 133, 2029–2044. [Google Scholar] [CrossRef]
- Yoshihara, E.; Wei, Z.; Lin, C.S.; Fang, S.; Ahmadian, M.; Kida, Y.; Tseng, T.; Dai, Y.; Yu, R.T.; Liddle, C.; et al. ERRγ is required for the metabolic maturation of therapeutically functional glucose-responsive β cells. Cell Metab. 2016, 23, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, J.; Kulkarni, R.N. ERRγ—A new player in β cell maturation. Cell Metab. 2016, 23, 765–767. [Google Scholar] [CrossRef]
- Eichner, L.J.; Giguère, V. Estrogen related receptors (ERRs): A new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion 2011, 11, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Nakatsuka, A. Mitochondrial dynamics and mitochondrial dysfunction in diabetes. Acta Med. Okayama 2016, 70, 151–158. [Google Scholar] [PubMed]
- Misra, J.; Kim, D.K.; Choi, H.S. ERRγ: A junior orphan with a senior role in metabolism. Trends Endocrinol. Metab. 2017, 28, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, K.; Sadana, P.; Chowdhury, F.; Gaillard, S.; Wang, F.; McDonnell, D.P.; Unterman, T.G.; Elam, M.B.; Park, E.A. Estrogen-related receptors stimulate pyruvate dehydrogenase kinase isoform 4 gene expression. J. Biol. Chem. 2006, 281, 39897–39906. [Google Scholar] [CrossRef]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef]
- Hu, S.; Kuwabara, R.; de Haan, B.J.; Smink, A.M.; de Vos, P. Acetate and butyrate improve β-cell metabolism and mitochondrial respiration under oxidative stress. Int. J. Mol. Sci. 2020, 21, 1542. [Google Scholar] [CrossRef]
- Devarakonda, S.; Gupta, K.; Chalmers, M.J.; Hunt, J.F.; Griffin, P.R.; Van Duyne, G.D.; Spiegelman, B.M. Disorder-to-order transition underlies the structural basis for the assembly of a transcriptionally active PGC-1α/ERRγ complex. Proc. Natl. Acad. Sci. USA 2011, 108, 18678–18683. [Google Scholar] [CrossRef]
- Takacs, M.; Petoukhov, M.V.; Atkinson, R.A.; Roblin, P.; Ogi, F.X.; Demeler, B.; Potier, N.; Chebaro, Y.; Dejaegere, A.; Svergun, D.I.; et al. The asymmetric binding of PGC-1α to the ERRα and ERRγ nuclear receptor homodimers involves a similar recognition mechanism. PLoS ONE 2013, 8, e67810. [Google Scholar] [CrossRef]
- TargetSacnHuman 8.0. Human ESRRG ENST00000361525.3. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000361525.3&taxid=9606&showcnc=0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Liu, N.; Cai, X.; Liu, T.; Zou, J.; Wang, L.; Wang, G.; Liu, Y.; Ding, X.; Zhang, B.; Sun, P.; et al. Hypoxia-inducible factor-1α mediates the expression of mature β cell-disallowed genes in hypoxia-induced β cell dedifferentiation. Biochem. Biophys. Res. Commun. 2020, 523, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, É.; Giguére, V. The multiple universes of estrogen-related receptor α and γ in metabolic control and related diseases. Acta Pharmacol. Sin. 2015, 36, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Eichner, L.J.; Perry, M.C.; Dufour, C.R.; Bertos, N.; Park, M.; St-Pierre, J.; Giguère, V. miR-378(∗) mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab. 2010, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; JunHui, L.; PeiFeng, L. Candidate genes mediated by estrogen-related receptor γ in pancreatic β cells. J. Biochem. Mol. Toxicol. 2019, 33, e22390. [Google Scholar] [CrossRef]
- Jung, Y.S.; Kim, Y.H.; Radhakrishnan, K.; Kim, J.; Lee, I.K.; Cho, S.J.; Kim, D.K.; Dooley, S.; Lee, C.H.; Choi, H.S. Orphan nuclear receptor ERRγ regulates hepatic TGF-β2 expression and fibrogenic response in CCl4-induced acute liver injury. Arch. Toxicol. 2021, 95, 3071–3084. [Google Scholar] [CrossRef]
- Kim, D.K.; Ryu, D.; Koh, M.; Lee, M.W.; Lim, D.; Kim, M.J.; Kim, Y.H.; Cho, W.J.; Lee, C.H.; Park, S.B.; et al. Orphan nuclear receptor estrogen-related receptor γ (ERRγ) is key regulator of hepatic gluconeogenesis. J. Biol. Chem. 2012, 287, 21628–21639. [Google Scholar] [CrossRef]
- Kim, D.K.; Gang, G.T.; Ryu, D.; Koh, M.; Kim, Y.N.; Kim, S.S.; Park, J.; Kim, Y.H.; Sim, T.; Lee, I.K.; et al. Inverse agonist of nuclear receptor ERRγ mediates antidiabetic effect through inhibition of hepatic gluconeogenesis. Diabetes 2013, 62, 3093–3102. [Google Scholar] [CrossRef]
- Yu, S.; Wang, X.; Ng, C.F.; Chen, S.; Chan, F.L. ERRgamma suppresses cell proliferation and tumor growth of androgen-sensitive and androgen-insensitive prostate cancer cells and its implication as a therapeutic target for prostate cancer. Cancer Res. 2007, 67, 4904–4914. [Google Scholar] [CrossRef]
- Dhawan, S.; Tschen, S.I.; Zeng, C.; Guo, T.; Hebrok, M.; Matveyenko, A.; Bhushan, A. DNA methylation directs functional maturation of pancreatic β cells. J. Clin. Investig. 2015, 125, 2851–2860. [Google Scholar] [CrossRef]
- Parveen, N.; Dhawan, S. DNA methylation patterning and the regulation of beta cell homeostasis. Front. Endocrinol. 2021, 12, 651258. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L. Nuclear receptor SHP inhibition of Dnmt1 expression via ERRγ. FEBS Lett. 2011, 585, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, C.; Kaname, T.; Shimizu-Okabe, C.; Takayama, C.; Tsutsui, M.; Matsushita, M.; Abe, K.; Masuzaki, H. Impact of brown rice-specific γ-oryzanol on epigenetic modulation of dopamine D2 receptors in brain striatum in high-fat-diet-induced obesity in mice. Diabetologia 2017, 60, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kubiura-Ichimaru, M.; Murakami, Y.; Bogutz, A.B.; Lefebvre, L.; Suetake, I.; Tajima, S.; Tada, M. DNMT1 regulates the timing of DNA methylation by DNMT3 in an enzymatic activity-dependent manner in mouse embryonic stem cells. PLoS ONE 2022, 17, e0262277. [Google Scholar] [CrossRef] [PubMed]
- Baier, S.R.; Nguyen, C.; Xie, F.; Wood, J.R.; Zempleni, J. MicroRNAs are absorbed in biologically meaningful amounts from nutritionally relevant doses of cow milk and affect gene expression in peripheral blood mononuclear cells, HEK-293 kidney cell cultures, and mouse livers. J. Nutr. 2014, 144, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Howard, K.M.; Jati Kusuma, R.; Baier, S.R.; Friemel, T.; Markham, L.; Vanamala, J.; Zempleni, J. Loss of miRNAs during processing and storage of cow’s (Bos taurus) milk. J. Agric. Food Chem. 2015, 63, 588–592. [Google Scholar] [CrossRef]
- Feng, X.; Chen, X.; Zheng, X.; Zhu, H.; Qi, Q.; Liu, S.; Zhang, H.; Che, J. Latest trend of milk derived exosomes: Cargos, functions, and applications. Front. Nutr. 2021, 8, 747294. [Google Scholar] [CrossRef]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef]
- Zhang, Z.; Cao, Y.; Zhai, Y.; Ma, X.; An, X.; Zhang, S.; Li, Z. MicroRNA-29b regulates DNA methylation by targeting Dnmt3a/3b and Tet1/2/3 in porcine early embryo development. Dev. Growth Differ. 2018, 60, 197–204. [Google Scholar] [CrossRef]
- TargetScanHuman 8.0. Human DNMT3A ENST00000380746.4. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000380746.4&taxid=9606&members=.&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Wang, L.; Zuercher, W.J.; Consler, T.G.; Lambert, M.H.; Miller, A.B.; Orband-Miller, L.A.; McKee, D.D.; Willson, T.M.; Nolte, R.T. X-ray crystal structures of the estrogen-related receptor-gamma ligand binding domain in three functional states reveal the molecular basis of small molecule regulation. J. Biol. Chem. 2006, 281, 37773–37781. [Google Scholar] [CrossRef]
- Hu, F.; Wang, M.; Xiao, T.; Yin, B.; He, L.; Meng, W.; Dong, M.; Liu, F. miR-30 promotes thermogenesis and the development of beige fat by targeting RIP140. Diabetes 2015, 64, 2056–2068. [Google Scholar] [CrossRef] [Green Version]
- Porat, S.; Weinberg-Corem, N.; Tornovsky-Babaey, S.; Schyr-Ben-Haroush, R.; Hija, A.; Stolovich-Rain, M.; Dadon, D.; Granot, Z.; Ben-Hur, V.; White, P.; et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 2011, 13, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman Release 8.8. Human TSC2 ENST00000568454.1. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000568454.1&taxid=9606&showcnc=.0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Madan, E.; Gogna, R.; Bhatt, M.; Pati, U.; Kuppusamy, P.; Mahdi, A.A. Regulation of glucose metabolism by p53: Emerging new roles for the tumor suppressor. Oncotarget 2011, 2, 948–957. [Google Scholar] [CrossRef]
- Van de Velde, S.; Hogan, M.F.; Montminy, M. mTOR links incretin signaling to HIF induction in pancreatic beta cells. Proc. Natl. Acad. Sci U S A 2011, 108, 16876–16882. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Bensellam, M.; Duvillié, B.; Rybachuk, G.; Laybutt, D.R.; Magnan, C.; Guiot, Y.; Pouysségur, J.; Jonas, J.C. Glucose-induced O2 consumption activates hypoxia inducible factors 1 and 2 in rat insulin-secreting pancreatic beta-cells. PLoS ONE 2012, 7, e29807. [Google Scholar]
- Redel, B.K.; Brown, A.N.; Spate, L.D.; Whitworth, K.M.; Green, J.A.; Prather, R.S. Glycolysis in preimplantation development is partially controlled by the Warburg effect. Mol. Reprod. Dev. 2012, 79, 262–271. [Google Scholar] [CrossRef]
- Krisher, R.L.; Prather, R.S. A role for the Warburg effect in preimplantation embryo development: Metabolic modification to support rapid cell proliferation. Mol. Reprod. Dev. 2012, 79, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.G.; Sturmey, R.G. Parallels between embryo and cancer cell metabolism. Biochem. Soc. Trans. 2013, 41, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, H.; Aulehla, A. Revisiting the role of metabolism during development. Development 2018, 145, dev131110. [Google Scholar] [CrossRef] [PubMed]
- Manzo, G. Similarities between embryo development and cancer process suggest new strategies for research and therapy of tumors: A new point of view. Front. Cell Dev. Biol. 2019, 7, 20. [Google Scholar] [CrossRef]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef]
- TargetScanHuman Release 8.0. Human HIF1AN ENST00000299163.6. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000299163.6&taxid=9606&showcnc=.0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of p53 in glucose metabolism. Curr. Opin. Cell Biol. 2010, 22, 186–191. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Yaney, G.C.; Schultz, V.; Cunningham, B.A.; Dunaway, G.A.; Corkey, B.E.; Tornheim, K. Phosphofructokinase isozymes in pancreatic islets and clonal beta-cells (INS-1). Diabetes 1995, 44, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Roche, E.; Assimacopoulos-Jeannet, F.; Witters, L.A.; Perruchoud, B.; Yaney, G.; Corkey, B.; Asfari, M.; Prentki, M. Induction by glucose of genes coding for glycolytic enzymes in a pancreatic beta-cell line (INS-1). J. Biol. Chem. 1997, 272, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Malaisse, W.J.; Malaisse-Lagae, F.; Sener, A. The glycolytic cascade in pancreatic islets. Diabetologia 1982, 23, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Parker, J.C.; Najafi, H.; Matschinsky, F.M. Control of glucose metabolism in pancreatic beta-cells by glucokinase, hexokinase, and phosphofructokinase. Model study with cell lines derived from beta-cells. Diabetes 1988, 37, 1524–1530. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Hatziapostolou, M.; Polytarchou, C.; Iliopoulos, D. miRNAs link metabolic reprogramming to oncogenesis. Trends Endocrinol. Metab. 2013, 24, 361–373. [Google Scholar] [CrossRef]
- Artner, I.; Hang, Y.; Mazur, M.; Yamamoto, T.; Guo, M.; Lindner, J.; Magnuson, M.A.; Stein, R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes 2010, 59, 2530–2539. [Google Scholar] [CrossRef]
- Nishimura, W.; Kondo, T.; Salameh, T.; El Khattabi, I.; Dodge, R.; Bonner-Weir, S.; Sharma, A. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev. Biol. 2006, 293, 526–539. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Artner, I.; Henderson, E.; Means, A.; Sander, M.; Stein, R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc. Natl. Acad. Sci. USA 2004, 101, 2930–2933. [Google Scholar] [CrossRef]
- Hang, Y.; Yamamoto, T.; Benninger, R.K.; Brissova, M.; Guo, M.; Bush, W.; Piston, D.W.; Powers, A.C.; Magnuson, M.; Thurmond, D.C.; et al. The MafA transcription factor becomes essential to islet β-cells soon after birth. Diabetes 2014, 63, 1994–2005. [Google Scholar] [CrossRef]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Chen, C.; He, H.B.; Hu, R.; Zhou, H.D.; Xie, H.; Zhu, W.; Dai, R.C.; Wu, X.P.; Liao, E.Y.; et al. miR-148a regulates osteoclastogenesis by targeting V-maf musculoaponeurotic fibrosarcoma oncogene homolog B. J. Bone Miner. Res. 2013, 28, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wei, Y.; Geißler, C.; Abschlag, K.; Corbalán Campos, J.; Hristov, M.; Möllmann, J.; Lehrke, M.; Karshovska, E.; Schober, A. Hyperlipidemia-induced microRNA-155-5p improves β-cell function by targeting Mafb. Diabetes 2017, 66, 3072–3084. [Google Scholar] [CrossRef] [PubMed]
- Conrad, E.; Dai, C.; Spaeth, J.; Guo, M.; Cyphert, H.A.; Scoville, D.; Carroll, J.; Yu, W.M.; Goodrich, L.V.; Harlan, D.M.; et al. The MAFB transcription factor impacts islet α-cell function in rodents and represents a unique signature of primate islet β-cells. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E91–E102. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Carnese, P.P.; Hennings, T.G.; Walker, E.M.; Russ, H.A.; Liu, J.S.; Giacometti, S.; Stein, R.; Hebrok, M. Loss of the transcription factor MAFB limits β-cell derivation from human PSCs. Nat. Commun. 2020, 11, 2742. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman Release 8.0. Human SLC2A1 ENST00000426263.3. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000426263.3&taxid=9606&members=.&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Brunham, L.R.; Kruit, J.K.; Pape, T.D.; Timmins, J.M.; Reuwer, A.Q.; Vasanji, Z.; Marsh, B.J.; Rodrigues, B.; Johnson, J.D.; Parks, J.S.; et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat. Med. 2007, 13, 340–347. [Google Scholar] [CrossRef]
- Kruit, J.K.; Wijesekara, N.; Westwell-Roper, C.; Vanmierlo, T.; de Haan, W.; Bhattacharjee, A.; Tang, R.; Wellington, C.L.; LütJohann, D.; Johnson, J.D.; et al. Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired β-cell function. Diabetes 2012, 61, 659–664. [Google Scholar] [CrossRef]
- Bodzioch, M.; Orsó, E.; Klucken, J.; Langmann, T.; Böttcher, A.; Diederich, W.; Drobnik, W.; Barlage, S.; Büchler, C.; Porsch-Ozcürümez, M.; et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999, 22, 347–351. [Google Scholar] [CrossRef]
- Koseki, M.; Matsuyama, A.; Nakatani, K.; Inagaki, M.; Nakaoka, H.; Kawase, R.; Yuasa-Kawase, M.; Tsubakio-Yamamoto, K.; Masuda, D.; Sandoval, J.C.; et al. Impaired insulin secretion in four Tangier disease patients with ABCA1 mutations. J. Atheroscler. Thromb. 2009, 16, 292–296. [Google Scholar] [CrossRef]
- Vergeer, M.; Brunham, L.R.; Koetsveld, J.; Kruit, J.K.; Verchere, C.B.; Kastelein, J.J.; Hayden, M.R.; Stroes, E.S. Carriers of loss-of-function mutations in ABCA1 display pancreatic beta-cell dysfunction. Diabetes Care 2010, 33, 869–874. [Google Scholar] [CrossRef]
- Kruit, J.K.; Wijesekara, N.; Fox, J.E.; Dai, X.Q.; Brunham, L.R.; Searle, G.J.; Morgan, G.P.; Costin, A.J.; Tang, R.; Bhattacharjee, A.; et al. Islet cholesterolaccumulation due to loss of ABCA1 leads to impaired exocytosis of insulin granules. Diabetes 2011, 60, 3186–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedeke, L.; Rotllan, N.; Canfrán-Duque, A.; Aranda, J.F.; Ramírez, C.M.; Araldi, E.; Lin, C.S.; Anderson, N.N.; Wagschal, A.; de Cabo, R.; et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat. Med. 2015, 21, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, S. Identification and comparison of exosomal microRNAs in the milk and colostrum of two different cow breeds. Gene 2020, 743, 144609. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, R.; Dai, S.; Zhang, X.; Li, X.; Bai, C. Role of TGF-β/Smad pathway in the transcription of pancreas-specific genes during beta cell differentiation. Front. Cell Dev. Biol. 2019, 7, 351, Erratum in: Front. Cell Dev. Biol. 2020, 8, 614840.. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Rane, S.G. TGF-β signaling in pancreatic islet β cell development and function. Endocrinology 2021, 162, bqaa233. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Wang, L.; Zhao, C.Y.; Lan, H.Y. Role of TGF-beta signaling in beta cell proliferation and function in diabetes. Biomolecules 2022, 12, 373. [Google Scholar] [CrossRef]
- Xiao, X.; Fischbach, S.; Song, Z.; Gaffar, I.; Zimmerman, R.; Wiersch, J.; Prasadan, K.; Shiota, C.; Guo, P.; Ramachandran, S.; et al. Transient suppression of TGFβ receptor signaling facilitates human islet transplantation. Endocrinology 2016, 157, 1348–1356. [Google Scholar] [CrossRef]
- Saleh, M.; Mohamed, N.A.; Sehrawat, A.; Zhang, T.; Thomas, M.; Wang, Y.; Kalsi, R.; Molitoris, J.; Prasadan, K.; Gittes, G.K. β-cell Smad2 null mice have improved β-cell function and are protected from diet-induced hyperglycemia. J. Biol. Chem. 2021, 297, 101235. [Google Scholar] [CrossRef]
- Dhawan, S.; Dirice, E.; Kulkarni, R.N.; Bhushan, A. Inhibition of TGF-β signaling promotes human pancreatic β-cell replication. Diabetes 2016, 65, 1208–1218. [Google Scholar] [CrossRef]
- Sheng, J.; Wang, L.; Tang, P.M.; Wang, H.L.; Li, J.C.; Xu, B.H.; Xue, V.W.; Tan, R.Z.; Jin, N.; Chan, T.F.; et al. Smad3 deficiency promotes beta cell proliferation and function in db/db mice via restoring Pax6 expression. Theranostics 2021, 11, 2845–2859. [Google Scholar] [CrossRef]
- Lee, J.H.; Mellado-Gil, J.M.; Bahn, Y.J.; Pathy, S.M.; Zhang, Y.E.; Rane, S.G. Protection from β-cell apoptosis by inhibition of TGF-β/Smad3 signaling. Cell Death Dis. 2020, 11, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Mu, J.; Wang, X.; Ye, X.; Si, L.; Ning, S.; Li, Z.; Li, Y. The repressive effect of miR-148a on TGF beta-SMADs signal pathway is involved in the glabridin-induced inhibition of the cancer stem cells-like properties in hepatocellular carcinoma cells. PLoS ONE 2014, 9, e96698. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Luo, Z.; Pan, Y.; Zheng, W.; Li, W.; Zhang, Z.; Xiong, P.; Xu, D.; Du, M.; Wang, B.; et al. H19/miR-148a/USP4 axis facilitates liver fibrosis by enhancing TGF-β signaling in both hepatic stellate cells and hepatocytes. J. Cell Physiol. 2019, 234, 9698–9710. [Google Scholar] [CrossRef] [PubMed]
- Heo, M.J.; Kim, Y.M.; Koo, J.H.; Yang, Y.M.; An, J.; Lee, S.K.; Lee, S.J.; Kim, K.M.; Park, J.W.; Kim, S.G. microRNA-148a dysregulation discriminates poor prognosis of hepatocellular carcinoma in association with USP4 overexpression. Oncotarget 2014, 5, 2792–2806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xing, M.; Wang, X.; Cao, W.; Wang, H. MiR-148a suppresses invasion and induces apoptosis of breast cancer cells by regulating USP4 and BIM expression. Int. J. Clin. Exp. Pathol. 2017, 10, 8361–8368. [Google Scholar] [PubMed]
- Hu, B.; Zhang, D.; Zhao, K.; Wang, Y.; Pei, L.; Fu, Q.; Ma, X. Spotlight on USP4: Structure, function, and regulation. Front. Cell Dev. Biol. 2021, 9, 595159. [Google Scholar] [CrossRef]
- Zhang, X.; Berger, F.G.; Yang, J.; Lu, X. USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO J. 2011, 30, 2177–2189. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; Drabsch, Y.; Gao, R.; Snaar-Jagalska, B.E.; Mickanin, C.; Huang, H.; Sheppard, K.A.; Porter, J.A.; Lu, C.X.; et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat. Cell Biol. 2012, 14, 717–726. [Google Scholar] [CrossRef]
- Sjöholm, A.; Hellerström, C. TGF-beta stimulates insulin secretion and blocks mitogenic response of pancreatic beta-cells to glucose. Am. J. Physiol. 1991, 260, C1046–C1051. [Google Scholar] [CrossRef]
- Lin, H.M.; Lee, J.H.; Yadav, H.; Kamaraju, A.K.; Liu, E.; Zhigang, D.; Vieira, A.; Kim, S.J.; Collins, H.; Matschinsky, F.; et al. Transforming growth factor-beta/Smad3 signaling regulates insulin gene transcription and pancreatic islet beta-cell function. J. Biol. Chem. 2009, 284, 12246–12257. [Google Scholar] [CrossRef]
- Zhong, F.; Jiang, Y. Endogenous pancreatic β cell regeneration: A potential strategy for the recovery of β cell deficiency in diabetes. Front. Endocrinol. 2019, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinno, C.; Cota, P.; Bastidas-Ponce, A.; Tarquis-Medina, M.; Lickert, H.; Bakhti, M. β-Cell maturation and identity in health and sisease. Int. J. Mol. Sci. 2019, 20, 5417. [Google Scholar] [CrossRef] [PubMed]
- Bohuslavova, R.; Smolik, O.; Malfatti, J.; Berkova, Z.; Novakova, Z.; Saudek, F.; Pavlinkova, G. NEUROD1 is required for the early α and β endocrine differentiation in the pancreas. Int. J. Mol. Sci. 2021, 22, 6713. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Guo, M.; Huang, S.; Stein, R. Insulin gene transcription is mediated by interactions between the p300 coactivator and PDX-1, BETA2, and E47. Mol. Cell Biol. 2002, 22, 412–420. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman Release 8.0. Human NEUROD1 ENST00000295108.3. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000295108.3&taxid=9606&showcnc=.0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Wang, J.; Elghazi, L.; Parker, S.E.; Kizilocak, H.; Asano, M.; Sussel, L.; Sosa-Pineda, B. The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Dev. Biol. 2004, 266, 178–189. [Google Scholar] [CrossRef]
- Doyle, M.J.; Sussel, L. Nkx2.2 regulates beta-cell function in the mature islet. Diabetes 2007, 56, 1999–2007. [Google Scholar] [CrossRef]
- TargetScanHuman Release 8.8. Human NKX2-2 ENST00000377142.4. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000377142.4&taxid=.9606&showcnc=0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- He, X.; Gao, F.; Hou, J.; Li, T.; Tan, J.; Wang, C.; Liu, X.; Wang, M.; Liu, H.; Chen, Y.; et al. Metformin inhibits MAPK signaling and rescues pancreatic aquaporin 7 expression to induce insulin secretion in type 2 diabetes mellitus. J. Biol. Chem. 2021, 297, 101002. [Google Scholar] [CrossRef]
- Kimura, T.; Kaneto, H. Metformin induces insulin secretion by preserving pancreatic aquaporin 7 expression in type 2 diabetes mellitus. J. Diabetes Investig. 2022, 13, 227–229. [Google Scholar] [CrossRef]
- Ngu, A.; Wang, S.; Wang, H.; Khanam, A.; Zempleni, J. Milk exosomes in nutrition and drug delivery. Am. J. Physiol. Cell Physiol. 2022, 322, C865–C874. [Google Scholar] [CrossRef]
- Leiferman, A.; Shu, J.; Grove, R.; Cui, J.; Adamec, J.; Zempleni, J. A diet defined by its content of bovine milk exosomes and their RNA cargos has moderate effects on gene expression, amino acid profiles and grip strength in skeletal muscle in C57BL/6 mice. J. Nutr. Biochem. 2018, 59, 123–128. [Google Scholar] [CrossRef]
- Wu, D.; Kittana, H.; Shu, J.; Kachman, S.D.; Cui, J.; Ramer-Tait, A.E.; Zempleni, J. Dietary depletion of milk exosomes and their microRNA cargos elicits a depletion of miR-200a-3p and elevated intestinal inflammation and chemokine (C-X-C motif) ligand 9 expression in Mdr1a-/- mice. Curr. Dev. Nutr. 2019, 3, nzz122. [Google Scholar] [CrossRef] [PubMed]
- Neu, J.; Hauser, N.; Douglas-Escobar, M. Postnatal nutrition and adult health programming. Semin. Fetal Neonatal Med. 2007, 12, 78–86. [Google Scholar] [CrossRef]
- Burdge, G.C.; Hanson, M.A.; Slater-Jefferies, J.L.; Lillycrop, K.A. Epigenetic regulation of transcription: A mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? Br. J. Nutr. 2007, 97, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.E.; Sebert, S.P.; Hyatt, M.A.; Budge, H. Nutritional programming of the metabolic syndrome. Nat. Rev. Endocrinol. 2009, 5, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Lillycrop, K.A. Nutrition, epigenetics, and developmental plasticity: Implications for understanding human disease. Annu. Rev. Nutr. 2010, 30, 315–339. [Google Scholar] [CrossRef]
- Attig, L.; Gabory, A.; Junien, C. Early nutrition and epigenetic programming: Chasing shadows. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 284–293. [Google Scholar] [CrossRef]
- Gabory, A.; Attig, L.; Junien, C. Epigenetic mechanisms involved in developmental nutritional programming. World J. Diabetes 2011, 2, 164–175. [Google Scholar] [CrossRef]
- Wiedmeier, J.E.; Joss-Moore, L.A.; Lane, R.H.; Neu, J. Early postnatal nutrition and programming of the preterm neonate. Nutr. Rev. 2011, 69, 76–82. [Google Scholar] [CrossRef]
- Vickers, M.H. Early life nutrition, epigenetics and programming of later life disease. Nutrients 2014, 6, 2165–2178. [Google Scholar] [CrossRef]
- Gabory, A.; Attig, L.; Junien, C. Developmental programming and epigenetics. Am. J. Clin. Nutr. 2011, 94, 1943S–1952S. [Google Scholar]
- Martínez, J.A.; Cordero, P.; Campión, J.; Milagro, F.I. Interplay of early-life nutritional programming on obesity, inflammation and epigenetic outcomes. Proc. Nutr. Soc. 2012, 71, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xiao, X.; Zhang, Q.; Yu, M. DNA methylation: The pivotal interaction between early-life nutrition and glucose metabolism in later life. Br. J. Nutr. 2014, 112, 1850–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, J.; Gustafson, K.M.; Carlson, S.E. Critical and sensitive periods in development and nutrition. Ann. Nutr. Metab. 2019, 75, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, N.; Alashkar Alhamwe, B.; Caraballo, L.; Ding, M.; Ferrante, A.; Garn, H.; Garssen, J.; Hii, C.S.; Irvine, J.; Llinás-Caballero, K.; et al. Perinatal and early-life nutrition, epigenetics, and allergy. Nutrients 2021, 13, 724. [Google Scholar] [CrossRef] [PubMed]
- Linnér, A.; Almgren, M. Epigenetic programming-the important first 1000 days. Acta Paediatr. 2020, 109, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Shiff, Y.E.; Reif, S.; Marom, R.; Shiff, K.; Reifen, R.; Golan-Gerstl, R. MiRNA-320a is less expressed and miRNA-148a more expressed in preterm human milk. J. Funct. Foods 2019, 57, 68–74. [Google Scholar] [CrossRef]
- Melnik, B.C.; Weiskirchen, R.; Schmitz, G. Milk exosomal microRNAs: Friends or foe?—A narrative review. ExRNA 2022. [Google Scholar] [CrossRef]
- Zhao, H.; Wen, G.; Huang, Y.; Yu, X.; Chen, Q.; Afzal, T.A.; Luong le, A.; Zhu, J.; Ye, S.; Zhang, L.; et al. MicroRNA-22 regulates smooth muscle cell differentiation from stem cells by targeting methyl CpG-binding protein 2. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 918–929, Erratum in: Arterioscler. Thromb. Vasc. Biol. 2015, 35, e24. Erratum in: Arterioscler. Thromb. Vasc. Biol. 2015, 35, e59. [Google Scholar] [CrossRef]
- Thibonnier, M.; Esau, C. Metabolic benefits of microRNA-22 inhibition. Nucleic Acid Ther. 2020, 30, 104–116. [Google Scholar] [CrossRef]
- Thibonnier, M.; Esau, C.; Ghosh, S.; Wargent, E.; Stocker, C. Metabolic and energetic benefits of microRNA-22 inhibition. BMJ Open Diabetes Res. Care 2020, 8, e001478. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, H.X.; Jena, P.K.; Sheng, L.; Ali, M.R.; Wan, Y.Y. miR-22 inhibition reduces hepatic steatosis via FGF21 and FGFR1 induction. JHEP Rep. 2020, 2, 100093. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Chen, X.; Huang, Z.; Chen, D.; Chen, H.; Luo, Y.; He, J.; Zheng, P.; Yu, J.; Yu, B. Resveratrol regulates muscle fiber type conversion via miR-22-3p and AMPK/SIRT1/PGC-1α pathway. J. Nutr. Biochem. 2020, 77, 108297. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Chen, X.; Huang, Z.; Chen, D.; Zheng, P.; He, J.; Chen, H.; Yu, J.; Luo, Y.; Yu, B. miR-22-3p regulates muscle fiber-type conversion through inhibiting AMPK/SIRT1/PGC-1α pathway. Anim. Biotechnol. 2021, 32, 254–261. [Google Scholar] [CrossRef]
- Lou, P.; Bi, X.; Tian, Y.; Li, G.; Kang, Q.; Lv, C.; Song, Y.; Xu, J.; Sheng, X.; Yang, X.; et al. MiR-22 modulates brown adipocyte thermogenesis by synergistically activating the glycolytic and mTORC1 signaling pathways. Theranostics 2021, 11, 3607–3623. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Wang, W.; Li, X.; Dong, T.; Zhang, Q.; Zhu, B.; Zhao, H.; Wu, S. MicroRNA-21-5p induces the metastatic phenotype of human cervical carcinoma cells in vitro by targeting the von Hippel-Lindau tumor suppressor. Oncol. Lett. 2018, 15, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; McDaniel, K.; Zhou, T.; Ramos-Lorenzo, S.; Wu, C.; Huang, L.; Chen, D.; Annable, T.; Francis, H.; Glaser, S.; et al. Knockout of microRNA-21 attenuates alcoholic hepatitis through the VHL/NF-κB signaling pathway in hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G385–G398. [Google Scholar] [CrossRef]
- Xu, H.; Ling, M.; Xue, J.; Dai, X.; Sun, Q.; Chen, C.; Liu, Y.; Zhou, L.; Liu, J.; Luo, F.; et al. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics 2018, 8, 5419–5433. [Google Scholar] [CrossRef]
- Sun, J.; Jiang, Z.; Li, Y.; Wang, K.; Chen, X.; Liu, G. Downregulation of miR-21 inhibits the malignant phenotype of pancreatic cancer cells by targeting VHL. Onco. Targets Ther. 2019, 12, 7215–7226. [Google Scholar] [CrossRef]
- Gebrie, A. Disease progression role as well as the diagnostic and prognostic value of microRNA-21 in patients with cervical cancer: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0268480. [Google Scholar] [CrossRef]
- Cantley, J.; Grey, S.T.; Maxwell, P.H.; Withers, D.J. The hypoxia response pathway and β-cell function. Diabetes Obes. Metab. 2010, 12, 159–167. [Google Scholar] [CrossRef]
- TargetScanHuman Release 8.0. Human TP53 ENST00000420246.2. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000420246.2&taxid=9606&members=.&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Floris, I.; Billard, H.; Boquien, C.Y.; Joram-Gauvard, E.; Simon, L.; Legrand, A.; Boscher, C.; Rozé, J.C.; Bolaños-Jiménez, F.; Kaeffer, B. MiRNA analysis by quantitative PCR in preterm human breast milk reveals daily fluctuations of hsa-miR-16-5p. PLoS ONE 2015, 10, e0140488. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhao, S. miRNA-16-5p inhibits the apoptosis of high glucose-induced pancreatic β cells via targeting of CXCL10: Potential biomarkers in type 1 diabetes mellitus. Endokrynol. Pol. 2020, 71, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Juchnicka, I.; Kuźmicki, M.; Niemira, M.; Bielska, A.; Sidorkiewicz, I.; Zbucka-Krętowska, M.; Krętowski, A.J.; Szamatowicz, J. miRNAs as predictive factors in early diagnosis of gestational diabetes mellitus. Front. Endocrinol. 2022, 13, 839344. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Ma, M.; Chen, B.; Li, Z.; Abdalla, B.A.; Nie, Q.; Zhang, X. MiR-16-5p targets SESN1 to regulate the p53 signaling pathway, affecting myoblast proliferation and apoptosis, and is involved in myoblast differentiation. Cell Death Dis. 2018, 9, 367. [Google Scholar] [CrossRef]
- TargetScanHuman Release 8.0. Human SESN2 ENST00000253063.3. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000253063.3&taxid=9606&showcnc=0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef]
- Lee, J.H.; Cho, U.S.; Karin, M. Sestrin regulation of TORC1: Is sestrin a leucine sensor? Sci. Signal. 2016, 9, re5. [Google Scholar] [CrossRef]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.L.; Karin, M.; Budanov, A.V. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef]
- Kim, J.S.; Ro, S.H.; Kim, M.; Park, H.W.; Semple, I.A.; Park, H.; Cho, U.S.; Wang, W.; Guan, K.L.; Karin, M.; et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci. Rep. 2015, 5, 9502. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Li, M.O. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 2014, 159, 122–133. [Google Scholar] [CrossRef]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef]
- Lutt, N.; Brunkard, J.O. Amino acid signaling for TOR in eukaryotes: Sensors, transducers, and a sustainable agricultural fuTORe. Biomolecules 2022, 12, 387. [Google Scholar] [CrossRef] [PubMed]
- Grasso, S.; Messina, A.; Saporito, N.; Reitano, G. Serum-insulin response to glucose and aminoacids in the premature infant. Lancet 1968, 2, 755–756. [Google Scholar] [CrossRef]
- Gidrewicz, D.A.; Fenton, T.R. A systematic review and meta-analysis of the nutrient content of preterm and term breast milk. BMC Pediatr. 2014, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Sahin, S.; Ozdemir, T.; Katipoglu, N.; Akcan, A.B.; Kaynak Turkmen, M. Comparison of changes in breast milk macronutrient content during the first month in preterm and term Infants. Breastfeed. Med. 2020, 15, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Entringer, S.; Buss, C.; Wadhwa, P.D. Prenatal stress and developmental programming of human health and disease risk: Concepts and integration of empirical findings. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Bozack, A.K.; Colicino, E.; Rodosthenous, R.; Bloomquist, T.R.; Baccarelli, A.A.; Wright, R.O.; Wright, R.J.; Lee, A.G. Associations between maternal lifetime stressors and negative events in pregnancy and breast milk-derived extracellular vesicle microRNAs in the programming of intergenerational stress mechanisms (PRISM) pregnancy cohort. Epigenetics 2021, 16, 389–404. [Google Scholar] [CrossRef]
- Sun, J.F.; Zhang, D.; Gao, C.J.; Zhang, Y.W.; Dai, Q.S. Exosome-mediated miR-155 transfer contributes to hepatocellular carcinoma cell proliferation by targeting PTEN. Med. Sci. Monit. Basic Res. 2019, 25, 218–228. [Google Scholar] [CrossRef]
- Siu, M.K.; Tsai, Y.C.; Chang, Y.S.; Yin, J.J.; Suau, F.; Chen, W.Y.; Liu, Y.N. Transforming growth factor-β promotes prostate bone metastasis through induction of microRNA-96 and activation of the mTOR pathway. Oncogene 2015, 34, 4767–4776. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Wang, L.; Harris, T.E.; Lawrence, J.C., Jr. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem. 2008, 283, 15619–15627. [Google Scholar] [CrossRef]
- Wiza, C.; Nascimento, E.B.; Ouwens, D.M. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1453–E1460. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Q.; Wen, Q.; Zheng, Y.; Lazarovici, P.; Jiang, H.; Lin, J.; Zheng, W. Proline-rich Akt substrate of 40kDa (PRAS40): A novel downstream target of PI3K/Akt signaling pathway. Cell Signal. 2012, 24, 17–24, Erratum in: Cell Signal. 2012, 24, 2226. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z. Targeting PRAS40 for multiple diseases. Drug Discov. Today 2016, 21, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Isganaitis, E.; Venditti, S.; Matthews, T.J.; Lerin, C.; Demerath, E.W.; Fields, D.A. Maternal obesity and the human milk metabolome: Associations with infant body composition and postnatal weight gain. Am. J. Clin. Nutr. 2019, 110, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.; Eriksson, J.G.; Broekman, B.F. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef]
- Rughani, A.; Friedman, J.E.; Tryggestad, J.B. Type 2 diabetes in youth: The role of early life exposures. Curr. Diab. Rep. 2020, 20, 45. [Google Scholar] [CrossRef]
- Lahti-Pulkkinen, M.; Bhattacharya, S.; Wild, S.H.; Lindsay, R.S.; Räikkönen, K.; Norman, J.E.; Bhattacharya, S.; Reynolds, R.M. Consequences of being overweight or obese during pregnancy on diabetes in the offspring: A record linkage study in Aberdeen, Scotland. Diabetologia 2019, 62, 1412–1419. [Google Scholar] [CrossRef]
- Perng, W.; Oken, E. Maternal obesity and associated offspring diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 630–632. [Google Scholar] [CrossRef]
- Isganaitis, E. Milky ways: Effects of maternal obesity on human milk composition and childhood obesity risk. Am. J. Clin. Nutr. 2021, 113, 772–774, Erratum in: Am. J. Clin. Nutr. 2021, 113, 1715. [Google Scholar] [CrossRef]
- Daniel, A.I.; Shama, S.; Ismail, S.; Bourdon, C.; Kiss, A.; Mwangome, M.; Bandsma, R.H.J.; O’Connor, D.L. Maternal BMI is positively associated with human milk fat: A systematic review and meta-regression analysis. Am. J. Clin. Nutr. 2021, 113, 1009–1022. [Google Scholar] [CrossRef]
- Shah, K.B.; Chernausek, S.D.; Garman, L.D.; Pezant, N.P.; Plows, J.F.; Kharoud, H.K.; Demerath, E.W.; Fields, D.A. Human milk exosomal microRNA: Associations with maternal overweight/obesity and infant body composition at 1 month of life. Nutrients 2021, 13, 1091. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Jiang, X.; Li, R.; Chen, M.; Song, W.; Li, X. The levels of human milk microRNAs and their association with maternal weight characteristics. Eur. J. Clin. Nutr. 2016, 70, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Metzger, B.E. Long-term outcomes in mothers diagnosed with gestational diabetes mellitus and their offspring. Clin. Obstet. Gynecol. 2007, 50, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, J. Through the looking glass: Gestational diabetes as a predictor of maternal and offspring long-term health. Diabetes Metab. Res. Rev. 2012, 28, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Yang, H. Gestational diabetes mellitus, programing and epigenetics. J. Matern. Fetal Neonatal Med. 2014, 27, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Chavey, A.; Ah Kioon, M.D.; Bailbé, D.; Movassat, J.; Portha, B. Maternal diabetes, programming of beta-cell disorders and intergenerational risk of type 2 diabetes. Diabetes Metab. 2014, 40, 323–330. [Google Scholar] [CrossRef]
- Damm, P.; Houshmand-Oeregaard, A.; Kelstrup, L.; Lauenborg, J.; Mathiesen, E.R.; Clausen, T.D. Gestational diabetes mellitus and long-term consequences for mother and offspring: A view from Denmark. Diabetologia 2016, 59, 1396–1399. [Google Scholar] [CrossRef]
- Franzago, M.; Fraticelli, F.; Stuppia, L.; Vitacolonna, E. Nutrigenetics, epigenetics and gestational diabetes: Consequences in mother and child. Epigenetics 2019, 14, 215–235. [Google Scholar] [CrossRef]
- Alejandro, E.U.; Mamerto, T.P.; Chung, G.; Villavieja, A.; Gaus, N.L.; Morgan, E.; Pineda-Cortel, M.R.B. Gestational diabetes mellitus: A harbinger of the vicious cycle of diabetes. Int. J. Mol. Sci. 2020, 21, 5003. [Google Scholar] [CrossRef]
- Bailbe, D.; Liu, J.; Gong, P.; Portha, B. Effect of postnatal nutritional environment due to maternal diabetes on beta cell mass programming and glucose intolerance risk in male and female offspring. Biomolecules 2021, 11, 179. [Google Scholar] [CrossRef]
- Wen, L.; Wu, Y.; Yang, Y.; Han, T.L.; Wang, W.; Fu, H.; Zheng, Y.; Shan, T.; Chen, J.; Xu, P.; et al. Gestational diabetes mellitus changes the metabolomes of human colostrum, transition milk and mature milk. Med. Sci. Monit. 2019, 25, 6128–6152. [Google Scholar] [CrossRef] [PubMed]
- Bardanzellu, F.; Puddu, M.; Fanos, V. The human breast milk metabolome in preeclampsia, gestational diabetes, and intrauterine growth restriction: Implications for child growth and development. J. Pediatr. 2020, 221S, S20–S28. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.B.; Fields, D.A.; Pezant, N.P.; Kharoud, H.K.; Gulati, S.; Jacobs, K.; Gale, C.A.; Kharbanda, E.O.; Nagel, E.M.; Demerath, E.W.; et al. Gestational diabetes mellitus is associated with altered abundance of exosomal microRNAs in human milk. Clin. Ther. 2022, 44, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.D.; Confair, A.; Warren, K.; Chandran, D. Levels of breast milk microRNAs and other non-coding RNAs are impacted by milk maturity and maternal diet. Front. Immunol. 2022, 12, 785217. [Google Scholar] [CrossRef]
- Kim, J.; Okla, M.; Erickson, A.; Carr, T.; Natarajan, S.K.; Chung, S. Eicosapentaenoic acid potentiates brown thermogenesis through FFAR4-dependent up-regulation of miR-30b and miR-378. J. Biol. Chem. 2016, 291, 20551–20562. [Google Scholar] [CrossRef]
- Lalanza, J.F.; Snoeren, E.M.S. The cafeteria diet: A standardized protocol and its effects on behavior. Neurosci. Biobehav. Rev. 2021, 122, 92–119. [Google Scholar] [CrossRef]
- Pomar, C.A.; Castro, H.; Picó, C.; Serra, F.; Palou, A.; Sánchez, J. Cafeteria diet consumption during lactation in rats, rather than obesity per se, alters miR-222, miR-200a, and miR-26a levels in milk. Mol. Nutr. Food Res. 2019, 63, e1800928. [Google Scholar] [CrossRef]
- Pomar, C.A.; Castillo, P.; Palou, A.; Palou, M.; Picó, C. Dietary improvement during lactation normalizes miR-26a, miR-222 and miR-484 levels in the mammary gland, but not in milk, of diet-induced obese rats. Biomedicines 2022, 10, 1292. [Google Scholar] [CrossRef]
- Tsukita, S.; Yamada, T.; Takahashi, K.; Munakata, Y.; Hosaka, S.; Takahashi, H.; Gao, J.; Shirai, Y.; Kodama, S.; Asai, Y.; et al. MicroRNAs 106b and 222 improve hyperglycemia in a mouse model of insulin-deficient diabetes via pancreatic β-cell proliferation. EBioMedicine 2017, 15, 163–172. [Google Scholar] [CrossRef]
- TargetScanHuman Release 8.0. Human CDKN1B ENST00000228872.4. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000228872.4&taxid=9606&members=.&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Uvnäs-Moberg, K.; Ekström-Bergström, A.; Berg, M.; Buckley, S.; Pajalic, Z.; Hadjigeorgiou, E.; Kotłowska, A.; Lengler, L.; Kielbratowska, B.; Leon-Larios, F.; et al. Maternal plasma levels of oxytocin during physiological childbirth—A systematic review with implications for uterine contractions and central actions of oxytocin. BMC Pregnancy Childbirth 2019, 19, 285. [Google Scholar] [CrossRef]
- Gutman-Ido, E.; Reif, S.; Musseri, M.; Schabes, T.; Golan-Gerstl, R. Oxytocin regulates the expression of selected colostrum-derived microRNAs. J. Pediatr. Gastroenterol. Nutr. 2022, 74, e8–e15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, J.; Li, J.; Geng, H.; Zhou, B.; Zhang, B.; Chen, H. miR-320/ELF3 axis inhibits the progression of breast cancer via the PI3K/AKT pathway. Oncol. Lett. 2020, 19, 3239–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, F.F.; An, T.; Zhang, Z.J.; Liu, Y.F.; Liu, H.X.; Pan, Y.Y.; Miao, J.N.; Zhao, D.D.; Yang, X.Y.; Zhang, D.W.; et al. Jiang Tang Xiao Ke granule play an anti-diabetic role in diabetic mice pancreatic tissue by regulating the mRNAs and microRNAs associated with PI3K-Akt signaling pathway. Front. Pharmacol. 2017, 8, 795. [Google Scholar] [CrossRef]
- Lopes, M.; Kutlu, B.; Miani, M.; Bang-Berthelsen, C.H.; Størling, J.; Pociot, F.; Goodman, N.; Hood, L.; Welsh, N.; Bontempi, G.; et al. Temporal profiling of cytokine-induced genes in pancreatic β-cells by meta-analysis and network inference. Genomics 2014, 103, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Prior, E.; Santhakumaran, S.; Gale, C.; Philipps, L.H.; Modi, N.; Hyde, M.J. Breastfeeding after cesarean delivery: A systematic review and meta-analysis of world literature. Am. J. Clin. Nutr. 2012, 95, 1113–1135. [Google Scholar] [CrossRef]
- Raymond, F.; Lefebvre, G.; Texari, L.; Pruvost, S.; Metairon, S.; Cottenet, G.; Zollinger, A.; Mateescu, B.; Billeaud, C.; Picaud, J.C.; et al. Longitudinal human milk miRNA composition over the first 3 mo of lactation in a cohort of healthy mothers delivering term infants. J. Nutr. 2022, 152, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Izadi, V.; Kelishadi, R.; Qorbani, M.; Esmaeilmotlagh, M.; Taslimi, M.; Heshmat, R.; Ardalan, G.; Azadbakht, L. Duration of breast-feeding and cardiovascular risk factors among Iranian children and adolescents: The CASPIAN III study. Nutrition 2013, 29, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Yew, S.S.Y.; Devenish, G.; Ha, D.; Do, L.; Scott, J. Duration of breastfeeding, but not timing of solid food, reduces the risk of overweight and obesity in children aged 24 to 36 months: Findings from an Australian cohort study. Int. J. Environ. Res. Public Health 2018, 15, 599. [Google Scholar] [CrossRef]
- Sherwood, W.B.; Bion, V.; Lockett, G.A.; Ziyab, A.H.; Soto-Ramírez, N.; Mukherjee, N.; Kurukulaaratchy, R.J.; Ewart, S.; Zhang, H.; Arshad, S.H.; et al. Duration of breastfeeding is associated with leptin (LEP) DNA methylation profiles and BMI in 10-year-old children. Clin. Epigenetics 2019, 11, 128. [Google Scholar] [CrossRef]
- Sherwood, W.B.; Kothalawala, D.M.; Kadalayil, L.; Ewart, S.; Zhang, H.; Karmaus, W.; Arshad, S.H.; Holloway, J.W.; Rezwan, F.I. Epigenome-wide association study reveals duration of breastfeeding is associated with epigenetic differences in children. Int. J. Environ. Res. Public Health 2020, 17, 3569. [Google Scholar] [CrossRef]
- Mallisetty, Y.; Mukherjee, N.; Jiang, Y.; Chen, S.; Ewart, S.; Arshad, S.H.; Holloway, J.W.; Zhang, H.; Karmaus, W. Epigenome-wide association of infant feeding and changes in DNA methylation from birth to 10 years. Nutrients 2020, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Walaszczyk, E.; Luijten, M.; Spijkerman, A.M.W.; Bonder, M.J.; Lutgers, H.L.; Snieder, H.; Wolffenbuttel, B.H.R.; van Vliet-Ostaptchouk, J.V. DNA methylation markers associated with type 2 diabetes, fasting glucose and HbA1c levels: A systematic review and replication in a case-control sample of the Lifelines study. Diabetologia 2018, 61, 354–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güngör, D.; Nadaud, P.; LaPergola, C.C.; Dreibelbis, C.; Wong, Y.P.; Terry, N.; Abrams, S.A.; Beker, L.; Jacobovits, T.; Järvinen, K.M.; et al. Infant milk-feeding practices and diabetes outcomes in offspring: A systematic review. Am. J. Clin. Nutr. 2019, 109, 817S–837S. [Google Scholar] [CrossRef] [PubMed]
- Kalra, B.; Gupta, Y.; Kalra, S. Breast feeding: Preventive therapy for type 2 diabetes. J. Pak. Med. Assoc. 2015, 65, 1134–1136. [Google Scholar]
- Pereira, P.F.; Alfenas Rde, C.; Araújo, R.M. Does breastfeeding influence the risk of developing diabetes mellitus in children? A review of current evidence. J. Pediatr. (Rio J.) 2014, 90, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Horta, B.L.; Loret de Mola, C.; Victora, C.G. Long-term consequences of breastfeeding on cholesterol, obesity, systolic blood pressure and type 2 diabetes: A systematic review and meta-analysis. Acta Paediatr. 2015, 104, 30–37. [Google Scholar] [CrossRef]
- Underwood, M.A. Human milk for the premature infant. Pediatr. Clin. North Am. 2013, 60, 189–207. [Google Scholar] [CrossRef]
- Miller, J.; Tonkin, E.; Damarell, R.A.; McPhee, A.J.; Suganuma, M.; Suganuma, H.; Middleton, P.F.; Makrides, M.; Collins, C.T. A systematic review and meta-analysis of human milk feeding and morbidity in very low birth weight infants. Nutrients 2018, 10, 707. [Google Scholar] [CrossRef]
- Perri, M.; Lucente, M.; Cannataro, R.; De Luca, I.F.; Gallelli, L.; Moro, G.; De Sarro, G.; Caroleo, M.C.; Cione, E. Variation in immune-related microRNAs profile in human milk amongst lactating women. Microrna 2018, 7, 107–114. [Google Scholar] [CrossRef]
- Smyczynska, U.; Bartlomiejczyk, M.A.; Stanczak, M.M.; Sztromwasser, P.; Wesolowska, A.; Barbarska, O.; Pawlikowska, E.; Fendler, W. Impact of processing method on donated human breast milk microRNA content. PLoS ONE 2020, 15, e0236126. [Google Scholar] [CrossRef]
- Leiferman, A.; Shu, J.; Upadhyaya, B.; Cui, J.; Zempleni, J. Storage of extracellular vesicles in human milk, and microRNA profiles in human milk exosomes and infant formulas. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Mutai, E.; Ngu, A.K.H.; Zempleni, J. Preliminary evidence that lectins in infant soy formula apparently bind bovine milk exosomes and prevent their absorption in healthy adults. BMC Nutr. 2022, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Osborn, D.A.; Sinn, J. Soy formula for prevention of allergy and food intolerance in infants. Cochrane Database Syst. Rev. 2006, 2006, CD003741. [Google Scholar] [CrossRef] [PubMed]
- Vandenplas, Y.; Hegar, B.; Munasir, Z.; Astawan, M.; Juffrie, M.; Bardosono, S.; Sekartini, R.; Basrowi, R.W.; Wasito, E. The role of soy plant-based formula supplemented with dietary fiber to support children’s growth and development: An expert opinion. Nutrition 2021, 90, 111278. [Google Scholar] [CrossRef]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filén, J.J.; Lahesmaa, R.; Norman, M.; Neve, E.P.; Scheynius, A.; Gabrielsson, S. Exosomes with immune modulatory features are present in human breast milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef]
- Melnik, B.C.; John, S.M.; Carrera-Bastos, P.; Schmitz, G. Milk: A postnatal imprinting system stabilizing FoxP3 expression and regulatory T cell differentiation. Clin. Transl. Allergy 2016, 6, 18. [Google Scholar] [CrossRef]
- Chen, X.; Li, B.; Luo, R.; Cai, S.; Zhang, C.; Cao, X. Analysis of the function of microRNA-375 in humans using bioinformatics. Biomed. Rep. 2017, 6, 561–566. [Google Scholar] [CrossRef]
- Yang, Y.Z.; Zhao, X.J.; Xu, H.J.; Wang, S.C.; Pan, Y.; Wang, S.J.; Xu, Q.; Jiao, R.Q.; Gu, H.M.; Kong, L.D. Magnesium isoglycyrrhizinate ameliorates high fructose-induced liver fibrosis in rat by increasing miR-375-3p to suppress JAK2/STAT3 pathway and TGF-β1/Smad signaling. Acta Pharmacol. Sin. 2019, 40, 879–894. [Google Scholar] [CrossRef]
- Chen, B.; Guo, S.; Yu, Z.; Feng, Y.; Hui, L. Downregulation of microRNA-375, combined with upregulation of its target gene Janus kinase 2, predicts unfavorable prognosis in patients with gastric cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 11106–11113. [Google Scholar]
- Miao, L.; Liu, K.; Xie, M.; Xing, Y.; Xi, T. miR-375 inhibits Helicobacter pylori-induced gastric carcinogenesis by blocking JAK2-STAT3 signaling. Cancer Immunol. Immunother. 2014, 63, 699–711. [Google Scholar] [CrossRef]
- Wang, T.; Chen, D.; Wang, P.; Xu, Z.; Li, Y. miR-375 prevents nasal mucosa cells from apoptosis and ameliorates allergic rhinitis via inhibiting JAK2/STAT3 pathway. Biomed. Pharmacother. 2018, 103, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, X.; Su, H. Overexpression of microRNA-375 impedes platelet-derived growth factor-induced proliferation and migration of human fetal airway smooth muscle cells by targeting Janus kinase 2. Biomed. Pharmacother. 2018, 98, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Luschnig, P.; Kienzl, M.; Roula, D.; Pilic, J.; Atallah, R.; Heinemann, A.; Sturm, E.M. The JAK1/2 inhibitor baricitinib suppresses eosinophil effector function and restricts allergen-induced airway eosinophilia. Biochem. Pharmacol. 2021, 192, 114690. [Google Scholar] [CrossRef] [PubMed]
- Cartron, A.M.; Nguyen, T.H.; Roh, Y.S.; Kwatra, M.M.; Kwatra, S.G. Janus kinase inhibitors for atopic dermatitis: A promising treatment modality. Clin. Exp. Dermatol. 2021, 46, 820–824. [Google Scholar] [CrossRef]
- Trivedi, P.M.; Graham, K.L.; Scott, N.A.; Jenkins, M.R.; Majaw, S.; Sutherland, R.M.; Fynch, S.; Lew, A.M.; Burns, C.J.; Krishnamurthy, B.; et al. Repurposed JAK1/JAK2 inhibitor reverses established autoimmune insulitis in NOD mice. Diabetes 2017, 66, 1650–1660. [Google Scholar] [CrossRef]
- Latreille, M.; Herrmanns, K.; Renwick, N.; Tuschl, T.; Malecki, M.T.; McCarthy, M.I.; Owen, K.R.; Rülicke, T.; Stoffel, M. miR-375 gene dosage in pancreatic β-cells: Implications for regulation of β-cell mass and biomarker development. J. Mol. Med. 2015, 93, 1159–1169. [Google Scholar] [CrossRef]
- Cheshmeh, S.; Nachvak, S.M.; Rezvani, N.; Saber, A. Effects of breastfeeding and formula feeding on the expression level of FTO, CPT1A and PPAR-α genes in healthy infants. Diabetes Metab. Syndr. Obes. 2020, 13, 2227–2237. [Google Scholar] [CrossRef]
- Kleinjan, M.; van Herwijnen, M.J.; Libregts, S.F.; van Neerven, R.J.; Feitsma, A.L.; Wauben, M.H. Regular industrial processing of bovine milk impacts the integrity and molecular composition of extracellular vesicles. J. Nutr. 2021, 151, 1416–1425. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Q.; Hou, J.; Huang, G.; Zhao, S.; Zheng, N.; Wang, J. Loss of bioactive microRNAs in cow’s milk by ultra-high-temperature treatment but not by pasteurization treatment. J. Sci. Food Agric. 2022, 102, 2676–2685. [Google Scholar] [CrossRef]
- Wiley, A.S. Cow milk consumption, insulin-like growth factor-I, and human biology: A life history approach. Am. J. Hum. Biol. 2012, 24, 130–138. [Google Scholar] [CrossRef]
- Hoppe, C.; Mølgaard, C.; Michaelsen, K.F. Cow’s milk and linear growth in industrialized and developing countries. Annu. Rev. Nutr. 2006, 26, 131–173. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. Lifetime impact of cow’s milk on overactivation of mTORC1: From fetal to childhood overgrowth, acne, diabetes, cancers, and neurodegeneration. Biomolecules 2021, 11, 404. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, L.E.; Brett, J.; Kelton, D.; Majowicz, S.E.; Snedeker, K.; Sargeant, J.M. A systematic review and meta-analysis of the effects of pasteurization on milk vitamins, and evidence for raw milk consumption and other health-related outcomes. J. Food Prot. 2011, 74, 1814–1832. [Google Scholar] [CrossRef] [PubMed]
- Committee on Infectious Diseases; Committee on Nutrition; American Academy of Pediatrics. Consumption of raw or unpasteurized milk and milk products by pregnant women and children. Pediatrics 2014, 133, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Lucey, J.A. Raw milk consumption: Risks and benefits. Nutr. Today 2015, 50, 189–193. [Google Scholar] [CrossRef]
- Michaëlsson, K.; Wolk, A.; Langenskiöld, S.; Basu, S.; Warensjö Lemming, E.; Melhus, H.; Byberg, L. Milk intake and risk of mortality and fractures in women and men: Cohort studies. BMJ 2014, 349, g6015. [Google Scholar] [CrossRef]
- Melnik, B.C.; Schmitz, G. Pasteurized non-fermented cow’s milk but not fermented milk is a promoter of mTORC1-driven aging and increased mortality. Ageing Res. Rev. 2021, 67, 101270. [Google Scholar] [CrossRef]
- International Dairy Federation, 2018. Heat Treatment of Milk—Overview, 001/2018-02. IDF Factsheet. 2018. Available online: http://www.ukidf.org/documents/Factsheet-001_2018_Heat-treatment-1-2_001.pdf (accessed on 4 June 2022).
- Mutai, E.; Ramer-Tait, A.E.; Zempleni, J. MicroRNAs in bovine milk exosomes are bioavailable in humans but do not elicit a robust pro-inflammatory cytokine response. ExRNA 2020, 2, 2. [Google Scholar] [CrossRef]
- Fromm, B.; Tosar, J.P.; Lu, Y.; Halushka, M.K.; Witwer, K.W. Human and cow have identical miR-21-5p and miR-30a-5p sequences, which are likely unsuited to study dietary uptake from cow milk. J. Nutr. 2018, 148, 1506–1507. [Google Scholar] [CrossRef]
- Go, H.; Jang, J.Y.; Kim, P.J.; Kim, Y.G.; Nam, S.J.; Paik, J.H.; Kim, T.M.; Heo, D.S.; Kim, C.W.; Jeon, Y.K. MicroRNA-21 plays an oncogenic role by targeting FOXO1 and activating the PI3K/AKT pathway in diffuse large B-cell lymphoma. Oncotarget 2015, 6, 15035–15049. [Google Scholar] [CrossRef]
- Luo, A.; Yan, H.; Liang, J.; Du, C.; Zhao, X.; Sun, L.; Chen, Y. MicroRNA-21 regulates hepatic glucose metabolism by targeting FOXO1. Gene 2017, 627, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.X.; Liu, Z.H.; Li, Z.J.; Li, C.; Deng, Y.F. miR-21 induces cell proliferation and suppresses the chemosensitivity in glioblastoma cells via downregulation of FOXO1. Int. J. Clin. Exp. Med. 2014, 7, 2060–2066. [Google Scholar] [PubMed]
- Wang, J.; Shen, L.; Hong, H.; Li, J.; Wang, H.; Li, X. Atrasentan alleviates high glucose-induced podocyte injury by the microRNA-21/forkhead box O1 axis. Eur. J. Pharmacol. 2019, 852, 142–150. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman Release 8.0. Human FOXO1 ENST00000379561.5. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000379561.5&taxid=9606&showcnc=.0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Dey, N.; Das, F.; Mariappan, M.M.; Mandal, C.C.; Ghosh-Choudhury, N.; Kasinath, B.S.; Choudhury, G.G. MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J. Biol. Chem. 2011, 286, 25586–25603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmi, V.; D’Adamo, M.; Menghini, R.; Cardellini, M.; Gentileschi, P.; Federici, M.; Sbraccia, P. MicroRNA 21 is up-regulated in adipose tissue of obese diabetic subjects. Nutr. Healthy Aging 2017, 4, 141–145. [Google Scholar] [CrossRef]
- Doghish, A.S.; Elsisi, A.M.; Amin, A.I.; Abulsoud, A.I. Circulating miR-148a-5p and miR-21-5p as novel diagnostic biomarkers in adult Egyptian male patients with metabolic syndrome. Can. J. Diabetes 2021, 45, 614–618. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef]
- Backe, M.B.; Novotny, G.W.; Christensen, D.P.; Grunnet, L.G.; Mandrup-Poulsen, T. Altering β-cell number through stable alteration of miR-21 and miR-34a expression. Islets 2014, 6, e27754. [Google Scholar] [CrossRef]
- Ibrahim, S.; Johnson, M.; Stephens, C.H.; Xu, J.; Moore, R.; Mariani, A.; Contreras, C.; Syed, F.; Mirmira, R.G.; Anderson, R.M.; et al. β-Cell pre-mir-21 induces dysfunction and loss of cellular identity by targeting transforming growth factor beta 2 (Tgfb2) and Smad family member 2 (Smad2) mRNAs. Mol. Metab. 2021, 53, 101289. [Google Scholar] [CrossRef]
- Frederiksen, B.; Kroehl, M.; Lamb, M.M.; Seifert, J.; Barriga, K.; Eisenbarth, G.S.; Rewers, M.; Norris, J.M. Infant exposures and development of type 1 diabetes mellitus: The Diabetes Autoimmunity Study in the Young (DAISY). JAMA Pediatr. 2013, 167, 808–815. [Google Scholar] [CrossRef]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef]
- Benslama, Y.; Dennouni-Medjati, N.; Dali-Sahi, M.; Meziane, F.Z.; Harek, Y. Childhood type 1 diabetes mellitus and risk factor of interactions between dietary cow’s milk intake and HLA-DR3/DR4 genotype. J. Biomol. Struct. Dyn. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Patelarou, E.; Girvalaki, C.; Brokalaki, H.; Patelarou, A.; Androulaki, Z.; Vardavas, C. Current evidence on the associations of breastfeeding, infant formula, and cow’s milk introduction with type 1 diabetes mellitus: A systematic review. Nutr. Rev. 2012, 70, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Verge, C.F.; Howard, N.J.; Irwig, L.; Simpson, J.M.; Mackerras, D.; Silink, M. Environmental factors in childhood IDDM A population-based, case-control study. Diabetes Care 1994, 17, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, S.M.; Läärä, E.; Hyppönen, E.; Reijonen, H.; Räsänen, L.; Aro, A.; Knip, M.; Ilonen, J.; Akerblom, H.K. Cow’s milk consumption, HLA-DQB1 genotype, and type 1 diabetes: A nested case-control study of siblings of children with diabetes. Childhood diabetes in Finland study group. Diabetes 2000, 49, 912–917. [Google Scholar] [CrossRef]
- Rosenbauer, J.; Herzig, P.; Giani, G. Early infant feeding and risk of type 1 diabetes mellitus-a nationwide population-based case-control study in pre-school children. Diabetes Metab. Res. Rev. 2008, 24, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Lamb, M.M.; Miller, M.M.; Seifert, J.A.; Frederiksen, B.; Kroehl, M.; Rewers, M.; Norris, J.M. The effect of childhood cow’s milk intake and HLA-DR genotype on risk of islet autoimmunity and type 1 diabetes: The Diabetes Autoimmunity Study in the Young. Pediatr. Diabetes 2015, 16, 31–38. [Google Scholar] [CrossRef]
- Assmann, T.S.; Recamonde-Mendoza, M.; De Souza, B.M.; Crispim, D. MicroRNA expression profiles and type 1 diabetes mellitus: Systematic review and bioinformatic analysis. Endocr. Connect. 2017, 6, 773–790. [Google Scholar] [CrossRef]
- Garcia-Contreras, M.; Shah, S.H.; Tamayo, A.; Robbins, P.D.; Golberg, R.B.; Mendez, A.J.; Ricordi, C. Plasma-derived exosome characterization reveals a distinct microRNA signature in long duration type 1 diabetes. Sci. Rep. 2017, 7, 5998. [Google Scholar] [CrossRef]
- Khanam, A.; Yu, J.; Zempleni, J. Class A scavenger receptor-1/2 facilitates the uptake of bovine milk exosomes in murine bone marrow-derived macrophages and C57BL/6J mice. Am. J. Physiol. Cell Physiol. 2021, 321, C607–C614. [Google Scholar] [CrossRef]
- Cosentino, C.; Regazzi, R. Crosstalk between macrophages and pancreatic β-cells in islet development, homeostasis and disease. Int. J. Mol. Sci. 2021, 22, 1765. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Li, J.; Ye, Z.; Yin, Z.; Sun, Q.; Liao, Z.; Li, G.; Deng, J.; Liu, L.; Yu, Y.; et al. MicroRNA-148a facilitates inflammatory dendritic cell differentiation and autoimmunity by targeting MAFB. JCI Insight 2020, 5, e133721. [Google Scholar] [CrossRef] [PubMed]
- Price, J.D.; Tarbell, K.V. The role of dendritic cell subsets and innate immunity in the pathogenesis of type 1 diabetes and other autoimmune diseases. Front. Immunol. 2015, 6, 288. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.U.; Khongorzul, P.; Raki, A.A.; Rajasekaran, A.; Gris, D.; Amrani, A. Dendritic cells and their immunotherapeutic potential for treating type 1 diabetes. Int. J. Mol. Sci. 2022, 23, 4885. [Google Scholar] [CrossRef]
- Jun, H.S.; Yoon, C.S.; Zbytnuik, L.; van Rooijen, N.; Yoon, J.W. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Exp. Med. 1999, 189, 347–358. [Google Scholar] [CrossRef]
- Huang, F.; Zhao, J.L.; Wang, L.; Gao, C.C.; Liang, S.Q.; An, D.J.; Bai, J.; Chen, Y.; Han, H.; Qin, H.Y. miR-148a-3p mediates Notch signaling to promote the differentiation and M1 activation of macrophages. Front. Immunol. 2017, 8, 1327. [Google Scholar] [CrossRef]
- Burg, A.R.; Tse, H.M. Redox-sensitive innate immune pathways during macrophage activation in type 1 diabetes. Antioxid. Redox Signal. 2018, 29, 1373–1398. [Google Scholar] [CrossRef]
- Zhang, C.; Han, X.; Yang, L.; Fu, J.; Sun, C.; Huang, S.; Xiao, W.; Gao, Y.; Liang, Q.; Wang, X.; et al. Circular RNA circPPM1F modulates M1 macrophage activation and pancreatic islet inflammation in type 1 diabetes mellitus. Theranostics 2020, 10, 10908–10924. [Google Scholar] [CrossRef] [PubMed]
- Sluijs, I.; Forouhi, N.G.; Beulens, J.W.; van der Schouw, Y.T.; Agnoli, C.; Arriola, L.; Balkau, B.; Barricarte, A.; Boeing, H.; Bueno-de-Mesquita, H.B.; et al. The amount and type of dairy product intake and incident type 2 diabetes: Results from the EPIC-InterAct Study. Am. J. Clin. Nutr. 2012, 96, 382–390. [Google Scholar]
- Brouwer-Brolsma, E.M.; Sluik, D.; Singh-Povel, C.M.; Feskens, E.J.M. Dairy product consumption is associated with pre-diabetes and newly diagnosed type 2 diabetes in the Lifelines Cohort Study. Br. J. Nutr. 2018, 119, 442–455. [Google Scholar] [CrossRef]
- Melnik, B.C. The pathogenic role of persistent milk signaling in mTORC1- and milk-microRNA-driven type 2 diabetes mellitus. Curr. Diabetes Rev. 2015, 11, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Schmitz, G. The impact of persistent milk consumption in the pathogenesis of type 2 diabetes mellitus. Funct. Foods Health Dis. 2019, 9, 629–647. [Google Scholar] [CrossRef]
- Melnik, B.C.; Schmitz, G. Milk consumption does not prevent but induces type 2 diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3200. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Schmitz, G. Exosomes of pasteurized milk: Potential pathogens of Western diseases. J. Transl. Med. 2019, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. Synergistic effects of milk-derived exosomes and galactose on α-synuclein pathology in Parkinson’s disease and type 2 diabetes mellitus. Int. J. Mol. Sci. 2021, 22, 1059. [Google Scholar] [CrossRef]
- La Sala, L.; Mrakic-Sposta, S.; Tagliabue, E.; Prattichizzo, F.; Micheloni, S.; Sangalli, E.; Specchia, C.; Uccellatore, A.C.; Lupini, S.; Spinetti, G.; et al. Circulating microRNA-21 is an early predictor of ROS-mediated damage in subjects with high risk of developing diabetes and in drug-naïve T2D. Cardiovasc. Diabetol. 2019, 18, 18. [Google Scholar] [CrossRef]
- Sims, E.K.; Lakhter, A.J.; Anderson-Baucum, E.; Kono, T.; Tong, X.; Evans-Molina, C. MicroRNA 21 targets BCL2 mRNA to increase apoptosis in rat and human beta cells. Diabetologia 2017, 60, 1057–1065. [Google Scholar] [CrossRef]
- Dalgaard, L.T.; Eliasson, L. An ‘alpha-beta’ of pancreatic islet microribonucleotides. Int. J. Biochem. Cell Biol. 2017, 88, 208–219. [Google Scholar] [CrossRef]
- Bolmeson, C.; Esguerra, J.L.; Salehi, A.; Speidel, D.; Eliasson, L.; Cilio, C.M. Differences in islet-enriched miRNAs in healthy and glucose intolerant human subjects. Biochem. Biophys. Res. Commun. 2011, 404, 16–22. [Google Scholar] [CrossRef]
- Nesca, V.; Guay, C.; Jacovetti, C.; Menoud, V.; Peyot, M.L.; Laybutt, D.R.; Prentki, M.; Regazzi, R. Identification of particular groups of microRNAs that positively or negatively impact on beta cell function in obese models of type 2 diabetes. Diabetologia 2013, 56, 2203–2212. [Google Scholar] [CrossRef]
- Zhao, E.; Keller, M.P.; Rabaglia, M.E.; Oler, A.T.; Stapleton, D.S.; Schueler, K.L.; Neto, E.C.; Moon, J.Y.; Wang, P.; Wang, I.M.; et al. Obesity and genetics regulate microRNAs in islets, liver, and adipose of diabetic mice. Mamm. Genome 2009, 20, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.; Rosenstierne, M.W.; Gaarn, L.W.; Bagge, A.; Pedersen, L.; Dahmcke, C.M.; Nielsen, J.H.; Dalgaard, L.T. Expression and localization of microRNAs in perinatal rat pancreas: Role of miR-21 in regulation of cholesterol metabolism. PLoS ONE 2011, 6, e25997. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, S.; Jayachandran, I.; Balasubramanyam, M.; Mohan, V.; Venkatesan, B.; Manickam, N. Sestrin2 regulates monocyte activation through AMPK-mTOR nexus under high-glucose and dyslipidemic conditions. J. Cell Biochem. 2018, 120, jcb28102. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, S.; Jayachandran, I.; Subramanian, S.C.; Anjana, R.M.; Balasubramanyam, M.; Mohan, V.; Venkatesan, B.; Manickam, N. Decreased sestrin levels in patients with type 2 diabetes and dyslipidemia and their association with the severity of atherogenic index. J. Endocrinol. Investig. 2021, 44, 1395–1405. [Google Scholar] [CrossRef]
- Mohany, K.M.; Al Rugaie, O.; Al-Wutayd, O.; Al-Nafeesah, A. Investigation of the levels of circulating miR-29a, miR-122, sestrin 2 and inflammatory markers in obese children with/without type 2 diabetes: A case control study. BMC Endocr. Disord. 2021, 21, 152. [Google Scholar] [CrossRef]
- TargetScan Human Release 8.0. Human SESN2 ENST00000253063.3. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000253063.3&taxid=9606&showcnc=.0&shownc=0&shownc_nc=&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Ozcan, U.; Ozcan, L.; Yilmaz, E.; Düvel, K.; Sahin, M.; Manning, B.D.; Hotamisligil, G.S. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell 2008, 29, 541–551. [Google Scholar] [CrossRef]
- Bartolomé, A.; Kimura-Koyanagi, M.; Asahara, S.; Guillén, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; García-Aguilar, A.; Koike, M.; et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef]
- Wang, J.; Yang, X.; Zhang, J. Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic β cells. Cell Signal. 2016, 28, 1099–1104. [Google Scholar] [CrossRef]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef]
- Pulito, C.; Mori, F.; Sacconi, A.; Goeman, F.; Ferraiuolo, M.; Pasanisi, P.; Campagnoli, C.; Berrino, F.; Fanciulli, M.; Ford, R.J.; et al. Metformin-induced ablation of microRNA 21-5p releases sestrin-1 and CAB39L antitumoral activities. Cell Discov. 2017, 3, 17022. [Google Scholar] [CrossRef]
- Khokhar, M.; Roy, D.; Bajpai, N.K.; Bohra, G.K.; Yadav, D.; Sharma, P.; Purohit, P. Metformin mediates microRNA-21 regulated circulating matrix metalloproteinase-9 in diabetic nephropathy: An in-silico and clinical study. Arch. Physiol. Biochem. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30. [Google Scholar] [CrossRef] [PubMed]
- TargetScanHuman Release 8.0. Human AQP7 ENST00000537089.1. Available online: https://www.targetscan.org/cgi-bin/targetscan/vert_80/view_gene.cgi?rs=ENST00000537089.1&taxid=9606&members=.&showcnc=0&shownc=0&showncf1=&showncf2=&subset=1 (accessed on 10 June 2022).
- Srinivasan, M.; Patel, M.S. Metabolic programming in the immediate postnatal period. Trends Endocrinol. Metab. 2008, 19, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Srinivasan, M. Metabolic programming in the immediate postnatal life. Ann. Nutr. Metab. 2011, 58, 18–28. [Google Scholar] [CrossRef]
- Cione, E.; Cannataro, R.; Gallelli, L.; De Sarro, G.; Caroleo, M.C. Exosome microRNAs in metabolic syndrome as tools for the early monitoring of diabetes and possible therapeutic options. Pharmaceuticals 2021, 14, 1257. [Google Scholar] [CrossRef]
- Ebert, M.S.; Sharp, P.A. Roles for microRNAs in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef]
- Guay, C.; Jacovetti, C.; Nesca, V.; Motterle, A.; Tugay, K.; Regazzi, R. Emerging roles of non-coding RNAs in pancreatic β-cell function and dysfunction. Diabetes Obes. Metab. 2012, 14, 12–21. [Google Scholar] [CrossRef]
- Özcan, S. microRNAs in pancreatic β-cell physiology. Adv. Exp. Med. Biol. 2015, 887, 101–117. [Google Scholar]
- Eliasson, L. The small RNA miR-375—A pancreatic islet abundant miRNA with multiple roles in endocrine beta cell function. Mol. Cell Endocrinol. 2017, 456, 95–101. [Google Scholar] [CrossRef]
- Brozzi, F. Role of microRNA in pancreatic beta cell function. Int. Rev. Cell Mol. Biol. 2021, 359, 257–286. [Google Scholar]
- Kaspi, H.; Pasvolsky, R.; Hornstein, E. Could microRNAs contribute to the maintenance of β cell identity? Trends Endocrinol. Metab. 2014, 25, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, A.; Rutter, G.A.; Latreille, M. MiRNAs in β-cell development, identity, and disease. Front. Genet. 2017, 7, 226. [Google Scholar] [CrossRef] [PubMed]
- Grieco, G.E.; Brusco, N.; Licata, G.; Fignani, D.; Formichi, C.; Nigi, L.; Sebastiani, G.; Dotta, F. The landscape of microRNAs in β cell: Between phenotype maintenance and protection. Int. J. Mol. Sci. 2021, 22, 803. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.S.; Stein, R.W. Evidence for loss in identity, de-differentiation, and trans-differentiation of islet β-cells in type 2 diabetes. Front. Genet. 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, C.; Jimenez-Gonzalez, M.; Song, W.J.; Hussain, M.A. The undoing and redoing of the diabetic β-cell. J. Diabetes Complicat. 2017, 31, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Moin, A.S.M.; Butler, A.E. Alterations in beta cell identity in type 1 and type 2 diabetes. Curr. Diab. Rep. 2019, 19, 83. [Google Scholar] [CrossRef] [PubMed]
- Spijker, H.S.; Song, H.; Ellenbroek, J.H.; Roefs, M.M.; Engelse, M.A.; Bos, E.; Koster, A.J.; Rabelink, T.J.; Hansen, B.C.; Clark, A.; et al. Loss of β-cell identity occurs in type 2 diabetes and is associated with islet amyloid deposits. Diabetes 2015, 64, 2928–2938. [Google Scholar] [CrossRef] [Green Version]
- Mucibabic, M.; Steneberg, P.; Lidh, E.; Straseviciene, J.; Ziolkowska, A.; Dahl, U.; Lindahl, E.; Edlund, H. α-Synuclein promotes IAPP fibril formation in vitro and β-cell amyloid formation in vivo in mice. Sci. Rep. 2020, 10, 20438. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, H.; Cheng, J.; Zhu, Y.; Xiao, Q.; Bai, Y.; Tao, J. Neuroprotective effects of lithium on a chronic MPTP mouse model of Parkinson’s disease via regulation of α synuclein methylation. Mol. Med. Rep. 2019, 19, 4989–4997. [Google Scholar] [CrossRef]
- Su, C.; Yang, X.; Lou, J. Geniposide reduces α-synuclein by blocking microRNA-21/lysosome-associated membrane protein 2A interaction in Parkinson disease models. Brain Res. 2016, 1644, 98–106. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013, 4, e545. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Bian, Z. MicroRNA-21 is a versatile regulator and potential treatment target in central nervous system disorders. Front. Mol. Neurosci. 2022, 15, 842288. [Google Scholar] [CrossRef] [PubMed]
- Popov, V.B.; Aytaman, A.; Alemán, J.O. Obesity: The forgotten pandemic. Am. J. Gastroenterol. 2022, 117, 7–10. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 5 June 2022).
- Almahmeed, B.; Shah, B.R.; Mukerji, G.; Ling, V.; Booth, G.L.; Feig, D.S. Effect of multiparity and ethnicity on the risk of development of diabetes: A large population-based cohort study. Diabet. Med. 2017, 34, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, L.; Hindi, S.; Sorenson, R.L.; German, M.S. β-Cell adaptation in pregnancy. Diabetes Obes. Metab. 2016, 18, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Al-Sofiani, M.E.; Ganji, S.S.; Kalyani, R.R. Body composition changes in diabetes and aging. J. Diabetes Complicat. 2019, 33, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, U.; Gannon, M. Type 2 diabetes and the aging pancreatic beta cell. Aging 2011, 3, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Halper, B.; Hofmann, M.; Oesen, S.; Franzke, B.; Stuparits, P.; Vidotto, C.; Tschan, H.; Bachl, N.; Strasser, E.M.; Quittan, M.; et al. Influence of age and physical fitness on miRNA-21, TGF-β and its receptors in leukocytes of healthy women. Exerc. Immunol. Rev. 2015, 21, 154–163. [Google Scholar]
- WHO. Caesarean Section Rates Continue to Rise, Amid Growing Inequalities in Access. Available online: https://www.who.int/news/item/16-06-2021-caesarean-section-rates-continue-to-rise-amid-growing-inequalities-in-access (accessed on 7 June 2022).
- Andersen, V.; Möller, S.; Jensen, P.B.; Møller, F.T.; Green, A. Caesarean delivery and risk of chronic inflammatory diseases (inflammatory bowel disease, rheumatoid arthritis, coeliac disease, and diabetes mellitus): A population based registry study of 2,699,479 births in Denmark during 1973–2016. Clin. Epidemiol. 2020, 12, 287–293. [Google Scholar] [CrossRef]
- Bryder, L. From breast to bottle: A history of modern infant feeding. Endeavour 2009, 33, 54–59. [Google Scholar] [CrossRef]
- Marriott, M.; Schoenthal, L. An experimental study of the use of unsweetened evaporated milk for the preparation of infant feeding formulas. Arch. Pediatr. 1929, 46, 135–148. [Google Scholar]
- Röszer, T. Mother-to-child signaling through breast milk biomolecules. Biomolecules 2021, 11, 1743. [Google Scholar] [CrossRef]
- Camacho-Morales, A.; Caba, M.; García-Juárez, M.; Caba-Flores, M.D.; Viveros-Contreras, R.; Martínez-Valenzuela, C. Breastfeeding contributes to physiological immune programming in the newborn. Front. Pediatr. 2021, 9, 744104. [Google Scholar] [CrossRef]
- Boor, K.J.; Wiedmann, M.; Murphy, S.; Alcaine, S. A 100-year review: Microbiology and safety of milk handling. J. Dairy Sci. 2017, 100, 9933–9951. [Google Scholar] [CrossRef]
- Currier, R.W.; Widness, J.A. A brief history of milk hygiene and its impact on infant mortality from 1875 to 1925 and implications for today: A review. J. Food Prot. 2018, 81, 1713–1722. [Google Scholar] [CrossRef]
- Askenase, P.W. Ancient evolutionary origin and properties of universally produced natural exosomes contribute to their therapeutic superiority compared to artificial nanoparticles. Int. J. Mol. Sci. 2021, 22, 1429. [Google Scholar] [CrossRef]
- Melnik, B.C.; Weiskirchen, R.; Stremmel, W.; Schmitz, G. Letter to the editor regarding “Dietary bovine milk miRNAs transported in extracellular vesicles are partially stable during GI digestion, are bioavailable and reach target tissues but need a minimum dose to impact on gene expression”. Eur. J. Nutr. 2022, 61, 1695–1696. [Google Scholar] [CrossRef]
- Ong, S.L.; Blenkiron, C.; Haines, S.; Acevedo-Fani, A.; Leite, J.A.S.; Zempleni, J.; Anderson, R.C.; McCann, M.J. Ruminant milk-derived extracellular vesicles: A nutritional and therapeutic opportunity? Nutrients 2021, 13, 2505. [Google Scholar] [CrossRef]
- García-Martínez, J.; Pérez-Castillo, Í.M.; Salto, R.; López-Pedrosa, J.M.; Rueda, R.; Girón, M.D. Beneficial effects of bovine milk exosomes in metabolic interorgan cross-talk. Nutrients 2022, 14, 1442. [Google Scholar] [CrossRef]
- Munagala, R.M.; Aqil, F.; Jeyabalan, J.; Gupta, R.C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016, 371, 48–61. [Google Scholar] [CrossRef]
- Aqil, F.; Munagala, R.; Jeyabalan, J.; Agrawal, A.K.; Kyakulaga, A.H.; Wilcher, S.A.; Gupta, R.C. Milk exosomes-Natural nanoparticles for siRNA delivery. Cancer Lett. 2019, 449, 186–195. [Google Scholar] [CrossRef]
- Del Pozo-Acebo, L.; Hazas, M.L.L.; Tomé-Carneiro, J.; Gil-Cabrerizo, P.; San-Cristobal, R.; Busto, R.; García-Ruiz, A.; Dávalos, A. Bovine milk-derived exosomes as a drug delivery vehicle for miRNA-based therapy. Int. J. Mol. Sci. 2021, 22, 1105. [Google Scholar] [CrossRef]
- Alsaweed, M.; Lai, C.T.; Hartmann, P.E.; Geddes, D.T.; Kakulas, F. Human milk cells contain numerous miRNAs that may change with milk removal and regulate multiple physiological processes. Int. J. Mol. Sci. 2016, 17, 956. [Google Scholar] [CrossRef]
- Leroux, C.; Chervet, M.L.; German, J.B. Perspective: Milk microRNAs as important players in infant physiology and development. Adv. Nutr. 2021, 12, 1625–1635. [Google Scholar] [CrossRef]
- Carrillo-Lozano, E.; Sebastián-Valles, F.; Knott-Torcal, C. Circulating microRNAs in breast milk and their potential impact on the infant. Nutrients 2020, 12, 3066. [Google Scholar] [CrossRef]
- Nguyen, T. Unravelling the mysteries of microRNA in breast milk. Nature 2020, 582, S12–S13. [Google Scholar] [CrossRef]
- Arntz, O.J.; Pieters, B.C.; Oliveira, M.C.; Broeren, M.G.; Bennink, M.B.; de Vries, M.; van Lent, P.L.; Koenders, M.I.; van den Berg, W.B.; van der Kraan, P.M.; et al. Oral administration of bovine milk derived extracellular vesicles attenuates arthritis in two mouse models. Mol. Nutr. Food Res. 2015, 59, 1701–1712. [Google Scholar] [CrossRef]
- Aarts, J.; Boleij, A.; Pieters, B.C.H.; Feitsma, A.L.; van Neerven, R.J.J.; Ten Klooster, J.P.; M’Rabet, L.; Arntz, O.J.; Koenders, M.I.; van de Loo, F.A.J. Flood control: How milk-derived extracellular vesicles can help to improve the intestinal barrier function and break the gut-joint axis in rheumatoid arthritis. Front. Immunol. 2021, 12, 703277. [Google Scholar] [CrossRef]
- Yun, B.; Maburutse, B.E.; Kang, M.; Park, M.R.; Park, D.J.; Kim, Y.; Oh, S. Short communication: Dietary bovine milk-derived exosomes improve bone health in an osteoporosis-induced mouse model. J. Dairy Sci. 2020, 103, 7752–7760. [Google Scholar] [CrossRef]
- Stremmel, W.; Weiskirchen, R.; Melnik, B.C. Milk exosomes prevent intestinal inflammation in a genetic mouse model of ulcerative colitis: A pilot experiment. Inflamm. Intest. Dis. 2020, 5, 117–123. [Google Scholar] [CrossRef]
- Ayyar, K.K.; Moss, A.C. Exosomes in intestinal inflammation. Front. Pharmacol. 2021, 12, 658505. [Google Scholar] [CrossRef]
- Mun, D.; Oh, S.; Kim, Y. Perspectives on bovine milk-derived extracellular vesicles for therapeutic applications in gut health. Food Sci. Anim. Resour. 2022, 42, 197–209. [Google Scholar] [CrossRef]
- Mecocci, S.; Ottaviani, A.; Razzuoli, E.; Fiorani, P.; Pietrucci, D.; De Ciucis, C.G.; Dei Giudici, S.; Franzoni, G.; Chillemi, G.; Cappelli, K. Cow milk extracellular vesicle effects on anin vitro model of intestinal inflammation. Biomedicines 2022, 10, 570. [Google Scholar] [CrossRef]
- Zhang, C.; Lu, X.; Hu, J.; Li, P.; Yan, J.; Ling, X.; Xiao, J. Bovine milk exosomes alleviate cardiac fibrosis via enhancing angiogenesis in vivo and in vitro. J. Cardiovasc. Transl. Res. 2022, 15, 560–570. [Google Scholar] [CrossRef]
- Melnik, B.C.; Weiskirchen, R.; Stremmel, W.; John, S.M.; Schmitz, G. Narrative review of bovine milk exosomes as a potential therapeutic option for hepatic fibrosis. Digest. Med. Res. 2022, 5, 14. [Google Scholar] [CrossRef]
- Askenase, P.W. Exosome carrier effects; resistance to digestion in phagolysosomes may assist transfers to targeted cells; II transfers of miRNAs are better analyzed via systems approach as they do not fit conventional reductionist stoichiometric concepts. Int. J. Mol. Sci. 2022, 23, 6192. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
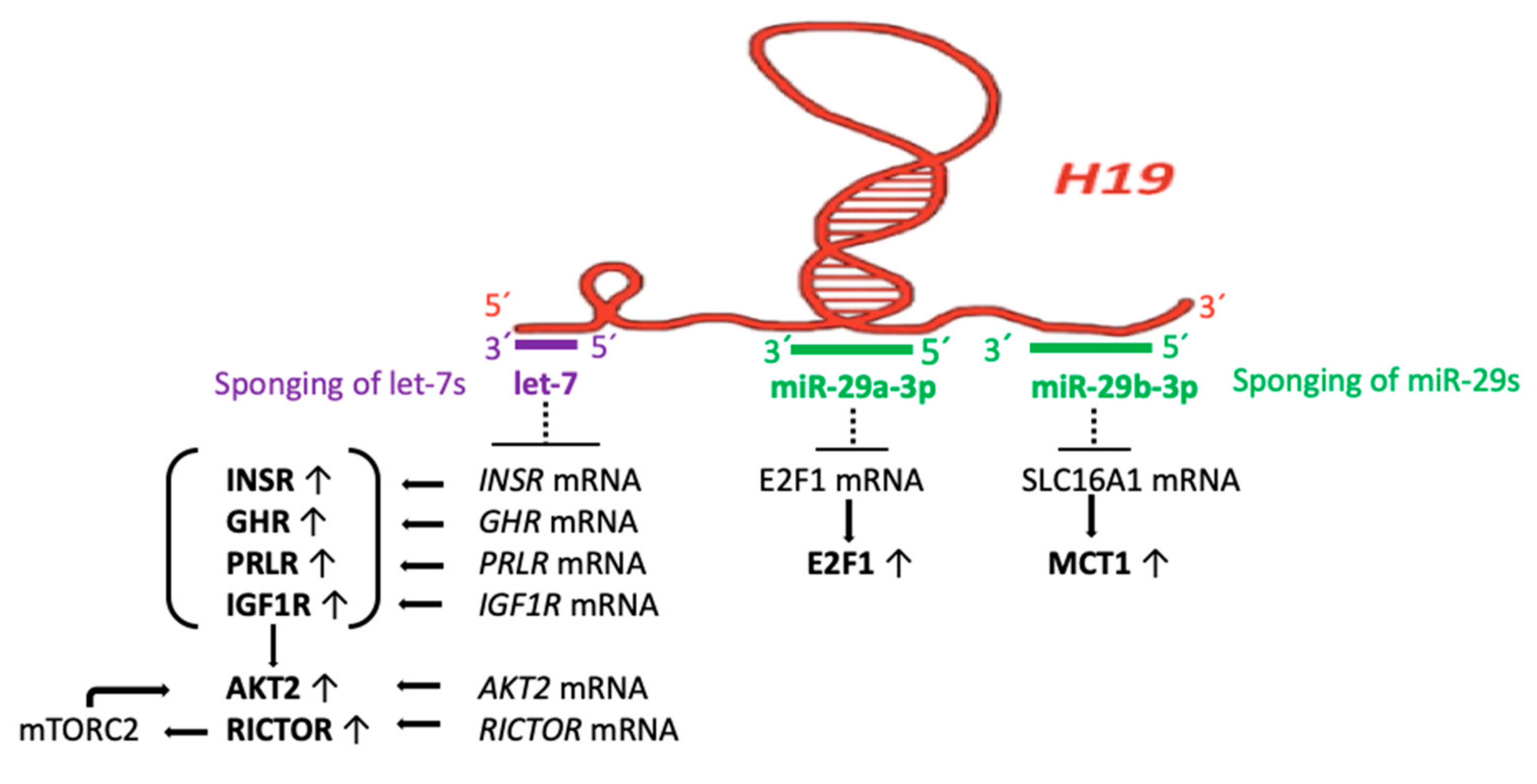{kind=link}
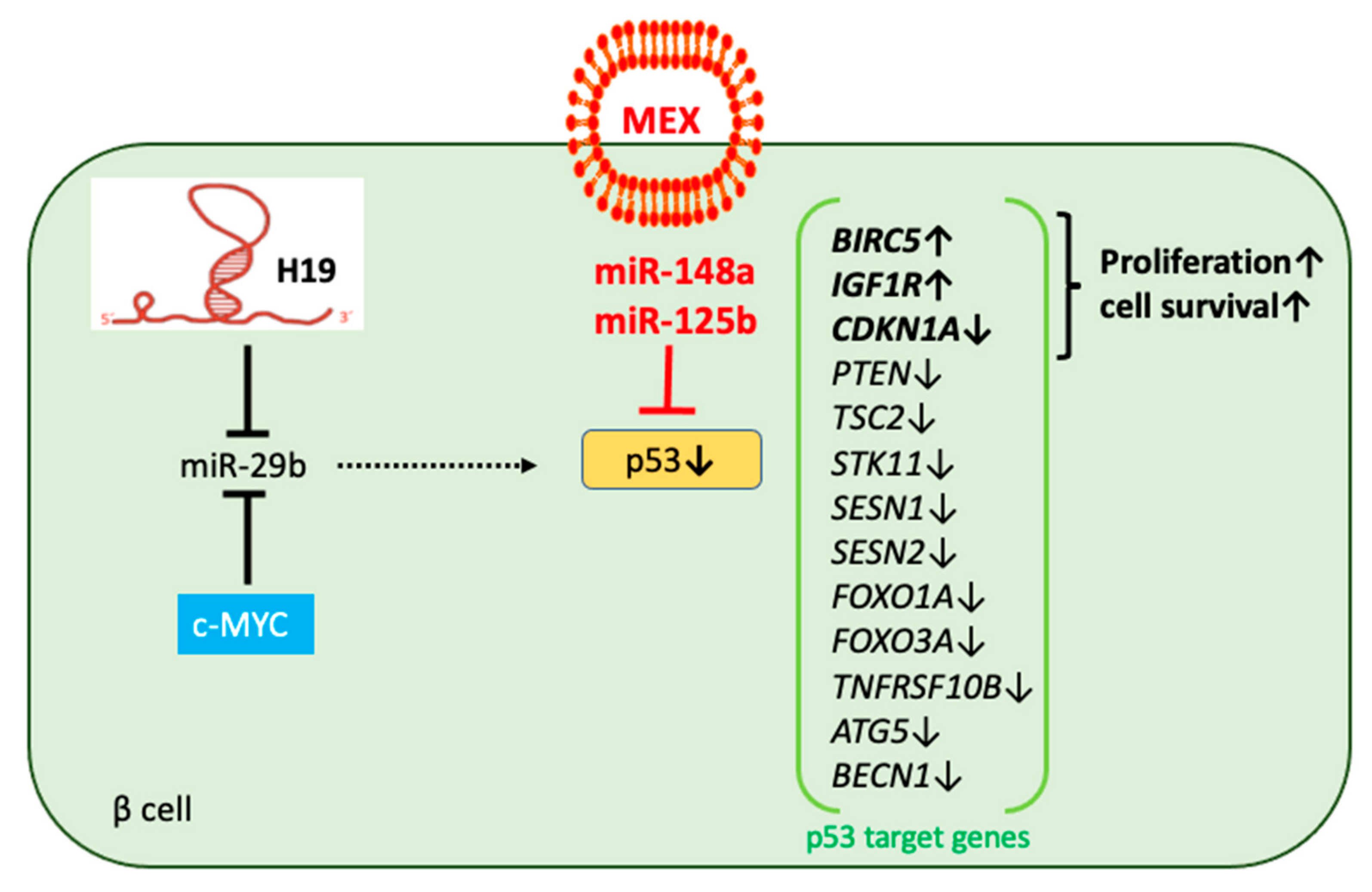{kind=link}
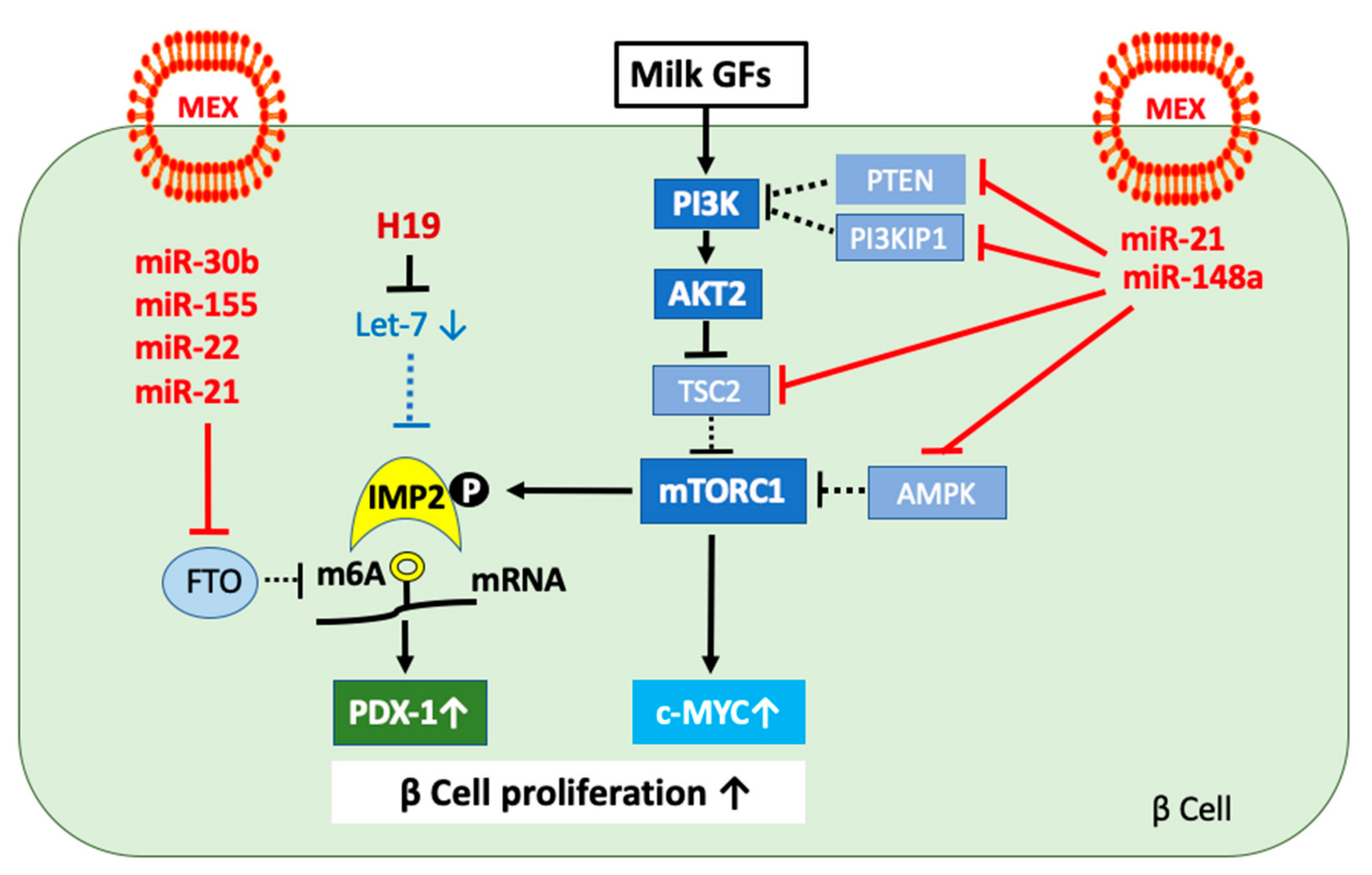{kind=link}
{kind=link}
{kind=link}
{kind=link}
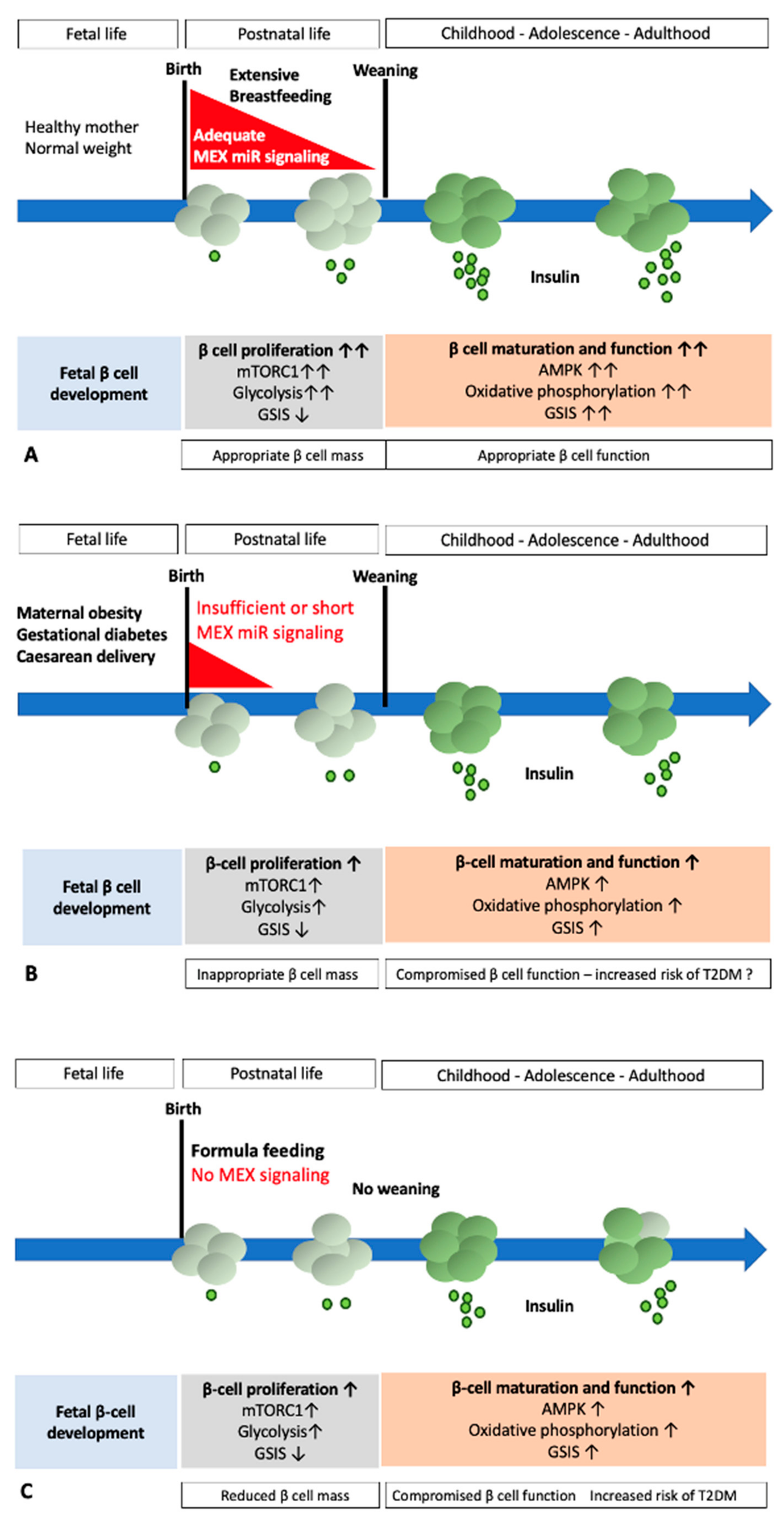{kind=link}
| miR-148a Target Genes | Predicted Gene-Regulatory Effects of MEX miR-148a | References |
|---|---|---|
| TP53 | Increased expression of survivin (BIRC5) and suppression of p21 (CDKN1A) stimulates β cell proliferation; decreased expression of TSC2 activates mTORC1; reduced sestrins (SESN1, SESN2) and LKB (STK11) expression attenuates AMPK activity enhancing mTORC1 activation; decreased expression of miR-29 with diminished suppression of MCT1; decreased expression of FoxO1, which controls β cell differentiation/identity | [132] |
| DNMT1 | Reduced expression of DNMT1 results in lower DNA methylation of gene promoter regions, increasing general transcription | [15,160] |
| PTEN | Reduced expression of PTEN activates PI3K and thus AKT-mTORC1 | [160] |
| MECP2 | Reduced expression of MECP2 attenuates DNMT1-mediated DNA methylation, enhancing gene expression | [183] |
| PRKAA1 | Reduced AMPK activity increases mTORC1 signaling | [231] |
| PRKAG2 | Reduced AMPK activity increases mTORC1 signaling | [232] |
| TSC2 | Reduced expression of TSC2 enhances RHEB-mediated mTORC1 activation | [282] |
| ESRRG | Reduced expression of ERRγ inhibits the mitochondrial function and oxidative phosphorylation required for GSIS | [258] |
| PPARGC1A | Suppression of mitochondrial biogenesis, oxidative phosphorylation, and GSIS | [241,242] |
| CPT1A | Reduced transfer of free fatty acids to mitochondrial oxidative phosphorylation | [245] |
| HIF1AN | enhances glycolysis for the promotion of β cell growth | [296] |
| MAFB | Reduced expression of MAFB results in reduced expression of GLUT1 and GLUT2 | [313] |
| NEUROD1 | Attenuation of β cell differentiation and insulin gene expression | [347] |
| SLC2A1 | Reduced expression of GLUT1 decreases glucose sensing | [317] |
| SMAD2 | Reduced SMAD2-mediated TGF-β signaling | [334] |
| MAX | Reduced c-MYC/MAX-mediated promoter activation of pyruvate kinase (PK) expression attenuates PK-controlled GSIS | [180] |
| ABCA1 | Impaired insulin granule function and insulin secretion | [324] |
| USP4 | Increased proteasomal degradation of TGFBR1 attenuates TGF-β signaling | [335,336,337] |
| AQP7 | Reduced AQP7-mediated insulin secretion | [351,352] |
| Impact Factors | Effects on MEX miR Concentrations | References |
|---|---|---|
| Maternal obesity | miR-30b | [417] |
| Gestational diabetes mellitus | miR-30b | [429] |
| Maternal stress during pregnancy | [402] | |
| Caesarean delivery | miR-125b | [153] |
| Oxytocin treatment | miR-30 miR-320 | [438] |
| Holder pasteurized human donor milk | [457] | |
| Preterm birth | miR-22 | [182,370] |
| Infant formula | (below detection limit) | [153,158,458] |
| Soy protein-based formula | Impaired intestinal MEX miR uptake | [459] |
| Raw cow milk | miR-21 | [158,475] |
| Pasteurized cow milk | miR-148a miR-21 miR-30d | [475,476] |
| Ultraheat-treated cow milk | [475,476] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melnik, B.C.; Schmitz, G. Milk Exosomal microRNAs: Postnatal Promoters of β Cell Proliferation but Potential Inducers of β Cell De-Differentiation in Adult Life. Int. J. Mol. Sci. 2022, 23, 11503. https://doi.org/10.3390/ijms231911503
Melnik BC, Schmitz G. Milk Exosomal microRNAs: Postnatal Promoters of β Cell Proliferation but Potential Inducers of β Cell De-Differentiation in Adult Life. International Journal of Molecular Sciences. 2022; 23(19):11503. https://doi.org/10.3390/ijms231911503
Chicago/Turabian StyleMelnik, Bodo C., and Gerd Schmitz. 2022. "Milk Exosomal microRNAs: Postnatal Promoters of β Cell Proliferation but Potential Inducers of β Cell De-Differentiation in Adult Life" International Journal of Molecular Sciences 23, no. 19: 11503. https://doi.org/10.3390/ijms231911503