Altered Cell Surface N-Glycosylation of Resting and Activated T Cells in Systemic Lupus Erythematosus

Abstract
:1. Introduction
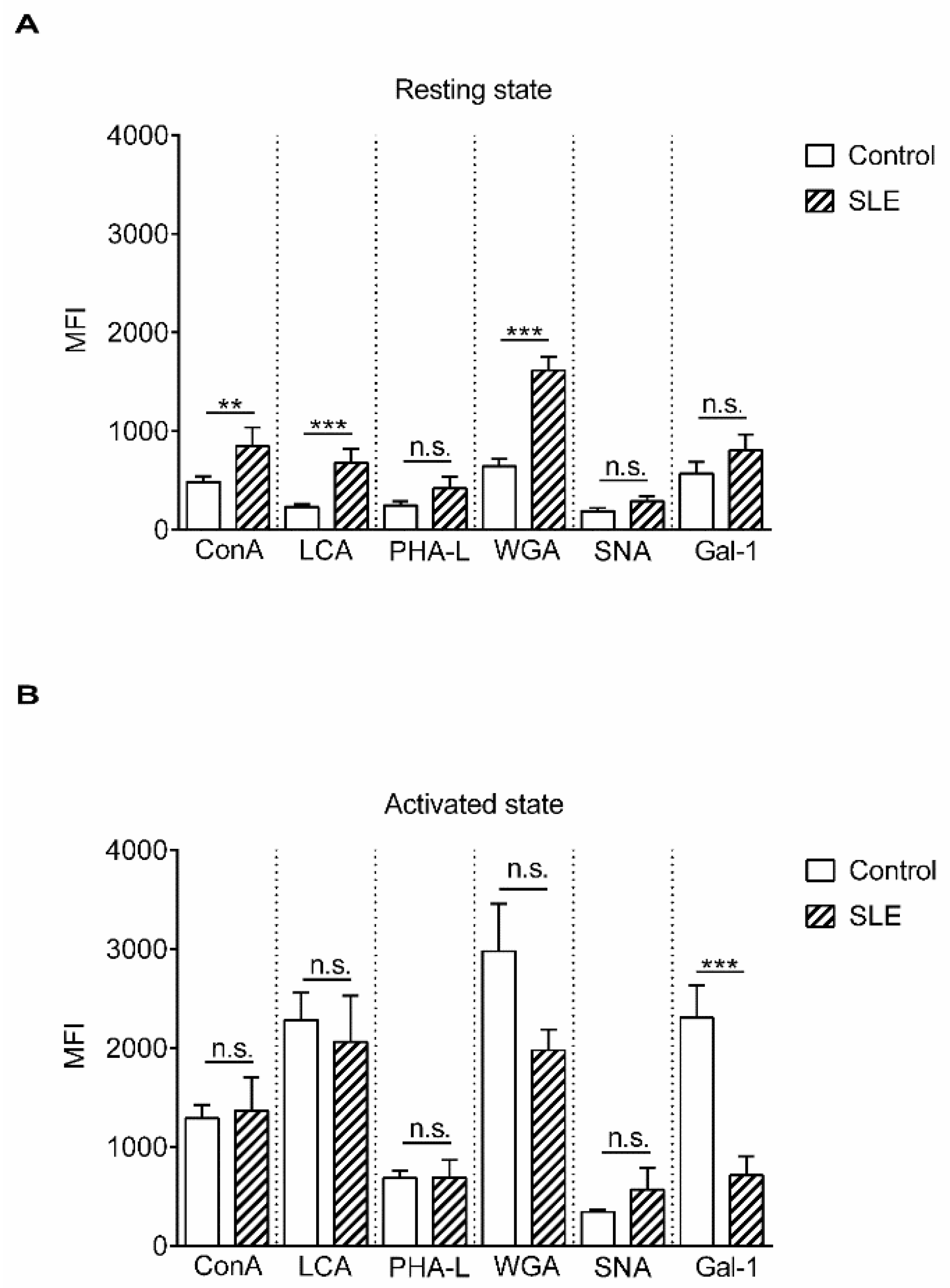
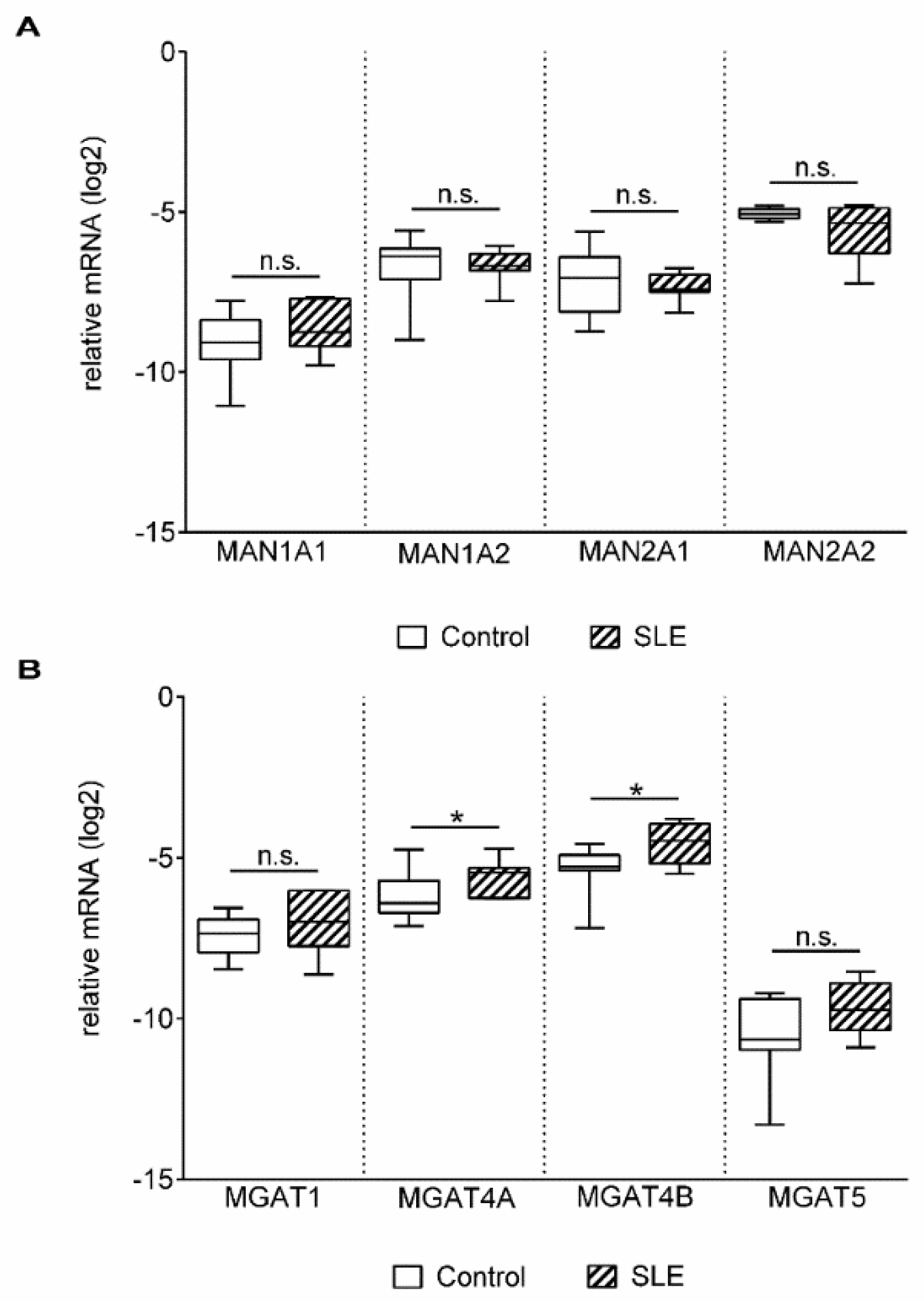
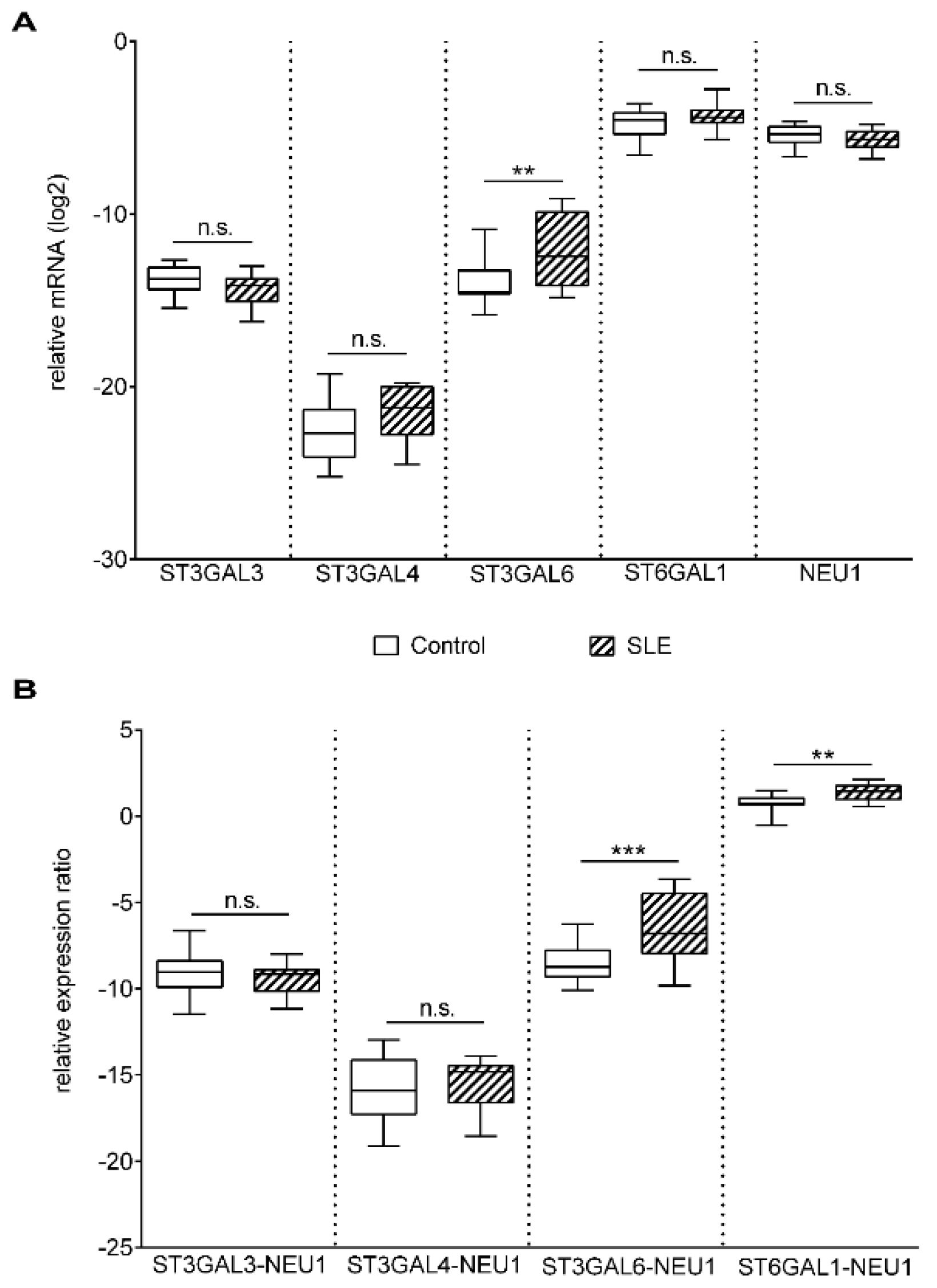
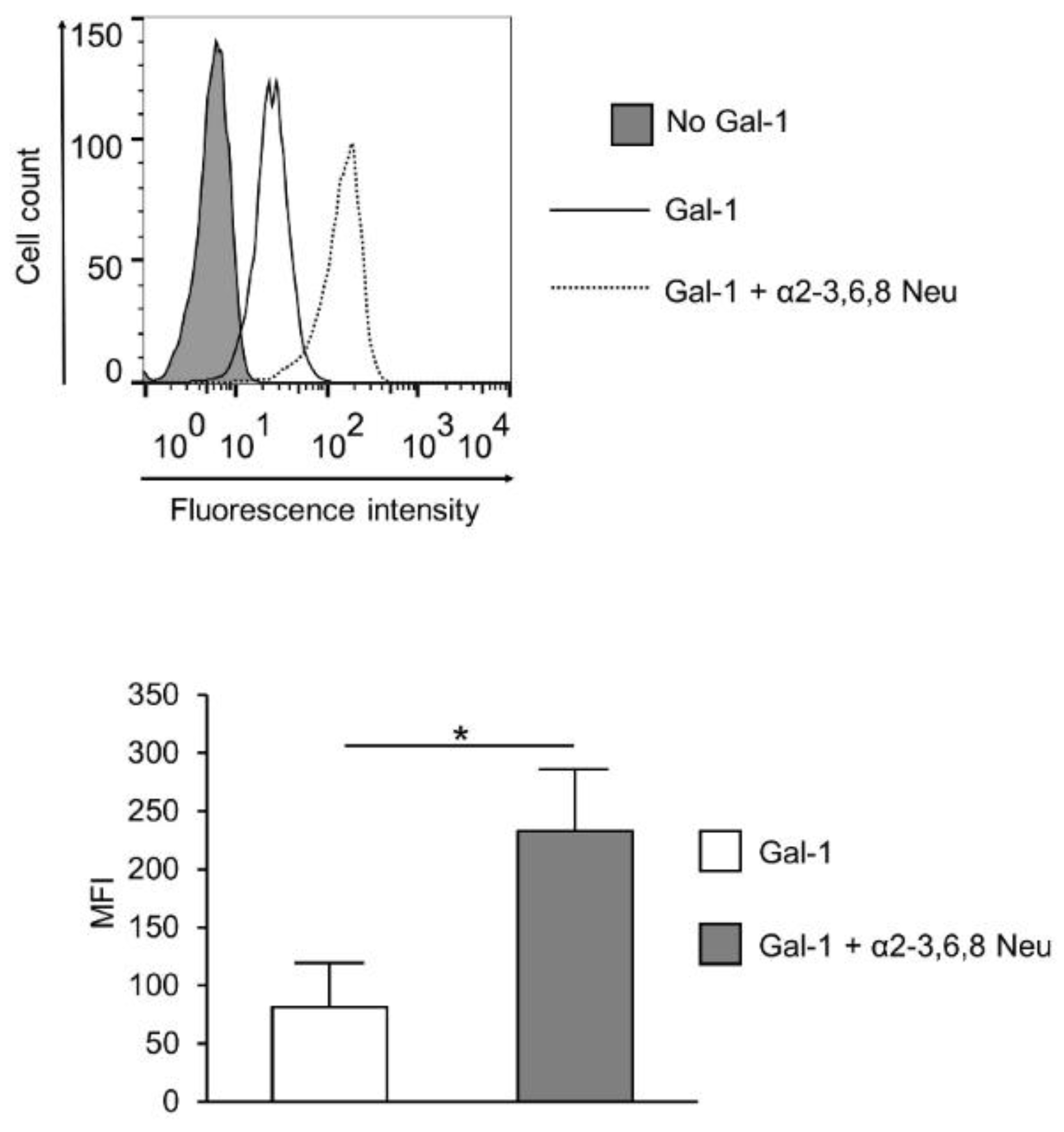
2. Results
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Patients
4.3. Cells
4.4. Lectin Binding Assay
4.5. Neuraminidase Treatment
4.6. Quantitative Real-Time PCR (qPCR)
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SLE | Systemic lupus erythematosus |
| Gal-1 | Galectin-1 |
| NEU | Neuraminidase |
| ConA | Concanavalin-A |
| LCA | Lens culinaris agglutinin |
| WGA | Wheat germ agglutinin |
| PHA | phytohaemagglutinin |
| PHA-L | Phaseolus vulgaris leukoagglutinin |
| SNA | Sambucus nigra agglutinin |
| MFI | Median fluorescence intensity |
| MGAT1–5- | Beta-N acetylglucosaminyltransferases |
| MAN | Mannosidase |
| MGAT | N-Acetyl glucosaminyltransferase |
| ST | Sialyltransferase |
| SLEDAI-2K | SLE disease activity index-2000 |
| Anti-dsDNA | Antibody to double-stranded DNA |
| PBMC | Peripheral blood mononuclear cell |
| PBS | Phosphate buffered saline |
| FACS | Fluorescence-activated cell sorting |
References
- Chachadi, V.B.; Cheng, H.; Klinkebiel, D.; Christman, J.K.; Cheng, P.W. 5-Aza-2′-deoxycytidine increases sialyl Lewis X on MUC1 by stimulating β-galactoside:α2,3-sialyltransferase 6 gene. Int. J. Biochem. Cell Biol. 2011, 43, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef] [PubMed]
- Axford, J.S. Glycosylation and rheumatic disease. Biochim. Biophys. Acta-Mol. Basis Dis. 1999, 1455, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Mackiewicz, A.; Mackiewicz, K. Glycoforms of serum alpha 1-acid glycoprotein as markers of inflammation and cancer. Glycoconj. J. 1995, 12, 241–247. [Google Scholar] [CrossRef]
- Sell, S. Progress in pathology cancer-associated carbohydrates identified by monoclonal antibodies. Hum. Pathol. 1990, 21, 1003–1019. [Google Scholar] [CrossRef]
- Gudelj, I.; Lauc, G.; Pezer, M. Immunoglobulin G glycosylation in aging and diseases. Cell. Immunol. 2018, 333, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Raychaudhuri, S.; Ruhaak, L.R.; Lebrilla, C.B. Glycans in the immune system and the altered glycan theory of autoimmunity: A critical review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef]
- Hauser, M.A.; Kindinger, I.; Laufer, J.M.; Späte, A.-K.; Bucher, D.; Vanes, S.L.; Krueger, W.A.; Wittmann, V.; Legler, D.F. Distinct CCR7 glycosylation pattern shapes receptor signaling and endocytosis to modulate chemotactic responses. J. Leukoc. Biol. 2016, 99, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Toscano, M.A.; Bianco, G.A.; Ilarregui, J.M.; Croci, D.O.; Correale, J.; Hernandez, J.D.; Zwirner, N.W.; Poirier, F.; Riley, E.M.; Baum, L.G.; et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 2007, 8, 825–834. [Google Scholar] [CrossRef]
- Bieberich, E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. Adv. Neurobiol. 2014, 9, 47–70. [Google Scholar] [PubMed] [Green Version]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A small protein with major functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef] [PubMed]
- Garin, M.I.; Chu, C.-C.; Golshayan, D.; Cernuda-Morollon, E.; Wait, R.; Lechler, R.I. Galectin-1: A key effector of regulation mediated by CD4+CD25+ T cells. Blood 2007, 109, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Motran, C.C.; Molinder, K.M.; Liu, S.D.; Poirier, F.; Miceli, M.C. Galectin-1 functions as a {Th}2 cytokine that selectively induces {Th}1 apoptosis and promotes {Th}2 function. Eur. J. Immunol. 2008, 38, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Ion, G.; Fajka-Boja, R.; Tóth, G.K.; Caron, M.; Monostori, É. Role of p56lck and ZAP70-mediated tyrosine phosphorylation in galectin-1-induced cell death. Cell Death Differ. 2005, 12, 1145–1147. [Google Scholar] [CrossRef]
- Ion, G.; Fajka-Boja, R.; Kovács, F.; Szebeni, G.; Gombos, I.; Czibula, Á.; Matkó, J.; Monostori, É. Acid sphingomyelinase mediated release of ceramide is essential to trigger the mitochondrial pathway of apoptosis by galectin-1. Cell Signal. 2006, 18, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Kovács-Sólyom, F.; Blaskó, A.; Fajka-Boja, R.; Katona, R.L.; Végh, L.; Novák, J.; Szebeni, G.J.; Krenács, L.; Uher, F.; Tubak, V.; et al. Mechanism of tumor cell-induced T-cell apoptosis mediated by galectin-1. Immunol. Lett. 2010, 127, 108–118. [Google Scholar] [CrossRef]
- Blaskó, A.; Fajka-Boja, R.; Ion, G.; Monostori, É. How does it act when soluble? Critical evaluation of mechanism of galectin-1 induced T-cell apoptosis. Acta Biol. Hung. 2011, 62, 106–111. [Google Scholar] [CrossRef]
- Novák, J.; Kriston-Pál, É.; Czibula, Á.; Deák, M.; Kovács, L.; Monostori, É.; Fajka-Boja, R. GM1 controlled lateral segregation of tyrosine kinase Lck predispose T-cells to cell-derived galectin-1-induced apoptosis. Mol. Immunol. 2014, 57, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, P.V.; Amano, M.; Mitoma, J.; Chan, J.; Said, J.; Fukuda, M.; Baum, L.G. Haploinsufficiency of C2GnT-I glycosyltransferase renders T lymphoma cells resistant to cell death. Blood 2006, 108, 2399–2406. [Google Scholar] [CrossRef]
- Deák, M.; Hornung, Á.; Novák, J.; Demydenko, D.; Szabó, E.; Czibula, Á.; Fajka-Boja, R.; Kriston-Pál, É.; Monostori, É.; Kovács, L. Novel role for galectin-1 in T-cells under physiological and pathological conditions. Immunobiology 2015, 220, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.D.; Etzler, M.E. Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Maupin, K.A.; Liden, D.; Haab, B.B. The fine specificity of mannose-binding and galactose-binding lectins revealed using outlier motif analysis of glycan array data. Glycobiology 2012, 22, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Tateno, H.; Nakamura-Tsuruta, S.; Hirabayashi, J. Comparative analysis of core-fucose-binding lectins from Lens culinaris and Pisumsativum using frontal affinity chromatography. Glycobiology 2009, 19, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.P.; Goldstein, I.J.; Flashner, M.; Ebisu, S. Interaction of Wheat Germ Agglutinin with Sialic Acid. Biochemistry 1979, 18, 5505–5511. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Ibanez, D.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J. Rheumatol. 2002, 29, 288–291. [Google Scholar] [PubMed]
- Schwarz, R.E.; Wojciechowicz, D.C.; Park, P.Y.; Paty, P.B. Phytohemagglutinin-L (PHA-L) lectin surface binding of N-linked β1-6 carbohydrate and its relationship to activated mutant ras in human pancreatic cancer cell-lines. Cancer Lett. 1996, 107, 285–291. [Google Scholar] [CrossRef]
- Fischer, E.; Brossmer, R. Sialic acid-binding lectins: Submolecular specificity and interaction with sialoglycoproteins and tumour cells. Glycoconj. J. 1995, 12, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Itakura, Y.; Nakamura-Tsuruta, S.; Kominami, J.; Tateno, H.; Hirabayashi, J. Sugar-binding profiles of chitin-binding lectins from the hevein family: A comprehensive study. Int. J. Mol. Sci. 2017, 18, 1160. [Google Scholar] [CrossRef] [PubMed]
- Pothukuchi, P.; Agliarulo, I.; Russo, D.; Rizzo, R.; Russo, F.; Parashuraman, S. Translation of genome to glycome: Role of the Golgi apparatus. FEBS Lett. 2019. [Google Scholar] [CrossRef]
- Comelli, E.M.; Head, S.R.; Gilmartin, T.; Whisenant, T.; Haslam, S.M.; North, S.J.; Wong, N.K.; Kudo, T.; Narimatsu, H.; Esko, J.D.; et al. A focused microarray approach to functional glycomics: Transcriptional regulation of the glycome. Glycobiology 2006, 16, 117–131. [Google Scholar] [CrossRef]
- Nairn, A.V.; York, W.S.; Harris, K.; Hall, E.M.; Pierce, J.M.; Moremen, K.W. Regulation of glycan structures in animal tissues. J. Biol. Chem. 2008, 283, 17298–17313. [Google Scholar] [CrossRef] [PubMed]
- Altheide, T.K.; Hayakawa, T.; Mikkelsen, T.S.; Diaz, S.; Varki, N.; Varki, A. System-wide genomic and biochemical comparisons of sialic acid biology among primates and rodents. J. Biol. Chem. 2006, 281, 25689–25702. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Carubelli, I.; Stamatos, N.M. Sialidase expression in activated human T lymphocytes influences production of IFN-γ. J. Leukoc. Biol. 2007, 81, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Katsiari, C.G.; Liossis, S.N.C.; Dimopoulos, A.M.; Charalambopoulos, D.V.; Mavrikakis, M.; Sfikakis, P.P. CD40L overexpression on T cells and monocytes from patients with systemic lupus erythematosus is resistant to calcineurin inhibition. Lupus 2002, 11, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Lesley, J. CD44 structure and function. Front. Biosci. 2016, 3, 616–630. [Google Scholar] [CrossRef]
- Guan, H.; Nagarkatti, P.S.; Nagarkatti, M. Role of CD44 in the differentiation of Th1 and Th2 cells: CD44-deficiency enhances the development of Th2 effectors in response to sheep RBC and chicken ovalbumin. J. Immunol. 2009, 183, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Harada, T.; Juang, Y.-T.; Kyttaris, V.C.; Wang, Y.; Zidanic, M.; Tung, K.; Tsokos, G.C. Phosphorylated ERM Is Responsible for Increased T Cell Polarization, Adhesion, and Migration in Patients with Systemic Lupus Erythematosus. J. Immunol. 2007, 178, 1938–1947. [Google Scholar] [CrossRef] [Green Version]
- Jury, E.C.; Flores-Borja, F.; Kabouridis, P.S. Lipid rafts in T cell signalling and disease. Semin. Cell Dev. Biol. 2007, 18, 608–615. [Google Scholar] [CrossRef] [Green Version]
- Shental-Bechor, D.; Levy, Y. Effect of glycosylation on protein folding: A close look at thermodynamic stabilization. Proc. Natl. Acad. Sci. USA 2008, 105, 8256–8261. [Google Scholar] [CrossRef] [Green Version]
- Polley, A.; Orłowski, A.; Danne, R.; Gurtovenko, A.A.; Bernardino de la Serna, J.; Eggeling, C.; Davis, S.J.; Róg, T.; Vattulainen, I. Glycosylation and Lipids Working in Concert Direct CD2 Ectodomain Orientation and Presentation. J. Phys. Chem. Lett. 2017, 8, 1060–1066. [Google Scholar] [CrossRef]
- Chen, H.-L.; Li, C.F.; Grigorian, A.; Tian, W.; Demetriou, M. T cell receptor signaling co-regulates multiple Golgi genes to enhance N-glycan branching. J. Biol. Chem. 2009, 284, 32454–32461. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.D.; Klein, J.; Van Dyken, S.J.; Marth, J.D.; Baum, L.G. T-cell activation results in microheterogeneous changes in glycosylation of CD45. Int. Immunol. 2007, 19, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comelli, E.M.; Sutton-Smith, M.; Yan, Q.; Amado, M.; Panico, M.; Gilmartin, T.; Whisenant, T.; Lanigan, C.M.; Head, S.R.; Goldberg, D.; et al. Activation of murine CD4+ and CD8+ T lymphocytes leads to dramatic remodeling of N-linked glycans. J. Immunol. 2006, 177, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Piantoni, S.; Regola, F.; Zanola, A.; Andreoli, L.; Dall’Ara, F.; Tincani, A.; Airo’, P. Effector T-cells are expanded in systemic lupus erythematosus patients with highdisease activity and damage indexes. Lupus 2018, 27, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, R.D.; Shen, X.; Illei, G.G.; Yarboro, C.H.; Prussin, C.; Hathcock, K.S.; Hodes, R.J.; Lipsky, P.E. Abnormal differentiation of memory T cells in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2184–2197. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Arthur, C.M.; Mehta, P.; Slanina, K.A.; Blixt, O.; Leffler, H.; Smith, D.F.; Cummings, R.D. Galectin-1, -2, and -3 exhibit differential recognition of sialylatedglycans and blood group antigens. J. Biol. Chem. 2008, 283, 10109–10123. [Google Scholar] [CrossRef] [PubMed]
- Dotan, N.; Altstock, R.T.; Schwarz, M.; Dukler, A. Anti-glycan antibodies as biomarkers for diagnosis and prognosis. Lupus 2006, 15, 442–450. [Google Scholar] [CrossRef]
- Harding, C.V.; Kihlberg, J.; Elofsson, M.; Magnusson, G.; Unanue, E.R. Glycopeptides bind MHC molecules and elicit specific T cell responses. J. Immunol. 1993, 151, 2419. [Google Scholar]
- Jensen, T.; Hansen, P.; Galli-Stampino, L.; Mouritsen, S.; Frische, K.; Meinjohanns, E.; Meldal, M.; Werdelin, O. Glycopeptide specific T cell hybridomas raised against an αGalNAc O-glycosylated self peptide are discriminating between highly related carbohydrate groups. Immunol. Lett. 1997, 56, 449. [Google Scholar] [CrossRef]
- Green, R.S.; Stone, E.L.; Tenno, M.; Lehtonen, E.; Farquhar, M.G.; Marth, J.D. Mammalian N-Glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity 2007, 27, 308–320. [Google Scholar] [CrossRef]
- Moremen, K.W. Golgi alpha-mannosidase II deficiency in vertebrate systems: Implications for asparagine-linked oligosaccharide processing in mammals. Biochim. Biophys. Acta 2002, 1573, 225–235. [Google Scholar] [CrossRef]
- Hochberg, M.C. Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997, 40, 1725. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Orbai, A.-M.; Alarcon, G.S.; Gordon, C.; Merrill, J.T.; Fortin, P.R.; Bruce, I.N.; Isenberg, D.; Wallace, D.J.; Nived, O.; et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Ramhorst, R.E.; Rubinstein, N.; Corigliano, A.; Daroqui, M.C.; Kier-Joffé, E.B.; Fainboim, L. Induction of allogenic T-cell hyporesponsiveness by galectin-1-mediated apoptotic and non-apoptotic mechanisms. Cell Death Differ. 2002, 9, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhou, H.; Song, X.; Shi, S.; Zhang, J.; Jia, L. Modification of sialylation is associated with multidrug resistance in human acute myeloid leukemia. Oncogene 2015, 34, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Tringali, C.; Lupo, B.; Cirillo, F.; Papini, N.; Anastasia, L.; Lamorte, G.; Colombi, P.; Bresciani, R.; Monti, E.; Tettamanti, G.; et al. Silencing of membrane-associated sialidase NEU3 diminishes apoptosis resistance and triggers megakaryocytic differentiation of chronic myeloid leukemic cells K562 through the increase of ganglioside GM3. Cell Death Differ. 2009, 16, 164–174. [Google Scholar] [CrossRef]
- Zhou, H.; Ma, H.; Wei, W.; Ji, D.; Song, X.; Sun, J.; Zhang, J.; Jia, L. B4GALT family mediates the multidrug resistance of human leukemia cells by regulating the hedgehog pathway and the expression of p-glycoprotein and multidrug resistance-associated protein 1. Cell Death Dis. 2013, 4, e654. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lectins | Abbreviation | Specificity | Reference |
|---|---|---|---|
| Concanavalin A | ConA | mannose, glucose (low affinity) | [21,22] |
| Lens culinaris agglutinin | LCA | core-fucosylated bi-antennary N-glycan | [22,23] |
| Wheat germ agglutinin | WGA | GlcNAc, sialic acid | [24,25] |
| Phaseolus vulgaris leucoagglutinin | PHA-L | β-1,6-branched tri- and tetra-antennary N-glycans | [26] |
| Sambucus nigra agglutinin | SNA | α-2,6-linked sialic acid | [27] |
| Galectin-1 | Gal-1 | LAcNAc | [28] |
| Enzyme Genes | Gene Symbol | Full Gene Name |
|---|---|---|
| Mannosidases | MAN1A1 | Mannosidase alpha class 1A member 1 |
| MAN1A2 | Mannosidase alpha class 1A member 2 | |
| MAN2A1 | Mannosidase alpha class 2A member 1 | |
| MAN2A2 | Mannosidase alpha class 2A member 2 | |
| N-Acetylglucosaminyltransferase | MGAT1 | Mannosyl (alpha-1,3-)-glycoprotein beta-1,2-N-acetylglucosaminyl-transferase |
| MGAT4A | Mannosyl (alpha-1,3-)-glycoprotein beta-1,4-N-acetylglucosaminyl-transferase isozyme A | |
| MGAT4B | Mannosyl (alpha-1,3-)-glycoprotein beta-1,4-N-acetylglucosaminyl-transferase isozyme B | |
| MGAT5 | Mannosyl (alpha-1,6-)-glycoprotein beta-1,6-N-Acetyl-glucosaminyltransferase | |
| Sialyltransferases | ST3GAL3 | ST3 beta-galactosidealpha-2,3-sialyltransferase 3 |
| ST3GAL4 | ST3 beta-galactosidealpha-2,3-sialyltransferase 4 | |
| ST3GAL6 | ST3 beta-galactosidealpha-2,3-sialyltransferase 6 | |
| ST6GAL1 | ST6 beta-galactosamidealpha-2,6-sialyltranferase 1 | |
| Neuraminidases | NEU1 | Neuraminidase 1 |
| Subject Characteristics | Age | Female/Male | Disease Activity Parameter |
|---|---|---|---|
| SLE | 42 (23–54) | 17/1 | |
| SLEDAI-2K | 14 (6–30) | ||
| anti-dsDNA (IU/mL) | 88 (2–220) | ||
| Control | 54 (31–75) | 17/2 |
| Name | Forward Primer | Reverse Primer |
|---|---|---|
| RPL27 | 5′-CGCAAAGCTGTCATCGTG-3′ | 5′-GTCACTTTGCGGGGGTAG-3′ |
| MAN1A1 | 5′-TTGGGCATTGCTGAATATGA-3′ | 5′-CAGAATACTGCTGCCTCCAGA-3′ |
| MAN1A2 | 5′-GGAGGCCTACTTGCAGCATA-3′ | 5′-GAGTTTCTCAGCCAATTGCAC-3′ |
| MAN2A1 | 5′-CCTGGAAATGTCCAAAGCA-3′ | 5′-GCGGAAATCATCTCCTAGTGG-3′ |
| MAN2A2 | 5′-TCCACCTGCTCAACCTACG-3′ | 5′-TGTAAGATGAGTGCGGTCTCC-3′ |
| MGAT1 | 5′-CGGAGCAGGCCAAGTTC-3′ | 5′-CCTTGCCCGCAGTCCTA-3′ |
| MGAT4A | 5′-CATAGCGGCAACCAAGAAC-3′ | 5′-TGCTTATTTCCAAACCTTCACTC-3′ |
| MGAT4B | 5′-CACTCTGCACTCGCTCATCT-3′ | 5′-CACTGCCGAAGTGTACTGTGA-3′ |
| MGAT5 | 5′-GCTCATCTGCGAGCCTTCT-3′ | 5′-TTGGCAGGTCACCTTGTACTT-3′ |
| ST3GAL3 | 5′-TATGCTTCAGCCTTGATG-3′ | 5′-TTGGTGACTGACAAGATGG-3′ |
| ST3GAL4 | 5′-ATGTTGGCTCTGGTCCTG-3′ | 5′-AGGAAGATGGGCTGATCC-3′ |
| ST3GAL6 | 5′-TCTATTGGGTGGCACCTGTGGAAA-3 | 5′-TGATGAAACCTCAGCAGAGAGGCA-3′ |
| ST6GAL1 | 5′-TGGGACCCATCTGTATACCACT-3′ | 5′-ATTGGGGTGCAGCTTACGAT-3′ |
| NEU1 | 5′-CCTGGATATTGGCACTGAA-3′ | 5′-CATCGCTGAGGAGACAGAAG-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabó, E.; Hornung, Á.; Monostori, É.; Bocskai, M.; Czibula, Á.; Kovács, L. Altered Cell Surface N-Glycosylation of Resting and Activated T Cells in Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2019, 20, 4455. https://doi.org/10.3390/ijms20184455
Szabó E, Hornung Á, Monostori É, Bocskai M, Czibula Á, Kovács L. Altered Cell Surface N-Glycosylation of Resting and Activated T Cells in Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2019; 20(18):4455. https://doi.org/10.3390/ijms20184455
Chicago/Turabian StyleSzabó, Enikő, Ákos Hornung, Éva Monostori, Márta Bocskai, Ágnes Czibula, and László Kovács. 2019. "Altered Cell Surface N-Glycosylation of Resting and Activated T Cells in Systemic Lupus Erythematosus" International Journal of Molecular Sciences 20, no. 18: 4455. https://doi.org/10.3390/ijms20184455