
Smad2/3 Activation Regulates Smad1/5/8 Signaling via a Negative Feedback Loop to Inhibit 3T3-L1 Adipogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TGF-β Ligands Differentially Affect 3T3-L1 Adipogenesis
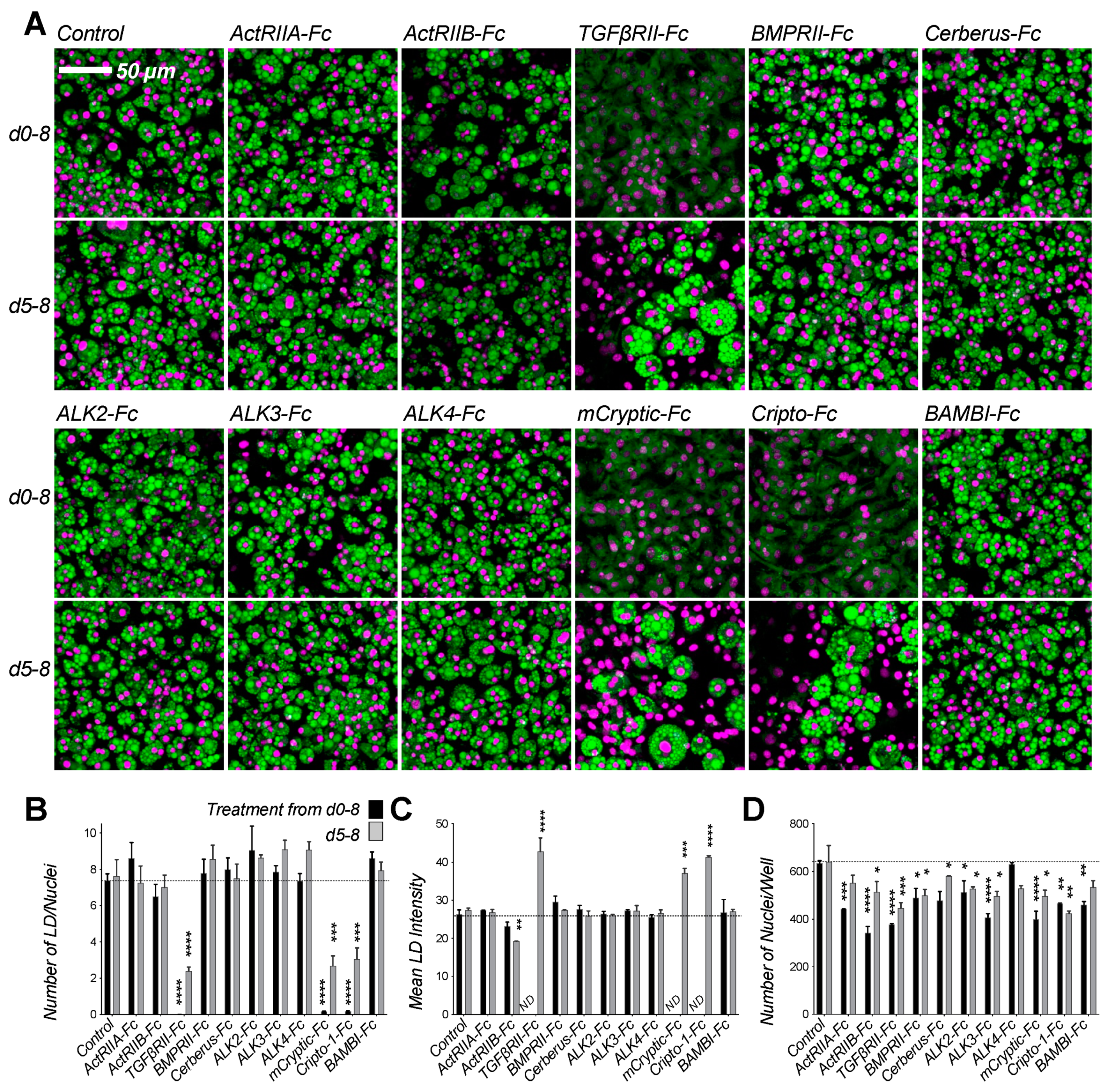
2.2. Screen Identifies Ligand Traps with Anti-Adipogenic Activity
2.3. Inhibitory Traps Suppress Adipogenic Gene Expression
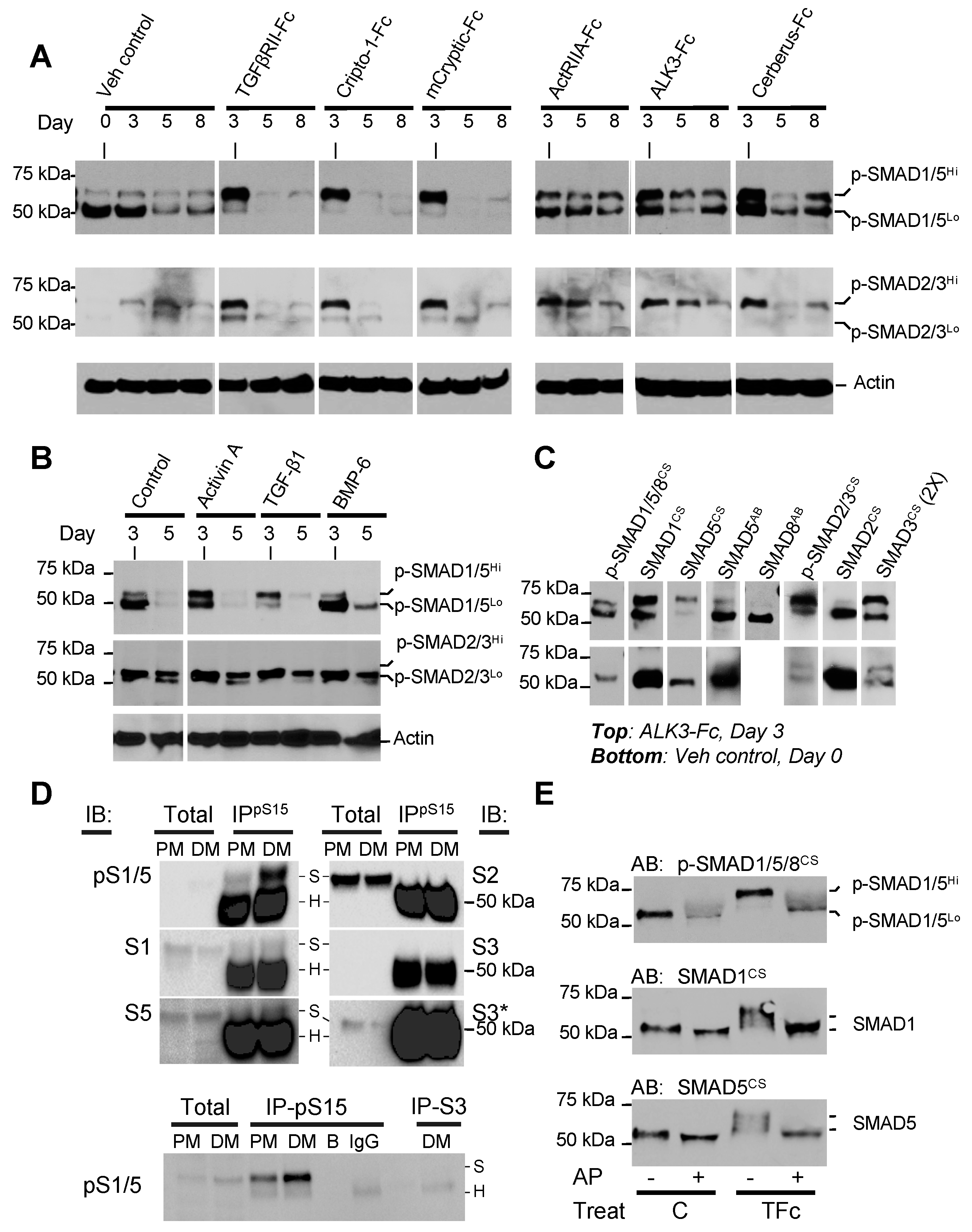
2.4. Anti-Adipogenic Ligands and Traps Alter SMAD1/5/8 Phosphorylation States
2.5. Adipogenesis Inhibitors Suppress SMAD1/5/8 Signaling
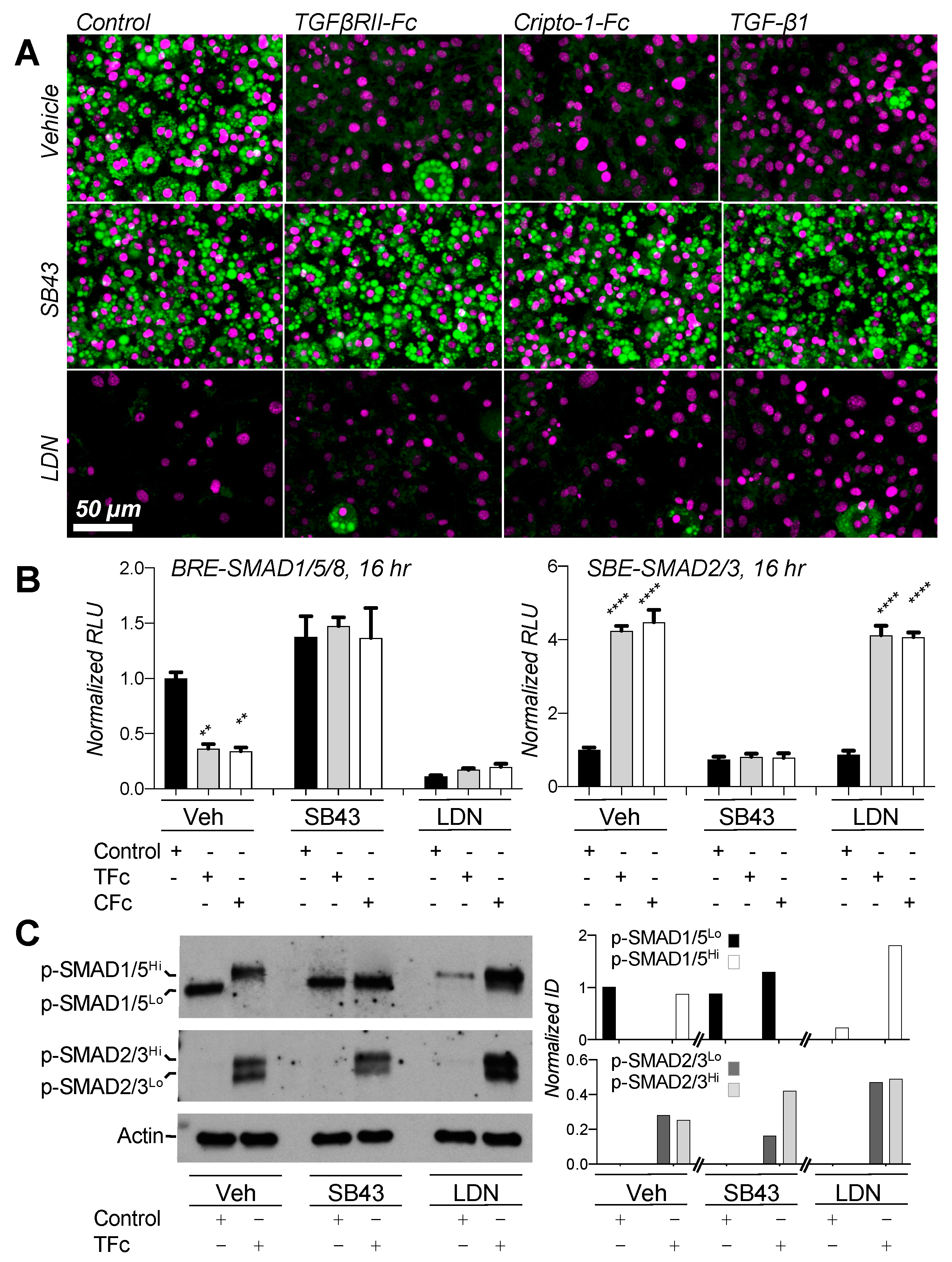
2.6. Small Molecule Inhibitors Suggest Negative Feedback Loop between SMAD2/3 and SMAD1/5/8 Pathways
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rutkowski, J.M.; Stern, J.H.; Scherer, P.E. The cell biology of fat expansion. J. Cell Biol. 2015, 208, 501–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, N.M. The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia, and hypertension. Arch. Intern. Med. 1989, 149, 1514–1520. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [CrossRef]
- Sun, K.; Scherer, P.E. Adipose Tissue Dysfunction: A Multistep Process. In Novel Insights into Adipose Cell Functions; Springer: Berlin/Heidelberg, Germany, 2010; pp. 67–75. [Google Scholar] [CrossRef]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef]
- Arner, P.; Andersson, D.P.; Thorne, A.; Wiren, M.; Hoffstedt, J.; Naslund, E.; Thorell, A.; Ryden, M. Variations in the size of the major omentum are primarily determined by fat cell number. J. Clin. Endocrinol. Metab. 2013, 98, E897–E901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, T.N.; Tuttle, L.J.; Bohnert, K.L.; Mueller, M.J.; Sinacore, D.R. Excessive adipose tissue infiltration in skeletal muscle in individuals with obesity, diabetes mellitus, and peripheral neuropathy: Association with performance and function. Phys. Ther. 2008, 88, 1336–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamrick, M.W.; McGee-Lawrence, M.E.; Frechette, D.M. Fatty Infiltration of Skeletal Muscle: Mechanisms and Comparisons with Bone Marrow Adiposity. Front. Endocrinol. 2016, 7, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Reviews. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.S.; Massaro, J.M.; Hoffmann, U.; Pou, K.M.; Maurovich-Horvat, P.; Liu, C.Y.; Vasan, R.S.; Murabito, J.M.; Meigs, J.B.; Cupples, L.A.; et al. Abdominal visceral and subcutaneous adipose tissue compartments: Association with metabolic risk factors in the Framingham Heart Study. Circulation 2007, 116, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.D.; Targher, G. Ectopic fat, insulin resistance, and nonalcoholic fatty liver disease: Implications for cardiovascular disease. Arter. Thromb. Vasc. Biol. 2014, 34, 1155–1161. [Google Scholar] [CrossRef] [Green Version]
- Laforest, S.; Labrecque, J.; Michaud, A.; Cianflone, K.; Tchernof, A. Adipocyte size as a determinant of metabolic disease and adipose tissue dysfunction. Crit. Rev. Clin. Lab. Sci. 2015, 52, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Okamura, T.; Hashimoto, Y.; Hamaguchi, M.; Obora, A.; Kojima, T.; Fukui, M. Ectopic fat obesity presents the greatest risk for incident type 2 diabetes: A population-based longitudinal study. Int. J. Obes. 2019, 43, 139–148. [Google Scholar] [CrossRef]
- Zamani, N.; Brown, C.W. Emerging roles for the transforming growth factor-{beta} superfamily in regulating adiposity and energy expenditure. Endocr. Rev. 2011, 32, 387–403. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J. Transforming growth factor beta superfamily regulation of adipose tissue biology in obesity. Biochim. Biophys Acta-Mol. Basis Dis. 2018, 1864, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Type beta transforming growth factor controls the adipogenic differentiation of 3T3 fibroblasts. Proc. Natl. Acad. Sci. USA 1985, 82, 8530–8534. [Google Scholar] [CrossRef] [Green Version]
- Sparks, R.L.; Allen, B.J.; Strauss, E.E. TGF-beta blocks early but not late differentiation-specific gene expression and morphologic differentiation of 3T3 T proadipocytes. J. Cell Physiol. 1992, 150, 568–577. [Google Scholar] [CrossRef]
- Choy, L.; Skillington, J.; Derynck, R. Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J. Cell Biol. 2000, 149, 667–682. [Google Scholar] [CrossRef]
- Kim, H.S.; Liang, L.; Dean, R.G.; Hausman, D.B.; Hartzell, D.L.; Baile, C.A. Inhibition of preadipocyte differentiation by myostatin treatment in 3T3-L1 cultures. Biochem. Biophys. Res. Commun. 2001, 281, 902–906. [Google Scholar] [CrossRef]
- Hirai, S.; Yamanaka, M.; Kawachi, H.; Matsui, T.; Yano, H. Activin A inhibits differentiation of 3T3-L1 preadipocyte. Mol. Cell Endocrinol. 2005, 232, 21–26. [Google Scholar] [CrossRef]
- Hoggard, N.; Cruickshank, M.; Moar, K.M.; Barrett, P.; Bashir, S.; Miller, J.D. Inhibin betaB expression in murine adipose tissue and its regulation by leptin, insulin and dexamethasone. J. Mol. Endocrinol. 2009, 43, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Guo, Y.; Liu, Y.; Wang, Y.; Zheng, R.; Ban, Y.; Peng, L.; Yuan, Q.; Liu, W. Growth differentiation factor 11 inhibits adipogenic differentiation by activating TGF-beta/Smad signalling pathway. Cell Prolif. 2019, 52, e12631. [Google Scholar] [CrossRef]
- Lee, M.J.; Pickering, R.T.; Shibad, V.; Wu, Y.; Karastergiou, K.; Jager, M.; Layne, M.D.; Fried, S.K. Impaired Glucocorticoid Suppression of TGFbeta Signaling in Human Omental Adipose Tissues Limits Adipogenesis and May Promote Fibrosis. Diabetes 2019, 68, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. USA 2004, 101, 9607–9611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Song, T.J.; Li, X.; Hu, L.; He, Q.; Liu, M.; Lane, M.D.; Tang, Q.Q. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. USA 2009, 106, 12670–12675. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Svensson, P.A.; Grimsby, J.; Rondinone, C.; Smith, U. BMP4 and BMP Antagonists Regulate Human White and Beige Adipogenesis. Diabetes 2015, 64, 1670–1681. [Google Scholar] [CrossRef] [Green Version]
- Modica, S.; Wolfrum, C. The dual role of BMP4 in adipogenesis and metabolism. Adipocyte 2017, 6, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, I.; Dorpholz, G.; Ott, C.E.; Kragesteen, B.; Schanze, N.; Lee, C.T.; Kohrle, J.; Mundlos, S.; Ruschke, K.; Knaus, P. BMPs as new insulin sensitizers: Enhanced glucose uptake in mature 3T3-L1 adipocytes via PPARgamma and GLUT4 upregulation. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Xu, Z.; Zhang, X.; Wang, L.; Gimble, J.M.; Lander, E.S.; Rosen, E.D. Comparative epigenomic analysis of murine and human adipogenesis. Cell 2010, 143, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Koster, J.; Volckmann, R.; Zwijnenburg, D.; Molenaar, P.; Versteeg, R. R2: Genomics analysis and visualization platform. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Pei, Z.; Yang, Y.; Kiess, W.; Sun, C.; Luo, F. Dynamic profile and adipogenic role of growth differentiation factor 5 (GDF5) in the differentiation of 3T3-L1 preadipocytes. Arch. Biochem. Biophys. 2014, 560, 27–35. [Google Scholar] [CrossRef]
- Zebisch, K.; Voigt, V.; Wabitsch, M.; Brandsch, M. Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal. Biochem. 2012, 425, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Rebbapragada, A.; Benchabane, H.; Wrana, J.L.; Celeste, A.J.; Attisano, L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol. Cell Biol. 2003, 23, 7230–7242. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.G.; Czepnik, M.; Goebel, E.J.; McCoy, J.C.; Vujic, A.; Cho, M.; Oh, J.; Aykul, S.; Walton, K.L.; Schang, G.; et al. Structural basis for potency differences between GDF8 and GDF11. BMC Biol. 2017, 15, 1–22. [Google Scholar] [CrossRef] [Green Version]
- de Caestecker, M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004, 15, 1–11. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townson, S.A.; Martinez-Hackert, E.; Greppi, C.; Lowden, P.; Sako, D.; Liu, J.; Ucran, J.A.; Liharska, K.; Underwood, K.W.; Seehra, J.; et al. Specificity and Structure of a High Affinity Activin Receptor-like Kinase 1 (ALK1) Signaling Complex. J. Biol. Chem. 2012, 287, 27313–27325. [Google Scholar] [CrossRef] [Green Version]
- Olsen, O.E.; Wader, K.F.; Misund, K.; Vatsveen, T.K.; Ro, T.B.; Mylin, A.K.; Turesson, I.; Stordal, B.F.; Moen, S.H.; Standal, T.; et al. Bone morphogenetic protein-9 suppresses growth of myeloma cells by signaling through ALK2 but is inhibited by endoglin. Blood Cancer J. 2014, 4, e196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, O.E.; Sankar, M.; Elsaadi, S.; Hella, H.; Buene, G.; Darvekar, S.R.; Misund, K.; Katagiri, T.; Knaus, P.; Holien, T. BMPR2 inhibits activin and BMP signaling via wild-type ALK2. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, B.; van Dinther, M.; Ten Dijke, P.; Inman, G.J. Autocrine bone morphogenetic protein-9 signals through activin receptor-like kinase-2/Smad1/Smad4 to promote ovarian cancer cell proliferation. Cancer Res. 2009, 69, 9254–9262. [Google Scholar] [CrossRef] [Green Version]
- Sako, D.; Grinberg, A.V.; Liu, J.; Davies, M.V.; Castonguay, R.; Maniatis, S.; Andreucci, A.J.; Pobre, E.G.; Tomkinson, K.N.; Monnell, T.E.; et al. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J. Biol. Chem. 2010, 285, 21037–21048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud’huin, M.; Solban, N.; Cornwall-Brady, M.; Sako, D.; Kawamoto, Y.; Liharska, K.; Lath, D.; Bouxsein, M.L.; Underwood, K.W.; Ucran, J.; et al. A soluble bone morphogenetic protein type IA receptor increases bone mass and bone strength. Proc. Natl. Acad. Sci. USA 2012, 109, 12207–12212. [Google Scholar] [CrossRef] [Green Version]
- Aykul, S.; Martinez-Hackert, E. New Ligand Binding Function of Human Cerberus and Role of Proteolytic Processing in Regulating Ligand-Receptor Interactions and Antagonist Activity. J. Mol. Biol. 2016, 428, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Martinez-Hackert, E. Transforming Growth Factor-beta Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J. Biol. Chem. 2016, 291, 10792–10804. [Google Scholar] [CrossRef] [Green Version]
- Aykul, S.; Parenti, A.; Chu, K.Y.; Reske, J.; Floer, M.; Ralston, A.; Martinez-Hackert, E. Biochemical and Cellular Analysis Reveals Ligand Binding Specificities, a Molecular Basis for Ligand Recognition, and Membrane Association-dependent Activities of Cripto-1 and Cryptic. J. Biol. Chem. 2017, 292, 4138–4151. [Google Scholar] [CrossRef] [Green Version]
- Gjidoda, A.; Tagore, M.; McAndrew, M.J.; Woods, A.; Floer, M. Nucleosomes are stably evicted from enhancers but not promoters upon induction of certain pro-inflammatory genes in mouse macrophages. PLoS ONE 2014, 9, e93971. [Google Scholar] [CrossRef]
- Rosen, E.D. The transcriptional basis of adipocyte development. Prostaglandins Leukot. Essent Fat. Acids 2005, 73, 31–34. [Google Scholar] [CrossRef]
- Daly, A.C.; Randall, R.A.; Hill, C.S. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell Biol. 2008, 28, 6889–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, L.; Derynck, R. Transforming growth factor-beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J. Biol. Chem. 2003, 278, 9609–9619. [Google Scholar] [CrossRef] [Green Version]
- Kretzschmar, M.; Doody, J.; Massague, J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [CrossRef]
- Pera, E.M.; Ikeda, A.; Eivers, E.; De Robertis, E.M. Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev. 2003, 17, 3023–3028. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, H.; Fuentealba, L.; Ikeda, A.; Reversade, B.; De Robertis, E.M. Default neural induction: Neuralization of dissociated Xenopus cells is mediated by Ras/MAPK activation. Genes Dev. 2005, 19, 1022–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapkota, G.; Alarcon, C.; Spagnoli, F.M.; Brivanlou, A.H.; Massague, J. Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol. Cell 2007, 25, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Kamato, D.; Burch, M.L.; Piva, T.J.; Rezaei, H.B.; Rostam, M.A.; Xu, S.; Zheng, W.; Little, P.J.; Osman, N. Transforming growth factor-beta signalling: Role and consequences of Smad linker region phosphorylation. Cell Signal. 2013, 25, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Aubin, J.; Davy, A.; Soriano, P. In vivo convergence of BMP and MAPK signaling pathways: Impact of differential Smad1 phosphorylation on development and homeostasis. Genes Dev. 2004, 18, 1482–1494. [Google Scholar] [CrossRef] [Green Version]
- Fuentealba, L.C.; Eivers, E.; Ikeda, A.; Hurtado, C.; Kuroda, H.; Pera, E.M.; De Robertis, E.M. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 2007, 131, 980–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Bubnoff, A.; Peiffer, D.A.; Blitz, I.L.; Hayata, T.; Ogata, S.; Zeng, Q.; Trunnell, M.; Cho, K.W. Phylogenetic footprinting and genome scanning identify vertebrate BMP response elements and new target genes. Dev. Biol. 2005, 281, 210–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonk, L.J.; Itoh, S.; Heldin, C.H.; ten Dijke, P.; Kruijer, W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem. 1998, 273, 21145–21152. [Google Scholar] [CrossRef] [Green Version]
- Inman, G.J.; Nicolas, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef]
- Cuny, G.D.; Yu, P.B.; Laha, J.K.; Xing, X.; Liu, J.F.; Lai, C.S.; Deng, D.Y.; Sachidanandan, C.; Bloch, K.D.; Peterson, R.T. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4388–4392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodeheffer, M.S.; Birsoy, K.; Friedman, J.M. Identification of white adipocyte progenitor cells in vivo. Cell 2008, 135, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.; McGee, S.L. 3T3-L1 adipocytes display phenotypic characteristics of multiple adipocyte lineages. Adipocyte 2015, 4, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Yeh, W.C.; Bierer, B.E.; McKnight, S.L. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc. Natl. Acad. Sci. USA 1995, 92, 11086–11090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, F.; Rivard, N.; Rudd, P.; Grondin, F.; Attisano, L.; Dubois, C.M. Cross-talk between the p42/p44 MAP kinase and Smad pathways in transforming growth factor beta 1-induced furin gene transactivation. J. Biol. Chem. 2001, 276, 33986–33994. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Alarcon, C.; Sapkota, G.; Rahman, S.; Chen, P.Y.; Goerner, N.; Macias, M.J.; Erdjument-Bromage, H.; Tempst, P.; Massague, J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell 2009, 36, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Burch, M.L.; Zheng, W.; Little, P.J. Smad linker region phosphorylation in the regulation of extracellular matrix synthesis. Cell. Mol. Life Sci. CMLS 2011, 68, 97–107. [Google Scholar] [CrossRef]
- Ramachandran, A.; Vizan, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Muller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-beta uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. eLife 2018, 7, e31756. [Google Scholar] [CrossRef]
- Gronroos, E.; Kingston, I.J.; Ramachandran, A.; Randall, R.A.; Vizan, P.; Hill, C.S. Transforming growth factor beta inhibits bone morphogenetic protein-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol. Cell Biol. 2012, 32, 2904–2916. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. Integration of Smad and MAPK pathways: A link and a linker revisited. Genes Dev. 2003, 17, 2993–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldridge, A.; Kouroupis, D.; Churchman, S.; English, A.; Ingham, E.; Jones, E. Assay validation for the assessment of adipogenesis of multipotential stromal cells--a direct comparison of four different methods. Cytotherapy 2013, 15, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervais, A.L.; Marques, M.; Gaudreau, L. PCRTiler: Automated design of tiled and specific PCR primer pairs. Nucleic Acids Res. 2010, 38, W308–W312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, J.M.; Boyce, F.M. High-throughput functional screening using a homemade dual-glow luciferase assay. J. Vis. Exp. JoVE 2014, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aykul, S.; Maust, J.; Thamilselvan, V.; Floer, M.; Martinez-Hackert, E. Smad2/3 Activation Regulates Smad1/5/8 Signaling via a Negative Feedback Loop to Inhibit 3T3-L1 Adipogenesis. Int. J. Mol. Sci. 2021, 22, 8472. https://doi.org/10.3390/ijms22168472
Aykul S, Maust J, Thamilselvan V, Floer M, Martinez-Hackert E. Smad2/3 Activation Regulates Smad1/5/8 Signaling via a Negative Feedback Loop to Inhibit 3T3-L1 Adipogenesis. International Journal of Molecular Sciences. 2021; 22(16):8472. https://doi.org/10.3390/ijms22168472
Chicago/Turabian StyleAykul, Senem, Jordan Maust, Vijayalakshmi Thamilselvan, Monique Floer, and Erik Martinez-Hackert. 2021. "Smad2/3 Activation Regulates Smad1/5/8 Signaling via a Negative Feedback Loop to Inhibit 3T3-L1 Adipogenesis" International Journal of Molecular Sciences 22, no. 16: 8472. https://doi.org/10.3390/ijms22168472