SAR-mediated Similarity Assessment of the Property Profile for New, Silicon-Based AChE/BChE Inhibitors

 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
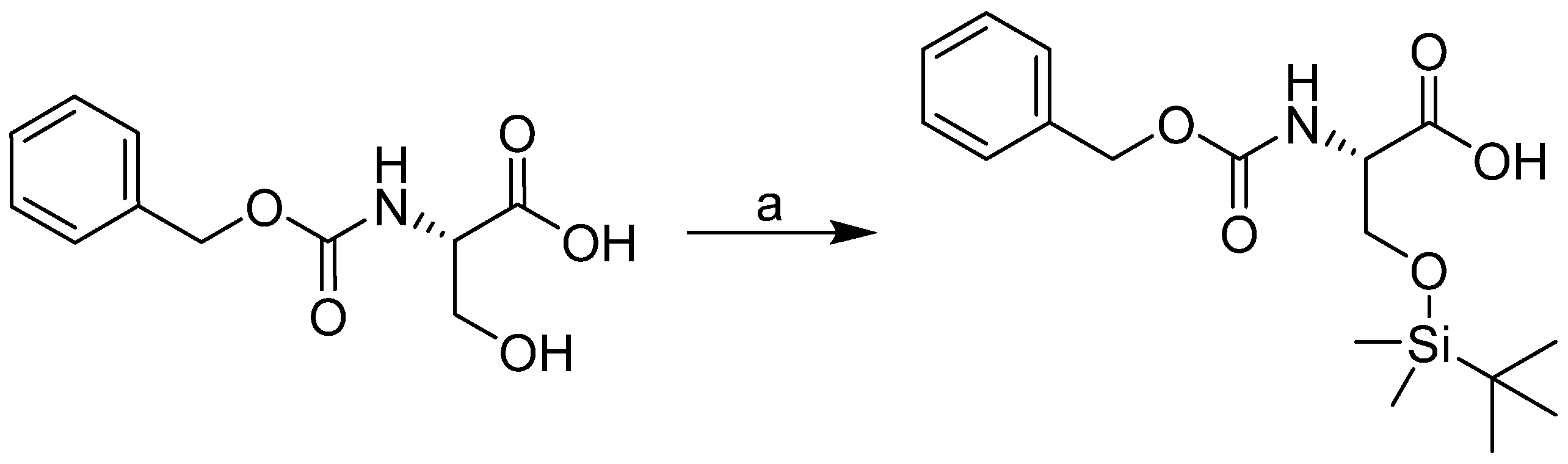
2.1. Design and Synthesis
2.2. Lipophilicity Measurement
2.3. In Vitro Evaluation of AChE and BChE-Inhibitory Profiles
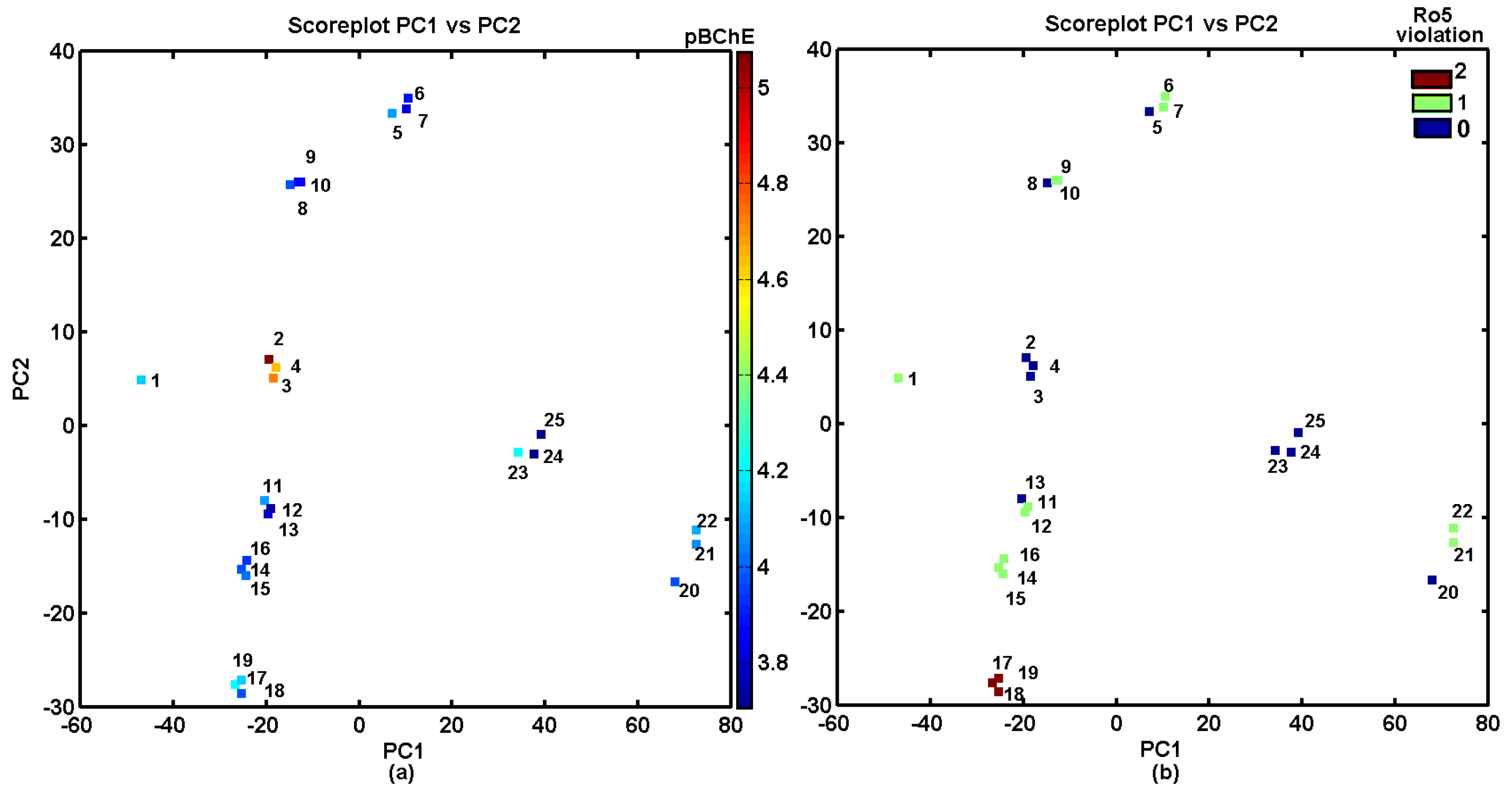
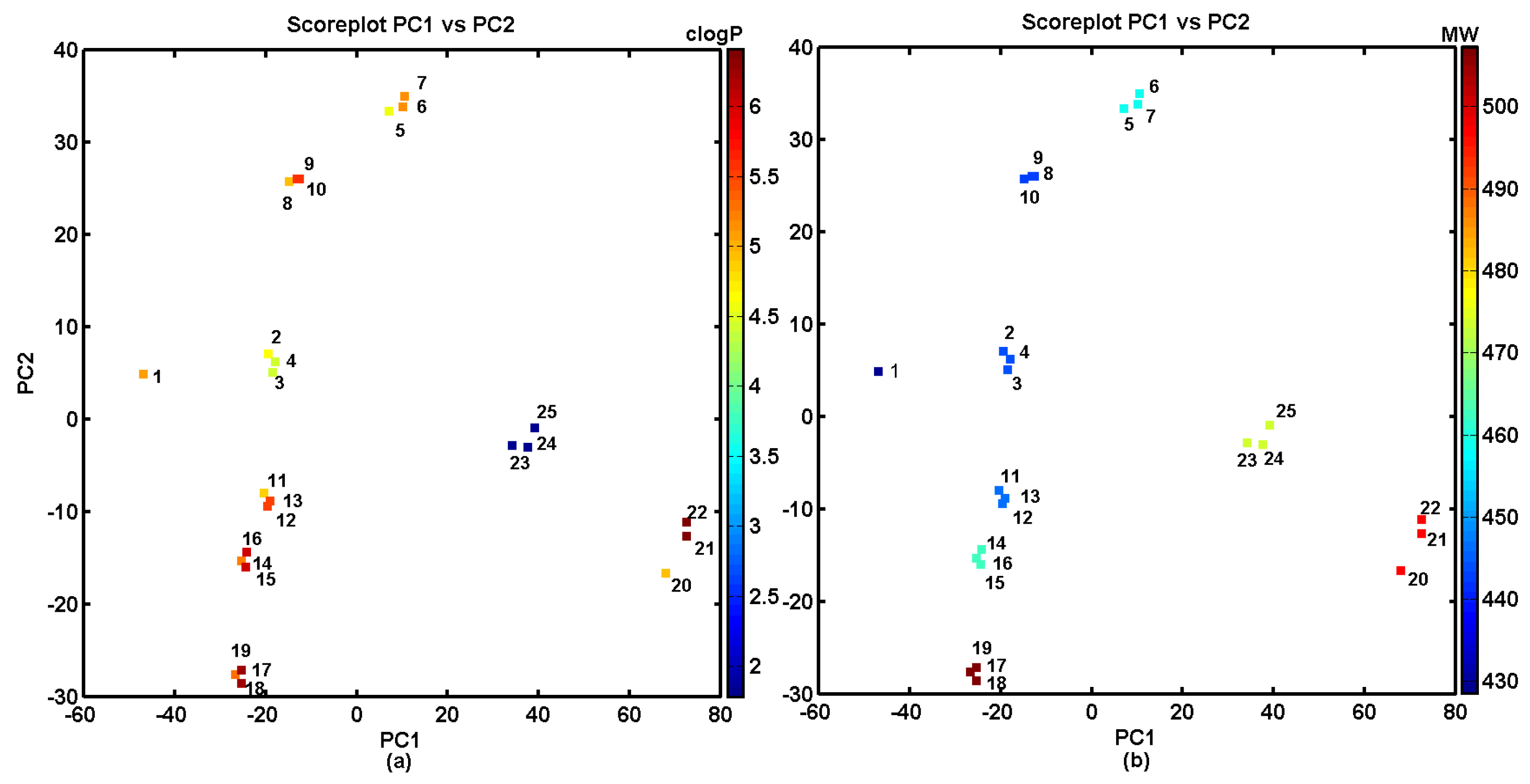
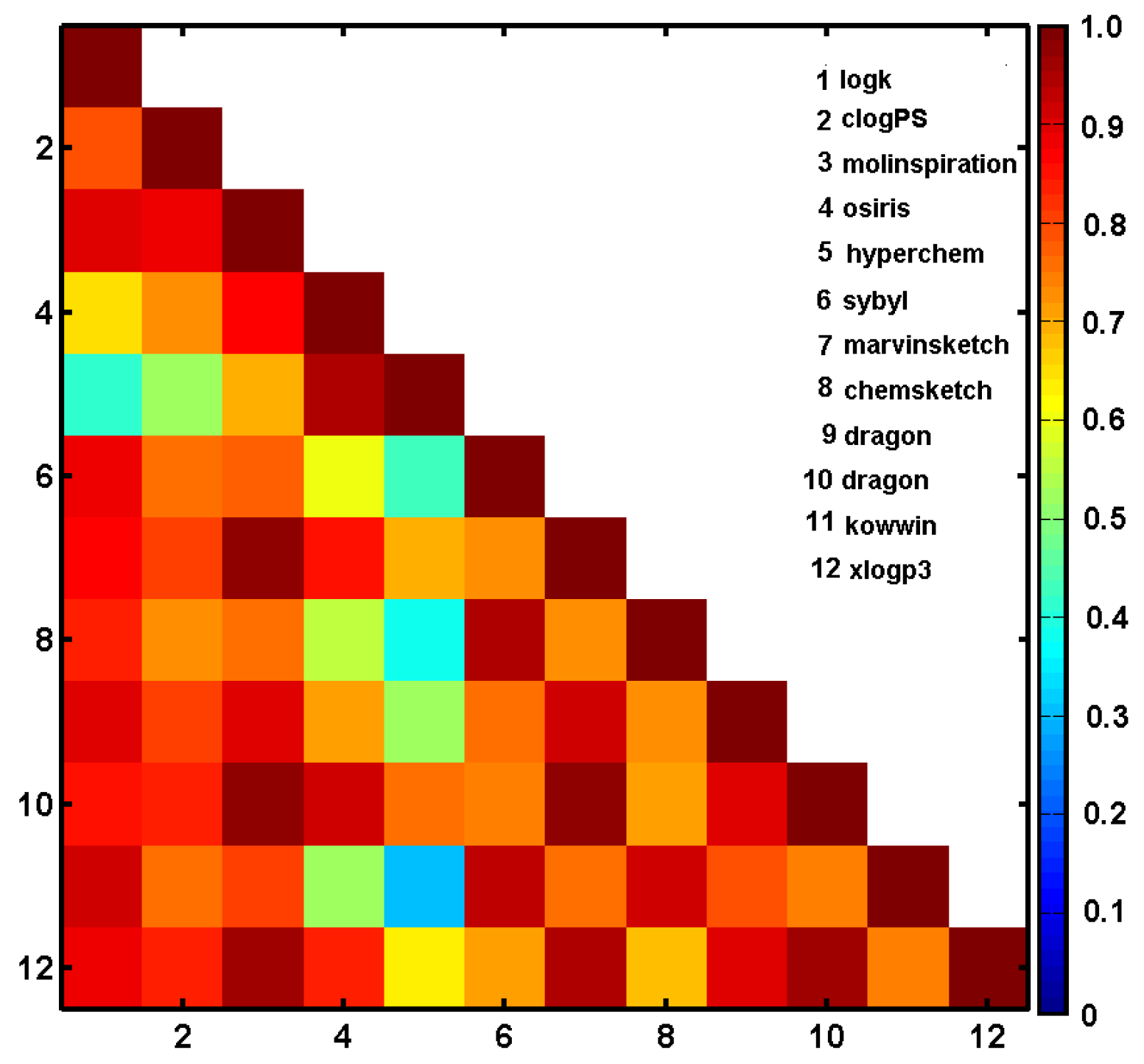
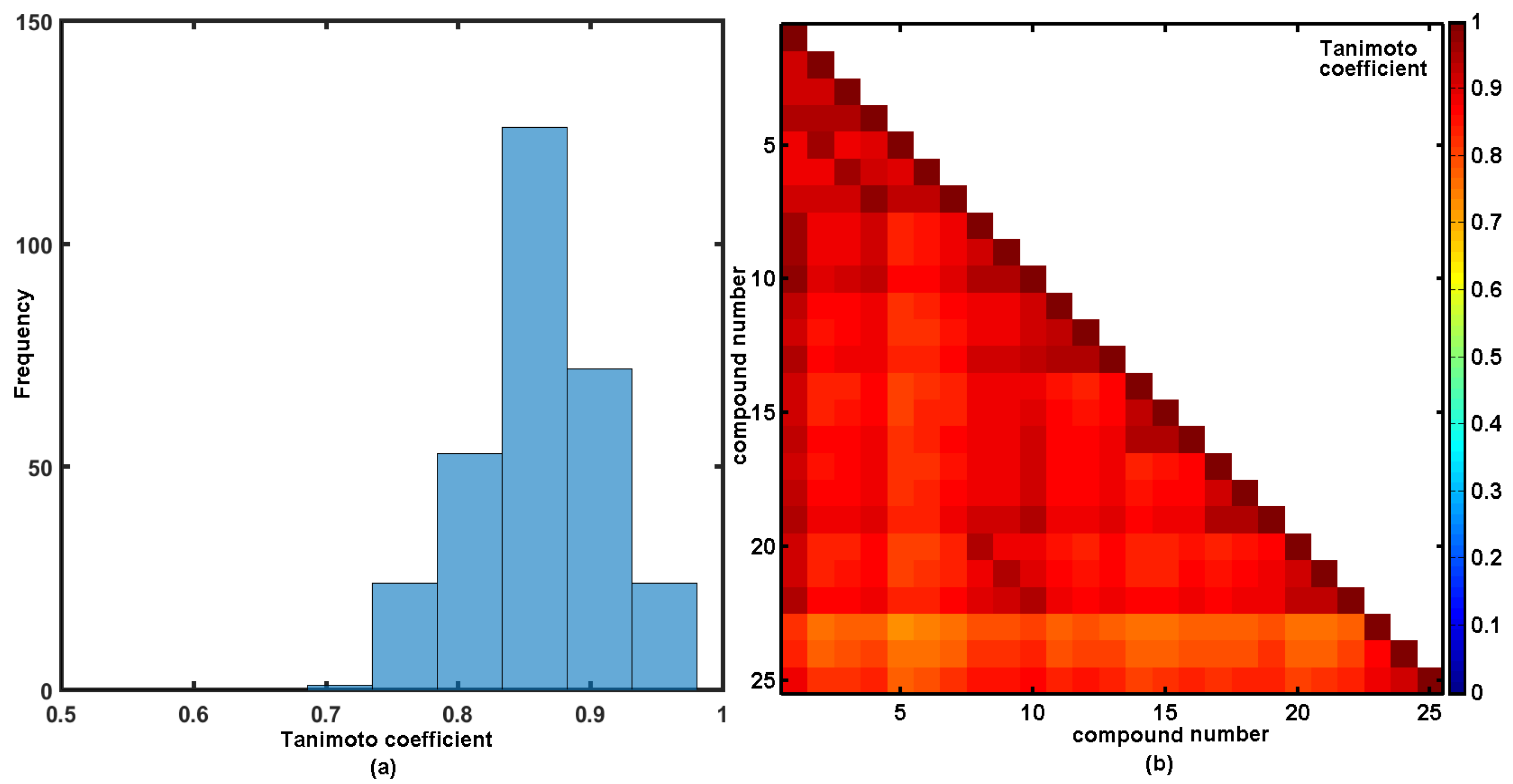
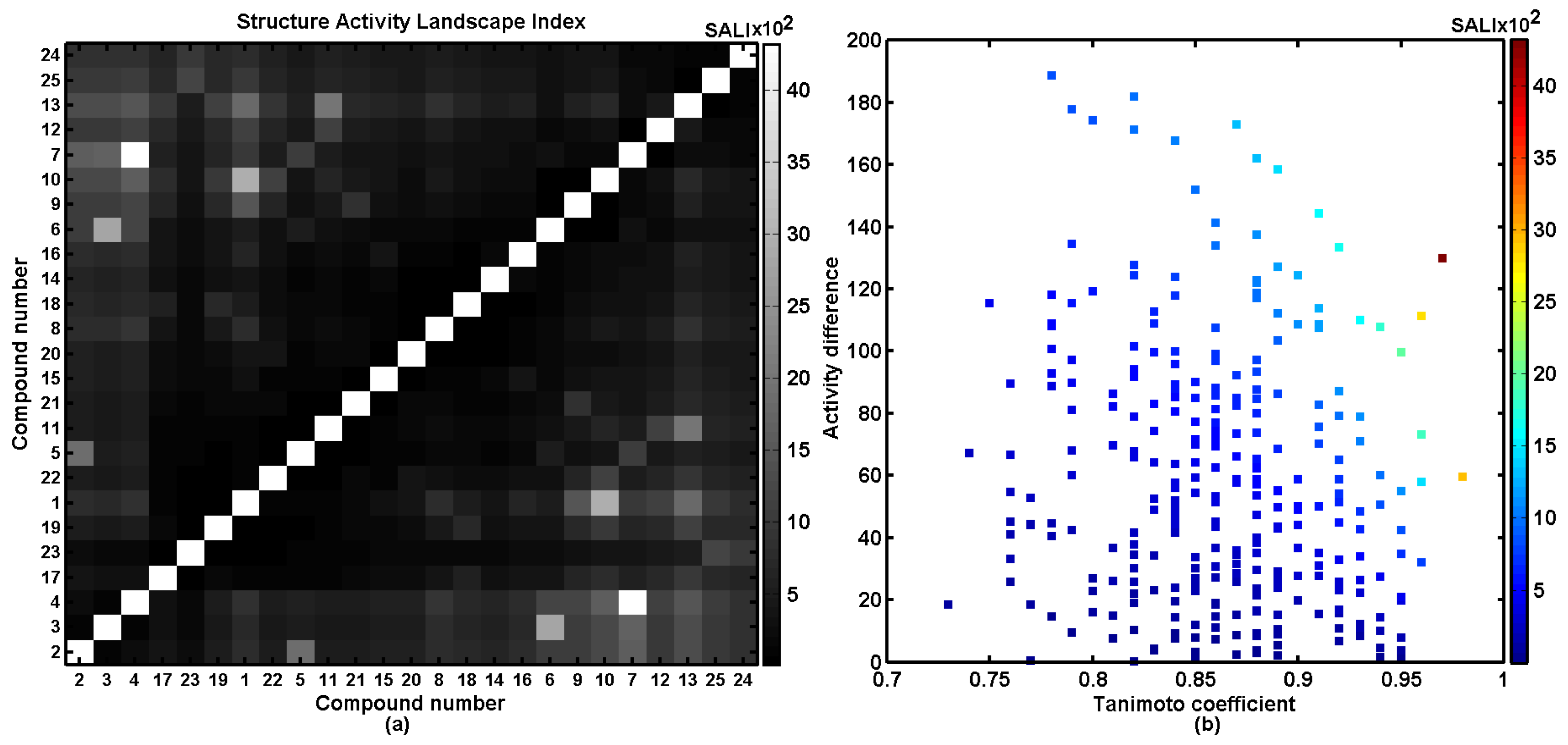
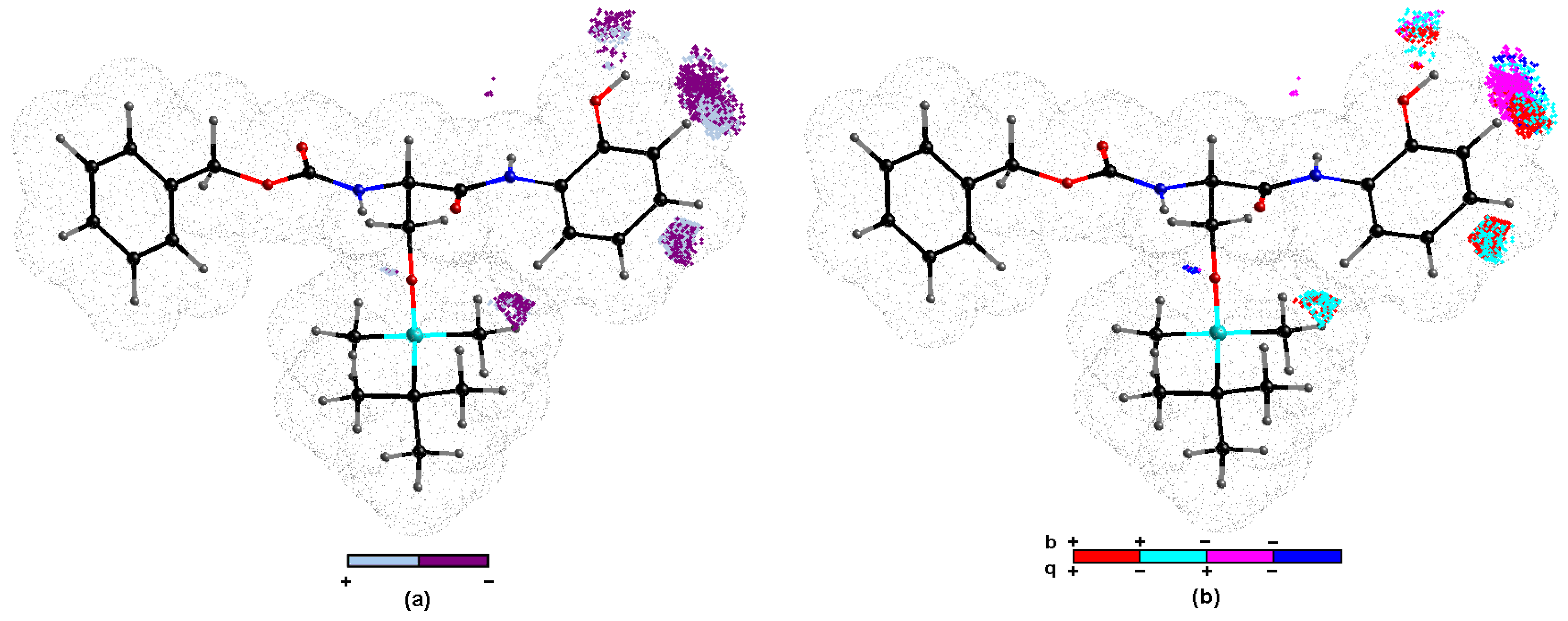
2.4. Similarity-Based Assessment of Property Profiles
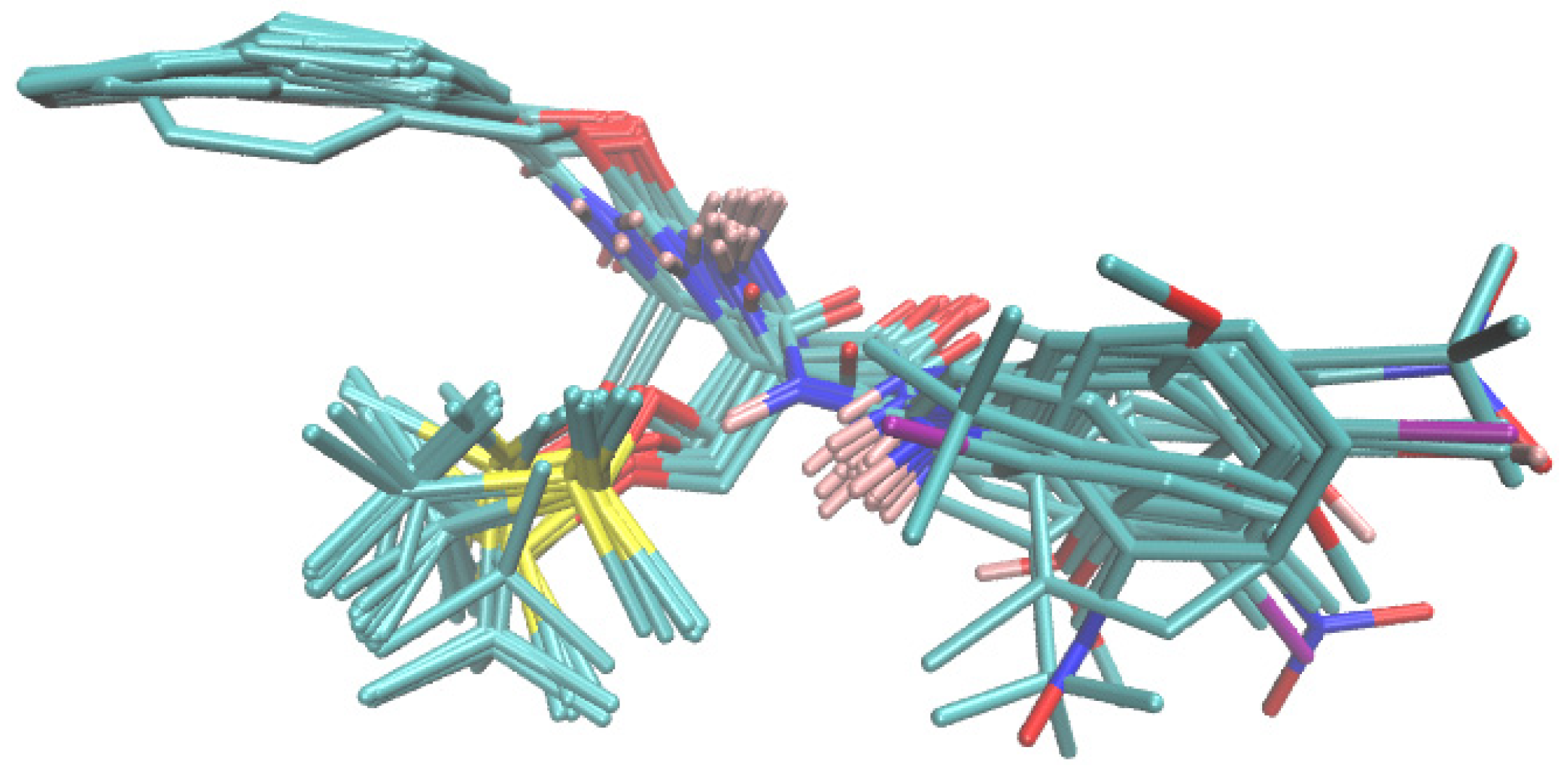
2.5. Probability-Oriented Pharmacophore Mapping
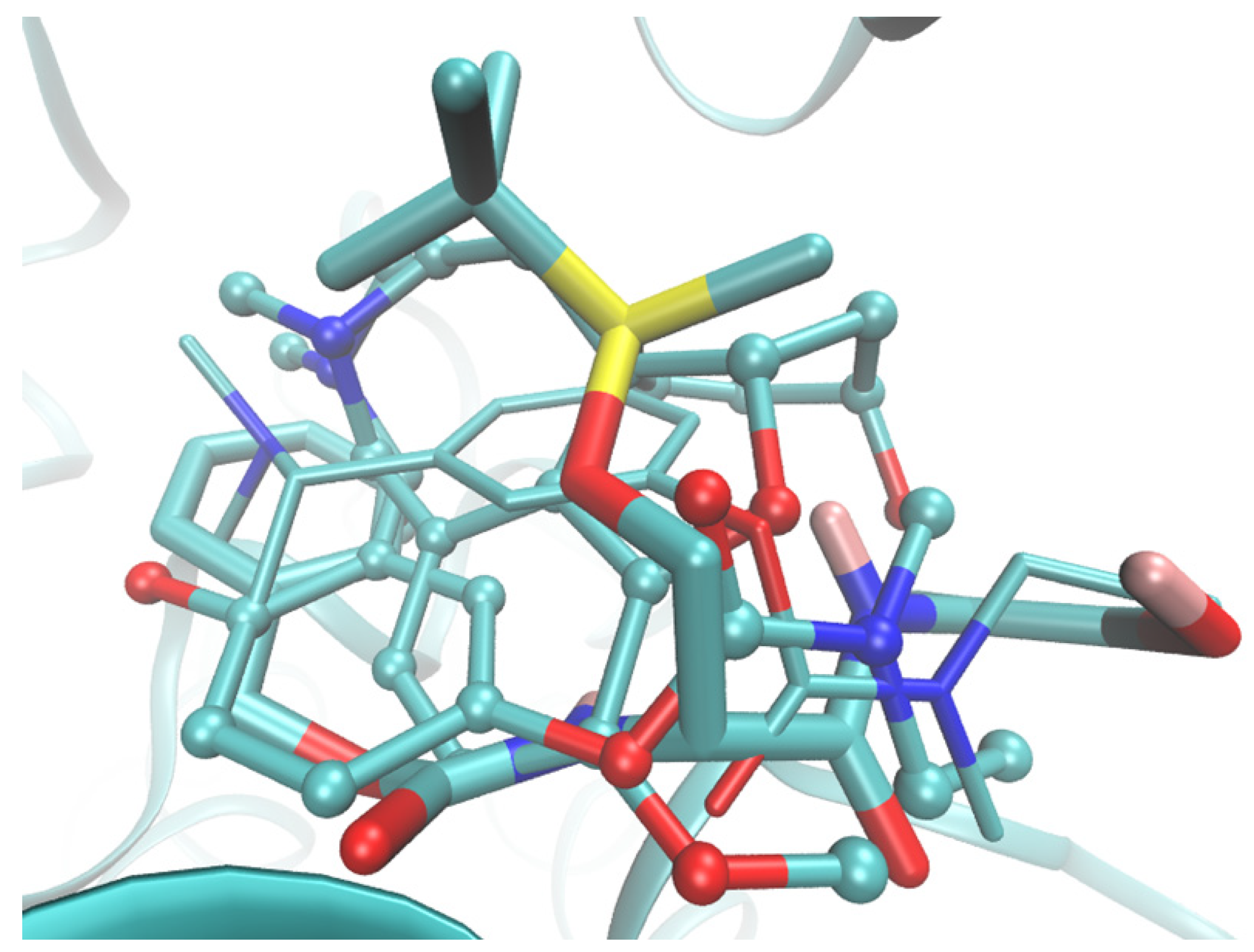
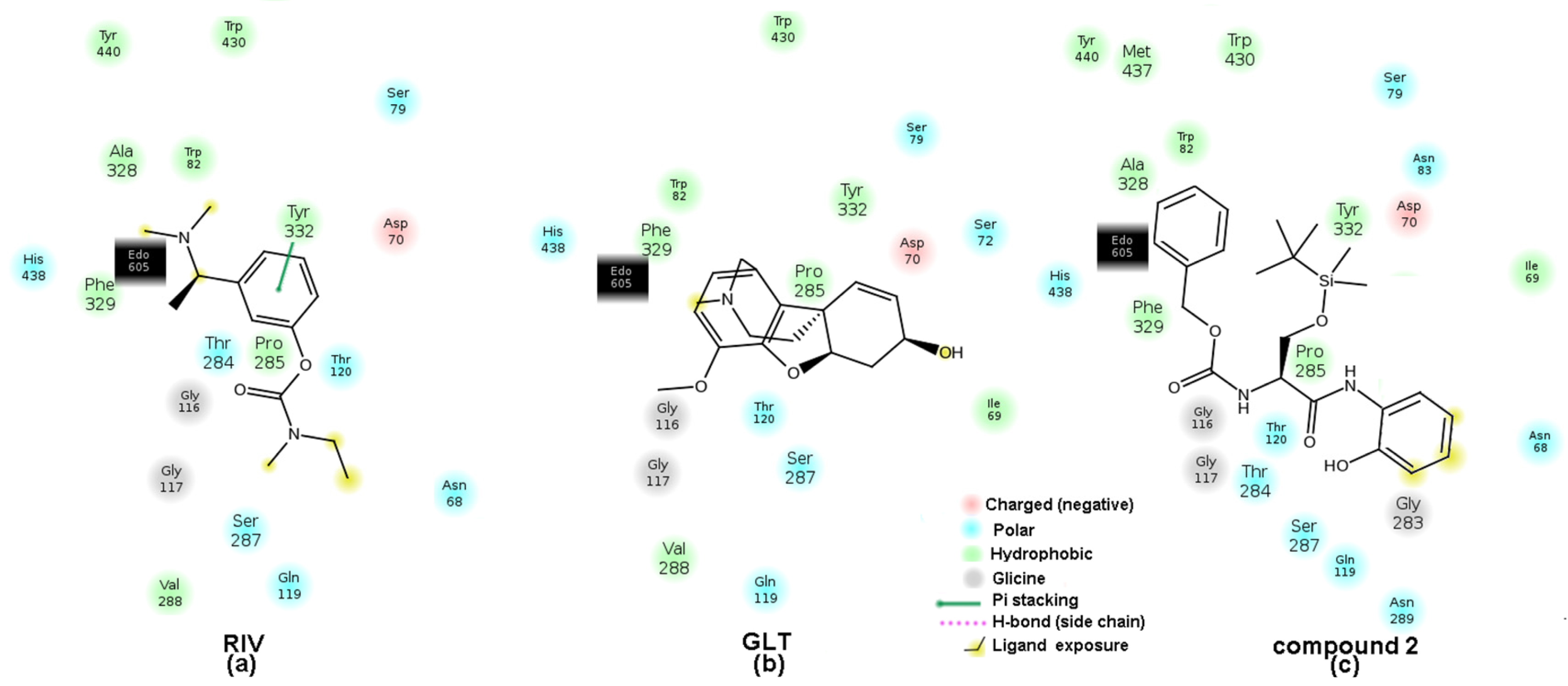
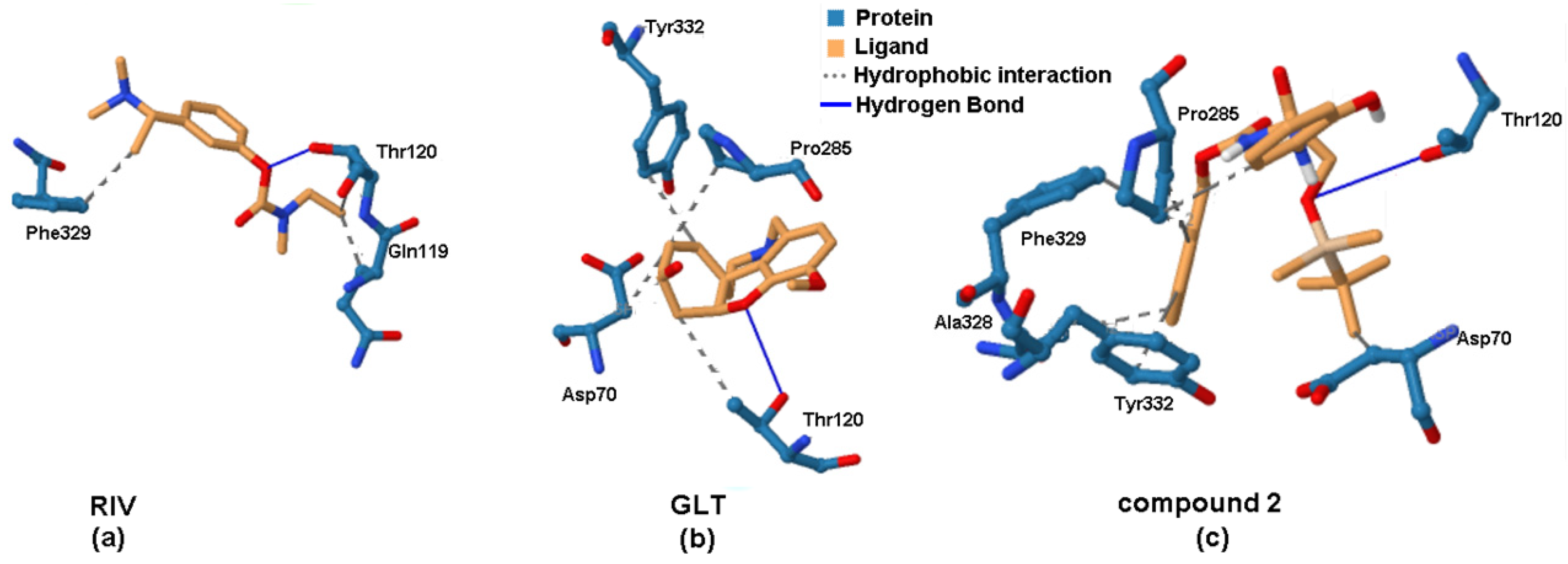
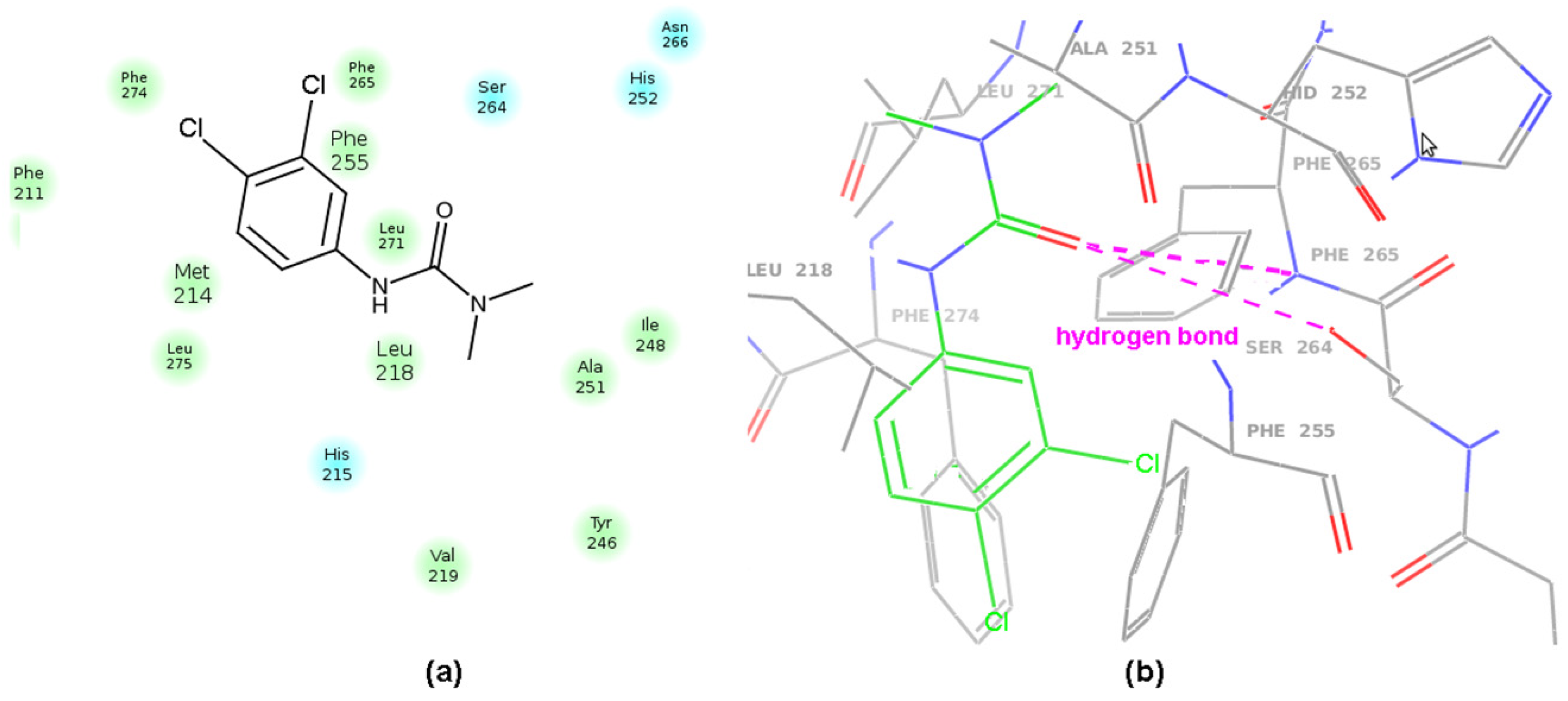
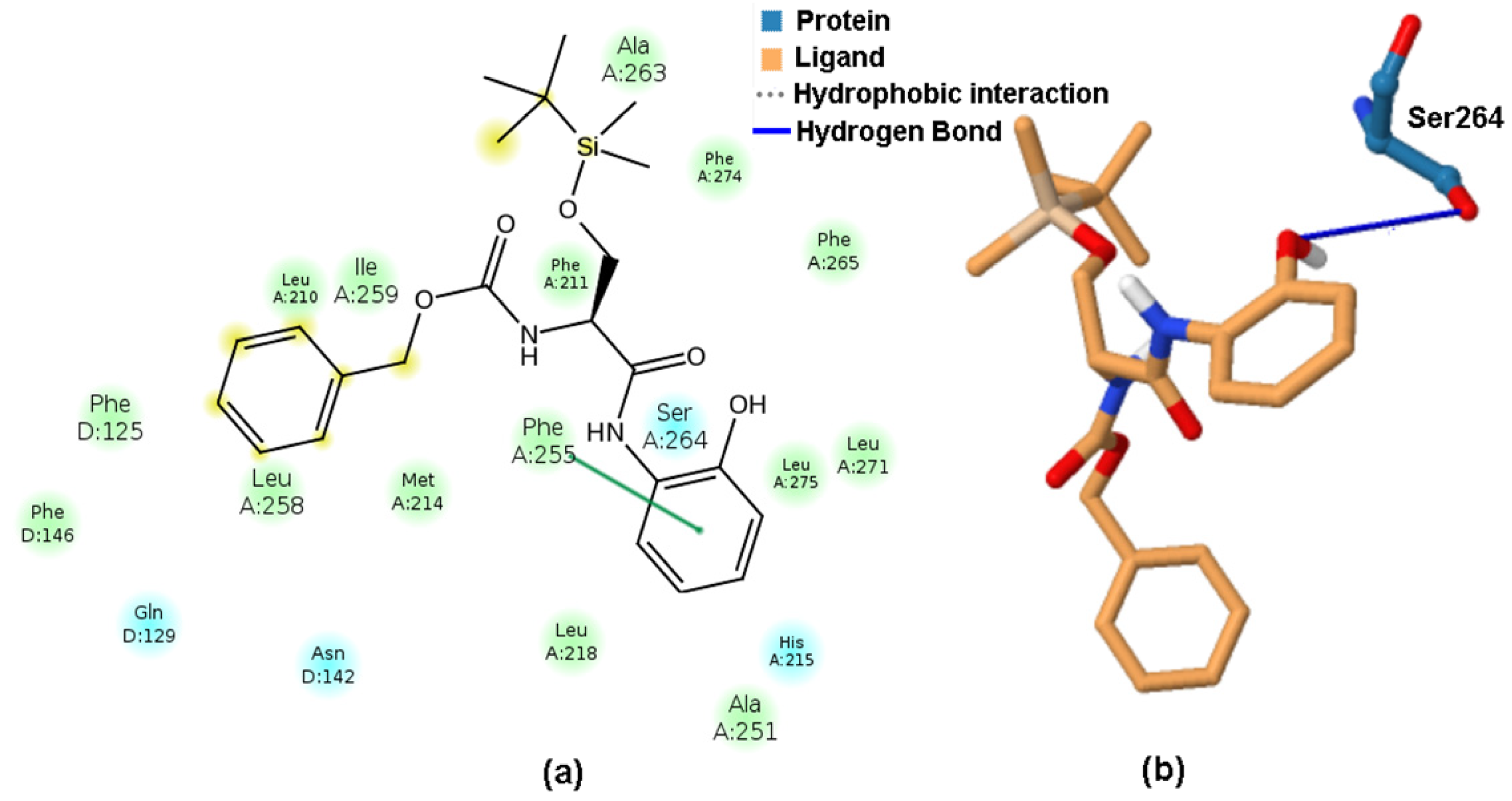
2.6. Molecular Docking Study of BChE and PET Activities
2.7. In Vitro Viability Assay
3. Materials and Methods
3.1. General Methods
3.2. Chemistry
3.2.1. General Procedure Used to Synthesize the Carbamates 1–24
3.2.2. General Procedure Used for the Synthesis of Carbamate 25
3.3. Lipophilicity Measurement
3.4. Evaluating In Vitro AChE and BChE-Inhibition Potencies
3.5. Study of Inhibition of Photosynthetic Electron Transport (PET) in Spinach Chloroplasts
3.6. In Vitro Viability Assay
3.7. Building the Model and Molecular Modelling
3.8. In Silico Lipophilicity Evaluation
3.9. Structure Activity Landscape Index
3.10. Principal Component Analysis and Partial Least Squares with Iterative Variable Elimination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| BChE | Butyrylcholinesterase |
| PSII | Photosystem II |
| CAMD | Computer Assisted Molecular Design |
| ADMET | Absorption Distribution Metabolism Excretion Toxicity |
| CoMSA | Comparative Molecular Surface Analysis |
| SMV | Stochastic Model Validation |
| RIV | Rivastigmine |
| GLT | Galanthamine |
| DCMU | 3-(3,4-dichlorophenyl)-1,1-dimethylurea |
| PQ | Plastoquinone |
| PCA | Principal Component Analysis |
| IVE-PLS | Iterative Variable Elimination Partial Least Squares |
| PLIP | Protein Ligand Interaction Profiler |
| PET | Photosynthetic Electron Transport |
References
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Devillers, J. Methods for building QSARs. Methods Mol. Biol. 2013, 930, 3–27. [Google Scholar] [PubMed]
- Bak, A.; Kozik, V.; Walczak, M.; Fraczyk, J.; Kaminski, Z.; Kolesinska, B.; Smolinski, A.; Jampilek, J. Towards intelligent drug design system: Application of artificial dipeptide receptor library in QSAR-oriented studies. Molecules 2018, 23, 1964. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2000. [Google Scholar]
- Hann, M.; Oprea, T. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef]
- Holliday, J.D.; Salim, N.; Whittle, M.; Willett, P. Analysis and display of the size dependence of chemical similarity coefficients. J. Chem. Inf. Comput. Sci. 2003, 43, 819–828. [Google Scholar] [CrossRef]
- Maggiora, G.M.; Shanmugasundaram, V. Molecular similarity measures. Methods Mol. Biol. 2011, 672, 39–100. [Google Scholar]
- Bak, A.; Magdziarz, T.; Kurczyk, A.; Serafin, K.; Polanski, J. Probing a chemical space for fragmental topology-activity landscapes (FRAGTAL): Application for diketo acid and catechol HIV integrase inhibitor offspring fragments. Comb. Chem. High Throughput Screen 2013, 16, 274–287. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Hann, M.M.; Keserü, G.M. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 2012, 11, 355–365. [Google Scholar] [CrossRef]
- Guha, R.; Van Drie, J.H. Structure – activity landscape index: Identifying and quantifying activity cliffs. J. Chem. Inf. Model. 2008, 48, 646–658. [Google Scholar] [CrossRef]
- Bajorath, J.; Peltason, L.; Wawer, M.; Guha, R.; Lajiness, M.S.; Van Drie, J.H. Navigating structure – activity landscapes. Drug Discov. Today 2009, 14, 698–705. [Google Scholar] [CrossRef] [PubMed]
- López-López, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity landscape and molecular modeling to explore the SAR of dual epigenetic inhibitors: A focus on G9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef] [PubMed]
- Guha, R.; Van Drie, J.H. Assessing how well a modeling protocol captures a structure – activity landscape. J. Chem. Inf. Model. 2008, 48, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Lemke, T.L.; Williams, D.A. Foye’s Principles of Medicinal Chemistry, 7th ed.; Lippincott Williams & Wilkins and Wolters Kluwer: Baltimore, MD, USA, 2013. [Google Scholar]
- Draber, W.; Tietjen, K.; Kluth, J.F.; Trebst, A. Herbicides in photosynthesis research. Angew. Chem. Int. Ed. 1991, 30, 1621–1633. [Google Scholar] [CrossRef]
- Bowyer, J.R.; Camilleri, P.; Vermaas, W.F.J. Herbicides, Topics in Photosynthesis; Baker, N.R., Percival, M.P., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; Volume 10, pp. 27–85. [Google Scholar]
- Imramovsky, A.; Pesko, M.; Kralova, K.; Vejsova, M.; Stolarikova, J.; Vinsova, J.; Jampilek, J. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy-N-[2-(arylamino)-1-alkyl-2-oxoethyl]benzamides. Molecules 2011, 16, 2414–2430. [Google Scholar] [CrossRef]
- Imramovsky, A.; Pesko, M.; Ferriz, J.M.; Kralova, K.; Vinsova, J.; Jampilek, J. Photosynthesis—Inhibiting efficiency of 4-chloro-2-(chlorophenylcarbamoyl)phenyl alkylcarbamates. Bioorg. Med. Chem. Lett. 2011, 21, 4564–4567. [Google Scholar] [CrossRef]
- Pauk, K.; Zadrazilova, I.; Imramovsky, A.; Vinsova, J.; Pokorna, M.; Masarikova, M.; Cizek, A.; Jampilek, J. New derivatives of salicylamides: Preparation and antimicrobial activity against various bacterial species. Bioorg. Med. Chem. 2013, 21, 6574–6581. [Google Scholar] [CrossRef]
- Zadrazilova, I.; Pospisilova, S.; Masarikova, M.; Imramovsky, A.; Monreal-Ferriz, J.; Vinsova, J.; Cizek, A.; Jampilek, J. Salicylanilide Carbamates: Promising antibacterial agents with high in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Pharm. Sci. 2015, 77, 197–207. [Google Scholar] [CrossRef]
- Peters, J.U. Polypharmacology – foe or friend. J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef]
- Pizova, H.; Havelkova, M.; Stepankova, S.; Bak, A.; Kauerova, T.; Kozik, V.; Oravec, M.; Imramovsky, A.; Kollar, P.; Bobal, P.; et al. Proline-based carbamates as cholinesterase inhibitors. Molecules 2017, 22, 1969. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Kozakiewicz, D.; Gajcy, K.; Strub, D.J.; Swietlicka, A.; Stepankova, S.; Imramovsky, A.; Polanski, J.; Smolinski, A.; et al. Novel benzene-based carbamates for AChE/BChE inhibition: Synthesis and ligand/structure-oriented SAR study. Int. J. Mol. Sci. 2019, 20, 1524. [Google Scholar] [CrossRef] [PubMed]
- Michnova, H.; Pospisilova, S.; Gonec, T.; Kapustikova, I.; Kollar, P.; Kozik, V.; Musiol, R.; Jendrzejewska, I.; Vanco, J.; Travnicek, Z.; et al. Bioactivity of methoxylated and methylated 1-hydroxynaphthalene-2-carboxanilides: Comparative molecular surface analysis. Molecules 2019, 24, 2991. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.E.; Perez, R.G.; Kobayashi, H. Cholinesterase inhibitor therapy in Alzheimer’s disease: The limits and tolerability of irreversible CNS-selective acetylcholinesterase inhibition in primates. J. Alzheimers Dis. 2017, 55, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.; Milovanovic, E.S.; Spremo-Potparevic, B.; Zivkovic, L.; Miliccivc, Z.; Stanimirovic, J.; Bogdanovic, N.; Isenovic, E.R. Treatment of Alzheimer’s disease: Classical therapeutic approach. Curr. Pharm. Anal. 2016, 12, 82–90. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: the advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging. 2017, 12, 697–707. [Google Scholar] [CrossRef]
- Jampilek, J.; Kralova, K. Nanotechnology based formulations for drug targeting to central nervous system. In Nanoparticulate Drug Delivery Systems; Keservani, R.K., Sharma, A.K., Eds.; Apple Academic Press & CRC Press: Warentown, NJ, USA, 2019; pp. 151–220. [Google Scholar]
- Wu, W.Y.; Dai, Y.C.; Li, N.G.; Dong, Z.X.; Gu, T.; Shi, Z.H.; Xue, X.; Tang, Y.P.; Duan, J.A. Novel multitarget-directed tacrine derivatives as potential candidates for the treatment of Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2017, 32, 572–587. [Google Scholar] [CrossRef]
- Lin, H.; Li, Q.; Gu, K.; Zhu, J.; Jiang, X.; Chen, Y.; Sun, H. Therapeutic agents in Alzheimer’s disease through a multi-targetdirected ligands strategy: recent progress based on tacrine core. Curr. Top. Med. Chem. 2017, 17, 3000–3016. [Google Scholar] [CrossRef]
- Hussein, W.; Saglik, B.N.; Levent, S.; Korkut, B.; Ilgın, S.; Ozkay, Y.; Kaplancıkli, Z.A. Synthesis and biological evaluation of new cholinesterase inhibitors for Alzheimer’s disease. Molecules 2018, 23, 2033. [Google Scholar] [CrossRef]
- Kumar, A.; Pintus, F.; Di Petrillo, A.; Medda, R.; Caria, P.; Matos, M.J.; Vina, D.; Pieroni, E.; Delogu, F.; Era, B.; et al. Novel 2-phenylbenzofuran derivatives as selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Sci. Rep. 2018, 8, 4424. [Google Scholar] [CrossRef]
- Acar Cevik, U.; Saglik, B.N.; Levent, S.; Osmaniye, D.; Kaya Cavusoglu, B.; Ozkay, Y.; Kaplancikli, Z.A. Synthesis and AChE-inhibitory activity of new benzimidazole derivatives. Molecules 2019, 24, 861. [Google Scholar] [CrossRef]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D.W. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J.; Kralova, K. Natural biopolymeric nanoformulations for brain drug delivery. In Nanocarriers for Brain Targetting: Principles and Applications; Keservani, R.K., Sharma, A.K., Kesharwani, R.K., Eds.; Apple Academic Press & CRC Press: Warentown, NJ, USA, 2019; pp. 131–203. [Google Scholar]
- Tischer, W.; Strotmann, H. Relationship between inhibitor binding by chloroplasts and inhibition of photosynthetic electron-transport. Biochim. Biophys. Acta 1977, 460, 113–125. [Google Scholar] [CrossRef]
- Trebst, A.; Draber, W. Structure activity correlations of recent herbicides in photosynthetic reactions. In Advances in Pesticide Science; Greissbuehler, H., Ed.; Pergamon Press: Oxford, UK, 1979; pp. 223–234. [Google Scholar]
- Izawa, S. Acceptors and donors for chloroplast electron transport. In Methods in Enzymology; Colowick, P., Kaplan, N.O., Eds.; Part C; Academic Press: London, UK, 1980; pp. 413–434. [Google Scholar]
- Lambreva, M.D.; Russo, D.; Polticelli, F.; Scognamiglio, V.; Antonacci, A.; Zobnina, V.; Campi, G.; Rea, G. Structure/function/dynamics of photosystem II plastoquinone binding sites. Curr. Protein Pept. Sci. 2014, 15, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, R.R.; de Andrade Barros, M.V.; Bressan, G.C.; Siqueira, R.P.; Dos Santos, F.S.; Bertazzini, M.; Kiralj, R.; Ferreira, M.M.C.; Forlani, G. Synthesis, theoretical studies, and effect on the photosynthetic electron transport of trifluoromethyl arylamides. Pest Manag. Sci. 2017, 73, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Broser, M.; Glöckner, C.; Gabdulkhakov, A.; Guskov, A.; Buchta, J.; Kern, J.; Müh, F.; Dau, H.; Saenger, W.; Zouni, A. Structural Basis of Cyanobacterial Photosystem II Inhibition by the Herbicide Terbutryn. J. Biol. Chem. 2011, 286, 15964–15972. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, R.; Hasegawa, K.; Takano, A.; Noguchi, T. Structures and Binding Sites of Phenolic Herbicides in the QB Pocket of Photosystem II. Biochemistry. 2010, 49, 5445–5454. [Google Scholar] [CrossRef]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef]
- Doores, K.J.; Gamblin, D.P.; Davis, B.G. Exploring and exploiting the therapeutic potential of glycoconjugates. Chem. Eur. Chem. 2007, 45, 2059–2072. [Google Scholar]
- Bobal, P.; Sujan, J.; Otevrel, J.; Imramovsky, A.; Padelkova, Z.; Jampilek, J. Microwave-assisted synthesis of new substituted anilides of quinaldic acid. Molecules 2012, 17, 1292–1306. [Google Scholar] [CrossRef]
- Gonec, T.; Bobal, P.; Sujan, J.; Pesko, M.; Guo, J.H.; Kralova, K.; Pavlacka, L.; Vesely, L.; Kreckova, E.; Kos, J.; et al. Investigating spectrum of biological activity of substituted quinoline-2-carboxamides and their isosteres. Molecules 2012, 17, 613–644. [Google Scholar] [CrossRef]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Bobal, P.; Kollar, P.; Cizek, A.; Kralova, K.; et al. Antimycobacterial and herbicidal activity of ring-substituted 1-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2013, 21, 6531–6541. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Nevin, E.; Soral, M.; Kushkevych, I.; Gonec, T.; Bobal, P.; Kollar, P.; Coffey, A.; O’Mahony, J.; Liptaj, T.; et al. Synthesis and antimycobacterial properties of ring-substituted 6-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2015, 23, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Pizova, H.; Bobal, P. An optimized and scalable synthesis of propylphosphonic anhydride for general use. Tetrahedron Lett. 2015, 56, 2014–2017. [Google Scholar] [CrossRef]
- Dighe, S.N.; De la Mora, E.; Chan, S.; Kantham, S.; McColl, G.; Miles, J.A.; Veliyath, S.K.; Sreenivas, B.Y.; Nassar, Z.D.; Silman, I.; et al. Rivastigmine and metabolite analogues with putative Alzheimer’s disease-modifying properties in a Caenorhabditis elegans model. Chem. Commun. 2019, 35, 1–14. [Google Scholar] [CrossRef]
- Kralova, K.; Masarovicova, E.; Jampilek, J. Plant responses to stress induced by toxic metals and their nanoforms. In Handbook of Plant and Crop Stress, 4th ed.; Pessarakli, M., Ed.; Taylor & Francis Group: Boca Raton, FL, USA, 2019; pp. 479–522. [Google Scholar]
- Liston, D.R.; Nielsen, J.A.; Villalobos, A.; Chapin, D.; Jones, S.B.; Hubbard, S.T.; Shalaby, I.A.; Ramirez, A.; Nason, D.; White, W.F. Pharmacology of selective acetylcholinesterase inhibitors: implications for use in Alzheimer’s disease. Eur. J. Pharmacol. 2004, 486, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Yu, Q.S.; Kulkarni, S.S.; Parrish, D.A.; Holloway, H.W.; Tweedie, D.; Shafferman, A.; Lahiri, D.K.; Brossi, A.; Greig, N.H. Inhibition of human acetyl- and butyrylcholinesterase by novel carbamates of (-)- and (+)-tetrahydrofurobenzofuran and methanobenzodioxepine. J. Med. Chem. 2006, 49, 2174–2185. [Google Scholar] [CrossRef] [PubMed]
- Ibrar, A.; Khan, A.; Ali, M.; Sarwar, R.; Mehsud, S.; Farooq, U.; Halimi, S.M.A.; Khan, I.; Al-Harrasi, A. Combined in vitro and in silico studies for the anticholinesterase activity and pharmacokinetics of coumarinyl thiazoles and oxadiazoles. Front. Chem. 2018, 6, 61. [Google Scholar] [CrossRef]
- Horakova, E.; Drabina, P.; Broy, B.; Stepánkova, S.; Vorcakova, K.; Kralovec, K.; Havelek, R.; Sedlak, M. Synthesis, characterization and in vitro evaluation of substituted N-(2-phenylcyclopropyl)carbamates as acetyl- and butyrylcholinesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, 173–179. [Google Scholar] [CrossRef]
- Kratky, M.; Stepánkova, S.; Vorcakova, K.; Vinsova, J. Investigation of salicylanilide and 4-chlorophenol-based N-monosubstituted carbamates as potential inhibitors of acetyl- and butyrylcholinesterase. Bioorg. Chem. 2018, 80, 668–673. [Google Scholar] [CrossRef]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2003, 46, 2310–2318. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Wiley-VCH Verlag GmbH & Co. KgaA: Germany, Weinheim, 2010. [Google Scholar]
- Peltason, L.; Bajorath, J. Systematic computational analysis of structure-activity relationships: concepts, challenges and recent advances. Future Med. Chem. 2009, 1, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. In silico estimation of basic activity-relevant parameters for a set of drug absorption promoters. SAR QSAR Env. Res. 2017, 28, 427–449. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. Hansch Analysis and Related Approaches; Wiley-VCH Verlag GmbH: Germany, Weinheim, 1993. [Google Scholar]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Kozik, V.; Malik, I.; Jampilek, J.; Smolinski, A. Probability-driven 3D pharmacophore mapping of antimycobacterial potential of hybrid molecules combining phenylcarbamoyloxy and N-arylpiperazine fragments. SAR QSAR Env. Res. 2018, 29, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, D. The quantitative analysis of drug–receptor interactions: A short history. Trends Pharm. Sci. 2006, 27, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Irwin, J.J. Docking screens: right for the right reasons? Curr. Top. Med. Chem. 2009, 9, 755–770. [Google Scholar] [CrossRef]
- Loll, B.; Kern, J.; Saenger, W.; Zouni, A.; Biesiadka, J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 2005, 438, 1040–1044. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, 443–447. [Google Scholar] [CrossRef]
- Broser, M.; Gabdulkhakov, A.; Kern, J.; Guskov, A.; Müh, F.; Saenger, W.; Zouni, A. Protein Structure and Folding: Crystal Structure of Monomeric Photosystem II from Thermosynechococcus elongatus at 3.6-Å Resolution. J. Biol. Chem. 2010, 285, 26255–26262. [Google Scholar] [CrossRef]
- Tanaka, A.; Fukushima, Y.; Kamiya, N. Two different structures of the oxygen-evolving complex in the same polypeptide frameworks of photosystem II. J. Am. Chem. Soc. 2017, 139, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- Otevrel, J.; Mandelova, Z.; Pesko, M.; Guo, J.; Kralova, K.; Sersen, F.; Vejsova, M.; Kalinowski, D.S.; Kovacevic, Z.; Coffey, A.; et al. Investigating the Spectrum of Biological Activity of Ring – Substituted Salicylanilides and Carbamoylphenylcarbamates. Molecules 2010, 15, 8122–8142. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Kos, J.; Pesko, M.; Dohanosova, J.; Oravec, M.; Liptaj, T.; Kralova, K.; Jampilek, J. Halogenated 1 – Hydroxynaphthalene – 2 – Carboxanilides Affecting Photosynthetic Electron Transport in Photosystem II. Molecules 2017, 22, 1709. [Google Scholar] [CrossRef] [PubMed]
- Skrzypek, A.; Matysiak, J.; Niewiadomy, A.; Bajda, M.; Szymański, P. Synthesis and biological evaluation of 1,3,4 – thiadiazole analogues as novel AchE and BuChE inhibitors. Eur. J. Med. Chem. 2013, 62, 311–319. [Google Scholar] [CrossRef]
- Chen, Y.C. Beware of docking. Trends Pharmacol Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Suffness, M.; Douros, J. Current status of the NCI plant and animal product program. J. Nat. Prod. 1982, 45, 1–14. [Google Scholar] [CrossRef]
- Pospisilova, S.; Kos, J.; Michnova, H.; Kapustikova, I.; Strharsky, T.; Oravec, M.; Moricz, A.M.; Bakonyi, J.; Kauerova, T.; Kollar, P.; et al. Synthesis and spectrum of biological activities of novel N-arylcinnamamides. Int. J. Mol. Sci. 2018, 19, 2318. [Google Scholar] [CrossRef]
- Kauerova, T.; Kos, J.; Gonec, T.; Jampilek, J.; Kollar, P. Antiproliferative and pro-apoptotic effect of novel nitro-substituted hydroxynaphthanilides on human cancer cell lines. Int. J. Mol. Sci. 2016, 17, 1219. [Google Scholar] [CrossRef]
- Doores, K.J.; Davis, B.G. Polar patch proteases as glycopeptiligases. Chem. Commun. 2005, 2, 168–170. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Ou, S.; Kwok, K.C.; Wang, Y.; Bao, H. An improved method to determine SH and –S–S– group content in soymilk protein. Food Chem. 2004, 88, 317–320. [Google Scholar] [CrossRef]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Z. für Nat. C 2004, 59, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Kralova, K.; Masarovičová, E.; Györyová, K. Inhibition of photosynthetic electron transport in spinach chloroplasts and Chlorella vulgaris and reduction of Sinapis alba L. growth by some ZN(II) compounds. Fresen. Environ. Bull. 2003, 12, 857–860. [Google Scholar]
- Zupan, J.; Gasteiger, J. Neural Networks and Drug Design for Chemists, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Likus-Cieslik, J.; Smolinski, A.; Pietrzykowski, M.; Bak, A. Sulphur contamination impact on seasonal and surface water chemistry on a reforested area of a former sulphur mine. Land Degrad. Dev. 2019, 30, 212–225. [Google Scholar] [CrossRef]
- Centner, V.; Massart, D.L.; de Noord, O.E.; de Jong, S.; Vandeginste, B.M.V.; Sterna, C. Elimination of uninformative variables for multivariate calibration. Anal. Chem. 1996, 68, 3851–3858. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
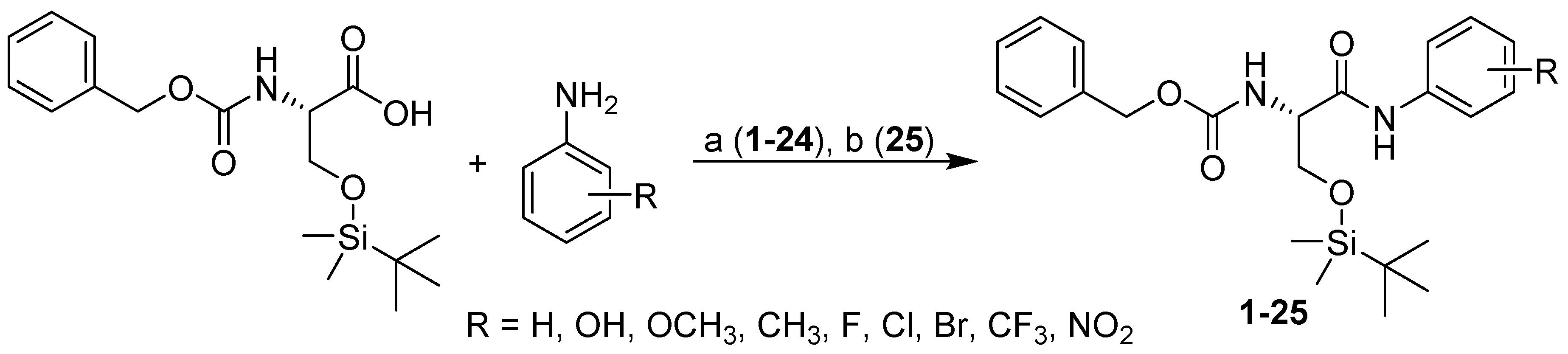{kind=link}

| No. | R | log kw | σ a | PET IC50 [µM] | AChE IC50 [µM] | BChE IC50 [µM] | SI b | WST IC50 [µM] |
|---|---|---|---|---|---|---|---|---|
| 1. | H | 6.52 ± 0.13 | 0 | 4.90 | 53.52 ± 0.40 | 73.29 ± 0.28 | 0.73 | >30 |
| 2. | 2-OH | 6.00 ± 0.04 | −0.38 | 0.48 | 42.15 ± 2.06 | 8.37 ± 0.03 | 5.04 | 3.08 ± 0.07 |
| 3. | 3-OH | 5.59 ± 0.03 | 0.12 | 4.94 | 59.70 ± 0.43 | 19.08 ± 0.17 | 3.13 | 3.47 ± 0.08 |
| 4. | 4-OH | 5.40 ± 0.03 | −0.37 | 483 | 36.22 ± 1.65 | 22.70 ± 0.24 | 1.60 | 6.43 ± 0.29 |
| 5. | 2-OCH3 | 5.85 ± 0.21 | −0.28 | >500 | 38.63 ± 1.81 | 81.47 ± 0.21 | 0.47 | – |
| 6. | 3-OCH3 | 6.26 ± 0.03 | 0.12 | 234 | 29.39 ± 0.44 | 130.30 ± 1.37 | 0.23 | >30 |
| 7. | 4-OCH3 | 6.09 ± 0.03 | −0.27 | >500 | 28.37 ± 0.74 | 152.54 ± 4.54 | 0.19 | >30 |
| 8. | 2-CH3 | 6.24 ± 0.02 | −0.17 | >500 | 46.41 ± 0.14 | 105.40 ± 2.78 | 0.44 | – |
| 9. | 3-CH3 | 6.61 ± 0.03 | −0.07 | 447 | 48.51 ± 3.77 | 131.22 ± 2.88 | 0.37 | – |
| 10. | 4-CH3 | 6.62 ± 0.03 | −0.17 | 452 | 45.04 ± 1.76 | 132.75 ± 2.46 | 0.34 | – |
| 11. | 2-F | 6.32 ± 0.02 | 0.06 | 51.5 | 43.03 ± 1.07 | 81.52 ± 4.81 | 0.53 | >30 |
| 12. | 3-F | 6.44 ± 0.02 | 0.34 | 49.2 | 45.10 ± 1.43 | 160.29 ± 7.89 | 0.28 | >30 |
| 13. | 4-F | 6.30 ± 0.02 | 0.06 | 22.9 | 62.65 ± 1.41 | 181.13 ± 10.05 | 0.35 | >30 |
| 14. | 2-Cl | 6.74 ± 0.03 | 0.22 | >500 | 47.15 ± 0.92 | 108.29 ± 1.74 | 0.44 | – |
| 15. | 3-Cl | 6.90 ± 0.02 | 0.37 | 332 | 48.12 ± 1.63 | 96.23 ± 5.29 | 0.50 | – |
| 16. | 4-Cl | 6.94 ± 0.03 | 0.23 | >500 | 42.56 ± 0.23 | 115.98 ± 0.60 | 0.68 | – |
| 17. | 2-Br | 6.82 ± 0.03 | 0.22 | 354 | 46.27 ± 1.09 | 62.60 ± 5.59 | 0.74 | – |
| 18. | 3-Br | 7.05 ± 0.03 | 0.39 | 390 | 54.37 ± 0.83 | 107.33 ± 0.43 | 0.51 | – |
| 19. | 4-Br | 7.16 ± 0.04 | 0.23 | 42.3 | 53.18 ± 0.01 | 72.58 ± 3.72 | 0.73 | >30 |
| 20. | 2-CF3 | 6.90 ± 0.04 | 0.51 | 453 | 38.08 ± 0.16 | 104.17 ± 0.36 | 0.37 | – |
| 21. | 3-CF3 | 7.32 ± 0.03 | 0.43 | 432 | 51.16 ± 1.03 | 88.87 ± 0.71 | 0.58 | – |
| 22. | 4-CF3 | 7.53 ± 0.02 | 0.51 | >500 | 42.79 ± 2.45 | 77.76 ± 0.97 | 0.55 | – |
| 23. | 2-NO2 | 6.53 ± 0.06 | 0.77 | 43.2 | 46.14 ± 2.62 | 63.12 ± 1.14 | 0.73 | >30 |
| 24. | 3-NO2 | 6.40 ± 0.04 | 0.71 | >500 | 49.42 ± 0.47 | 197.00 ± 11.28 | 0.25 | – |
| 25. | 4-NO2 | 6.58 ± 0.04 | 0.78 | 253 | 26.35 ± 0.84 | 190.35 ± 6.61 | 0.14 | >30 |
| DCMU | – | – | – | 2.1 | – | – | – | – |
| RIV | – | – | – | – | 50.10 ± 3.08 | 19.95 ± 0.31 | 1.46 | – |
| GLT | – | – | – | – | 4.0 ± 0.13 | 7.96 ± 0.59 | 0.56 | – |
| Comp. | Mobile phase |
|---|---|
| 1 | n-hexane/ethyl acetate (6:1) |
| 2 | 2% TEA in 10% methanol in dichloromethane |
| 3 | 2% TEA in 10% methanol in dichloromethane |
| 4 | 2% TEA in 10% methanol in dichloromethane |
| 5 | n-hexane/ethyl acetate (4:1) |
| 6 | n-hexane/ethyl acetate (4:1) |
| 7 | n-hexane/ethyl acetate (4:1) |
| 8 | n-hexane/ethyl acetate (4:1) |
| 9 | n-hexane/ethyl acetate (6:1) |
| 10 | n-hexane/ethyl acetate (4:1) |
| 11 | 1% DEA in n-hexane/ethyl acetate (6:1) |
| 12 | 1% DEA in n-hexane/ethyl acetate (6:1) |
| 13 | 1% DEA in n-hexane/ethyl acetate (6:1) |
| 14 | 8% TEA in n-hexane/ethyl acetate (7:1) |
| 15 | n-hexane/ethyl acetate 9:1 → 6:1 → 4:1 |
| 16 | 1% DEA in n-hexane/ethyl acetate (6:1) |
| 17 | 12% TEA in n-hexane/ethyl acetate (5:1) |
| 18 | 1% DEA in n-hexane/ethyl acetate (6:1) |
| 19 | 1% DEA in n-hexane/ethyl acetate (6:1) |
| 20 | 1% DEA in n-hexane/ethyl acetate (6:1) |
| 21 | 4% TEA in dichloromethane/n-hexane (5:1) |
| 22 | 1% DEA in n-hexane/ethyl acetate (6:1) |
| 23 | 1% DEA in n-hexane/ethyl acetate (2:1 → 3:1) |
| 24 | n-hexane/ethyl acetate (6:1 → 4:1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bak, A.; Pizova, H.; Kozik, V.; Vorcakova, K.; Kos, J.; Treml, J.; Odehnalova, K.; Oravec, M.; Imramovsky, A.; Bobal, P.; et al. SAR-mediated Similarity Assessment of the Property Profile for New, Silicon-Based AChE/BChE Inhibitors. Int. J. Mol. Sci. 2019, 20, 5385. https://doi.org/10.3390/ijms20215385
Bak A, Pizova H, Kozik V, Vorcakova K, Kos J, Treml J, Odehnalova K, Oravec M, Imramovsky A, Bobal P, et al. SAR-mediated Similarity Assessment of the Property Profile for New, Silicon-Based AChE/BChE Inhibitors. International Journal of Molecular Sciences. 2019; 20(21):5385. https://doi.org/10.3390/ijms20215385
Chicago/Turabian StyleBak, Andrzej, Hana Pizova, Violetta Kozik, Katarina Vorcakova, Jiri Kos, Jakub Treml, Klara Odehnalova, Michal Oravec, Ales Imramovsky, Pavel Bobal, and et al. 2019. "SAR-mediated Similarity Assessment of the Property Profile for New, Silicon-Based AChE/BChE Inhibitors" International Journal of Molecular Sciences 20, no. 21: 5385. https://doi.org/10.3390/ijms20215385