
A Walk in the Memory, from the First Functional Approach up to Its Regulatory Role of Mitochondrial Bioenergetic Flow in Health and Disease: Focus on the Adenine Nucleotide Translocator


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondria: Not Just an Energy Production Site
2.1. Two Membranes and Two Spaces with Specific Functions
2.2. As Students in a Bioenergetics Laboratory
2.2.1. Those Were Times When
2.2.2. Becoming Familiar with the Mitochondrion
2.2.3. There Are Those Who Go and There Are Those Who Come: Traffic across the Mitochondrial Membranes
2.2.4. Methods Used to Follow the Mitochondrial Trafficking of Metabolites
2.3. ADP and ATP Move Back and Forth within the Mitochondria
- (1)
- ADP is exchanged against intramitochondrial ATP with a one-to-one stoichiometry when mitochondria are actively respiring;
- (2)
- The only physiological substrates are ADP and ATP; surprisingly in their free forms, i.e., Mg-ADP and Mg-ATP, they are not recognized by the carrier;
- (3)
- Very high selectivity for the substrates: It does not transport AMP;
- (4)
- The highly charged and very hydrophilic nature of the large substrates;
- (5)
- The strong regulation by high electrical forces of the mitochondrial membrane potential of the highly charged substrates ADP3− and ATP4−;
- (6)
- The kinetic parameters of the carrier are consistent with the mitochondrial ATP production and the cell nucleotide concentrations under physiological conditions;
- (7)
- The high levels of the underlying carrier in mitochondria to cope with the high demand for ATP in most aerobic cells;
- (8)
- The unique existence of highly specific inhibitors, as represented first by atractyloside (ATR) and then by other atractylogenin glucosides, in particular by carboxyatractyloside (CAT), greatly facilitating the characterization of the transport sites;
- (9)
- Another type inhibitor was represented by bongkrekic acid (BKA), which became a key element to elucidating the transport mechanism of transport of the ADP/ATP carrier at the molecular level;
- (10)
- Stabilized by these inhibitors toward detergents, in particular by CAT and thanks to its high levels in heart mitochondria, the ANT protein became the first carrier protein to be isolated in the native state;
- (11)
- The translocation mechanism of ANT was further elucidated and the involved conformation changes were demonstrated;
- (12)
- The first rational reconstitution of a carrier into vesicles was achieved, with the isolated ANT, and the transport parameters were determined in detail, in particular the differences between ADP and ATP and the control by the membrane potential and lastly;
- (13)
- The complete primary structure was determined by amino acid sequencing with large amounts of purified ANT protein.,. The elucidation of the complete primary structures of ANT in 1982 [28] and UCP1 in 1985 [29] by Klingenberg’s group, together with the sequence of the phosphate carrier, led to the initial definition of the homologous family of mitochondrial solute transporters.
3. ANT: Its Essential Features
3.1. A Brief History of ANT
3.2. ANT Exchanges Cytosolic ADP for Newly Synthesized Mitochondrial ATP
3.3. Some Data about ANT Structure
3.3.1. Cardiolipin Associates Strongly with the Mitochondrial ADP/ATP Carrier
3.4. Does ANT Work as a Dimer or Monomer?
3.5. ANT Isoforms
3.6. ANT: A Multitasking Protein
3.6.1. ANT Is Part of mRC Supercomplexes
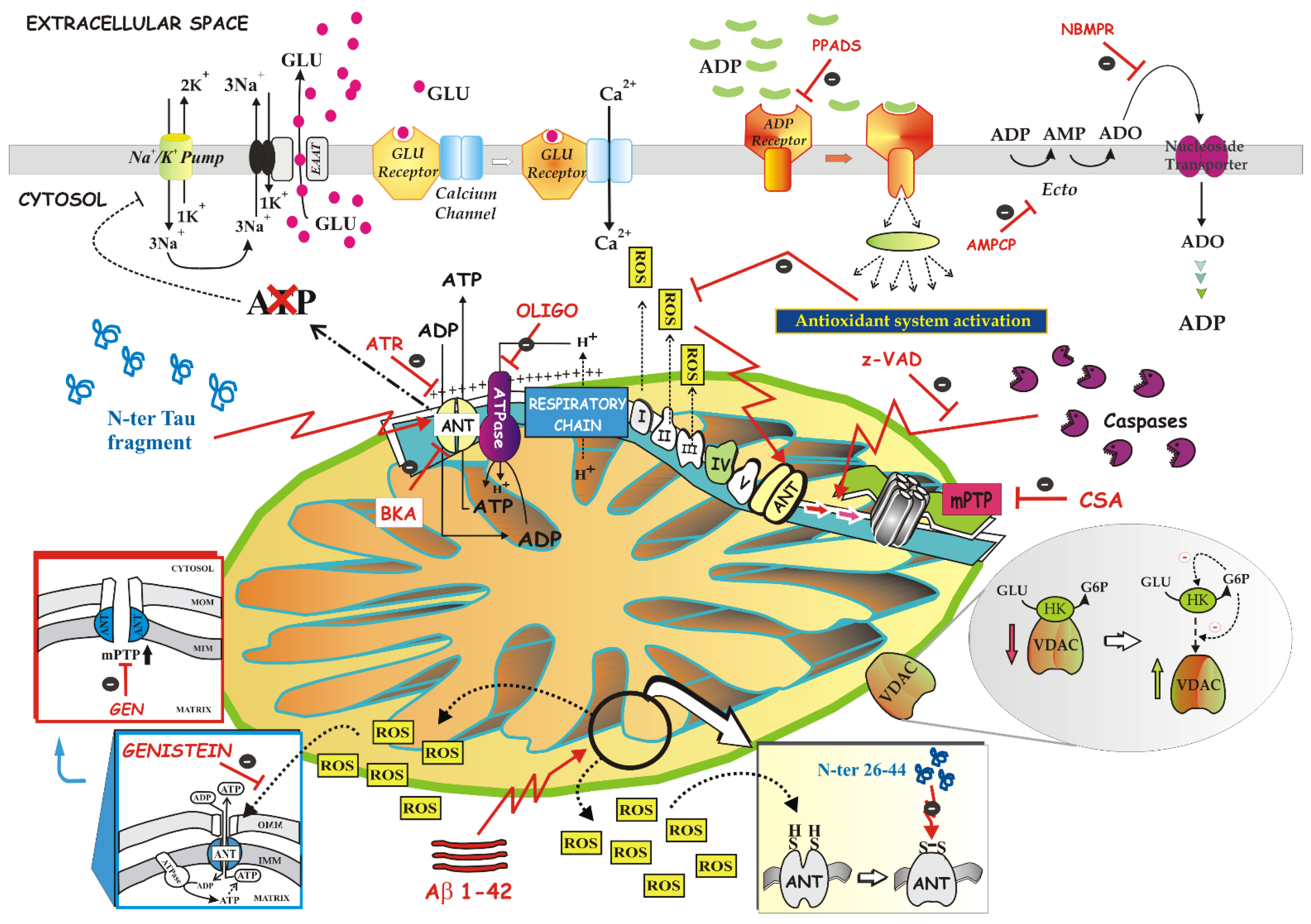
3.6.2. ANT Is a Component of mPTP
3.6.3. ANT Promotes Mitophagy
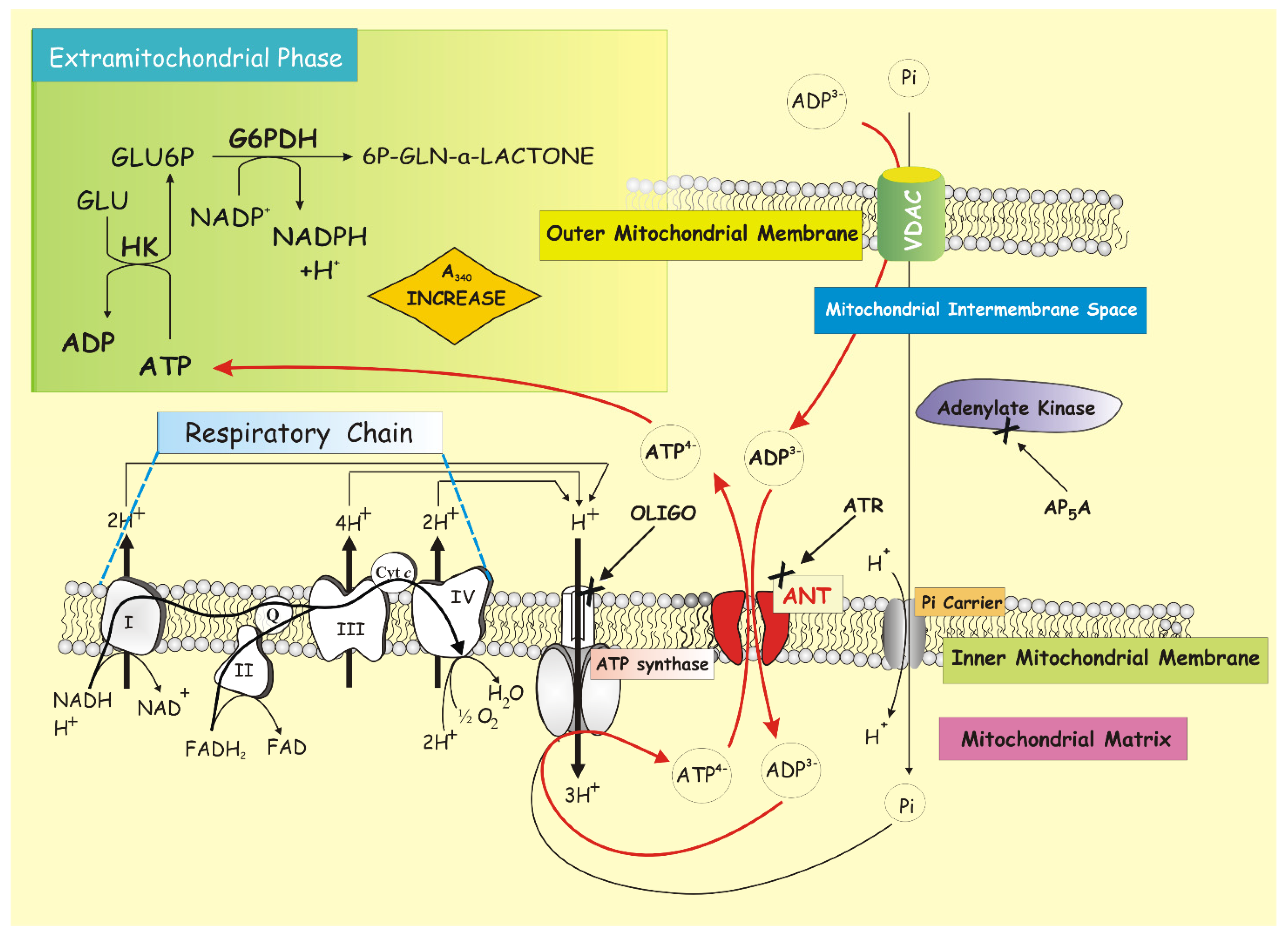
3.7. ANT Kinetic Determination by Using the ATP Detecting System
4. ANT: In Health and in Disease
4.1. Mutations or Altered Gene Expression Associate ANT with Human Diseases
4.2. ANT: Its Metabolic Role in Various Diseases
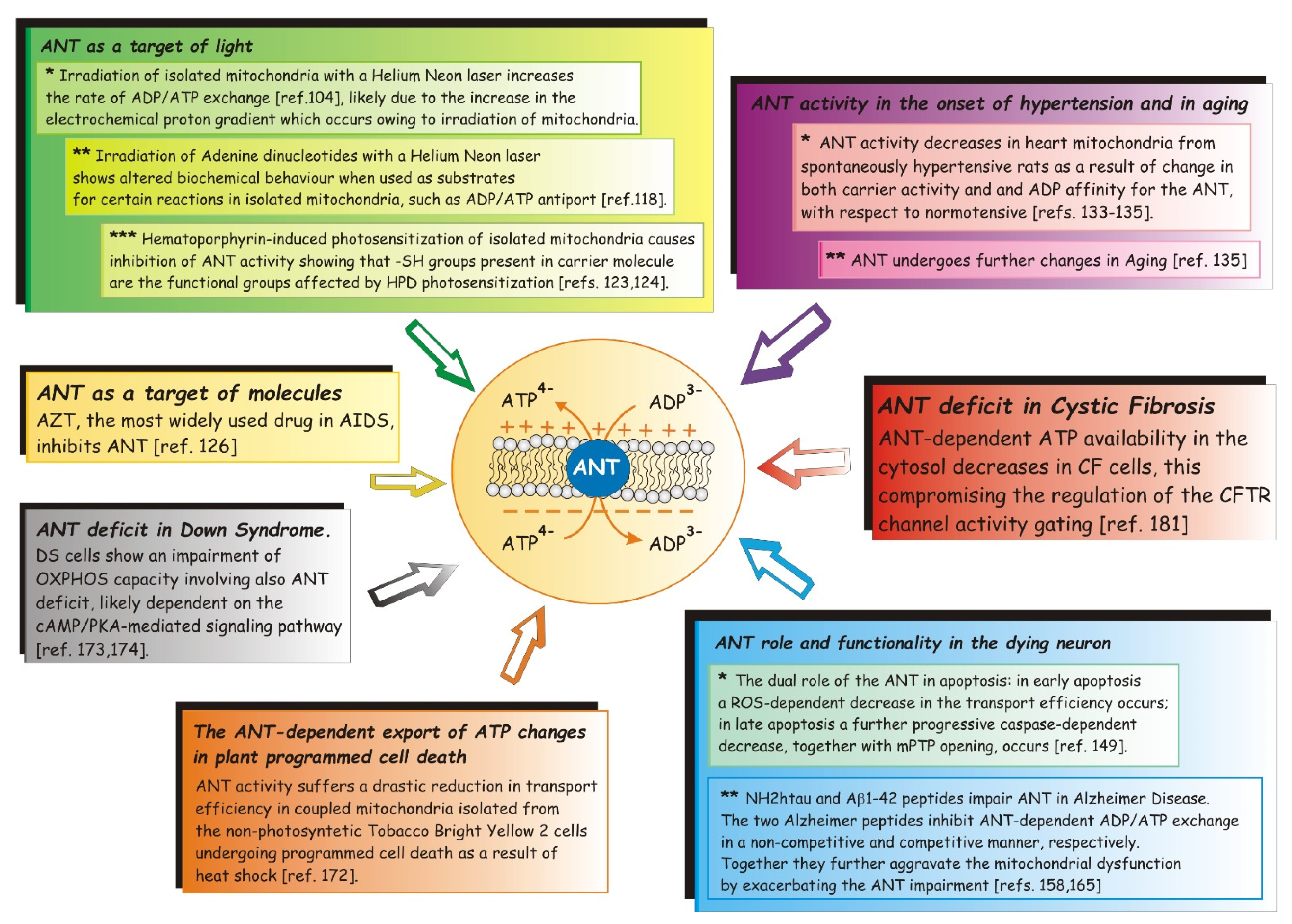
4.2.1. ANT as a Target of Light
4.2.2. ANT as a Target of Molecules
4.2.3. The Rate of ATP Export Declines with the Onset of Hypertension and in Aging
4.2.4. Apoptosis and Necrosis: ANT Role and Functionality in the Dying Neuron
4.2.4.1. The Dual Role of the Mitochondrial ANT in Apoptosis
4.2.4.2. NH2htau and Aβ1-42 Peptides Impair Mitochondrial ANT-1 in Alzheimer’s Disease
4.2.5. The ANT-Dependent Export of ATP Changes in Plant-Programmed Cell Death (PCD)
4.2.6. ANT Deficit in Down Syndrome and in Cystic Fibrosis
5. Closing Remarks
Funding
Conflicts of Interest
Abbreviations
| Aβ | β-amyloid peptide |
| AD | Alzheimer’s disease |
| ADK | adenylate kinase |
| adPEO | autosomal dominant progressive external ophthalmoplegia |
| AIDS | acquired immunodeficiency syndrome |
| ANT | adenine nucleotide translocator |
| Ap5A | P1,P5-di(adenosine-50)penta-phosphate |
| ATP D.S. | ATP detecting system |
| ATR | atractyloside |
| AZT | 3′-azido-3′-deoxythymidine |
| BKA | bongkrekic acid |
| CAT | carboxyatractyloside |
| CF | cystic fibrosis |
| CL | cardiolipin |
| CM | cristae membranes |
| CsA | cyclosporin A |
| CyP-D | cyclophilin D |
| Cyt c | cytochrome c |
| DIDS | 4,40-diisothiocyanostilbene-2,20-disulfonic acid |
| DS | Down syndrome |
| G6PDH | glucose-6-phosphate dehydrogenase |
| HK | hexokinase |
| HSF | human skin fibroblasts |
| IBM | inner boundary membrane |
| IMM | inner mitochondrial membrane |
| MLV | mitochondria left ventricles |
| mPTP | mitochondrial permeability transition pore |
| mRC | mitochondrial respiratory chain |
| NMDA | N-methyl-D-aspartate |
| OLIGO | oligomycin |
| OMM | outer mitochondrial membrane |
| ORN | ornithine |
| OXPHOS | oxidative phosphorylation |
| PCD | programmed cell death |
| PF II | photofrin II |
| PINK1 | phosphatase and tensin homolog-induced kinase 1 |
| PKA | protein kinase A |
| RCS | respiratory chain supercomplex |
| RLM | rat liver mitochondria |
| ROS | reactive oxygen species |
| SHRs | spontaneously hypertensive rats |
| SOD | superoxide dismutase |
| TIM | translocase of the inner membrane |
| TBY-2 | Tobacco Bright Yellow- 2 |
| VDAC | voltage dependent anion channel |
| WKYRs | Wistar Kyoto rats |
References
- Palade, G.E. An electron microscope study of the mitochondrial structure. J. Histochem. Cytochem. 1953, 1, 188–211. [Google Scholar] [CrossRef]
- Vogel, F.; Bornhovd, C.; Neupert, W.; Reichert, A.S. Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 2006, 175, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Renken, C.; Martone, M.E.; Young, S.J.; Ellisman, M.; Frey, T. Electron tomography of neuronal mitochondria: Three-dimensional structure and organization of cristae and membrane contacts. J. Struct. Biol. 1997, 119, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Appelhans, T.; Busch, K.B. Dynamic imaging of mitochondrial membrane proteins in specific sub-organelle membrane locations. Biophys. Rev. 2017, 9, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlov, E.; Grigoriev, S.M.; Dejean, L.M.; Zweihorn, C.L.; Mannella, C.A.; Kinnally, K.W. The mitochondrial channel VDAC has a cation-selective open state. Biochim. Biophys. Acta 2005, 1710, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Lipper, C.H.; Stofleth, J.T.; Bai, F.; Sohn, Y.-S.; Roy, S.; Mittler, R.; Nechushtai, R.; Onuchic, J.N.; Jennings, P.A. Redox-dependent gating of VDAC by mitoNEET. Proc. Natl. Acad. Sci. USA 2019, 116, 19924–19929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobba, A.; Amadoro, G.; La Piana, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Glucose-6-phosphate tips the balance in modulating apoptosis in cerebellar granule cells. FEBS Lett. 2015, 589, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, V.; Griffin, L.; Towbin, J.; Gelb, B.; Worley, K.; McCabe, E.R. Porin interaction with hexokinase and glycerol kinase: Metabolic microcompartmentation at the outer mitochondrial membrane. Biochem. Med. Metab. Biol. 1991, 45, 271–291. [Google Scholar] [CrossRef]
- Golshani-Hebroni, S.G.; Bessman, S.P. Hexokinase binding to mitochondria: A basis for proliferative energy metabolism. J. Bioenerg. Biomembr. 1997, 29, 331–338. [Google Scholar] [CrossRef]
- Klingenberg, M.; Riccio, P.; Aquila, H. Isolation of the ADP, ATP carrier as the carboxyatractylate protein complex from mitochondria. Biochim. Biophys. Acta. 1978, 503, 193–210. [Google Scholar] [CrossRef]
- Krämer, R.; Aquila, H.; Klingenberg, M. Isolation of the unliganded adenosine 5′-diphosphate, adenosine 5′-triphosphate Carrier-linked binding protein and incorporation into the membranes of liposomes. Biochemistry 1977, 16, 4949–4953. [Google Scholar] [CrossRef]
- Krämer, R.; Klingenberg, M. Electrophoretic control of reconstituted adenine nucleotide translocation. Biochemistry 1982, 21, 1082–1089. [Google Scholar] [CrossRef]
- LaNoue, K.F.; Schoolwerth, A.C. Metabolite transport in mitochondria. Annu. Rev. Biochem. 1979, 48, 871–922. [Google Scholar] [CrossRef] [PubMed]
- LaNoue, K.F.; Schoolwerth, A.C. Metabolic transport in mammalian mitochondria. In Bioenergetics; Elsevier: Amsterdam, The Netherlands, 1984; pp. 221–261. [Google Scholar]
- Passarella, S.; Atlante, A.; Valenti, D.; de Bari, L. The role of mitochondrial transport in energy metabolism. Mitochondrion 2003, 2, 319–343. [Google Scholar] [CrossRef]
- Gamble, J.G.; Lehninger, A.L. Transport of ornithine and citrulline across the mitochondrial membrane. J. Biol. Chem. 1973, 248, 610–618. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Identification and purification of the ornithine/citrulline carrier from rat liver mitochondria. Eur. J. Biochem. 1992, 207, 449–454. [Google Scholar] [CrossRef]
- Passarella, S.; Atlante, A.; Quagliariello, E. Metabolite transport in rat kidney mitochondria: Ornithine/phosphate translocator. Biochem. Biophys. Res. Commun. 1989, 158, 870–879. [Google Scholar] [CrossRef]
- Passarella, S.; Atlante, A.; Quagliariello, E. Ornithine/phosphate antiport in rat kidney mitochondria. Some characteristics of the process. Eur. J. Biochem. 1990, 193, 221–227. [Google Scholar] [CrossRef]
- Palmieri, F.; Klingenberg, M. Direct methods for measuring metabolite transport and distribution in mitochondria. Methods Enzymol. 1979, 56, 279–301. [Google Scholar]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, J.B.; Haarhoff, K.N. Biochemistry of Mitochondria; Slater, E.C., Kaniuga, Z., Wojtczak, L., Eds.; Academic Press: London, UK; New York, NY, USA, 1967; p. 75. [Google Scholar]
- Atlante, A.; Passarella, S.; Giannattasio, S.; Quagliariello, E. Fumarate permeation in rat liver mitochondria: Fumarate/malate and fumarate/phosphate translocators. Biochem. Biophys. Res. Commun. 1985, 132, 8–18. [Google Scholar] [CrossRef]
- Atlante, A.; Passarella, S.; Minervini, G.M.; Quagliariello, E. Glutamine transport in normal and acidotic rat kidney mitochondria. Arch. Biochem. Biophys. 1994, 315, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.B.; Crofts, A.R. Gramicidin and ion transport in isolated liver mitochondria. Biochem. J. 1965, 95, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunji, E.R.; Aleksandrova, A.; King, M.S.; Majd, H.; Ashton, V.L.; Cerson, E.; Springett, R.; Kibalchenko, M.; Tavoulari, S.; Crichton, P.G.; et al. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta 2016, 1863, 2379–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquila, H.; Misra, D.; Eulitz, M.; Klingenberg, M. Complete amino acid sequence of the ADP/ATP carrier from beef heart mitochondria. Hoppe Seylers Z. Physiol. Chem. 1982, 363, 345–349. [Google Scholar]
- Aquila, H.; Link, T.A.; Klingenberg, M. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane. EMBO J. 1985, 4, 2369–2376. [Google Scholar] [CrossRef]
- Siekevitz, P.; Potter, V.R. Biochemical structure of mitochondria. II. Radioactive labeling of intra-mitochondrial nucleotides during oxidative phosphorylation. J. Biol. Chem. 1955, 215, 237–255. [Google Scholar] [CrossRef]
- Pressman, B.C. Intramitochondrial nucleotides. I. Some factors affecting net interconversions of adenine nucleotides. J. Biol. Chem. 1958, 232, 967–978. [Google Scholar] [CrossRef]
- Bruni, A.; Contessa, A.R.; Luciani, S. Atractyloside as inhibitor of energy-transfer reactions in liver mitochondria. Biochim. Biophys. Acta 1962, 60, 301–311. [Google Scholar] [CrossRef]
- Vignais, P.V.; Vignais, P.M.; Stanislas, E. Action of potassium atractylate on oxidative phosphorylation in mitochondria and in submitochondrial particles. Biochim. Biophys. Acta 1962, 60, 284–300. [Google Scholar] [CrossRef]
- Nury, H.; Dahout-Gonzalez, C.; Trézéguet, V.; Lauquin, G.J.M.; Brandolin, G.; Pebay-Peyroula, E. Relations Between Structure and Function of the Mitochondrial ADP/ATP Carrier. Ann. Rev. Biochem. 2006, 75, 713–774. [Google Scholar] [CrossRef] [Green Version]
- Henderson, P.J.; Lardy, H.A. Bongkrekic acid: An inhibitor of the adenine nucleotide translocase of mitochondria. J. Biol. Chem. 1970, 245, 1319–1326. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Bround, M.J.; Bersb, D.M.; Molkentina, J.D. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J. Mol. Cell. Cardiol. 2020, 144, A3–A13. [Google Scholar] [CrossRef]
- Klingenberg, M. Ligand-protein interaction in biomembrane carriers. The induced transition fit of transport catalysis. Biochemistry 2005, 44, 8563–8570. [Google Scholar] [CrossRef]
- Daniele, C.; Dahamna, S.; Firuzi, O.; Sekfali, N.; Saso, L.; Mazzanti, G. Atractylis gummifera L. poisoning: An ethnopharmacological review. J. Ethnopharmacol. 2005, 97, 175–181. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, M.; Grebe, K.; Scherer, B. The binding of atractylate and carboxyatractylate to mitochondria. Eur. J. Biochem. 1975, 52, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Lumbach, G.; Cox, H.; Berends, W.J. Elucidation of the chemical structure of bongkrekic acid—I: Isolation, purification and properties of bongkrekic acid. Tetrahedron 1970, 26, 5993–5999. [Google Scholar] [CrossRef]
- Krämer, R.; Klingenberg, M. Reconstitution of inhibitor binding properties of the isolated adenosine 5′-diphosphate, adenosine 5′-triphosphate carrier-linked binding protein. Biochemistry 1977, 16, 4954–4961. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Klingenberg, M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 1985, 24, 3821–3826. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Nuscher, B. Specific cardiolipin binding interferes with labeling of sulfhydryl residues in the adenosine diphosphate/adenosine triphosphate carrier protein from beef heart mitochondria. Biochemistry 1996, 35, 15784–15790. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Stockl, A.; Schlame, M.; Beyer, K.; Klingenberg, M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J. Biol. Chem. 1994, 269, 1940–1944. [Google Scholar] [CrossRef]
- Claypool, S.M. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta. 2009, 1788, 2059–2068. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 2005, 280, 29403–29408. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.L.; Ruprecht, J.R.; Kunji, E.R.S.; Robinson, A.J. Cardiolipin dynamics and binding to conserved residues in the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1035–1045. [Google Scholar] [CrossRef]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Nury, H.; Dahout-Gonzalez, C.; Trézéguet, V.; Lauquin, G.; Brandolin, G.; Pebay-Peyroula, E. Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett. 2005, 579, 6031–6036. [Google Scholar] [CrossRef]
- Senoo, N.; Kandasamy, S.; Ogunbona, O.B.; Baile, M.G.; Lu, Y.; Claypool, S.M. Cardiolipin, conformation, and respiratory complex-dependent oligomerization of the major mitochondrial ADP/ATP carrier in yeast. Sci. Adv. 2020, 6, eabb0780. [Google Scholar] [CrossRef]
- Han, X.; Yang, J.; Cheng, H.; Yang, K.; Abendschein, D.R.; Gross, R.W. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 2005, 44, 16684–16694. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Koumura, T.; Nakajima, R.; Nomura, K.; Nakagawa, Y. Protection from inactivation of the adenine nucleotide translocator during hypoglycaemia-induced apoptosis by mitochondrial phospholipid hydroperoxide glutathione peroxidase. Biochem. J. 2003, 371, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunji, E.R.S.; Ruprecht, J.J. The mitochondrial ADP/ATP carrier exists and functions as a monomer. Biochem. Soc. Trans. 2020, 48, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Subramaniam, K.; Pertuiset, C.; Pervaiz, S. Adenine nucleotide translocase family: Four isoforms for apoptosis modulation in cancer. Oncogene 2011, 30, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Forrest, M.D. Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy. bioRxiv 2015. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Pierri, C.L. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar]
- Levy, S.E.; Chen, Y.-S.; Graham, B.H.; Wallace, D.C. Expression and sequence analysis of the mouse adenine nucleotide translocase 1 and 2 genes. Gene 2000, 254, 57–66. [Google Scholar] [CrossRef]
- Chevrollier, A.; Loiseau, D.; Reynier, P.; Stepien, G. Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim. Biophys. Acta 2011, 1807, 562–567. [Google Scholar] [CrossRef] [Green Version]
- Barath, P.; Luciakova, K.; Hodny, Z.; Li, R.; Nelson, B.D. The growth-dependent expression of the adenine nucleotide translocase-2 (ANT2) gene is regulated at the level of transcription and is a marker of cell proliferation. Exp. Cell Res. 1999, 248, 583–588. [Google Scholar] [CrossRef]
- Doerner, A.; Pauschinger, M.; Badorff, A.; Noutsias, M.; Giessen, S.; Schulze KBilger, J.; Rauch, U.; Schultheiss, H.P. Tissue-specific transcription pattern of the adenine nucleotide translocase isoforms in humans. FEBS Lett. 1997, 414, 258–262. [Google Scholar]
- Dolce, V.; Scarcia, P.; Iacopetta, D.; Palmieri, F. A fourth ADP/ATP carrier isoform in man: Identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 2005, 579, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.H.; Brower, J.V.; Resnick, J.L.; Oh, S.P.; Terada, N. Adenine nucleotide translocase 4 is expressed within embryonic ovaries and dispensable during oogenesis. Reprod. Sci. 2015, 22, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.K.; Schubert, A.; Rocks, O.; Grimm, S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J. Cell Biol. 1999, 147, 1493–1502. [Google Scholar] [CrossRef]
- Zamora, M.; Merono, C.; Vinas, O.; Mampel, T. Recruitment of NF-kappaB into mitochondria is involved in adenine nucleotide translocase 1 (ANT1)-induced apoptosis. J. Biol. Chem. 2004, 279, 38415–38423. [Google Scholar] [CrossRef]
- Belzacq, A.S.; Vieira, H.L.; Verrier, F.; Vandecasteele, G.; Cohen, I.; Prevost, M.C.; Larquet, E.; Pariselli, F.; Petit, P.X.; Kahn, A.; et al. Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Res. 2003, 63, 541–546. [Google Scholar]
- Zamora, M.; Granell, M.; Mampel, T.; Vinas, O. Adenine nucleotide translocase 3 (ANT3) overexpression induces apoptosis in cultured cells. FEBS Lett. 2004, 563, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Chevrollier, A.; Loiseau, D.; Chabi, B.; Renier, G.; Douay, O.; Malthièry, Y.; Stepien, G. ANT2 isoform required for cancer cell glycolysis. J. Bioenerg. Biomembr. 2005, 37, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Lemasters, J.J. ATP/ADP ratio, the missed connection between mitochondria and the Warburg effect. Mitochondrion 2014, 19, 78–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunji, E.R.S. The role and structure of mitochondrial carriers. FEBS Lett. 2004, 564, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Nuskova, H.; Mracek, T.; Mikulova, T.; Vrbacky, M.; Kovarova, N.; Kovalcikova, J.; Pecina, P.; Houštěk, J. Mitochondrial ATP synthasome: Expression and structural interaction of its components. Biochem. Biophys. Res. Commun. 2015, 464, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.W.; Acoba, M.G.; Selvaraju, K.; Huang, T.C.; Nirujogi, R.S.; Sathe, G.; Pandey, A.; Claypool, S.M. Human adenine nucleotide translocases physically and functionally interact with respirasomes. Mol. Biol. Cell 2017, 28, 1489–1506. [Google Scholar] [CrossRef]
- Parodi-Rullán, R.M.; Chapa-Dubocq, X.; Guzmán-Hernández, R.; Jang, S.; Torres-Ramos, C.A.; Ayala-Peña, S.; Javadov, S. The Role of Adenine Nucleotide Translocase in the Assembly of Respiratory Supercomplexes in Cardiac Cells. Cells 2019, 8, 1247. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Fournier, N.; Ducet, G.; Crevat, A. Action of cyclosporine on mitochondrial calcium fluxes. J. Bioenerg. Biomembr. 1987, 19, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Naumova, N.; Šachl, R. Regulation of Cell Death by Mitochondrial Transport Systems of Calcium and Bcl-2 Proteins. Membranes 2020, 10, 299. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, M.; Andrade-Navarro, M.A.; Gross, A. Mitochondrial carriers and pores: Key regulators of the mitochondrial apoptotic program? Apoptosis 2007, 12, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, A.W.; Halestrap, A.P. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2008, 1777, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Davidson, A.M. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990, 268, 153–160. [Google Scholar]
- Belzacq, A.S.; Vieira, H.L.; Kroemer, G.; Brenner, C. The adenine nucleotide translocator in apoptosis. Biochimie 2002, 84, 167–176. [Google Scholar] [CrossRef]
- Bernardi, P. Mechanisms for Ca 2+-dependent permeability transition in mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 2743–2744. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Bernardi, P. Measurement of membrane permeability and the mitochondrial permeability transition. Methods Cell Biol. 2020, 155, 369–379. [Google Scholar]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP Translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and Cyp D. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP synthase C-subunit-deficient mitochondria have a small cyclosporine A-sensitive channel, but lack the permeability transition pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brustovetsky, N. The Role of Adenine Nucleotide Translocase in the Mitochondrial Permeability Transition. Cells 2020, 9, 2686. [Google Scholar] [CrossRef] [PubMed]
- Um, J.H.; Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Supinski, G.S.; Schroder, E.A.; Callahan, L.A. Mitochondria and Critical Illness. Chest 2020, 157, 310–322. [Google Scholar] [CrossRef]
- Lim, K.L.; Dawson, V.L.; Dawson, T.M. Parkin-mediated lysine 63-linked polyubiquitination: A link to protein inclusions formation in Parkinson’s and other conformational diseases? Neurobiol. Aging. 2006, 27, 524–529. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. The art of mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Wang, W.-J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ATP/ADP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER–Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, R.; Klingenberg, M. Enhancement of reconstituted ADP, ATP exchange activity by phosphatidylethanolamine and by anionic phospholipids. FEBS Lett. 1980, 119, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Krämer, R.; Klingenberg, M. Reconstitution of adenine nucleotide transport with purified ADP, ATP-carrier protein. FEBS Lett. 1977, 82, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Krämer, R.; Klingenberg, M. Modulation of the reconstituted adenine nucleotide exchange by membrane potential. Biochemistry 1980, 19, 556–560. [Google Scholar] [CrossRef]
- Krämer, R.; Klingenberg, M. Reconstitution of adenine nucleotide transport from beef heart mitochondria. Biochemistry 1979, 18, 4209–4215. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Catterall, W.A. The use of thin-layer chromatography on poly(ethyleneimine)cellulose to facilitate assays of ATP-ADP exchange, ATP-Pi exchange, adenylate kinase, and nucleoside diphosphokinase activity. Methods Enzymol. 1979, 55, 283–289. [Google Scholar]
- Hartwick, R.A.; Brown, P.R. The performance of microparticle chemically-bonded anion-exchange resins in the analysis of nucleotides. J. Chromatogr. 1975, 112, 650–662. [Google Scholar] [CrossRef]
- Lemasters, I.J.; Hackenbrock, C.R. Continuous measurement of adenosine triphosphate with firefly luciferase luminescence. Methods Enzymol. 1979, 56, 530–544. [Google Scholar]
- Block, M.R.; Boulay, F.; Brandolin, G.; Dupont, Y.; Lauquin, G.J.M.; Vignais, P.V. Fluorescent probes of the mitochondrial ADP/ATP carrier protein. Methods Enzymol. 1986, 125, 639–649. [Google Scholar]
- Wibom, R.; Lundin, A.; Hultman, E. A sensitive method for measuring ATP-formation in rat muscle mitochondria. Scand. J. Clin. Lab. Investig. 1990, 50, 143–152. [Google Scholar] [CrossRef]
- Passarella, S.; Ostuni, A.; Atlante, A.; Quagliariello, E. Increase in the ADP/ATP exchange in rat liver mitochondria irradiated in vitro by helium-neon laser. Biochem. Biophys. Res. Commun. 1988, 156, 978–986. [Google Scholar] [CrossRef]
- Lienhard, G.E.; Secemski, I.I. P1,P5-di (adenosine-5¢) pentaphosphate, a potent multisubstrate inhibitor of adenylate kinase. J. Biol. Chem. 1973, 248, 1121–1123. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Klingenberg, M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 1996, 35, 8483–8488. [Google Scholar] [CrossRef] [PubMed]
- Malorni, W.; Farrace, M.; Matarrese, P.; Tinari, A.; Ciarlo, L.; Mousavi-Shafaei, P.; D’eletto, M.; Di Giacomo, G.; Melino, G.; Palmieri, L.; et al. The adenine nucleotide translocator 1 acts as a type 2 transglutaminase substrate: Implications for mitochondrial dependent apoptosis. Cell Death Differ. 2009, 16, 1480–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, C.; Grimm, S. The permeability transition pore complex and cancer cell death. Oncogene 2006, 25, 4744–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallerne, C.; Touat, Z.; Chen, Z.; Martel, C.; Mayola, E.; Buron, N.; Le Bras, M.; Jacotot, E.; Borgne-Sanchez, A.; Lemoine, A.; et al. The fourth isoform of the adenine nucleotide translocator inhibits mitochondrial apoptosis in cancer cells. Int. J. Biochem. Cell Biol. 2010, 42, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.J. Adenine Nucleotide Translocase, Mitochondrial Stress, and Degenerative Cell Death. Oxid. Med. Cell Longev. 2013, 2013, 146860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordens, E.Z.; Palmieri, L.; Huizing, M.; van den Heuvel, L.P.; Sengers, R.C.; Dörner, A.; Ruitenbeek, W.; Trijbels, F.J.; Valsson, J.; Sigfusson, G.; et al. Adenine nucleotide translocator 1 deficiency associated with Sengers syndrome. Ann. Neurol. 2002, 52, 95–99. [Google Scholar] [CrossRef]
- Sharer, J.D. The adenine nucleotide translocase type 1 (ANT1): A new factor in mitochondrial disease. IUBMB Life 2005, 57, 607–614. [Google Scholar] [CrossRef]
- Padberg, G.W.; Lunt, P.W.; Koch, M.; Fardeau, M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscular Disord. 1991, 1, 231–234. [Google Scholar] [CrossRef]
- Kliment, C.R.; Nguyen, J.M.K.; Kaltreider, M.J.; Lu, Y.W.; Claypool, S.M.; Radder, J.E.; Sciurba, F.C.; Zhang, Y.; Gregory, A.D.; Iglesias, P.A.; et al. Adenine nucleotide translocase regulates airway epithelial metabolism, surface hydration and ciliary function. J. Cell Sci. 2021, 134, jcs257162. [Google Scholar] [CrossRef]
- Salet, C.; Passarella, S.; Quagliariello, E. Effects of selective irradiation on mammalian mitochondria. Photochem. Photobiol. 1987, 45, 433–438. [Google Scholar] [CrossRef]
- Passarella, S.; Casamassima, E.; Molinari, S.; Pastore, D.; Quagliariello, E.; Catalano, I.M.; Cingolani, A. Increase of proton electrochemical potential and ATP synthesis in rat liver mitochondria irradiated in vitro by helium-neon laser. FEBS Lett. 1984, 175, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Passarella, S.; Molinari, S.; Casamassima, E.; Pastore, D.; Quagliariello, E.; Catalano, I.M.; Cingolani, A. Bioelettrochemistry II; Milazzo, G., Blank, M., Eds.; Plenum Press: London, UK, 1987; pp. 275–299. [Google Scholar]
- Gagliardi, S.; Atlante, A.; Passarella, S. A novel property of adenine nucleotides: Sensitivity to helium-neon laser in mitochondrial reactions. Biochem. Mol. Biol. Int. 1997, 41, 449–460. [Google Scholar] [CrossRef]
- Salet, C. Hematoporphyrin and hematoporphyrin-derivative photosensitisation of mitochondria. Biochimie 1986, 68, 865–868. [Google Scholar] [CrossRef]
- Dougherty, T.J. Photodynamic therapy (PDT) of malignant tumors. Crit Rev. Oncol. Hematol. 1984, 2, 83–116. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic Therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilf, R. Mitochondria are targets of photodynamic therapy. J. Bioenerg. Biomembr. 2007, 39, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Passarella, S.; Quagliariello, E.; Moreno, G.; Salet, C. Hematoporphyrin derivative (Photofrin II) photosensitization of isolated mitochondria. Inhibition of ADP/ATP translocator. J. Photochem. Photobiol. B Biol. 1989, 4, 35–46. [Google Scholar] [CrossRef]
- Atlante, A.; Passarella, S.; Quagliariello, E.; Moreno, G.; Salet, C. Carrier thiols are targets of photofrin II photosensitization of isolated rat liver mitochondria. J. Photochem. Photobiol. B Biol. 1990, 7, 21–32. [Google Scholar] [CrossRef]
- Rougee, M.; Bensasson, R.V.; Land, E.J.; Pariente, R. Deactivation of singlet molecular oxygen by thiols and related compounds, possible protectors against skin photosensitivity. Photochem. Photobiol. 1988, 47, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Barile, M.; Valenti, D.; Passarella, S.; Quagliariello, E. 3′-Azido-3′-deoxythmidine uptake into isolated rat liver mitochondria and impairment of ADP/ATP translocator. Biochem. Pharmacol. 1997, 53, 913–920. [Google Scholar] [CrossRef]
- Valenti, D.; Atlante, A.; Barile, M.; Passarella, S. Mitochondria as agents of disease and as drug targets. In Mitochondrial Pharmacology and Toxicology; António, J.M., Moreno, P., Oliveira, J., Palmeira, C.M., Eds.; Transworld Research Network Publisher: Trivandrum, Kerala, India, 2006; ISBN 81-7895-207-6. [Google Scholar]
- Chen, C.; Vazquez-Padua, M.; Cheng, Y. Effect of antihuman immunodeficiency virus nucleoside analogs on mitochondrial DNA and its implication for delayed toxicity. Mol. Pharmacol. 1991, 39, 625–628. [Google Scholar] [PubMed]
- Hobbs, G.A.; Keilbaugh, S.A.; Simpson, M.V. The friend murine erythroleukemia cell, a model system for studying the association between bone marrow toxicity induced by 3’-azido-3’dideoxythymidine and inhibition of mtDNA replication. Biochem. Pharmacol. 1992, 43, 1397–1400. [Google Scholar] [CrossRef]
- Pesole, G. What is a gene? An updated operational definition. Gene 2008, 417, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Xia, L.; Wang, L.-S.; Shi, J.-Z.; Zheng, Y.; Chen, W.-L.; Zhang, L.; Liu, Z.-G.; Chen, G.-Q.; Fang, N.-Y. Differential protein expression in hypertrophic heart with and without hypertension in spontaneously hypertensive rats. Proteomics 2006, 6, 1948–1956. [Google Scholar] [CrossRef]
- Zamorano-León, J.J.; Modrego, J.; Mateos-Cáceres, P.J.; Macaya, C.; Martín-Fernández, B.; Miana, M.; de las Heras, N.; Cachofeiro, V.; Lahera, V.; López-Farré, A.J. A proteomic approach to determine changes in proteins involved in the myocardial metabolism in left ventricles of spontaneously hypertensive rats. Cell. Physiol. Biochem. 2006, 25, 347–358. [Google Scholar] [CrossRef]
- Atlante, A.; Seccia, T.M.; Pierro, P.; Vulpis, V.; Marra, E.; Pirrelli, A.; Passarella, S. ATP synthesis and export in heart left ventricle mitochondria from spontaneously hypertensive rat. Int. J. Mol. Med. 1998, 1, 709–716. [Google Scholar] [CrossRef]
- Seccia, T.M.; Atlante, A.; Vulpis, V.; Marra, E.; Passarella, S.; Pirrelli, A. Mitochondrial energy metabolism in the left ventricular tissue of spontaneously hypertensive rats: Abnormalities in both adenine nucleotide and phosphate translocators and enzyme adenylate-kinase and creatine-phosphokinase activities. Clin. Exp. Hypertens. 1998, 20, 345–358. [Google Scholar] [CrossRef]
- Atlante, A.; Seccia, T.M.; Marra, E.; Passarella, S. The rate of ATP export in the extramitochondrial phase via the adenine nucleotide translocator changes in aging in mitochondria isolated from heart left ventricle of either normotensive or spontaneously hypertensive rats. Mech. Ageing Dev. 2011, 132, 488–495. [Google Scholar] [CrossRef]
- Portman, M.A. Adenine nucleotide translocator in heart. Mol. Genet. Metab. 2000, 71, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.; Tschöpe, C.; Sterner-Kock, A.; Westermann, D.; Heringer-Walther, S.; Riad, A.; Nikolic, A.; Wang, Y.; Ebermann, L.; Siems, W.-E.; et al. Accelerated mitochondrial adenosine diphosphate/adenosine triphosphate transport improves hypertension-induced heart disease. Circulation 2007, 23, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.A.; Das, A.M. Control of mitochondrial ATP synthesis in the heart. Biochem. J. 1991, 280, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Schulze, K.; Dörner, A.; Schultheiss, H.-P. Mitochondrial function in heart failure. Heart Fail. Rev. 1999, 4, 229–244. [Google Scholar] [CrossRef]
- Schultheiss, H.-P. Dysfunction of the ADP/ATP carrier as a causative factor for the disturbance of the myocardial energy metabolism in dilated cardiomyopathy. Basic Res. Cardiol. 1992, 87, 311–320. [Google Scholar]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Age dependent impairment of mitochondrial function in rat heart tissue. Effect of pharmacological agents. Ann. N.Y. Acad. Sci. 1996, 768, 252–263. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: Role of cardiolipin. FEBS Lett. 1997, 406, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Nohl, H. Age-dependent changes in the structure-function correlation of ADP/ATP-translocating mitochondrial membranes. Gerontology 1982, 28, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three Dimensions of the Amyloid Hypothesis: Time, Space, and “Wingmen”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Bobba, A.; de Bari, L.; Fontana, F.; Calissano, P.; Marra, E.; Passarella, S. Caspase-dependent alteration of the ADP/ATP translocator triggers the mitochondrial permeability transition which is not required for the low-potassium-dependent apoptosis of cerebellar granule cells. J. Neurochem. 2006, 97, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Bobba, A.; Amadoro, G.; La Piana, G.; Calissano, P.; Atlante, A. Glycolytic enzyme upregulation and numbness of mitochondrial activity characterize the early phase of apoptosis in cerebellar granule cells. Apoptosis 2015, 20, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Bobba, A.; Amadoro, G.; Azzariti, A.; Pizzuto, R.; Atlante, A. Extracellular ADP prevents neuronal apoptosis via activation of cell antioxidant enzymes and protection of mitochondrial ANT-1. Biochim. Biophys. Acta 2014, 1837, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; Bobba, A.; Paventi, G.; Pizzuto, R.; Passarella, S. Genistein and daidzein prevent low potassium-dependent apoptosis of cerebellar granule cells. Biochem. Pharmacol. 2010, 79, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta 2008, 1777, 1289–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florenzano, F.; Corsetti, V.; Ciasca, G.; Ciotti, M.T.; Pittaluga, A.; Olivero, A.; Feligioni, M.; Iannuzzi, F.; Latina, V.; Sciacca, M.F.; et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 2017, 8, 64745–64778. [Google Scholar] [CrossRef] [Green Version]
- Amadoro, G.; Serafino, A.L.; Barbato, C.; Ciotti, M.T.; Sacco, A.; Calissano, P.; Canu, N. Role of N-terminal tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ. 2004, 11, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Amadoro, G.; Ciotti, M.T.; Costanzi, M.; Cestari, V.; Calissano, P.; Canu, N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2892–2897. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Yan, S.S. Mitochondrial medicine for neurodegenerative diseases. Int. J. Biochem. Cell Biol. 2010, 42, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Bobba, A.; Atlante, A.; de Bari, L.; Passarella, S.; Marra, E. Apoptosis and cytochrome c release in cerebellar granule cells. In Vivo 2004, 18, 335–344. [Google Scholar]
- Nakagawa, Y. Initiation of apoptotic signal by the peroxidation of cardiolipin of mitochondria. Ann. N. Y. Acad. Sci. 2004, 1011, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Jiang, C.; Li, L.; Wu, C.; Chen, Q.; Liu, S.S. A study on permeability transition pore opening and cytochrome c release from mitochondria, induced by caspase-3 in vitro. FEBS Lett. 2002, 510, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; Gagliardi, S.; Marra, E.; Calissano, P. Neuronal apoptosis in rats is accompanied by rapid impairment of cellular respiration and is prevented by scavengers of reactive oxygen species. Neurosci Lett. 1998, 245, 127–130. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Israelson, A.; Brdiczka, D.; Sheu, S.S. The voltage-dependent anion channel (VDAC): Function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 2006, 12, 2249–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharstuhl, A.; Mutsaers, H.A.; Pennings, S.W.; Russel, F.G.; Wagener, F.A. Involvement of VDAC, Bax and ceramides in the efflux of AIF from mitochondria during curcumin-induced apoptosis. PLoS ONE 2009, 4, e6688. [Google Scholar] [CrossRef] [PubMed]
- Horáková, L. Flavonoids in prevention of diseases with respect to modulation of Ca-pump function. Interdiscip. Toxicol. 2011, 4, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atlante, A.; Amadoro, G.; Bobba, A.; Latina, V. Functional Foods: An Approach to Modulate Molecular Mechanisms of Alzheimer’s Disease. Cells 2020, 9, 2347. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.S.; Ksiezak-Reding, H. Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur. J. Biochem. 1995, 233, 9–17. [Google Scholar] [CrossRef]
- Cente, M.; Filipcik, P.; Pevalova, M.; Novak, M. Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. Eur. J. Neurosci. 2006, 24, 1085–1090. [Google Scholar] [CrossRef]
- Klingenberg, M. Molecular aspects of the adenine nucleotide carrier from mitochondria. Arch. Biochem. Biophys. 1989, 270, 1–14. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH2-tau fragment and A_ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Bobba, A.; Amadoro, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Dissecting the molecular mechanism by which NH2htau and Aβ1-42 peptides impair mitochondrial ANT-1 in Alzheimer disease. Biochim. Biophys. Acta. 2013, 1827, 848–860. [Google Scholar] [CrossRef] [Green Version]
- Fonyo, A. SH-group reagents as tools in the study of mitochondrial anion transport. J. Bioenerg. Biomembr. 1978, 10, 171–194. [Google Scholar] [CrossRef]
- Vignais, P.V.; Vignais, P.M. Effect of SH reagents on atractyloside binding to mitochondria and ADP translocation. Potentiation by ADP and its prevention by uncoupler FCCP. FEBS Lett. 1972, 26, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Van Doorn, W.G.; Woltering, E.J. Many ways to exit? Cell death categories in plants. Trends Plant. Sci. 2005, 10, 117–122. [Google Scholar] [CrossRef]
- Lam, E.; Kato, N.; Lawton, M. Programmed cell death, mitochondria and the plant hypersensitive response. Nature 2001, 411, 848–853. [Google Scholar] [CrossRef]
- Valenti, D.; Vacca, R.A.; de Pinto, M.C.; De Gara, L.; Marra, E.; Passarella, S. In the early phase of programmed cell death in Tobacco Bright Yellow 2 cells the mitochondrial adenine nucleotide translocator, adenylate kinase and nucleoside diphosphate kinase are impaired in a reactive oxygen species-dependent manner. Biochim. Biophys. Acta. 2007, 1767, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; Tullo, A.; Caratozzolo, M.F.; Merafina, R.S.; Scartezzini, P.; Marra, E.; Vacca, R.A. Impairment of F1F0-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem. J. 2010, 431, 299–310. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta. 2016, 1862, 1093–1104. [Google Scholar] [CrossRef]
- Brodlie, M.; Haq, I.J.; Roberts, K.; Elborn, J.S. Targeted therapies to improve CFTR function in cystic fibrosis. Genome Med. 2015, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef] [Green Version]
- De Stefano, D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; De Gregorio, F.; Salvadori, L.; Grassia, R.; Leone, C.A.; Rosa, G.D.; et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014, 10, 2053–2074. [Google Scholar] [CrossRef] [Green Version]
- Fernandez Fernandez, E.; De Santi, C.; De Rose, V.; Greene, C.M. CFTR dysfunction in cystic fibrosis and chronic obstructive pulmonary disease. Expert Rev. Respir Med. 2018, 12, 483–492. [Google Scholar] [CrossRef]
- Favia, M.; de Bari, L.; Bobba, A.; Atlante, A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. J. Clin. Med. 2019, 8, 1890. [Google Scholar] [CrossRef] [Green Version]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, gating, and regulation of the cftr anion channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef]
- Atlante, A.; Favia, M.; Bobba, A.; Guerra, L.; Casavola, V.; Reshkin, S.J. Characterization of mitochondrial function in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. J. Bioenerg. Biomembr. 2016, 48, 197–210. [Google Scholar] [CrossRef]
- Hwang, T.C.; Kirk, K.L. The CFTR ion channel: Gating, regulation and anion permeation. Cold Spring Harb. Perspect. Med. 2013, 3, a009498. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atlante, A.; Valenti, D. A Walk in the Memory, from the First Functional Approach up to Its Regulatory Role of Mitochondrial Bioenergetic Flow in Health and Disease: Focus on the Adenine Nucleotide Translocator. Int. J. Mol. Sci. 2021, 22, 4164. https://doi.org/10.3390/ijms22084164
Atlante A, Valenti D. A Walk in the Memory, from the First Functional Approach up to Its Regulatory Role of Mitochondrial Bioenergetic Flow in Health and Disease: Focus on the Adenine Nucleotide Translocator. International Journal of Molecular Sciences. 2021; 22(8):4164. https://doi.org/10.3390/ijms22084164
Chicago/Turabian StyleAtlante, Anna, and Daniela Valenti. 2021. "A Walk in the Memory, from the First Functional Approach up to Its Regulatory Role of Mitochondrial Bioenergetic Flow in Health and Disease: Focus on the Adenine Nucleotide Translocator" International Journal of Molecular Sciences 22, no. 8: 4164. https://doi.org/10.3390/ijms22084164