Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
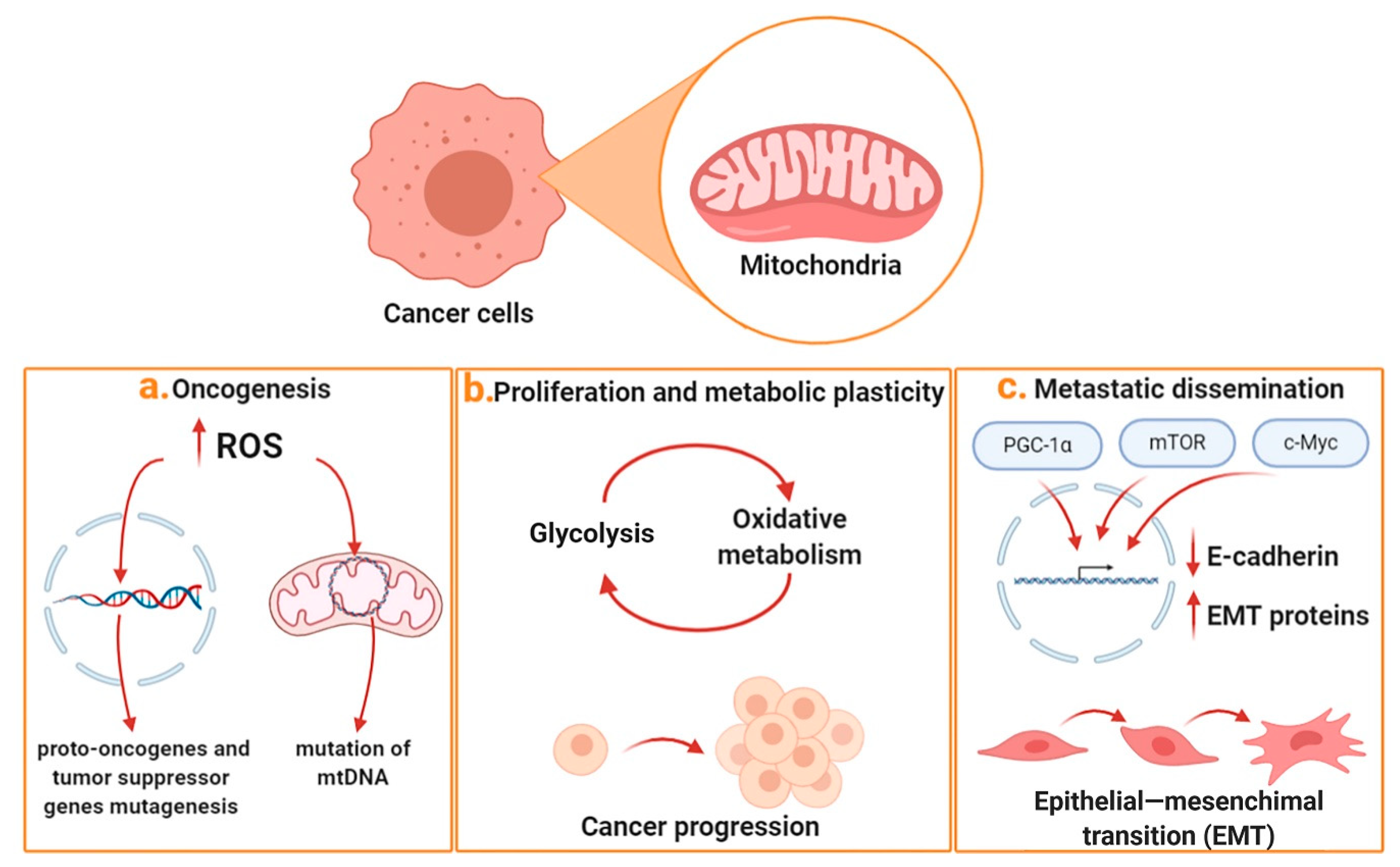
:1. Mitochondria and Cancer
2. Targeting Mitochondrial Metabolism in Cancer
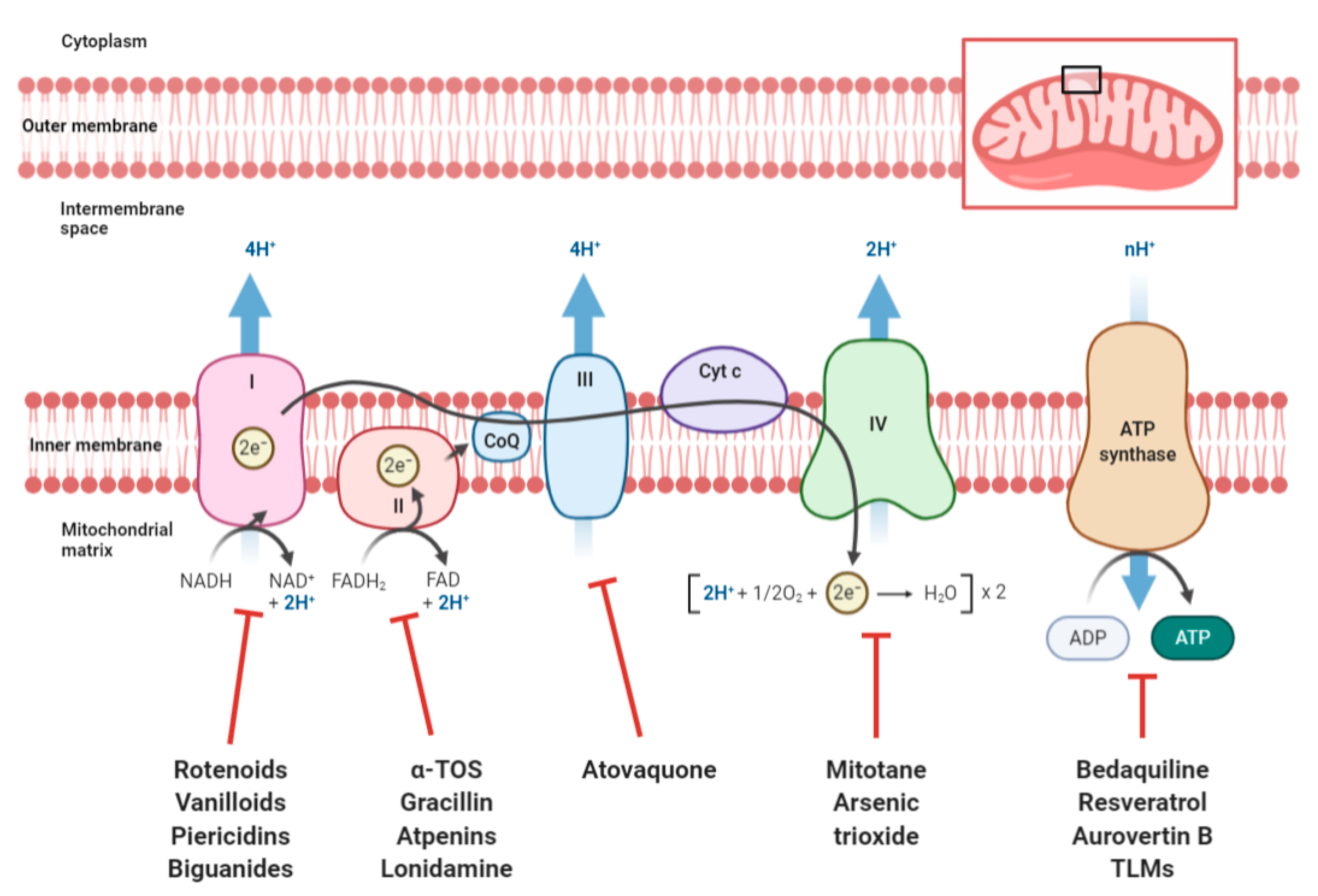
2.1. Targeting OXPHOS
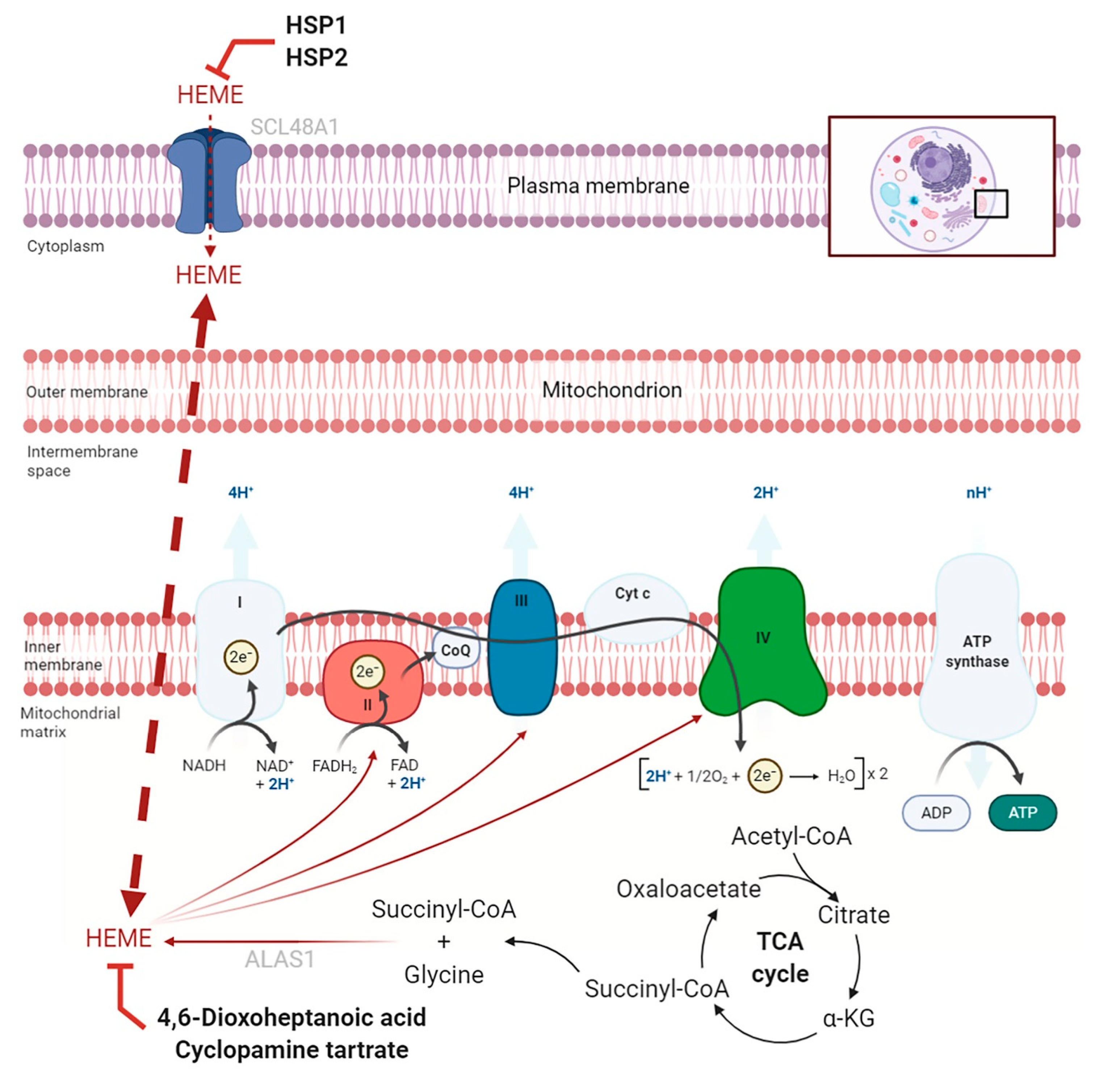
2.2. Targeting Heme
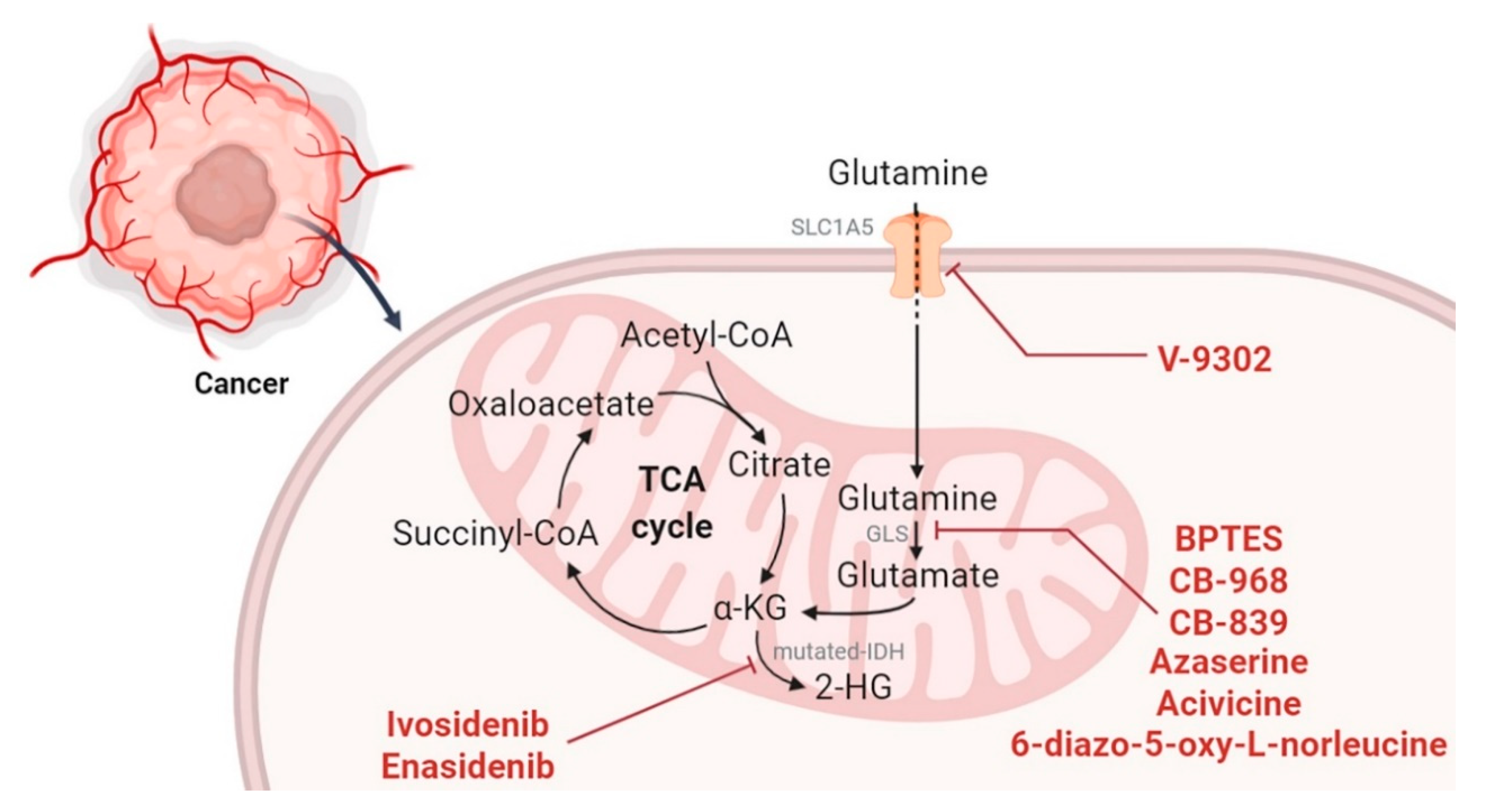
2.3. Targeting the TCA Cycle and Glutaminolysis
2.4. Targeting Mitochondrial Biogenesis
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| NAD+ | Nicotinamide adenine dinucleotide (oxidized form) |
| NADPH | Nicotinamide adenine dinucleotide (reduced form) |
| NADPH | Nicotinamide adenine dinucleotide phosphate (reduced form) |
| FDA | Food and Drug Administration |
| SLC25A1 | solute carrier family 25 member 1 (citrate transporter) |
| SLC1A5 | solute carrier family 1 member 5 (neutral amino acid transporter) |
| AML | acute myeloid leukemia |
| PI3K | Phosphoinositide 3-kinase |
| AKT | Protein kinase B |
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.Y.; Muller, W.J. Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Sharma, L.K.; Bai, Y.J. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009, 19, 802–815. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, R.R.; Patel, K.; Putnam, W.C.; Kapur, P.; Rakheja, D. Oncometabolites: A new paradigm for oncology, metabolism, and the clinical laboratory. Clin. Chem. 2017, 63, 1812–1820. [Google Scholar] [CrossRef] [Green Version]
- Morin, A.; Letouzé, E.; Gimenez-Roqueplo, A.P.; Favier, J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int. J. Cancer 2014, 135, 2237–2248. [Google Scholar] [CrossRef]
- Catalina-Rodriguez, O.; Kolukula, V.K.; York Tomita, A.P.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; Avantaggiati, M.L. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [Green Version]
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells with Tolerance to Cycling Severe Hypoxia. Front. Oncol. 2018, 8, 170. [Google Scholar] [CrossRef]
- Fernandez, H.R.; Gadre, S.M.; Tan, M.; Graham, G.T.; Mosaoa, R.; Ongkeko, M.S.; Kim, K.A.; Riggins, R.B.; Parasido, E.; Petrini, I.; et al. The mitochondrial citrate carrier, SLC25A1, drives stemness and therapy resistance in non-small cell lung cancer. Cell Death Differ. 2018, 25, 1239–1258. [Google Scholar] [CrossRef] [Green Version]
- Ochoa-Ruiz, E.; Diaz-Ruiz, R. Anaplerosis in cancer: Another step beyond the warburg effect. Am. J. Mol. Boil. 2012, 2, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Wei, B.; Lu, C.; Li, P.; Chen, L. Glutaminase sustains cell survival via the regulation of glycolysis and glutaminolysis in colorectal cancer. Oncol. Lett. 2017, 14, 3117–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 2019, 8, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, A.N.; Higashi, R.M.; Fan, T.W.-M. Metabolic reprogramming in tumors: Contributions of the tumor microenvironment. Genes Dis. 2020, 7, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front. Oncol. 2018, 8, 117. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Boil. 2017, 27, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.; Witkiewicz, A.K.; Howell, A.; Philp, N.J.; Pestell, R.G.; Lisanti, M.P. Mitochondrial metabolism in cancer metastasis. Cell Cycle 2012, 11, 1445–1454. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Thomson, T.M.; Balcells, C.; Cascante, M. Metabolic Plasticity and Epithelial-Mesenchymal Transition. J. Clin. Med. 2019, 8, 967. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Scimè, A. Metabolic Regulation of Epithelial to Mesenchymal Transition: Implications for Endocrine Cancer. Front. Endocrinol. 2019, 10, 773. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Cheryan, V.T.; Xu, L.; Rishi, A.K.; Reddy, K.B. Myc mediates cancer stem-like cells and EMT changes in triple negative breast cancers cells. PLoS ONE 2017, 12, e0183578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.B.; Cho, M.K.; Lee, W.Y.; Kang, K.W. Overexpression of c-myc induces epithelial mesenchymal transition in mammary epithelial cells. Cancer Lett. 2010, 293, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Roshan, M.K.; Soltani, A.; Soleimani, A.; Kahkhaie, K.R.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Yoriki, K.; Mori, T.; Kokabu, T.; Matsushima, H.; Umemura, S.; Tarumi, Y.; Kitawaki, J. Estrogen-related receptor alpha induces epithelial-mesenchymal transition through cancer-stromal interactions in endometrial cancer. Sci. Rep. 2019, 9, 6697. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Sohoni, S.; Ghosh, P.; Wang, T.; Kalainayakan, S.P.; Vidal, C.; Dey, S.; Konduri, P.C.; Zhang, L. Elevated Heme Synthesis and Uptake Underpin Intensified Oxidative Metabolism and Tumorigenic Functions in Non–Small Cell Lung Cancer Cells. Cancer Res. 2019, 79, 2511–2525. [Google Scholar] [CrossRef] [Green Version]
- Yadav, N.; Kumar, S.; Marlowe, T.; Chaudhary, A.; Kumar, R.; Wang, J.; O’malley, J.; Boland, P.; Jayanthi, S.; Kumar, T.K.S.; et al. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015, 6, e1969. [Google Scholar] [CrossRef]
- Dey, R.; Moraes, C.T. Lack of Oxidative Phosphorylation and Low Mitochondrial Membrane Potential Decrease Susceptibility to Apoptosis and Do Not Modulate the Protective Effect of Bcl-xLin Osteosarcoma Cells. J. Boil. Chem. 2000, 275, 7087–7094. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, M.; Tóth, F.; Brindisi, M.; Sotgia, F.; Lisanti, M.P. Deferiprone (DFP) Targets Cancer Stem cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells 2020, 9, 1529. [Google Scholar] [CrossRef]
- Urra, F.A.; Córdova-Delgado, M.; Lapier, M.; Orellana-Manzano, A.; Acevedo-Arévalo, L.; Pessoa-Mahana, H.; González-Vivanco, J.M.; Martínez-Cifuentes, M.; Ramírez-Rodríguez, O.; Millas-Vargas, J.P.; et al. Small structural changes on a hydroquinone scaffold determine the complex I inhibition or uncoupling of tumoral oxidative phosphorylation. Toxicol. Appl. Pharmacol. 2016, 291, 46–57. [Google Scholar] [CrossRef]
- Degli Esposti, M. Inhibitors of NADH–ubiquinone reductase: An overview. Biochim. Biophys. Acta (BBA) Bioenerg. 1998, 1364, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Urra, F.A.; Muñoz, F.; Lovy, A.; Cárdenas, C. The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 2017, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Palorini, R.; Simonetto, T.; Cirulli, C.; Chiaradonna, F. Mitochondrial Complex I Inhibitors and Forced Oxidative Phosphorylation Synergize in Inducing Cancer Cell Death. Int. J. Cell Boil. 2013, 2013, 1–14. [Google Scholar] [CrossRef]
- Schöckel, L.; Glasauer, A.; Basit, F.; Bitschar, K.; Truong, H.; Erdmann, G.; Algire, C.; Hägebarth, A.; Willems, P.H.; Kopitz, C.; et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Naguib, A.; Mathew, G.; Reczek, C.R.; Watrud, K.; Ambrico, A.; Herzka, T.; Salas, I.C.; Lee, M.F.; El-Amine, N.; Zheng, W.; et al. Mitochondrial Complex I Inhibitors Expose a Vulnerability for Selective Killing of Pten-Null Cells. Cell Rep. 2018, 23, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Kluckova, K.; Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Neuzil, J. Mitochondrial complex II, a novel target for anti-cancer agents. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1827, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Gruber, J.; Staniek, K.; Krewenka, C.; Moldzio, R.; Patel, A.; Böhmdorfer, S.; Rosenau, T.; Gille, L. Tocopheramine succinate and tocopheryl succinate: Mechanism of mitochondrial inhibition and superoxide radical production. Bioorg. Med. Chem. 2014, 22, 684–691. [Google Scholar] [CrossRef]
- Min, H.-Y.; Jang, H.-J.; Park, K.H.; Hyun, S.Y.; Park, S.J.; Kim, J.H.; Son, J.; Kang, S.S.; Lee, H.-Y. The natural compound gracillin exerts potent antitumor activity by targeting mitochondrial complex II. Cell Death Dis. 2019, 10, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Kawada, M.; Momose, I.; Someno, T.; Tsujiuchi, G.; Ikeda, D. New atpenins, NBRI23477 A and B, inhibit the growth of human prostate cancer cells. J. Antibiot. 2009, 62, 243–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Huwaimel, B.; Verma, K.; Miller, J.; Germain, T.M.; Kinarivala, N.; Pappas, D.; Brookes, P.S.; Trippier, P.C. Synthesis and Antineoplastic Evaluation of Mitochondrial Complex II (Succinate Dehydrogenase) Inhibitors Derived from Atpenin A5. ChemMedChem 2017, 12, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Miyadera, H.; Shiomi, K.; Ui, H.; Yamaguchi, Y.; Masuma, R.; Tomoda, H.; Miyoshi, H.; Osanai, A.; Kita, K.; Ōmura, S. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc. Natl. Acad. Sci. USA 2003, 100, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Shestov, A.A.; Worth, A.J.; Nath, K.; Nelson, D.S.; Leeper, D.B.; Glickson, J.D.; Blair, I.A. Inhibition of Mitochondrial Complex II by the Anticancer Agent Lonidamine. J. Boil. Chem. 2015, 291, 42–57. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.; Holden, S.; Herman, T.; Frei, E., 3rd. Modulation of alkylating agents by lonidamine in vivo. Semin. Oncol. 1991, 18, 7. [Google Scholar]
- Nath, K.; Nelson, D.S.; Heitjan, D.F.; Leeper, D.B.; Zhou, R.; Glickson, J.D. Lonidamine induces intracellular tumor acidification and ATP depletion in breast, prostate and ovarian cancer xenografts and potentiates response to doxorubicin. NMR Biomed. 2015, 28, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Srivastava, S.K. Atovaquone: An Antiprotozoal Drug Suppresses Primary and Resistant Breast Tumor Growth by Inhibiting HER2/β-Catenin Signaling. Mol. Cancer Ther. 2019, 18, 1708–1720. [Google Scholar] [CrossRef]
- Gao, X.; Liu, X.; Shan, W.; Liu, Q.; Wang, C.; Zheng, J.; Yao, H.; Tang, R.; Zheng, J. Anti-malarial atovaquone exhibits anti-tumor effects by inducing DNA damage in hepatocellular carcinoma. Am. J. Cancer Res. 2018, 8, 1697–1711. [Google Scholar]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [Green Version]
- Paragliola, R.M.; Torino, F.; Papi, G.; Locantore, P.; Pontecorvi, A.; Corsello, S.M. Role of Mitotane in Adrenocortical Carcinoma—Review and State of the art. Eur. Endocrinol. 2018, 14, 62–66. [Google Scholar] [CrossRef]
- Hescot, S.; Slama, A.; Lombes, A.; Paci, A.; Remy, H.; Leboulleux, S.; Chadarevian, R.; Trabado, S.; Amazit, L.; Young, J.; et al. Mitotane alters mitochondrial respiratory chain activity by inducing cytochrome c oxidase defect in human adrenocortical cells. Endocr. Relat. Cancer 2013, 20, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.J.; Graham, B.; Dugo, E.; Berhaneselassie-Sumner, B.; Ndebele, K.; Tchounwou, P.B. Arsenic Trioxide Induces Apoptosis via Specific Signaling Pathways in HT-29 Colon Cancer Cells. J. Cancer Sci. Ther. 2016, 9, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Sadaf, N.; Kumar, N.; Ali, M.; Ali, V.; Bimal, S.; Haque, R.; Bimal, S. Arsenic trioxide induces apoptosis and inhibits the growth of human liver cancer cells. Life Sci. 2018, 205, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Diepart, C.; Karroum, O.; Magat, J.; Feron, O.; Verrax, J.; Calderon, P.B.; Grégoire, V.; Leveque, P.; Stockis, J.; Dauguet, N.; et al. Arsenic Trioxide Treatment Decreases the Oxygen Consumption Rate of Tumor Cells and Radiosensitizes Solid Tumors. Cancer Res. 2011, 72, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-J.; Yang, M.-H.; Zheng, J.-C.; Li, B.; Nie, W. Arsenic trioxide inhibits cancer stem-like cells via down-regulation of Gli1 in lung cancer. Am. J. Transl. Res. 2016, 8, 1133–1143. [Google Scholar]
- Esparza-Moltó, P.B.; Cuezva, J.M. The Role of Mitochondrial H+-ATP Synthase in Cancer. Front. Oncol. 2018, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Isidoro, A.; Martínez, M.; Fernández, P.L.; Ortega, Á.D.; Santamaría, G.; Chamorro, M.; Reed, J.C.; Cuezva, J.M. Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem. J. 2004, 378, 17–20. [Google Scholar] [CrossRef] [Green Version]
- García-Ledo, L.; Nuevo-Tapioles, C.; Cuevas-Martín, C.; Martínez-Reyes, I.; Soldevilla, B.; González-Llorente, L.; Cuezva, J.M. Overexpression of the ATPase Inhibitory Factor 1 Favors a Non-metastatic Phenotype in Breast Cancer. Front. Oncol. 2017, 7, 69. [Google Scholar] [CrossRef]
- Sgarbi, G.; Barbato, S.; Costanzini, A.; Solaini, G.; Baracca, A. The role of the ATPase inhibitor factor 1 (IF1) in cancer cells adaptation to hypoxia and anoxia. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 99–109. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, Q.; Xiao, G.; Yang, S.; Di, C.; Si, J.; Zhou, R.; Ye, Y.; Zhang, Y.; Zhang, H. Selective ATP hydrolysis inhibition in F1Fo ATP synthase enhances radiosensitivity in non-small-cell lung cancer cells (A549). Oncotarget 2017, 8, 53602–53612. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging 2016, 8, 1593–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Li, F.; Wang, X.; Li, C.; Meng, Q.; Wang, C.; Huang, J.; Chen, S.; Zhu, Z. Antibiotic bedaquiline effectively targets growth, survival and tumor angiogenesis of lung cancer through suppressing energy metabolism. Biochem. Biophys. Res. Commun. 2018, 495, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Gledhill, J.R.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc. Natl. Acad. Sci. USA 2007, 104, 13632–13637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.-C.; Chang, H.-Y.; Hsu, C.-H.; Kuo, W.-H.; Chang, K.-J.; Juan, H.-F. Targeting Therapy for Breast Carcinoma by ATP Synthase Inhibitor Aurovertin B. J. Proteome Res. 2008, 7, 1433–1444. [Google Scholar] [CrossRef]
- Frattaruolo, L.; Fiorillo, M.; Brindisi, M.; Curcio, R.; Dolce, V.; Lacret, R.; Truman, A.W.; Sotgia, F.; Lisanti, M.P.; Cappello, A.R. Thioalbamide, A Thioamidated Peptide from Amycolatopsis alba, Affects Tumor Growth and Stemness by Inducing Metabolic Dysfunction and Oxidative Stress. Cells 2019, 8, 1408. [Google Scholar] [CrossRef] [Green Version]
- Dahlem, C.; Siow, W.X.; Lopatniuk, M.; Tse, W.K.; Kessler, S.M.; Kirsch, S.H.; Hoppstädter, J.; Vollmar, A.M.; Müller, R.; Luzhetskyy, A.; et al. Thioholgamide A, a New Anti-Proliferative Anti-Tumor Agent, Modulates Macrophage Polarization and Metabolism. Cancers 2020, 12, 1288. [Google Scholar] [CrossRef]
- Takase, S.; Kurokawa, R.; Kondoh, Y.; Honda, K.; Suzuki, T.; Kawahara, T.; Ikeda, H.; Dohmae, N.; Osada, H.; Shin-Ya, K.; et al. Mechanism of Action of Prethioviridamide, an Anticancer Ribosomally Synthesized and Post-Translationally Modified Peptide with a Polythioamide Structure. ACS Chem. Boil. 2019, 14, 1819–1828. [Google Scholar] [CrossRef]
- Turdo, A.; Porcelli, G.; D’Accardo, C.; Franco, S.D.; Verona, F.; Forte, S.; Giuffrida, D.; Memeo, L.; Todaro, M.; Stassi, G. Metabolic Escape Routes of Cancer Stem Cells and Therapeutic Opportunities. Cancers 2020, 12, 1436. [Google Scholar] [CrossRef]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.-V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Boil. 2019, 25, 101076. [Google Scholar] [CrossRef]
- Lee, M.; Hirpara, J.L.; Eu, J.-Q.; Sethi, G.; Wang, L.; Goh, B.-C.; Wong, A.L.-A. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Boil. 2019, 25, 101073. [Google Scholar] [CrossRef]
- Lee, H.-J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.-J.; Settleman, J. Drug Resistance via Feedback Activation of Stat3 in Oncogene-Addicted Cancer Cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.L.; Hirpara, J.L.; Pervaiz, S.; Eu, J.-Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.-J.; Yan, L.; Zhang, J.; Zhang, W.-D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef]
- Wong, A.; Soo, R.A.; Tan, D.; Lee, S.C.; Lim, J.; Marban, P.; Kong, L.R.; Lee, Y.; Wang, L.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Azuma, M.; Kabe, Y.; Kuramori, C.; Kondo, M.; Yamaguchi, Y.; Handa, H. Adenine nucleotide translocator transports haem precursors into mitochondria. PLoS ONE 2008, 3, e3070. [Google Scholar] [CrossRef] [Green Version]
- Hooda, J.; Cadinu, D.; Alam, M.M.; Shah, A.; Cao, T.M.; Sullivan, L.A.; Brekken, R.; Zhang, L. Enhanced heme function and mitochondrial respiration promote the progression of lung cancer cells. PLoS ONE 2013, 8, e63402. [Google Scholar] [CrossRef] [Green Version]
- Kalainayakan, S.P.; FitzGerald, K.E.; Konduri, P.C.; Vidal, C.; Zhang, L. Essential roles of mitochondrial and heme function in lung cancer bioenergetics and tumorigenesis. Cell Biosci. 2018, 8, 56. [Google Scholar] [CrossRef]
- Ghosh, P.; Vidal, C.; Dey, S.; Zhang, L. Mitochondria Targeting as an Effective Strategy for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 3363. [Google Scholar] [CrossRef]
- Lee, P.J.; Woo, S.J.; Yoo, H.M.; Cho, N.; Kim, H.P. Differential Mechanism of ATP Production Occurs in Response to Succinylacetone in Colon Cancer Cells. Molecules 2019, 24, 3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinbach, E.C.; Ebert, P.S. Effects of succinylacetone on growth and respiration of L1210 leukemia cells. Cancer Lett. 1985, 26, 253–259. [Google Scholar] [CrossRef]
- Ebert, P.S.; Hess, R.A.; Frykholm, B.C.; Tschudy, D.P. Succinylacetone, a potent inhibitor of heme biosynthesis: Effect on cell growth, heme content and δ-aminolevulinic acid dehydratase activity of malignant murine erythroleukemia cells. Biochem. Biophys. Res. Commun. 1979, 88, 1382–1390. [Google Scholar] [CrossRef]
- Ye, W.; Zhang, L. Heme controls the expression of cell cycle regulators and cell growth in HeLa cells. Biochem. Biophys. Res. Commun. 2004, 315, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Kalainayakan, S.P.; Ghosh, P.; Dey, S.; Fitzgerald, K.E.; Sohoni, S.; Konduri, P.C.; Garrossian, M.; Liu, L.; Zhang, L. Cyclopamine tartrate, a modulator of hedgehog signaling and mitochondrial respiration, effectively arrests lung tumor growth and progression. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Urban, D.J.; Martinez, N.J.; Davis, M.I.; Brimacombe, K.R.; Cheff, D.M.; Lee, T.D.; Henderson, M.J.; Titus, S.A.; Pragani, R.; Rohde, J.M.; et al. Assessing inhibitors of mutant isocitrate dehydrogenase using a suite of pre-clinical discovery assays. Sci. Rep. 2017, 7, 12758. [Google Scholar] [CrossRef] [Green Version]
- Fujii, T.; Khawaja, M.R.; DiNardo, C.D.; Atkins, J.T.; Janku, F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov. Med. 2016, 21, 373–380. [Google Scholar]
- Chen, J.; Yang, J.; Cao, P. The Evolving Landscape in the Development of Isocitrate Dehydrogenase Mutant Inhibitors. Mini Rev. Med. Chem. 2016, 16, 1344–1358. [Google Scholar] [CrossRef]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M.; et al. Discovery and Evaluation of Clinical Candidate IDH305, a Brain Penetrant Mutant IDH1 Inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef]
- Dao10, K.-H.; Kantarjian11, H.M.; Kelly12, P.; Sweeney12, J.; Watson12, C.; Mohamed12, H.; Cortes11, J.E. Phase 1 study of the IDH1m inhibitor FT-2102 as a single agent in patients with IDH1m acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Headache 2018, 5, 1453. [Google Scholar]
- Chaturvedi, A.; Gupta, C.; Gabdoulline, R.; Borchert, N.M.; Goparaju, R.; Kaulfuss, S.; Görlich, K.; Schottmann, R.; Othman, B.; Welzenbach, J.; et al. Synergistic activity of IDH1 inhibitor BAY1436032 with azacitidine in IDH1 mutant acute myeloid leukemia. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.; Herbst, L.; Pusch, S.; Klett, L.; Goparaju, R.; Stichel, D.; Kaulfuss, S.; Panknin, O.; Zimmermann, K.; Toschi, L.; et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia 2017, 31, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, A.; Deng, X.; Khuri, F.R.; Owonikoko, T.K.; Mohammed, M.A. Altered Glutamine Metabolism and Therapeutic Opportunities for Lung Cancer. Clin. Lung Cancer 2014, 15, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Márquez, J.; Alonso, F.J.; Matés, J.M.; Segura, J.A.; Martín-Rufián, M.; Campos-Sandoval, J.A.; Márquez, J.D. Glutamine Addiction In Gliomas. Neurochem. Res. 2017, 42, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Chen, M.; Tao, Z.; Gao, S.; Li, Y.; Cao, Y.; Lu, C.; Zou, X. Effects of targeting SLC1A5 on inhibiting gastric cancer growth and tumor development in vitro and in vivo. Oncotarget 2017, 8, 76458–76467. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 2012, 19, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Yang, T.; Peng, Z.; Xiao, H.; Jiang, N.; Zhang, L.; Dickerson, C.; Wu, P.; Pan, Q. SLC1A5 Silencing Inhibits Esophageal Cancer Growth via Cell Cycle Arrest and Apoptosis. Cell. Physiol. Biochem. 2018, 48, 397. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.-K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Trovati Maciel, T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.J.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Song, M.; Kim, S.-H.; Im, C.Y.; Hwang, H.-J. Recent Development of Small Molecule Glutaminase Inhibitors. Curr. Top. Med. Chem. 2018, 18, 432–443. [Google Scholar] [CrossRef]
- Xu, X.; Meng, Y.; Li, L.; Xu, P.; Wang, J.; Li, Z.; Bian, J. Overview of the Development of Glutaminase Inhibitors: Achievements and Future Directions. J. Med. Chem. 2018, 62, 1096–1115. [Google Scholar]
- Matés, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Márquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2020; pp. 34–43. [Google Scholar]
- Guo, L.; Zhou, B.; Liu, Z.; Xu, Y.; Lu, H.; Xia, M.; Guo, E.; Shan, W.; Chen, G.; Wang, C. Blockage of glutaminolysis enhances the sensitivity of ovarian cancer cells to PI3K/mTOR inhibition involvement of STAT3 signaling. Tumor Boil. 2016, 37, 11007–11015. [Google Scholar] [CrossRef]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.-C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Boysen, G.; Jamshidi-Parsian, A.; Davis, M.A.; Siegel, E.R.; Simecka, C.M.; Kore, R.A.; Dings, R.P.; Griffin, R.J. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int. J. Radiat. Boil. 2019, 95, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, N.M.; McCullough, C.; Shanmugavelandy, S.; Lee, J.; Lee, Y.; Dutta, P.; McHenry, J.; Nguyen, L.; Norton, W.; Jones, L.W.; et al. Metabolic Differences in Glutamine Utilization Lead to Metabolic Vulnerabilities in Prostate Cancer. Sci. Rep. 2017, 7, 16159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackner, L.L.; Nunnari, J. Small molecule inhibitors of mitochondrial division: Tools that translate basic biological research into medicine. Chem. Boil. 2010, 17, 578–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.-W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Boil. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; O’Connell, J.T.; Herrera, K.N.G.; Wikman, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Chinen, L.T.D.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [Green Version]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Evers, B.M. mTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Gravel, S.-P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Widlund, H.R.; Puigserver, P. PGC-1 coactivators: Shepherding the mitochondrial biogenesis of tumors. Trends Cancer 2016, 2, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Han, Y.-S.; Lee, S.H. PGC-1α controls mitochondrial biogenesis in drug-resistant colorectal cancer cells by regulating endoplasmic reticulum stress. Int. J. Mol. Sci. 2019, 20, 1707. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Zhao, J.; Sakamuru, S.; Zhang, L.; Huang, R.; Witt, K.L.; Merrick, B.A.; Teng, C.T.; Xia, M. Identification of compounds that inhibit estrogen-related receptor alpha signaling using high-throughput screening assays. Molecules 2019, 24, 841. [Google Scholar] [CrossRef] [Green Version]
- Brindisi, M.; Fiorillo, M.; Frattaruolo, L.; Sotgia, F.; Lisanti, M.P.; Cappello, A.R. Cholesterol and Mevalonate: Two Metabolites Involved in Breast Cancer Progression and Drug Resistance through the ERRα Pathway. Cells 2020, 9, 1819. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Chen, Z.-J.; Jiang, G.-M.; Zhang, K.-S.; Liu, Q.; Liang, S.-W.; Zhou, Y.; Huang, H.-B.; Du, J.; Wang, H. Inverse agonist of estrogen-related receptor α suppresses the growth of triple negative breast cancer cells through ROS generation and interaction with multiple cell signaling pathways. Oncotarget 2016, 7, 12568–12581. [Google Scholar] [CrossRef] [Green Version]
- Casaburi, I.; Avena, P.; De Luca, A.; Chimento, A.; Sirianni, R.; Malivindi, R.; Rago, V.; Fiorillo, M.; Domanico, F.; Campana, C.; et al. Estrogen related receptor α (ERRα) a promising target for the therapy of adrenocortical carcinoma (ACC). Oncotarget 2015, 6, 25135–25148. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Wang, J.; Wang, Y.; Kwok, T.-T.; Kong, S.-K.; Wong, C. Estrogen-related receptor α (ERRα) inverse agonist XCT-790 induces cell death in chemotherapeutic resistant cancer cells. Chem. Interact. 2009, 181, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Kokabu, T.; Mori, T.; Matsushima, H.; Yoriki, K.; Kataoka, H.; Tarumi, Y.; Kitawaki, J. Antitumor effect of XCT790, an ERRα inverse agonist, on ERα-negative endometrial cancer cells. Cell. Oncol. 2019, 42, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Carabet, L.A.; Rennie, P.S.; Cherkasov, A. Therapeutic inhibition of Myc in cancer. Structural bases and computer-aided drug discovery approaches. Int. J. Mol. Sci. 2019, 20, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Motta, L.L.; Ledaki, I.; Purshouse, K.; Haider, S.; De Bastiani, M.A.; Baban, D.; Morotti, M.; Steers, G.; Wigfield, S.; Bridges, E.; et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 2016, 36, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Vogt, P.K.; Boger, D.L.; Lunec, J. Disruption of the MYC transcriptional function by a small-molecule antagonist of MYC/MAX dimerization. Oncol. Rep. 2008, 19, 825–830. [Google Scholar] [CrossRef]
- Huang, M.-J.; Cheng, Y.-c.; Liu, C.-R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc reveals new mechanisms to inhibit the MYC oncogene. Mol. Cell. Boil. 2019, 39, 00248-19. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, E.C.; Lang, M.; Rais-Bahrami, S.; Gupta, G.N.; Wei, D.; Yang, Y.; Sourbier, C.; Srinivasan, R. Preclinical efficacy of dual mTORC1/2 inhibitor AZD8055 in renal cell carcinoma harboring a TFE3 gene fusion. BCM Cancer 2019, 19, 917. [Google Scholar] [CrossRef] [Green Version]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Cui, J.-F.; Chen, M.-B.; Liu, C.-Y.; Liu, F.; Zhang, Q.-D.; Zou, J.; Lu, P.-H. The preclinical evaluation of the dual mTORC1/2 inhibitor INK-128 as a potential anti-colorectal cancer agent. Cancer Biol. Ther. 2015, 16, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Dou, J.; Yu, Y.; Zhao, Y.; Fan, Y.; Cheng, J.; Xu, X.; Liu, W.; Guan, S.; Chen, Z.; et al. mTOR ATP-competitive inhibitor INK128 inhibits neuroblastoma growth via blocking mTORC signaling. Apoptosis 2015, 20, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagwat, S.V.; Gokhale, P.C.; Crew, A.P.; Cooke, A.; Yao, Y.; Mantis, C.; Kahler, J.; Workman, J.; Bittner, M.; Dudkin, L.; et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: Distinct from rapamycin. Mol. Cancer Ther. 2011, 10, 1394–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Xu, C.; Kirubakaran, S.; Zhang, X.; Hur, W.; Liu, Y.; Kwiatkowski, N.P.; Wang, J.; Westover, K.D.; Gao, P.; et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res. 2013, 73, 2574–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Silva, D.; Tu, Y.-T.; Amunts, A.; Fontanesi, F.; Barrientos, A. Mitochondrial ribosome assembly in health and disease. Cell Cycle 2015, 14, 2226–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, T.D.; Cook, D.R.; Choi, M.Y.; Li, M.Z.; Haigis, K.M.; Elledge, S.J. A role for mitochondrial translation in promotion of viability in K-Ras mutant cells. Cell Rep. 2017, 20, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The origin and diversification of mitochondria. Curr. Boil. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [Green Version]
- Dijk, S.N.; Protasoni, M.; Elpidorou, M.; Kroon, A.M.; Taanman, J.-W. Mitochondria as target to inhibit proliferation and induce apoptosis of cancer cells: The effects of doxycycline and gemcitabine. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Son, K.; Fujioka, S.; Iida, T.; Furukawa, K.; Fujita, T.; Yamada, H.; Chiao, P.J.; Yanaga, K. Doxycycline induces apoptosis in PANC-1 pancreatic cancer cells. Anticancer. Res. 2009, 29, 3995–4003. [Google Scholar]
- Lin, C.C.; Lo, M.C.; Moody, R.R.; Stevers, N.O.; Tinsley, S.L.; Sun, D. Doxycycline targets aldehyde dehydrogenase-positive breast cancer stem cells. Oncol. Rep. 2018, 39, 3041–3047. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Abbas, M.N.; Kausar, S.; Yang, J.; Li, L.; Tan, L.; Cui, H. Biological functions and molecular mechanisms of antibiotic tigecycline in the treatment of cancers. Int. J. Mol. Sci. 2019, 20, 3577. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Jain, S.; Rahimi, H. Evaluating the efficacy of Tigecycline to target multiple cancer-types: A Review. STEM Fellowsh. J. 2019, 4, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xu, N.; He, B.; Pan, C.; Lan, Y.; Zhou, H.; Liu, X. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J. Exp. Clin. Cancer Res. 2017, 36, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Liu, W.; Huang, Q.; Wang, J.; Wang, Y.; Li, H.; Fu, X. Tigecycline as a dual inhibitor of retinoblastoma and angiogenesis via inducing mitochondrial dysfunctions and oxidative damage. Sci. Rep. 2018, 8, 11747. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Roncella, M.; Di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an inhibitor of mitochondrial biogenesis, effectively reduces cancer stem cells (CSCs) in early breast cancer patients: A clinical pilot study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- McKee, E.; Ferguson, M.; Bentley, A.; Marks, T.A. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob. Agents Chemother. 2006, 50, 2042–2049. [Google Scholar] [CrossRef] [Green Version]
- Nagiec, E.E.; Wu, L.; Swaney, S.M.; Chosay, J.G.; Ross, D.E.; Brieland, J.K.; Leach, K.L. Oxazolidinones inhibit cellular proliferation via inhibition of mitochondrial protein synthesis. Antimicrob. Agents Chemother. 2005, 49, 3896–3902. [Google Scholar] [CrossRef] [Green Version]
- Hedaya, O.M.; Mathew, P.M.; Mohamed, F.H.; Phillips, O.A.; Luqmani, Y.A. Antiproliferative activity of a series of 5-(1H-1, 2, 3-triazolyl) methyl-and 5-acetamidomethyl-oxazolidinone derivatives. Mol. Med. Rep. 2016, 13, 3311–3318. [Google Scholar] [CrossRef] [Green Version]
- Armentano, B.; Curcio, R.; Brindisi, M.; Mancuso, R.; Rago, V.; Ziccarelli, I.; Frattaruolo, L.; Fiorillo, M.; Dolce, V.; Gabriele, B.; et al. 5-(Carbamoylmethylene)-oxazolidin-2-ones as a promising class of heterocycles inducing apoptosis triggered by increased ros levels and mitochondrial dysfunction in breast and cervical cancer. Biomedicines 2020, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Ozsvari, B.; Fiorillo, M.; Bonuccelli, G.; Cappello, A.R.; Frattaruolo, L.; Sotgia, F.; Trowbridge, R.; Foster, R.; Lisanti, M.P. Mitoriboscins: Mitochondrial-based therapeutics targeting cancer stem cells (CSCs), bacteria and pathogenic yeast. Oncotarget 2017, 8, 67457–67472. [Google Scholar] [CrossRef] [Green Version]
- Grandemange, S.; Herzig, S.; Martinou, J.-C. Mitochondrial dynamics and cancer. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2009; pp. 50–56. [Google Scholar]
- Peiris-Pagès, M.; Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget 2018, 9, 13254–13275. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Wang, G.; Chwa, J.; Oh, M.E.; Abeywardana, T.; Yang, Y.; Wang, Q.A.; Jiang, L. Mitochondrial division inhibitor (mdivi-1) decreases oxidative metabolism in cancer. Br. J. Cancer 2020, 122, 1288–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Dasgupta, A.; Chen, K.H.; Neuber-Hess, M.; Patel, J.; Hurst, T.E.; Mewburn, J.D.; Lima, P.D.; Alizadeh, E.; Martin, A.; et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020, 34, 1447–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 6014. https://doi.org/10.3390/ijms21176014
Frattaruolo L, Brindisi M, Curcio R, Marra F, Dolce V, Cappello AR. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. International Journal of Molecular Sciences. 2020; 21(17):6014. https://doi.org/10.3390/ijms21176014
Chicago/Turabian StyleFrattaruolo, Luca, Matteo Brindisi, Rosita Curcio, Federica Marra, Vincenza Dolce, and Anna Rita Cappello. 2020. "Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment" International Journal of Molecular Sciences 21, no. 17: 6014. https://doi.org/10.3390/ijms21176014