


2,5-Di-tert-butyl-2,5-diethylpyrrolidine-1-oxyls: Where Is a Reasonable Limit of Sterical Loading for Higher Resistance to Reduction?
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Procedure for 2,2-Dimethylpentan-3-one (7)
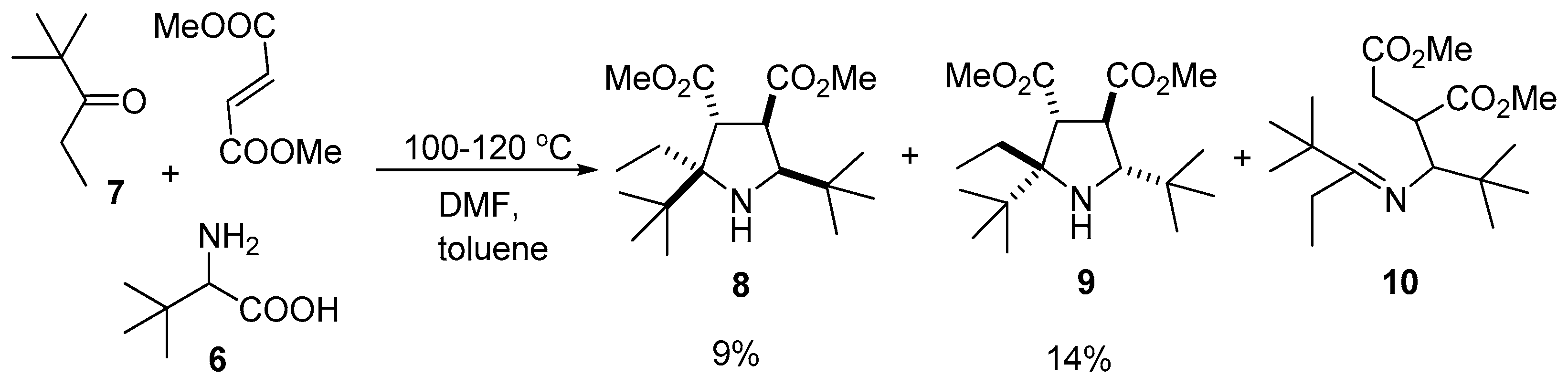
3.2.2. Condensation of 2,2-Dimethylpentan-3-one (7), 2-Amino-3,3-dimethylbutanoic Acid (6) and Dimethyl fumarate
3.2.3. Procedure for the Synthesis of (2R(S),3R(S),4R(S),5S(R))-2,5-Di-tert-butyl-3,4-bis(hydroxymethyl)-2-ethylpyrrolidine (11)
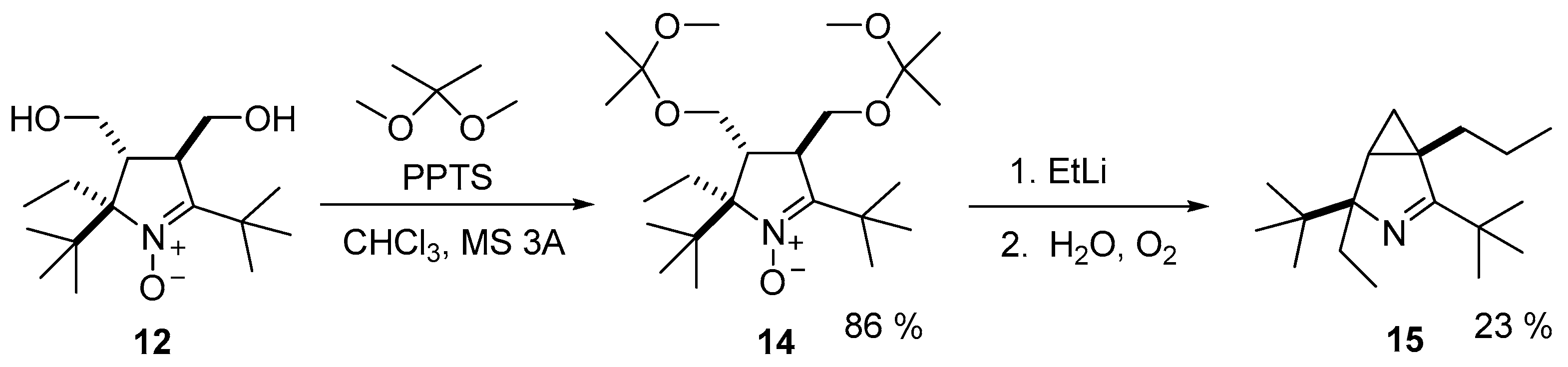
3.2.4. Procedure for the Synthesis of (2R(S),3R(S),4R(S))-2,5-Di-tert-butyl-2-ethyl-3,4-bis(hydroxymethyl)-3,4-dihydro-2H-pyrrole 1-oxide (12)
3.2.5. Procedure for the Synthesis of (2S(R),3R(S),4R(S))-2,5-Di-tert-butyl-2-ethyl-3,4-bis(methoxycarbonyl)-3,4-dihydro-2H-pyrrole 1-oxide (16)
3.2.6. Procedure for the Synthesis of (2R(S),3R(S),4R(S))-2,5-di-tert-butyl-2-ethyl-3,4-bis(((2-methoxypropan-2-yl)oxy)methyl)-3,4-dihydro-2H-pyrrole 1-oxide (14)
3.2.7. Preparation of Ethyllithium Solution
3.2.8. Procedure for the Synthesis of (2R(S),3R(S),4R(S),5S(R))-2,2,5-Triethyl-5-tert-butyl-3,4-bis(hydroxymethyl)-pyrrolidine-1-oxyl (1a)
3.2.9. Procedure for the Synthesis of (1S(R),4R(S))-2,4-Di-tert-butyl-4-ethyl-1-propyl-3-azabicyclo[3.1.0]hex-2-ene (15)
3.2.10. Procedure for the Synthesis of 2,5-Di-tert-butyl-2-ethyl-3-(methoxycarbonyl)-3,4-dihydro-2H-pyrrole 1-oxides (17) and (18)
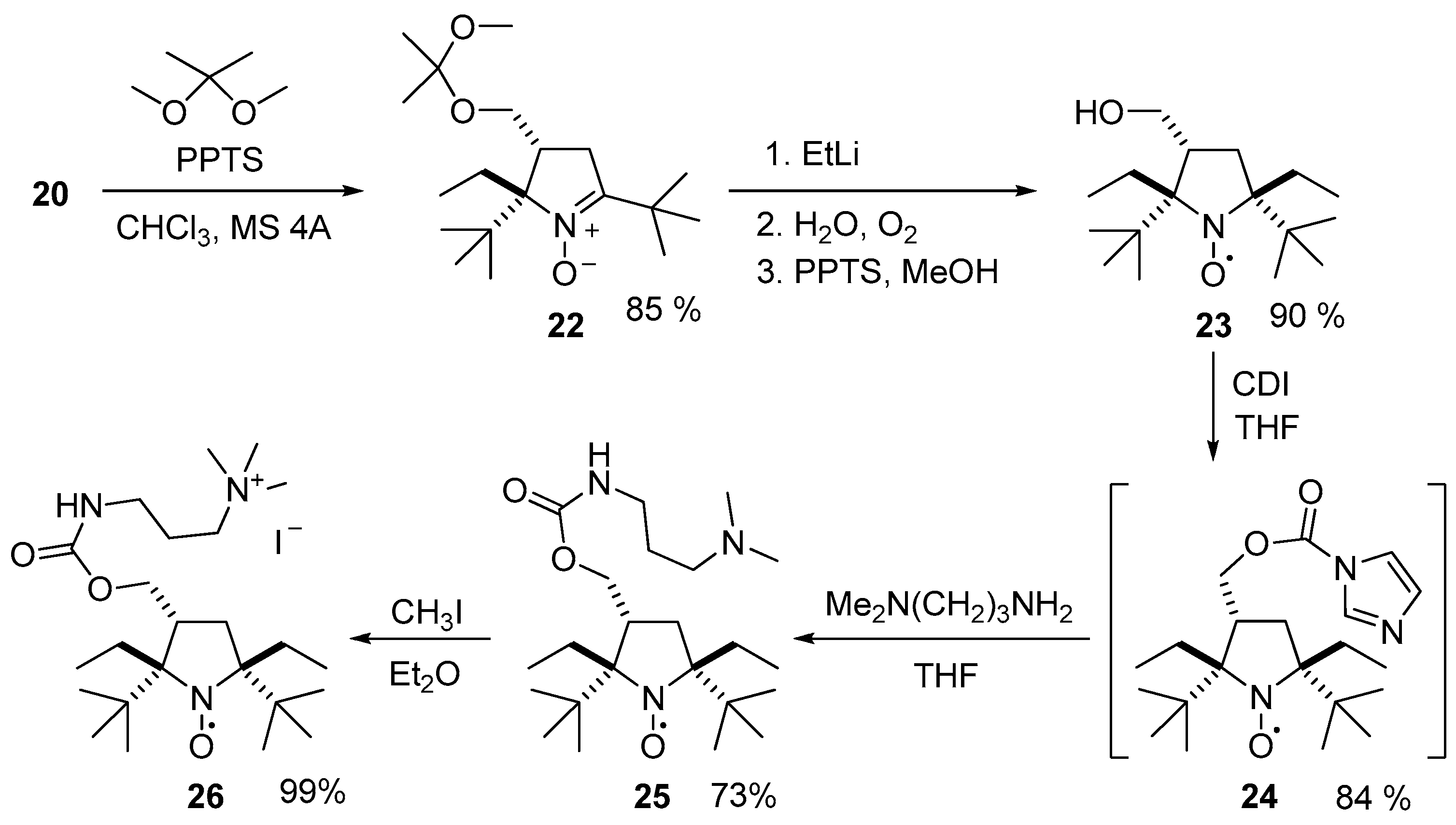
3.2.11. Procedure for the Synthesis of 2,5-Di-tert-butyl-2-ethyl-3-(hydroxymethyl)-3,4-dihydro-2H-pyrrole 1-oxides (20, 21)
3.2.12. Procedure for the Synthesis of (2S(R),3R(S))-2,5-Di-tert-butyl-2,5-diethyl-3-(hydroxymethyl)pyrrolidin-1-oxyl (23)
3.2.13. Procedure for the Synthesis of ((2S(R),3R(S),5S(R))-2,5-Di-tert-butyl-2,5-diethyl-3-((((3-(trimethylammonio)propyl)carbamoyl)oxy)methyl)pyrrolidin-1-oxyl) Monoiodide (26)
3.3. EPR Measurements and Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A1. Correlation Experiments 1H–13C HMBC and 1H–1H NOESY for 8 and 9
Appendix A2. Correlation Experiments 1H–13C HMBC and 1H–1H NOESY for 12
Appendix A3. 1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC and 1H–1H NOESY Correlation Experiments for 15
Appendix A4. 1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC and 1H–1H NOESY Correlation Experiments for 26 after Reduction with Zn/CF3COOH
References
- Likhtenshtein, G.I. Nitroxides; Springer International Publishing: Cham, Switzerland, 2020; Volume 292, ISBN 978-3-030-34821-2. [Google Scholar]
- Ouari, O.; Gigmes, D. Nitroxides: Synthesis, Properties and Applications; Royal Society of Chemistry: London, UK, 2021; ISBN 978-1-78801-752-7. [Google Scholar]
- Berliner, L.J.; Grampp, G.; Rassmusen, K.; Vorobiev, A.K.; Chumakova, N.A.; Kokorin, A.I.; Dzikovski, B.; Freed, J.; Tamura, R.; Uchida, Y.; et al. Nitroxides—Theory, Experiment and Applications; IntechOpen: London, UK, 2012; ISBN 978-9535107224. [Google Scholar]
- Leifert, D.; Studer, A. Organic Synthesis Using Nitroxides. Chem. Rev. 2023, 123, 10302–10380. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, M.; Gwozdzinski, K. Nitroxides as Antioxidants and Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 2490. [Google Scholar] [CrossRef] [PubMed]
- Galazzo, L.; Bordignon, E. Electron Paramagnetic Resonance Spectroscopy in Structural-Dynamic Studies of Large Protein Complexes. Prog. Nucl. Magn. Reson. Spectrosc. 2023, 134–135, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lilly Thankamony, A.S.; Wittmann, J.J.; Kaushik, M.; Corzilius, B. Dynamic Nuclear Polarization for Sensitivity Enhancement in Modern Solid-State NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102–103, 120–195. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.; Isse, A.A.; Barbon, A.; Altomare, L.; Hyppönen, V.; Rosa, J.; Olsson, V.; Kettunen, M.; Melone, L. Redox Properties and in Vivo Magnetic Resonance Imaging of Cyclodextrin-Polynitroxides Contrast Agents. ChemPhysChem 2023, 24, e202300100. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, H.R.; Lessard, B.H. Nitroxide-Mediated Polymerization: A Versatile Tool for the Engineering of Next Generation Materials. ACS Appl. Polym. Mater. 2020, 2, 5327–5344. [Google Scholar] [CrossRef]
- Fegy, K.; Lescop, C.; Luneau, D.; Rey, P. Magnetic Materials Based on Nitronyl Nitroxide Radicals Complexes: From Mononuclear Building Blocks to One- and Two-Dimensional Compounds. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. A 1999, 334, 521–532. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, K.; Yamauchi, Y.; Oyaizu, K.; Jia, Z. Nitroxide Radical Polymers for Emerging Plastic Energy Storage and Organic Electronics: Fundamentals, Materials, and Applications. Mater Horiz. 2021, 8, 803–829. [Google Scholar] [CrossRef]
- Nakazawa, S.; Nishida, S.; Ise, T.; Yoshino, T.; Mori, N.; Rahimi, R.D.; Sato, K.; Morita, Y.; Toyota, K.; Shiomi, D.; et al. A Synthetic Two-Spin Quantum Bit: G-Engineered Exchange-Coupled Biradical Designed for Controlled-NOT Gate Operations. Angew. Chem. Int. Ed. 2012, 51, 9860–9864. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Yamada, K.-I.; Yamasaki, T.; Sadasue, H.; Sakai, K.; Utsumi, H. Development of Novel Nitroxyl Radicals for Controlling Reactivity with Ascorbic Acid. Free Radic. Res. 2009, 43, 565–571. [Google Scholar] [CrossRef]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and Reduction Kinetics of Sterically Shielded Pyrrolidine Nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Paletta, J.T.; Berg, K.; Reinhart, E.; Rajca, S.; Rajca, A. Synthesis of Unnatural Amino Acids Functionalized with Sterically Shielded Pyrroline Nitroxides. Org. Lett. 2014, 16, 5298–5300. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, S.A.; Glazachev, Y.I.; Gatilov, Y.V.; Chernyak, E.I.; Salnikov, G.E.; Kirilyuk, I.A. Synthesis of 3,4-Bis(Hydroxymethyl)-2,2,5,5-Tetraethylpyrrolidin-1-Oxyl via 1,3-Dipolar Cycloaddition of Azomethine Ylide to Activated Alkene. J. Org. Chem. 2018, 83, 5392–5397. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. A Bioresistant Nitroxide Spin Label for In-Cell EPR Spectroscopy: In Vitro and In Oocytes Protein Structural Dynamics Studies. Angew. Chem. Int. Ed. 2018, 57, 1366–1370. [Google Scholar] [CrossRef]
- Bleicken, S.; Assafa, T.E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Bordignon, E. Gem-Diethyl Pyrroline Nitroxide Spin Labels: Synthesis, EPR Characterization, Rotamer Libraries and Biocompatibility. ChemistryOpen 2019, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Evans, E.G.B.; Jang, H.S.; Zhang, S.; Zhang, H.; Rajca, A.; Gordon, S.E.; Zagotta, W.N.; Stoll, S.; Mehl, R.A. Ultrafast Bioorthogonal Spin-Labeling and Distance Measurements in Mammalian Cells Using Small, Genetically Encoded Tetrazine Amino Acids. J. Am. Chem. Soc. 2023, 145, 14608–14620. [Google Scholar] [CrossRef] [PubMed]
- Ovcherenko, S.S.; Chinak, O.A.; Chechushkov, A.V.; Dobrynin, S.A.; Kirilyuk, I.A.; Krumkacheva, O.A.; Richter, V.A.; Bagryanskaya, E.G. Uptake of Cell-Penetrating Peptide RL2 by Human Lung Cancer Cells: Monitoring by Electron Paramagnetic Resonance and Confocal Laser Scanning Microscopy. Molecules 2021, 26, 5442. [Google Scholar] [CrossRef]
- Emoto, M.; Mito, F.; Yamasaki, T.; Yamada, K.-I.; Sato-Akaba, H.; Hirata, H.; Fujii, H. A Novel Ascorbic Acid-Resistant Nitroxide in Fat Emulsion Is an Efficient Brain Imaging Probe for in Vivo EPR Imaging of Mouse. Free Radic. Res. 2011, 45, 1325–1332. [Google Scholar] [CrossRef]
- Soikkeli, M.; Kettunen, M.I.; Nivajärvi, R.; Olsson, V.; Rönkkö, S.; Laakkonen, J.P.; Lehto, V.-P.; Kavakka, J.; Heikkinen, S. Assessment of the Relaxation-Enhancing Properties of a Nitroxide-Based Contrast Agent TEEPO-Glc with In Vivo Magnetic Resonance Imaging. Contrast Media Mol. Imaging 2019, 2019, 5629597. [Google Scholar] [CrossRef]
- Dobrynin, S.; Kutseikin, S.; Morozov, D.; Krumkacheva, O.; Spitsyna, A.; Gatilov, Y.; Silnikov, V.; Angelovski, G.; Bowman, M.K.; Kirilyuk, I.; et al. Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents. Molecules 2020, 25, 1709. [Google Scholar] [CrossRef]
- Komarov, D.A.; Ichikawa, Y.; Yamamoto, K.; Stewart, N.J.; Matsumoto, S.; Yasui, H.; Kirilyuk, I.A.; Khramtsov, V.V.; Inanami, O.; Hirata, H. In Vivo Extracellular PH Mapping of Tumors Using Electron Paramagnetic Resonance. Anal. Chem. 2018, 90, 13938–13945. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Mito, F.; Ito, Y.; Pandian, S.; Kinoshita, Y.; Nakano, K.; Murugesan, R.; Sakai, K.; Utsumi, H.; Yamada, K. Structure−Reactivity Relationship of Piperidine Nitroxide: Electrochemical, ESR and Computational Studies. J. Org. Chem. 2011, 76, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Kirilyuk, I.A.; Bobko, A.A.; Semenov, S.V.; Komarov, D.A.; Irtegova, I.G.; Grigor’ev, I.A.; Bagryanskaya, E. Effect of Sterical Shielding on the Redox Properties of Imidazoline and Imidazolidine Nitroxides. J. Org. Chem. 2015, 80, 9118–9125. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Naganuma, J.; Gotoh, H. A Theoretical, Dynamical Evaluation Method of the Steric Hindrance in Nitroxide Radicals Using Transition States of Model Reactions. Sci. Rep. 2019, 9, 20339. [Google Scholar] [CrossRef]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically Shielded Spin Labels for In-Cell EPR Spectroscopy: Analysis of Stability in Reducing Environment. Free Radic. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef]
- Panda, S.S.; Aziz, M.N.; Stawinski, J.; Girgis, A.S. Azomethine Ylides—Versatile Synthons for Pyrrolidinyl-Heterocyclic Compounds. Molecules 2023, 28, 668. [Google Scholar] [CrossRef]
- Najera, C.; Sansano, J. Azomethine Ylides in Organic Synthesis. Curr. Org. Chem. 2003, 7, 1105–1150. [Google Scholar] [CrossRef]
- Dubey, S.; Pal, A.; Roy, S.; Sasmal, S.; Tamrakar, A.; Jana, R.; Das, T. Recent Advances in the (3+2) Cycloaddition of Azomethine Ylide. New J. Chem. 2023, 47, 8997–9034. [Google Scholar] [CrossRef]
- Toledo, H.; Amar, M.; Bar, S.; Iron, M.A.; Fridman, N.; Tumanskii, B.; Shimon, L.J.W.; Botoshansky, M.; Szpilman, A.M. Synthesis and Stability of Cyclic α-Hydrogen Nitroxides. Org. Biomol. Chem. 2015, 13, 10726–10733. [Google Scholar] [CrossRef]
- Zhurko, I.F.; Dobrynin, S.; Gorodetskii, A.A.; Glazachev, Y.I.; Rybalova, T.V.; Chernyak, E.I.; Asanbaeva, N.; Bagryanskaya, E.G.; Kirilyuk, I.A. 2-Butyl-2-Tert-Butyl-5,5-Diethylpyrrolidine-1-Oxyls: Synthesis and Properties. Molecules 2020, 25, 845. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Gulman, M.M.; Morozov, D.A.; Zhurko, I.F.; Taratayko, A.I.; Sotnikova, Y.S.; Glazachev, Y.I.; Gatilov, Y.V.; Kirilyuk, I.A. Synthesis of Sterically Shielded Nitroxides Using the Reaction of Nitrones with Alkynylmagnesium Bromides. Molecules 2022, 27, 7626. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, S.A.; Usatov, M.S.; Zhurko, I.F.; Morozov, D.A.; Polienko, Y.F.; Glazachev, Y.I.; Parkhomenko, D.A.; Tyumentsev, M.A.; Gatilov, Y.V.; Chernyak, E.I.; et al. A Simple Method of Synthesis of 3-Carboxy-2,2,5,5-Tetraethylpyrrolidine-1-Oxyl and Preparation of Reduction-Resistant Spin Labels and Probes of Pyrrolidine Series. Molecules 2021, 26, 5761. [Google Scholar] [CrossRef] [PubMed]
- Taratayko, A.I.; Glazachev, Y.I.; Eltsov, I.V.; Chernyak, E.I.; Kirilyuk, I.A. 3,4-Unsubstituted 2-Tert-Butyl-Pyrrolidine-1-Oxyls with Hydrophilic Functional Groups in the Side Chains. Molecules 2022, 27, 1922. [Google Scholar] [CrossRef] [PubMed]
- Nesvadba, P. Beyond TEMPO. Synthesis of Cyclic Sterically Highly Hindered Nitroxides and Alkoxyamines and their Industrial Applications. In Proceedings of the 4th International Conference on Nitroxide Radicals: Synthesis, Properties and Implications of Nitroxides (SPIN-2005), Novosibirsk, Russia, 20–24 September 2005; Book of Abstracts: Novosibirsk, Russia, 2005; p. 26. [Google Scholar]
- Polienko, Y.F.; Dobrynin, S.A.; Lomanovich, K.A.; Brovko, A.O.; Bagryanskaya, E.G.; Kirilyuk, I.A. Origin of Long-Range Hyperfine Couplings in the EPR Spectra of 2,2,5,5-Tetraethylpyrrolidine-1-Oxyls. ACS Omega 2023, 8, 38723–38732. [Google Scholar] [CrossRef] [PubMed]
- Grigg, R.; Idle, J.; McMeekin, P.; Surendrakumar, S.; Vipond, D. X=Y-ZH Systems as Potential 1,3-Dipoles. Part 12. Mechanism of Formation of Azomethine Ylides via the Decarboxylative Route from α-Amino Acids. J. Chem. Soc. Perkin Trans. 1988, 1, 2703–2713. [Google Scholar] [CrossRef]
- Pandey, G.; Banerjee, P.; Gadre, S.R. Construction of Enantiopure Pyrrolidine Ring System via Asymmetric [3+2]-Cycloaddition of Azomethine Ylides. Chem. Rev. 2006, 106, 4484–4517. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, C.-J.; Lin, Z. Cu(I)/TF-BiphamPhos Catalyzed Reactions of Alkylidene Bisphosphates and Alkylidene Malonates with Azomethine Ylides: Michael Addition versus 1,3-Dipolar Cycloaddition. Organometallics 2012, 31, 7870–7876. [Google Scholar] [CrossRef]
- Cella, J.A.; Kelley, J.A.; Kenehan, E.F. Nitroxide-Catalyzed Oxidation of Alcohols Using m-Chloroperbenzoic Acid. New Method. J. Org. Chem. 1975, 40, 1860–1862. [Google Scholar] [CrossRef]
- Khoroshunova, Y.V.; Morozov, D.A.; Taratayko, A.I.; Gladkikh, P.D.; Glazachev, Y.I.; Kirilyuk, I.A. Synthesis of 1-Azaspiro [4.4]Nonan-1-Oxyls via Intramolecular 1,3-Dipolar Cycloaddition. Beilstein J. Org. Chem. 2019, 15, 2036–2042. [Google Scholar] [CrossRef]
- Edeleva, M.V.; Kirilyuk, I.A.; Zubenko, D.P.; Zhurko, I.F.; Marque, S.R.A.; Gigmes, D.; Guillaneuf, Y.; Bagryanskaya, E.G. Kinetic Study of H-atom Transfer in Imidazoline-, Imidazolidine-, and Pyrrolidine-based Alkoxyamines: Consequences for Nitroxide-mediated Polymerization. J. Polym. Sci. A Polym. Chem. 2009, 47, 6579–6595. [Google Scholar] [CrossRef]
- Mikhail, S.; Usatov Sergey, A.; Dobrynin Yuliya, F.; Polienko Denis, A.; Morozov Yurii, I.; Glazachev Sergey, V.; An’kov Tatiana, G.; Tolstikova Yuri, V.; Gatilov Irina, Y.; Bagryanskaya, E.A.; et al. Kirilyuk Hydrophilic reduction-resistant spin labels of pyrrolidine and pyrroline series from 3,4-bis-hydroxymethyl-2,2,5,5-tetraethylpyrrolidine-1-oxyl. Int. J. Mol. Sci. 2023. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C. Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Mosher, H.S.; Mooney, W.T. The Reduction of Trimethylacetonitrile with Grignard Reagents. J. Am. Chem. Soc. 1951, 73, 3948–3949. [Google Scholar] [CrossRef]
- Whitmore, F.C.; Noll, C.I.; Meunier, V.C. Synthesis of Aliphatic t-Butyl Ketones. J. Am. Chem. Soc. 1939, 61, 683–684. [Google Scholar] [CrossRef]
- Whitmore, F.C.; Meyer, R.E.; Pedlow, G.W.; Popkin, A.H. The Reducing Action of Primary Grignard Reagents with Trimethylacetyl Chloride. J. Am. Chem. Soc. 1938, 60, 2788–2789. [Google Scholar] [CrossRef]
- Sigma-Aldrich, Co. LLC. (Spectral Data Were Obtained from John Wiley & Sons, Inc.). Spectrum ID SLSH_013293. Available online: https://scifinder-n.cas.org/searchDetail/substance/658ba464dee62c69bf19b871/substanceSpectra (accessed on 7 December 2023).
- Enamine Ltd., Spectrum ID EN300-69443. Available online: https://scifinder-n.cas.org/searchDetail/substance/658ba39edee62c69bf19b017/substanceSpectra (accessed on 7 December 2023).
- Eid, C.N.; Konopelski, J.P. Acylketene Acetals in Organic Synthesis. Tetrahedron 1991, 47, 975–992. [Google Scholar] [CrossRef]
- Burchat, A.F.; Chong, J.M.; Nielsen, N. Titration of Alkyllithiums with a Simple Reagent to a Blue Endpoint. J. Organomet. Chem. 1997, 542, 281–283. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhurko, I.F.; Dobrynin, S.A.; Glazachev, Y.I.; Gatilov, Y.V.; Kirilyuk, I.A. 2,5-Di-tert-butyl-2,5-diethylpyrrolidine-1-oxyls: Where Is a Reasonable Limit of Sterical Loading for Higher Resistance to Reduction? Molecules 2024, 29, 599. https://doi.org/10.3390/molecules29030599
Zhurko IF, Dobrynin SA, Glazachev YI, Gatilov YV, Kirilyuk IA. 2,5-Di-tert-butyl-2,5-diethylpyrrolidine-1-oxyls: Where Is a Reasonable Limit of Sterical Loading for Higher Resistance to Reduction? Molecules. 2024; 29(3):599. https://doi.org/10.3390/molecules29030599
Chicago/Turabian StyleZhurko, Irina F., Sergey A. Dobrynin, Yurii I. Glazachev, Yuri V. Gatilov, and Igor A. Kirilyuk. 2024. "2,5-Di-tert-butyl-2,5-diethylpyrrolidine-1-oxyls: Where Is a Reasonable Limit of Sterical Loading for Higher Resistance to Reduction?" Molecules 29, no. 3: 599. https://doi.org/10.3390/molecules29030599