
One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates
by
 , and
, and
Su Been Kim
1,†,
Santanu Maiti
1,†,
Eun Sun Park
1,
Ga Young Kim
1,
Yunji Choun
1,
Soon Kil Ahn
2,
Jae Kwang Kim
3 and
Jinho Kim
1,* 1
Department of Chemistry and Research Institute of Basic Sciences, Incheon National University, Incheon 22012, Republic of Korea
2
Institute for New Drug Development, Division of Life Sciences, Incheon National University, Incheon 22012, Republic of Korea
3
Division of Life Sciences, Incheon National University, Incheon 22012, Republic of Korea
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2023, 28(9), 3815; https://doi.org/10.3390/molecules28093815
Submission received: 30 March 2023
/
Revised: 19 April 2023
/
Accepted: 27 April 2023
/
Published: 29 April 2023
(This article belongs to the Special Issue Green and Highly Efficient One-Pot Synthesis and Catalysis)
Abstract
:The framework of 1,3,4-oxadiazine is crucial for numerous bioactive molecules, but only a limited number of synthetic methods have been reported for its production. In 2015, Wang’s group developed a 4-(dimethylamino)pyridine (DMAP)-catalyzed [2 + 4] cycloaddition of allenoates with N-acyldiazenes, which provided an atom-efficient route for 1,3,4-oxadiazines. However, the practicality of this method was limited by the instability of N-acyldiazenes as starting materials. Building upon our ongoing research about the aerobic oxidation of hydrazides and their synthetic applications, we hypothesized that aerobic oxidative cycloadditions using acylhydrazides instead of N-acyldiazenes may provide a more practical synthetic route for 1,3,4-oxadiazines. In this manuscript, we describe a one-pot synthetic protocol for 1,3,4-oxadiazines from acylhydrazides and allenoates. The developed one-pot protocol consists of aerobic oxidations of acylhydrazides into N-acyldiazenes using NaNO2 and HNO3, followed by the DMAP-catalyzed cycloaddition of allenoate with the generated N-acyldiazenes. A variety of 1,3,4-oxadiazines were produced in good to high yields. In addition, the practicality of the developed method was demonstrated by a gram-scale synthesis of 1,3,4-oxadiazine.
1. Introduction
Azo compounds are one of the most versatile organic molecules in organic synthesis [1,2,3,4,5]. Azo compounds such as diethyl azodicarboxylate (DEAD) play a crucial role to facilitate the stereoselective conversion of alcohols in the Mitsunobu reaction [6,7,8,9,10]. In addition, the azo compounds can be employed as oxidants in dehydrogenations [11,12,13,14,15,16] as well as cross dehydrogenative couplings [17,18,19,20,21,22]. It was also found that azo compounds can serve as a directing group for C-H activation [23].
Among various azo compounds, N-acyldiazenes have recently emerged as attractive participants in cycloadditions for the construction of biologically important N-heterocycles [24]. A variety of synthetic methods for pyrazolidinones [25], 1,3,4-oxadiazin-6-ones [26,27,28,29,30], Blatter radicals [31], 1,3,4-oxadiazines [32,33], 1,3,4-oxadiazoles [34], isatin derivatives [35], 3-spiropyrazole-2-oxindoles [36], and dihydrobenzo[e]indoles [37] have been developed through cycloadditions between the N-acyldiazenes and corresponding partners.
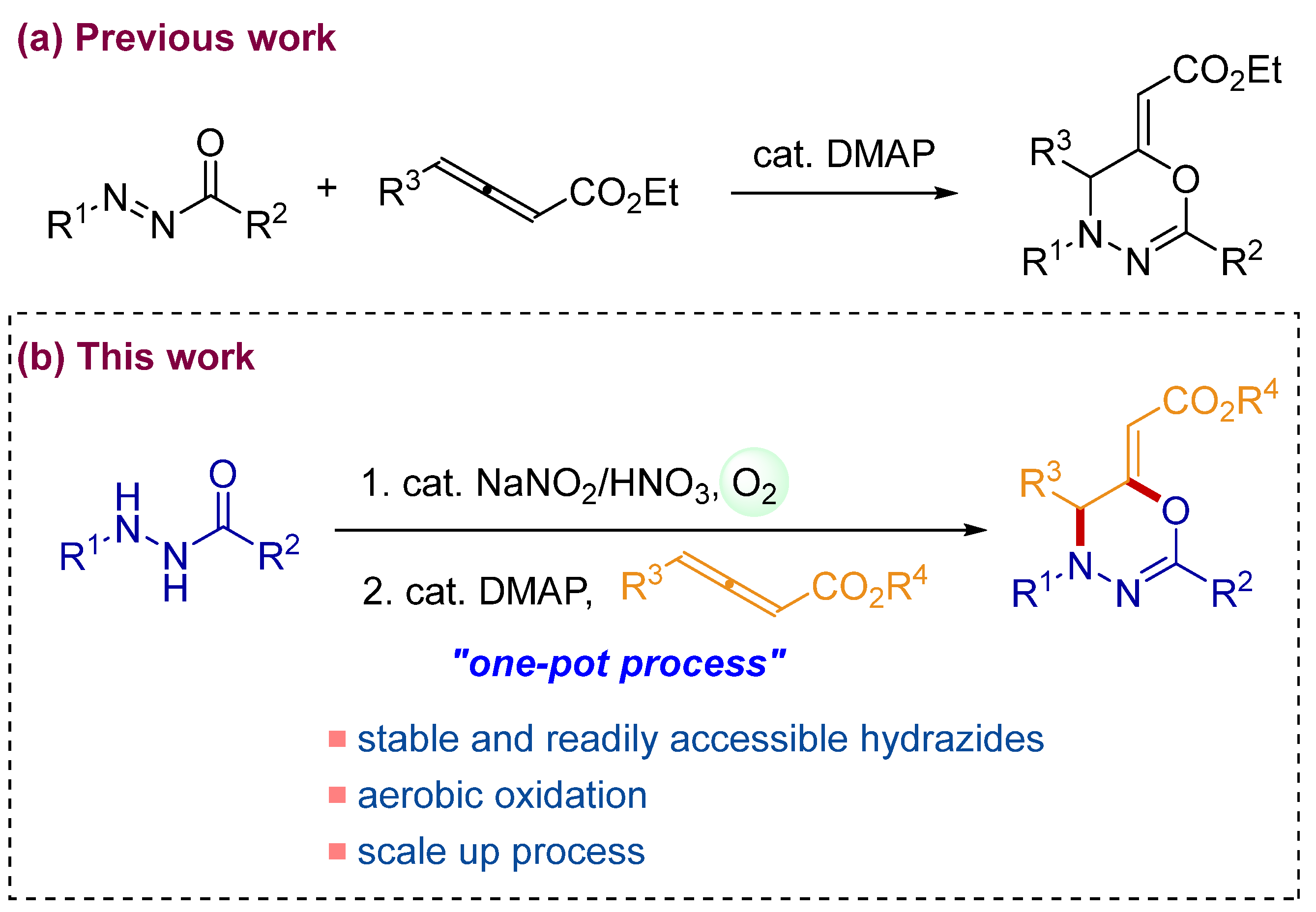
In 2015, Wang’s group developed an atom-efficient synthetic route for 1,3,4-oxadiazines using a [2 + 4] cycloaddition of allenoates with N-acyldiazenes catalyzed by 4-(dimethylamino)pyridine (DMAP) (Scheme 1a) [38]. Subsequently, Li and coworkers reported that the asymmetric synthesis of 1,3,4-oxadiazines from allenoates and N-acyldiazenes was achieved by chiral DMAP analogs as catalysts (70–87% enantiomeric excess) [39]. While these methods are promising for the synthesis of biologically important 1,3,4-oxadiazines, the use of less-stable N-acyldiazenes as starting materials requires laborious preparation and careful storage. Therefore, the development of simpler and more practical protocols for 1,3,4-oxadiazines using stable precursors of azo compounds is highly desirable.
Our group has investigated the aerobic oxidations of acylhydrazides to N-acyldiazenes [40,41,42] and their synthetic applications in organic transformations [43,44]. Building upon our studies in this research area, we envisioned that aerobic oxidative cycloaddition using acylhydrazides instead of N-acyldiazenes may provide more practical and green synthetic routes to 1,3,4-oxadiazines [45,46,47,48,49]. Because the acylhydrazides are relatively more stable than N-acyldiazenes, and only water is produced during the aerobic oxidations of the acylhydrazides, the laborious preparations and purifications of less-stable N-acyldiazenes are not required in our envisaged strategy. Herein, we report a one-pot route to 1,3,4-oxadiazines through the aerobic oxidation of acylhydrazides and the DMAP-catalyzed cycloaddition of the in situ generated N-acyldiazenes with allenoates (Scheme 1b).
2. Results
We initially examined the direct aerobic oxidative cyclization of N′-phenylbenzohydrazide (1a) and benzyl allenoate (2a) using the CuCl/DMAP system (Table 1, entry 1) [40]. We presumed that the used DMAP facilitates not only the aerobic oxidation of acylhydrazide to N-acyldiazene but also cyclization between the generated azo compound and allenoate; however, only 14% of 1,3,4-oxadiazine 3a was produced. The use of the NOx catalytic system (NaNO2 and HNO3) with DMAP showed a promising result [41] compared to other catalytic systems such as Fe(Pc) and Mn(Pc) (Table 1, entries 2–4) [44,50]. Despite implementing several optimizations in the direct aerobic oxidative cyclization approach, however, there was no notable improvement in the yield of 3a.
Next, we tested the feasibility of a one-pot synthesis of 3a from 1a and 2a (Table 2). Full conversions of 1a to benzoyl-2-phenyldiazene 4a were observed in the aerobic oxidations catalyzed by the previously reported catalytic systems such as CuCl/DMAP, Fe(Pc), and Mn(Pc) in 2 h; however, the following DAMP-catalyzed cycloadditions between 2a and the generated 4a were less reactive (entries 1–3). We assumed that the remaining metal catalysts probably hampered the desired DMAP-catalyzed cycloaddition. Interestingly, it was found that the one-pot sequential protocol consisting of aerobic oxidation using the NOx catalytic system followed by DMAP-catalyzed cycloaddition produced the desired 3a with a good yield (entry 4). This result indicated that the used NaNO2/HNO3 reagents and the byproducts formed during aerobic oxidation are compatible with the second step, DMAP-catalyzed cyclization of the generated 4a and 2a. With the NOx catalytic system, other bases and solvents were screened. The use of pyridine as a base showed an inferior result to DMAP (entry 5), and no reaction was observed when 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) was employed as a base (entry 6). Various solvents, such as CH3CN, CH2Cl2, and 1,4-dioxane, were tested as reaction media, but they resulted in lower yields compared to toluene (entries 7–9). The one-pot synthesis in an eco-friendly solvent such as EtOH was sluggish (entry 10). We aimed to minimize the amounts of DAMP and 2a and determined that the use of 30 mol % of DMAP and 1.2 equivalents of 2a was sufficient to facilitate the developed one-pot 1,3,4-oxadiazine synthesis (entry 11).
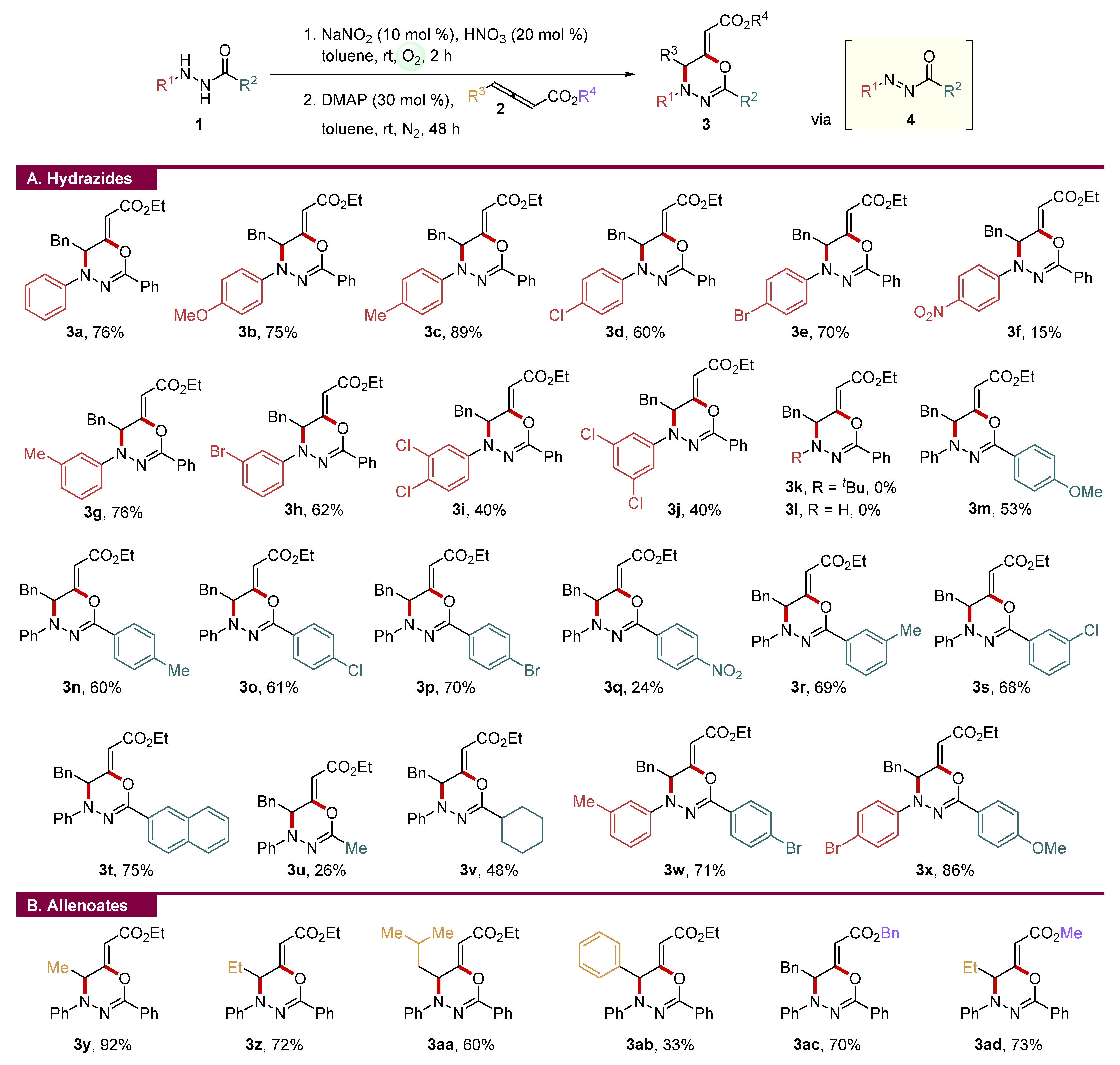
The optimized one-pot reaction conditions were then tested with various acylhydrazides to gain insight into the substrate scope, and the results are described in Figure 1A. First, the reactivities of acylhydrazides bearing the para-substituted phenyl group at the R1 position were studied. Electron-rich acylhydrazides or halogen-substituted acylhydrazides produced the corresponding 1,3,4-oxadiazines in good to high yields (3b–3e). The present one-pot protocol was successful in preparing 4-nitro phenyl substituted 1,3,4-oxadiazine 3f, which was not accessible using the previous method [38], albeit in a low yield. Both meta-substituted phenyl hydrazides and di-substituted phenyl hydrazides were successfully employed for the synthesis of 1,3,4-oxadiazines (3g–3j). However, several problematic substrates were observed in the developed one-pot protocol. For example, aliphatic acylhydrazides such as N′-propybenzohydrazide, N′-isopropylbenzohydrazide, and N′-tert-butylbenzohydrazide did not produce the desired 1,3,4-oxadiazine in spite of the full conversion to the corresponding N-acyldiazenes (3k) [51]. Non-substituted benzoyl hydrazide was also tested, but the desired 1,3,4-oxadiazines could not be accessible, presumably due to decomposition of the unstable azo intermediate (3l).
Following that, the substrate scope of the R2 position was investigated. In general, various para- or meta-substituted benzoyl hydrazides smoothly underwent the developed one-pot protocol regardless of the electronic environments (3m–3s). The 1,3,4-oxadiazine with a naphthalene moiety was synthesized with 75% yield through the developed one-pot protocol (3t). It is noteworthy that the synthesis of 1,3,4-oxadiazines with aliphatic groups at the R2 position could be synthesized by the present one-pot method, producing 3u with 26% yield and 3v with 48% yield. Other multi-substituted 1,3,4-oxadiazines could be prepared with good to high yields (3w and 3x).
The substrate scope of the γ-substituted allenoates was also investigated (Figure 1B) [52]. In addition to the benzyl substituent at γ-position, other aliphatic substituents such as methyl, ethyl, and isobutyl were successfully employed, and the corresponding 1,3,4-oxadiazines were produced with good yields (3y–3aa). The one-pot cyclization using the allenoate-bearing phenyl group at the γ-position had a poor yield in spite of the full conversion of allenoate (3ab). It was revealed that the present one-pot protocol was not significantly influenced by the ester group of allenoate (3ac and 3ad).
In order to showcase the practicality and efficiency of the present one-pot protocol, we carried out a gram-scale reaction using 1a (1.1 g, 5.0 mmol) and 2a (1.2 g, 6.0 mmol) under slightly modified conditions (Scheme 2). The desired 1,3,4-oxadiazine 3a was obtained with 72% yield (1.4 g, 3.6 mmol) without a significant decrease in reactivity. The previous reports only achieved small-scale (0.2 mmol) synthesis of 1,3,4-oxadiazines, possibly because of the challenging isolation and purification of N-acyldiazenes. Therefore, our developed one-pot method for 1,3,4-oxadiazine from acylhydrazides offers a practical and efficient approach for the synthesis of 1,3,4-oxadiazines on a larger scale.
The proposed mechanism of the developed one-pot synthesis 1,3,4-oxadiazines is depicted in Figure 2. Initially, the allenoate 2 was activated by DMAP and zwitterionic intermediate A was generated. The conjugate addition of the generated A to N-acyldiazene 4, which was produced by the NOx-catalyzed aerobic oxidation of acylhydrazide 1, provided intermediate B. Then, intramolecular 1,4-addition followed by elimination yielded the desired 1,3,4-oxadiazine 3 and DMAP catalyst.
3. Materials and Methods
3.1. General Information
All commercially available compounds and solvents were purchased and used as received, unless otherwise noted. Analytical thin-layer chromatography (TLC) was performed on precoated silica gel 60 F254 plates. TLC visualization was achieved by the use of UV light (254 nm) and treatment with phosphomolybdic acid, p-anisaldehyde, KMnO4, or Vanillin stain followed by heating. Flash chromatography was performed using silica gel (particle size 40−63 μm, 230−400 mesh). 1H and 13C NMR spectra were recorded using 300 MHz NMR (300 MHz for 1H, 75 MHz for 13C) or 400 MHz NMR (400 MHz for 1H, 101 MHz for 13C). Chemical shift values are given in parts per million relative to internal TMS (0.00 ppm for 1H) or CDCl3 (77.06 ppm for 13C). The following abbreviations were used to describe peak splitting patterns when appropriate: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, dd = double of doublet, dt = double of triplet, td = triple of doublet, tt = triple of triplet. Coupling constants, J, were reported in hertz unit (Hz). High-resolution mass spectra were obtained from the Korea Basic Science Institute (Daegu) by using the EI method and magnetic sector mass analyzer. Melting points were determined on a digital melting point apparatus, and temperatures were uncorrected.
3.2. Preparation of Acylhydrazides and Allenoates
3.2.1. Preparation of Acylhydrazides (1a–1j, 1l–1u, and 1w–1x) [49]
To a 50 mL round-bottom flask equipped with a magnetic stir bar, hydrazine hydrochloride (5.0 mmol) and CH2Cl2 (5.0 mL) were added. The solution was cooled to 0 °C, and pyridine (11.0 mmol, 2.2 equiv) was added. Then, acyl chloride (5.5 mmol, 1.1 equiv) was added dropwise. The reaction mixture was stirred at room temperature for 4 h. The mixture was diluted with CH2Cl2 and washed with 1.0 M HCl aqueous solution three times; then, the combined organic layer was dried over MgSO4, filtered, and concentrated on a rotary evaporator. Recrystallization with EtOH yielded the desired acylhydrazide.
3.2.2. Preparation of N′-(tert-butyl)benzohydrazide (1k) [51]
To a 100 mL round-bottom flask equipped with a magnetic stir bar, tert-butylhydrazine hydrochloride (10.0 mmol), Et3N (22.0 mmol, 2.2 equiv), and CH2Cl2 (20.0 mL) were added. The solution was cooled to 0 °C, and benzoyl chloride (10.0 mmol, 1.0 equiv) was added dropwise. The reaction mixture was stirred overnight at room temperature. The mixture was washed with water three times; then, the combined organic layer was dried over MgSO4, filtered, and concentrated on a rotary evaporator. Recrystallization with EtOH yielded the desired N′-(tert-butyl)benzohydrazide.
3.2.3. Preparation of N′-phenylcyclohexanecarbohydrazide (1v)
A 100 mL flame-dried round-bottom flask, which was equipped with a magnetic stir bar and charged with phenylhydrazine hydrochloride (12.0 mmol), was evacuated and backfilled with nitrogen (this process was repeated three times). After CH2Cl2 (30.0 mL) was added, the solution was cooled to 0 °C. To the reaction mixture, pyridine (24.0 mmol, 2.0 equiv) was added slowly, and then cyclohexanecarbonyl chloride (13.2 mmol, 1.1 equiv) was added dropwise. The reaction mixture was stirred at room temperature for 5 h. The mixture was diluted with CH2Cl2 and washed with 4.0 M HCl aqueous solution three times; then, the combined organic layer was dried over MgSO4, filtered, and concentrated on a rotary evaporator. Recrystallization with 1:4 EtOAc/Hx yielded the desired acylhydrazide.
3.2.4. Preparation of Allenoates [52]
A 100 mL round-bottom flask, which was equipped with a magnetic stir bar and charged with triphenylphosphorane (10.0 mmol), was evacuated and backfilled with nitrogen (this process was repeated three times). After CH2Cl2 (40 mL) and trimethylamine (11.0 mmol, 1.1 equiv) were added, acyl chloride (11.0 mmol, 1.1 equiv) was added dropwise at 0 °C. The reaction mixture was stirred at room temperature overnight. The mixture was filtered by a short pad of silica and concentrated on a rotary evaporator. The pure allenoates were obtained by column chromatography.
Ethyl 6-methylhepta-2,3-dienoate (for 3aa); colorless oil, EtOAc/Hx = 1:10, 1H NMR (300 MHz, CDCl3) δ 5.61–5.52 (m, 2H), 4.20–4.12 (m, 2H), 2.07–1.99 (m, 2H), 1.74 (dt, J = 13.3, 6.7 Hz, 1H), 1.27 (t, J = 7.1 Hz, 3H), 0.96 (dd, J = 6.6, 1.6 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 212.6, 166.2, 93.7, 87.6, 60.6, 36.8, 28.2, 22.1, 21.9, 14.2; HRMS (EI) m/z calcd. For C10H16O2 [M]+: 168.1150, found 168.1152.
Methyl hexa-2,3-dienoate (for 3ad); colorless oil, EtOAc/Hx = 1:10, 1H NMR (400 MHz, CDCl3) δ 5.69 (q, J = 6.3 Hz, 1H), 5.62 (dt, J = 6.2, 3.4 Hz, 1H), 3.74 (s, 3H), 2.24–2.08 (m, 2H), 1.08 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 212.2, 166.7, 97.1, 88.5, 51.9, 20.8, 13.0; HRMS (EI) m/z calcd. For C7H10O2 [M]+: 126.0681, found 126.0680.
3.3. General Procedure for One-Pot Synthesis of 1,3,4-Oxadiazines
One 10 mL flame-dried test tube (Tube A), which was equipped with a magnetic stir bar and charged with acylhydrazide (0.3 mmol) and NaNO2 (0.03 mmol, 10 mol %), was evacuated and backfilled with oxygen (this process was repeated three times). After toluene (1.0 mL), HNO3 (0.06 mmol, 20 mol %), and additional toluene (0.5 mL) were added in sequence, the reaction mixture was stirred for 2 h. The other 10 mL flame-dried test tube (Tube B), which was equipped with a magnetic stir bar, was evacuated and backfilled with nitrogen (this process was repeated three times). Allenoate (0.36 mmol, 1.2 equiv) in toluene (0.5 mL) was added to Tube B. Then, the reaction mixture in Tube A was added to Tube B using a syringe. By using toluene (0.5 mL), Tube A was washed, and the solution was transferred to Tube B. After the combined mixture in Tube B was stirred at room temperature for 0.5 h, DMAP (0.09 mmol, 30 mol %) in toluene (0.5 mL) was added. After 48 h, the reaction mixture in Tube B was diluted by adding CH2Cl2 and washed with a saturated aqueous solution of Na2CO3. Two layers were separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layer was dried over MgSO4, filtered, and concentrated on a rotary evaporator. The residue was purified by column chromatography to yield 1,3,4-oxadiazines.
3.4. Characterizations of the Newly Synthesized 1,3,4-Oxadiazines
(Z)-Ethyl 2-(5-benzyl-4-(4-methoxyphenyl)-2-phenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3b); yellow oil, EtOAc/Hx = 1:10, 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 6.9 Hz, 2H), 7.45 (d, J = 6.4 Hz, 3H), 7.29–7.14 (m, 8H), 6.92 (d, J = 8.8 Hz, 2H), 4.72 (s, 1H), 4.59 (dd, J = 9.2, 4.6 Hz, 1H), 4.25–4.15 (m, 2H), 3.82 (s, 3H), 2.98–2.84 (m, 2H), 1.30 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.5, 154.6, 153.3, 141.8, 138.8, 136.5, 130.1, 129.8, 129.5, 128.7, 128.5, 127.0, 125.6, 116.1, 114.7, 98.0, 60.1, 57.4, 55.7, 33.5, 14.3; HRMS (EI) m/z calcd. For C27H26N2O4 [M]+: 442.1893, found 442.1895.
(Z)-Ethyl 2-(5-benzyl-4-(4-nitrophenyl)-2-phenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3f); yellow oil, EtOAc/Hx = 1:10, 1H NMR (400 MHz, CDCl3) δ 8.06–7.96 (m, 4H), 7.49 (d, J = 7.0 Hz, 3H), 7.20–7.04 (m, 7H), 6.65 (dd, J = 8.2, 5.8 Hz, 1H), 5.73 (s, 1H), 4.22–4.14 (m, 2H), 3.10–2.99 (m, 2H), 1.29 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 166.2, 158.3, 149.6, 144.2, 140.0, 135.2, 130.9, 129.9, 129.1, 128.6, 128.5, 127.3, 125.8, 125.5, 112.3, 99.0, 60.6, 50.6, 35.2, 14.3; HRMS (EI) m/z calcd. For C26H23N3O5 [M]+: 457.1638, found 457.1635.
(Z)-Ethyl 2-(5-benzyl-2-phenyl-4-(m-tolyl)-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3g); yellow solid, EtOAc/Hx = 1:10, mp 102–103 °C, 1H NMR (400 MHz, CDCl3) δ 8.18–8.04 (m, 2H), 7.48–7.43 (m, 3H), 7.30–7.25 (m, 2H), 7.24–7.17 (m, 3H), 7.15 (s, 1H), 7.10 (s, 1H), 7.03 (dd, J = 8.2, 2.2 Hz, 1H), 6.82–6.76 (m, 1H), 4.75 (s, 1H), 4.67 (dd, J = 9.4, 4.8 Hz, 1H), 4.35–4.14 (m, 2H), 3.11–2.84 (m, 2H), 2.37 (s, 3H), 1.41–1.23 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 164.5, 153.0, 144.6, 141.8, 139.3, 136.4, 130.1, 129.9, 129.5, 129.2, 128.7, 128.5, 127.1, 125.7, 121.9, 115.0, 111.2, 98.2, 60.1, 56.3, 34.0, 21.9, 14.4; HRMS (EI) m/z calcd. For C27H26N2O3 [M]+: 426.1943, found 426.1941.
(Z)-Ethyl 2-(5-benzyl-4-(3-bromophenyl)-2-phenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3h); yellow oil, EtOAc/Hx = 1:10, 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 4.2 Hz, 2H), 7.47 (s, 3H), 7.43 (s, 1H), 7.28 (s, 2H), 7.23 (d, J = 7.4 Hz, 1H), 7.16 (d, J = 7.3 Hz, 3H), 7.10–7.04 (m, 2H), 4.83 (s, 1H), 4.68–4.63 (m, 1H), 4.22 (d, J = 6.4 Hz, 2H), 3.01–2.89 (m, 2H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 164.3, 152.6, 145.7, 142.4, 135.8, 130.5, 130.2, 129.6, 129.5, 128.7, 128.5, 127.3, 125.8, 123.5, 123.4, 117.1, 112.0, 98.6, 60.2, 55.9, 34.3, 14.3; HRMS (EI) m/z calcd. For C26H23BrN2O3 [M]+: 490.0892, found 490.0895.
(Z)-Ethyl 2-(5-benzyl-2-(4-nitrophenyl)-4-phenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3q); orange solid, EtOAc/Hx =1:10, mp 75–76 °C, 1H NMR (400 MHz, CDCl3) δ 8.31–8.19 (m, 4H), 7.43–7.34 (m, 2H), 7.34–7.24 (m, 4H), 7.23–7.13 (m, 3H), 7.03 (t, J = 7.2 Hz, 1H), 4.88 (s, 1H), 4.77 (dd, J = 9.1, 4.7 Hz, 1H), 4.27–4.17 (m, 2H), 3.07–2.89 (m, 2H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 164.2, 152.1, 148.2, 143.8, 139.7, 135.9, 135.7, 129.5, 129.5, 128.7, 127.3, 126.1, 123.8, 121.8, 114.2, 99.0, 60.3, 56.0, 34.7, 14.3; HRMS (EI) m/z calcd. For C26H23N3O5 [M]+: 457.1638, found 457.1639.
(Z)-Ethyl 2-(5-benzyl-4-phenyl-2-(m-tolyl)-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3r); yellow oil, EtOAc/Hx = 1:10, 1H NMR (300 MHz, CDCl3) δ 7.94 (d, J = 7.5 Hz, 2H), 7.39–7.32 (m, 3H), 7.31–7.20 (m, 6H), 7.20–7.14 (m, 2H), 7.00–6.91 (m, 1H), 4.74 (s, 1H), 4.68 (dd, J = 9.5, 4.9 Hz, 1H), 4.20 (dd, J = 7.1, 5.6 Hz, 2H), 3.01–2.84 (m, 2H), 2.45 (s, 3H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.6, 153.0, 144.6, 142.2, 138.2, 136.4, 130.9, 130.0, 129.5, 129.4, 128.8, 128.5, 127.2, 126.2, 123.1, 121.0, 114.2, 98.4, 60.2, 56.3, 33.9, 21.6, 14.4; HRMS (EI) m/z calcd. For C27H26N2O3 [M]+: 426.1943, found 426.1939.
(Z)-Ethyl 2-(5-benzyl-2-(3-chlorophenyl)-4-phenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3s); yellow oil, EtOAc/Hx = 1:10, 1H NMR (300 MHz, CDCl3) δ 8.07 (q, J = 1.3 Hz, 1H), 7.99 (dd, J = 5.5, 1.8 Hz, 1H), 7.42–7.33 (m, 4H), 7.32–7.21 (m, 5H), 7.18–7.13 (m, 2H), 6.99 (d, J = 7.2 Hz, 1H), 4.78 (s, 1H), 4.71 (dd, J = 9.3, 4.8 Hz, 1H), 4.21 (dd, J = 7.1, 5.0 Hz, 2H), 3.02–2.84 (m, 2H), 1.32 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.4, 152.4, 144.2, 140.6, 136.1, 134.6, 131.8, 129.8, 129.8, 129.5, 129.4, 128.7, 127.2, 125.6, 123.8, 121.3, 114.1, 98.8, 60.3, 56.2, 34.1, 14.4; HRMS (EI) m/z calcd. For C26H23ClN2O3 [M]+: 446.1397, found 446.1400.
(Z)-Ethyl 2-(5-benzyl-2-(naphthalen-2-yl)-4-phenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3t); yellow solid, EtOAc/Hx = 1:10, mp 66–67 °C, 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.25 (d, J = 8.6 Hz, 1H), 8.05–7.98 (m, 1H), 7.96–7.85 (m, 2H), 7.63–7.52 (m, 2H), 7.46–7.18 (m, 9H), 7.02 (t, J = 6.8 Hz, 1H), 4.82 (s, 1H), 4.76 (dd, J = 9.3, 4.6 Hz, 1H), 4.28 (dd, J = 12.4, 6.9 Hz, 2H), 3.07–2.94 (m, 2H), 1.39 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.6, 152.9, 144.5, 142.1, 136.3, 134.1, 133.1, 129.5, 129.4, 128.8, 128.7, 128.2, 127.8, 127.4, 127.2, 126.9, 126.5, 125.4, 123.0, 121.1, 114.1, 98.5, 60.2, 56.3, 34.1, 14.5; HRMS (EI) calcd, for C30H26N2O3 [M]+: 462.1943, found: 462.1947.
(Z)-Ethyl 2-(5-benzyl-2-cyclohexyl-4-phenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3v); yellow oil, EtOAc/Hx = 1:10, 1H NMR (300 MHz, CDCl3) δ 7.33–7.26 (m, 3H), 7.26–7.21 (m, 2H), 7.19–7.11 (m, 4H), 6.91 (d, J = 7.2 Hz, 1H), 4.61 (s, 1H), 4.54 (dd, J = 9.3, 5.0 Hz, 1H), 4.25–4.11 (m, 2H), 2.90–2.74 (m, 2H), 2.60–2.44 (m, 1H), 2.14–2.05 (m, 2H), 1.85 (d, J = 10.6 Hz, 2H), 1.67 (dd, J = 27.1, 13.4 Hz, 3H), 1.41–1.29 (m, 3H), 1.28–1.24 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 164.4, 153.8, 149.1, 144.9, 136.4, 129.5, 129.3, 128.6, 127.0, 120.4, 113.9, 97.6, 59.9, 56.1, 40.7, 32.8, 29.7, 25.9, 25.7, 14.3; HRMS (EI) m/z calcd. For C26H30N2O3 [M]+: 418.2256, found 418.2254.
(Z)-Ethyl 2-(5-benzyl-2-(4-bromophenyl)-4-(m-tolyl)-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3w); yellow solid, EtOAc/Hx = 1:10, mp 95–96 °C, 1H NMR (400 MHz, CDCl3) δ 8.06–7.89 (m, 2H), 7.64–7.52 (m, 2H), 7.29 (d, J = 7.9 Hz, 2H), 7.23 (d, J = 7.8 Hz, 2H), 7.16 (d, J = 7.3 Hz, 2H), 7.09 (s, 1H), 7.04 (d, J = 8.3 Hz, 1H), 6.80 (d, J = 7.4 Hz, 1H), 4.78 (s, 1H), 4.69 (dd, J = 9.5, 4.7 Hz, 1H), 4.31–4.11 (m, 2H), 3.01–2.85 (m, 2H), 2.39 (s, 3H), 1.29 (dd, J = 7.9, 6.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.4, 152.8, 144.3, 141.0, 139.3, 136.2, 131.6, 129.5, 129.2, 129.0, 128.8, 128.7, 127.2, 124.2, 122.1, 115.0, 111.2, 98.4, 60.1, 56.2, 34.2, 21.9, 14.3; HRMS (EI) m/z calcd. For C27H25BrN2O3 [M]+: 504.1049, found 504.1050.
(Z)-Ethyl 2-(5-isobutyl-2,4-diphenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3aa); yellow oil, EtOAc/Hx = 1:10, 1H NMR (300 MHz, CDCl3) δ 8.14–8.05 (m, 2H), 7.47–7.39 (m, 3H), 7.37–7.31 (m, 2H), 7.29–7.23 (m, 2H), 6.95 (tt, J = 7.2, 1.2 Hz, 1H), 5.20 (s, 1H), 4.61 (dd, J = 10.9, 3.9 Hz, 1H), 4.30–4.22 (m, 2H), 1.74–1.62 (m, 2H), 1.35 (t, J = 7.1 Hz, 4H), 1.04 (d, J = 6.4 Hz, 3H), 0.90 (d, J = 6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.7, 154.0, 144.6, 141.4, 130.0, 129.8, 129.3, 128.4, 125.6, 120.8, 113.8, 97.5, 60.2, 52.2, 35.1, 24.7, 23.5, 21.4, 14.4; HRMS (EI) m/z calcd. For C23H26N2O3 [M]+: 378.1943, found 378.1945.
(Z)-Ethyl 2-(2,4,5-triphenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3ab); yellow oil, EtOAc/Hx = 1:10, 1H NMR (300 MHz, CDCl3) δ 8.10–8.01 (m, 2H), 7.42–7.37 (m, 3H), 7.34–7.22 (m, 9H), 7.00–6.89 (m, 1H), 5.71 (s, 1H), 5.52 (s, 1H), 4.26 (q, J = 7.1 Hz, 2H), 1.35 (t, J = 7.2 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 164.7, 153.9, 144.9, 141.5, 134.6, 130.0, 129.8, 129.3, 129.1, 128.5, 128.4, 126.8, 125.6, 121.0, 113.9, 98.2, 60.4, 57.0, 14.4; HRMS (EI) m/z calcd. For C25H22N2O3 [M]+: 398.1630, found 398.1634.
(Z)-Benzyl 2-(5-benzyl-2,4-diphenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3ac); yellow oil, EtOAc/Hx =1:10, 1H NMR (300 MHz, CDCl3) δ 8.04–7.98 (m, 2H), 7.44–7.31 (m, 10H), 7.29–7.19 (m, 5H), 7.18–7.13 (m, 2H), 6.96 (tt, J = 7.1, 1.3 Hz, 1H), 5.23–5.11 (m, 2H), 4.80 (s, 1H), 4.70 (dd, J = 9.5, 4.9 Hz, 1H), 3.03–2.81 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 164.2, 153.4, 144.5, 141.9, 136.2, 136.0, 129.8, 129.5, 129.4, 128.7, 128.6, 128.4, 128.3, 128.2, 127.1, 125.7, 121.0, 114.1, 98.0, 66.1, 56.2, 33.9.; HRMS (EI) m/z calcd. For C31H26N2O3 [M]+: 474.1943, found 474.1944.
(Z)-Methyl 2-(5-ethyl-2,4-diphenyl-4H-1,3,4-oxadiazin-6(5H)-ylidene)acetate (3ad); yellow oil, EtOAc/Hx = 1:10, 1H NMR (300 MHz, CDCl3) δ 8.10–7.99 (m, 2H), 7.47–7.23 (m, 7H), 6.94 (tt, J = 7.0, 1.3 Hz, 1H), 5.20 (s, 1H), 4.43 (dd, J = 8.0, 6.6 Hz, 1H), 3.79 (s, 3H), 1.83–1.65 (m, 2H), 0.99 (t, J = 7.5 Hz, 3H); δ 13C NMR (75 MHz, CDCl3) δ 165.0, 154.1, 144.7, 141.4, 130.0, 129.7, 129.3, 128.4, 125.6, 120.7, 113.9, 97.2, 55.4, 51.4, 20.4, 10.7; HRMS (EI) m/z calcd. For C20H20N2O3 [M]+: 336.1474, found 336.1471.
3.5. 1H and 13C NMR Spectra
For the 1H and 13C NMR spectra, see Supplementary Materials.
4. Conclusions
In conclusion, we have developed a practical and green one-pot synthesis of 1,3,4-oxadiazines from acylhydrazides. The newly developed one-pot protocol consists of aerobic oxidations of acylhydrazides into N-acyldiazenes using a NOx catalytic system, followed by the DMAP-catalyzed cycloaddition of allenoate with the generated N-acyldiazenes. The present method was able to utilize various acylhydrazides to generate 1,3,4-oxadiazines with good to high yields. Interestingly, the electron-deficient phenyl-substituted 1,3,4-oxadiazines, which could not be synthesized by the previous method using N-acyldiazene, were able to be synthesized by the present one-pot method. However, aliphatic acylhydrazides displayed limited substrate scope. The practicality of the one-pot synthesis of 1,3,4-oxadiazines from acylhydrazides was demonstrated by the gram-scale experiment, which was not achieved by the previous synthesis of 1,3,4-oxadiazines from N-acyldiazenes. The synthesis of other heterocycles using N-acyldiazenes or acylhydrazides is underway in our laboratory.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules28093815/s1, Preparation of acylhydrazides and allenoates, detailed optimizations, experimental procedures, spectroscopic data, and copies of 1H and 13C NMR spectra.
Author Contributions
Conceptualization, J.K., S.B.K. and S.M.; methodology, S.B.K. and S.M.; data curation, S.B.K., S.M., E.S.P., G.Y.K., Y.C., S.K.A. and J.K.K.; writing, J.K., S.B.K. and S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2021R1A2C4002062). This work also was supported by the 2017 research program funded by the Institute of Convergence Science and Technology, Incheon National University, Republic of Korea.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Sample Availability
Not available.
References
- Hunger, K. Industrial Dyes: Chemistry, Properties, Applications; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar] [CrossRef]
- Merino, E. Synthesis of azobenzenes: The coloured pieces of molecular materials. Chem. Soc. Rev. 2011, 40, 3835–3853. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Moroz, E.; Castagner, B.; Leroux, J.-C. Activatable Cell Penetrating Peptide−Peptide Nucleic Acid Conjugate via Reduction of Azobenzene PEG Chains. J. Am. Chem. Soc. 2014, 136, 12868–12871. [Google Scholar] [CrossRef]
- Feringa, B.L.; van Delden, R.A.; Koumura, N.; Geertsema, E.M. Chiroptical Molecular Switches. Chem. Rev. 2000, 100, 1789–1816. [Google Scholar] [CrossRef] [PubMed]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Mitsunobu, O.; Yamada, M.; Mukaiyama, T. Preparation of Esters of Phosphoric Acid by the Reaction of Trivalent Phosphorus Compounds with Diethyl Azodicarboxylate in the Presence of Alcohols. Bull. Chem. Soc. Jpn. 1967, 40, 935–939. [Google Scholar] [CrossRef]
- Mitsunobu, O.; Yamada, M. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1981, 1–28. [Google Scholar] [CrossRef]
- But, T.Y.S.; Toy, P.H. The Mitsunobu Reaction: Origin, Mechanism, Improvements, and Applications. Chem. Asian J. 2007, 2, 1340–1355. [Google Scholar] [CrossRef]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, F.; Suzuki, K.; Nitta, Y. A New Hydrogen-Abstracting Reaction with Diethyl Azodicarboxylate. J. Am. Chem. Soc. 1966, 88, 2328–2329. [Google Scholar] [CrossRef]
- Yoneda, F.; Suzuki, K.; Nitta, Y. A New Hydrogen-Abstracting Reaction with Diethyl Azodicarboxylate. J. Org. Chem. 1967, 32, 727–729. [Google Scholar] [CrossRef]
- Cao, H.T.; Grée, R. DEAD-(cat) ZnBr2 an efficient system for the oxidation of alcohols to carbonyl compounds. Tetrahedron Lett. 2009, 50, 1493–1494. [Google Scholar] [CrossRef]
- Stone, M.T. An Improved Larock Synthesis of Quinolines via a Heck Reaction of 2-Bromoanilines and Allylic Alcohols. Org. Lett. 2011, 13, 2326–2329. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Mourya, M.; Guin, D.; Joshi, Y.C.; Dobhal, M.P.; Basak, A.K. Diisopropyl azodicarboxylate mediated selective dehydrogenation of 2-amino-3-cyano 4H-chromenes. Tetrahedron Lett. 2017, 58, 1727–1732. [Google Scholar] [CrossRef]
- Bang, S.B.; Kim, J. Efficient dehydrogenation of 1,2,3,4-tetrahydroquinolines mediated by dialkyl azodicarboxylates. Synth. Commun. 2018, 48, 1291–1298. [Google Scholar] [CrossRef]
- Xu, X.; Li, X. Copper/Diethyl Azodicarboxylate Mediated Regioselective Alkynylation of Unactivated Aliphatic Tertiary Methylamine with Terminal Alkyne. Org. Lett. 2009, 11, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.N.; Singh, P.; Kaur, A.; Singh, P. C-1 Alkynylation of N-Methyltetrahydroisoquinolines through CDC: A Direct Access to Phenethylisoquinoline Alkaloids. Synlett 2012, 23, 760–764. [Google Scholar] [CrossRef]
- Huang, W.; Ni, C.; Zhao, Y.; Hu, J. DIAD-mediated metal-free cross dehydrogenative coupling between tertiary amines and α-fluorinated sulfones. New J. Chem. 2013, 37, 1684–1687. [Google Scholar] [CrossRef]
- Singh, K.N.; Kessar, S.V.; Singh, P.; Singh, P.; Kaur, M.; Batra, A. Transition-Metal-Free Arylation of N-Alkyl-tetrahydroisoquinolines under Oxidative Conditions: A Convenient Synthesis of C1-Arylated Tetrahydroisoquinoline Alkaloids. Synthesis 2014, 46, 2644–2650. [Google Scholar] [CrossRef]
- Suga, T.; Iizuka, S.; Akiyama, T. Versatile and highly efficient oxidative C(sp3)–H bond functionalization of tetrahydroisoquinoline promoted by bifunctional diethyl azodicarboxylate (DEAD): Scope and mechanistic insights. Org. Chem. Front. 2016, 3, 1259–1264. [Google Scholar] [CrossRef]
- Kim, Y.H.; Gil, M.G.; Kim, D.Y. Diethyl Azodicarboxylate-promoted Oxidative Coupling Reaction of N-Phenyl Tetrahydroisoquinoline with β-Keto Acids. Bull. Korean Chem. Soc. 2017, 38, 1499–1502. [Google Scholar] [CrossRef]
- Sharma, S.; Han, S.H.; Han, S.; Ji, W.; Oh, J.; Lee, S.-Y.; Oh, J.S.; Jung, Y.H.; Kim, I.S. Rh(III)-Catalyzed Direct Coupling of Azobenzenes with α-Diazo Esters: Facile Synthesis of Cinnolin-3(2H)-ones. Org. Lett. 2015, 17, 2852–2855. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Scheidt, K.A. Direct Amination of Homoenolates Catalyzed by N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2008, 130, 2740–2741. [Google Scholar] [CrossRef]
- Huang, X.-L.; He, L.; Shao, P.-L.; Ye, S. [4+2] Cycloaddition of Ketenes with N-Benzoyldiazenes Catalyzed by N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. 2009, 48, 192–195. [Google Scholar] [CrossRef]
- Morrill, L.C.; Lebl, T.; Slawin, A.M.Z.; Smith, A.D. Catalytic asymmetric α-amination of carboxylic acids using isothioureas. Chem. Sci. 2012, 3, 2088–2093. [Google Scholar] [CrossRef]
- Taylor, J.E.; Daniels, D.S.B.; Smith, A.D. Asymmetric NHC-Catalyzed Redox α-Amination of α-Aroyloxyaldehydes. Org. Lett. 2013, 15, 6058–6061. [Google Scholar] [CrossRef]
- Morrill, L.C.; Smith, S.M.; Slawin, A.M.Z.; Smith, A.D. Isothiourea-Mediated Asymmetric Functionalization of 3-Alkenoic Acids. J. Org. Chem. 2014, 79, 1640–1655. [Google Scholar] [CrossRef]
- Yang, L.; Wang, F.; Lee, R.; Lv, Y.; Huang, K.-W.; Zhong, G. Asymmetric NHC-Catalyzed Aza-Diels–Alder Reactions: Highly Enantioselective Route to α-Amino Acid Derivatives and DFT Calculations. Org. Lett. 2014, 16, 3872–3875. [Google Scholar] [CrossRef]
- Savva, A.C.; Mirallai, S.I.; Zissimou, G.A.; Berezin, A.A.; Demetriades, M.; Kourtellaris, A.; Constantinides, C.P.; Nicolaides, C.; Trypiniotis, T.; Koutentis, P.A. Preparation of Blatter Radicals via Aza-Wittig Chemistry: The Reaction of N-Aryliminophosphoranes with 1-(Het)aroyl-2-aryldiazenes. J. Org. Chem. 2017, 82, 7564–7575. [Google Scholar] [CrossRef]
- Bigotto, A.; Forchiassin, M.; Fisaliti, A.; Russo, C. Reactions of cyclohexanone enamines with asymmeric diimides. Tetrahedron Lett. 1979, 20, 4761–4764. [Google Scholar] [CrossRef]
- Forchiassin, M.; Risaliti, A.; Russo, C. 1,3,4-Oxadiazine derivatives from cyclohexanone enamines and asymmetric diimides: Possibility of ring-chain tautomerism in such heterocyclic system. Tetrahedron 1981, 37, 2921–2928. [Google Scholar] [CrossRef]
- Zhou, R.; Han, L.; Zhang, H.; Liu, R.; Li, R. A Deoxygenative [4+1] Annulation Involving N-Acyldiazenes for an Efficient Synthesis of 2,2,5-Trisubstituted 1,3,4-Oxadiazole Derivatives. Adv. Synth. Catal. 2017, 359, 3977–3982. [Google Scholar] [CrossRef]
- Ma, C.; Zhou, J.-Y.; Zhang, Y.-Z.; Mei, G.-J.; Shi, F. Catalytic Asymmetric [2+3] Cyclizations of Azlactones with Azonaphthalenes. Angew. Chem. Int. Ed. 2018, 57, 5398–5402. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Meng, L.-G.; Peng, T.; Zhu, L.; Wang, L. 4-Dimethylaminopyridine-Catalyzed Regioselective [3+2] Cycloaddition of Isatin-Derived Morita−Baylis−Hillman Adducts with Azo Esters: A Simple Protocol to Access 3-Spiropyrazole-2-oxindoles. Adv. Synth. Catal. 2018, 360, 3176–3180. [Google Scholar] [CrossRef]
- Li, C.; Xu, D.-N.; Ma, C.; Mei, G.-J.; Shi, F. Diastereo- and Enantioselective Construction of Dihydrobenzo[e]indole Scaffolds via Catalytic Asymmetric [3 + 2] Cycloannulations. J. Org. Chem. 2018, 83, 9190–9200. [Google Scholar] [CrossRef]
- Zhang, Q.; Meng, L.-G.; Zhang, J.; Wang, L. DMAP-Catalyzed [2+4] Cycloadditions of Allenoates with N-Acyldiazenes: Direct Method to 1,3,4-Oxadiazine Derivatives. Org. Lett. 2015, 17, 3272–3275. [Google Scholar] [CrossRef]
- Guo, X.; Chen, X.; Cheng, Y.; Chang, X.; Li, X.; Li, P. Organocatalytic enantioselective [2 + 4]-annulation of γ-substituted allenoates with N-acyldiazenes for the synthesis of optically active 1,3,4-oxadiazines. Org. Biomol. Chem. 2021, 19, 1727–1731. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J. Aerobic Oxidation of Alkyl 2-Phenylhydrazinecarboxylates Catalyzed by CuCl and DMAP. J. Org. Chem. 2018, 83, 1673–1679. [Google Scholar] [CrossRef]
- Jo, G.; Kim, M.H.; Kim, J. A practical route to azo compounds by metal-free aerobic oxidation of arylhydrazides using an NOx system. Org. Chem. Front. 2020, 7, 834–839. [Google Scholar] [CrossRef]
- Kim, J.; Lee, D.H.; Kim, J. Cu-Catalyzed Aerobic Oxidative Azo-Ene Cyclization. Adv. Synth. Catal. 2021, 363, 4728–4733. [Google Scholar] [CrossRef]
- Jung, D.; Kim, M.H.; Kim, J. Cu-Catalyzed Aerobic Oxidation of Di-tert-butyl Hydrazodicarboxylate to Di-tert-butyl Azodicarboxylate and Its Application on Dehydrogenation of 1,2,3,4-Tetrahydroquinolines under Mild Conditions. Org. Lett. 2016, 18, 6300–6303. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Jang, S.H.; Yim, T.; Kim, J. Oxidation Potential Tunable Organic Molecules and Their Catalytic Application to Aerobic Dehydrogenation of Tetrahydroquinolines. Org. Lett. 2018, 20, 6436–6439. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Ren, L.; Hou, J.; Yu, W.; Chang, J. Annulation Reactions of In-Situ-Generated N-(Het)aroyldiazenes with Isothiocyanates Leading to 2-Imino-1,3,4-oxadiazolines. Org. Lett. 2019, 21, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Q.; Zhao, Q.; Yu, W.; Chang, J. Synthesis of 2-Imino-1,3,4-thiadiazoles from Hydrazides and Isothiocyanates via Sequential Oxidation and P(NMe2)3-Mediated Annulation Reactions. Org. Lett. 2020, 22, 4378–4382. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Q.; Yi, X.; Zhao, Z.; Yu, W.; Chang, J. Synthesis of 2-Imino-1,3,4-Selenadiazoles via Tributylphosphine-Mediated Annulation of N-Aroyldiazenes with Isoselenocyanates. Adv. Synth. Catal. 2021, 363, 4894–4898. [Google Scholar] [CrossRef]
- Kim, S.B.; Baek, S.E.; Lim, J.H.; Kim, J. One-pot synthesis of 2-imino-1,3,4-thiadiazolines from acylhydrazides and isothiocyanates. Bull. Korean Chem. Soc. 2022, 43, 1014–1018. [Google Scholar] [CrossRef]
- Lim, J.H.; Baek, S.E.; Lad, B.; Kim, J. Synthesis of 2-Imino-1,3,4-oxadiazolines from Acylhydrazides and Isothiocyanates via Aerobic Oxidation and DMAP-Mediated Annulation Sequence. ACS Omega 2022, 7, 28148–28159. [Google Scholar] [CrossRef]
- Hirose, D.; Taniguchi, T.; Ishibashi, H. Recyclable Mitsunobu Reagents: Catalytic Mitsunobu Reactions with an Iron Catalyst and Atmospheric Oxygen. Angew. Chem. Int. Ed. 2013, 52, 4613–4617. [Google Scholar] [CrossRef]
- Fang, T.; Tan, O.; Ding, Z.; Liu, B.; Bin Xu, B. Pd-Catalyzed Oxidative Annulation of Hydrazides with Isocyanides: Synthesis of 2-Amino-1,3,4-oxadiazoles. Org. Lett. 2014, 16, 2342–2345. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, L.; Ma, R.; Song, H.; He, Z. Phosphane-Catalyzed [3 + 2] Annulation of Allenoates with Aldehydes: A Simple and Efficient Synthesis of 2-Alkylidenetetrahydrofurans. Chem. Eur. J. 2009, 15, 8698–8702. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of 1,3,4-oxadiazines from allenoates. (a) Previously reported DMAP-catalyzed cycloaddition of allenoates with N-acyldiazenes. (b) Our developed one-pot synthesis of 1,3,4-oxadiazine from acylhydrazides and allenoates.
Scheme 1.
Synthesis of 1,3,4-oxadiazines from allenoates. (a) Previously reported DMAP-catalyzed cycloaddition of allenoates with N-acyldiazenes. (b) Our developed one-pot synthesis of 1,3,4-oxadiazine from acylhydrazides and allenoates.

Figure 1.
Substrate scope of acylhydrazides and allenoates a,b. a Reaction conditions: 1 (0.3 mmol), NaNO2 (10 mol %), and HNO3 (20 mol %) in toluene (1.5 mL) under O2 at room temperature for 2 h were used for the first step. The reaction mixture of the first step was added to DMAP (30 mol %) and 2 (0.36 mmol) in toluene (1.5 mL) under N2 at room temperature; then, the reaction mixture of the second step was stirred for 48 h. b Isolated yield.
Figure 1.
Substrate scope of acylhydrazides and allenoates a,b. a Reaction conditions: 1 (0.3 mmol), NaNO2 (10 mol %), and HNO3 (20 mol %) in toluene (1.5 mL) under O2 at room temperature for 2 h were used for the first step. The reaction mixture of the first step was added to DMAP (30 mol %) and 2 (0.36 mmol) in toluene (1.5 mL) under N2 at room temperature; then, the reaction mixture of the second step was stirred for 48 h. b Isolated yield.

Scheme 2.
Scaled-up process for the one-pot synthesis of 3a.

Figure 2.
Proposed mechanism of the developed one-pot protocol.


{kind=link}
{kind=link}
{kind=link}
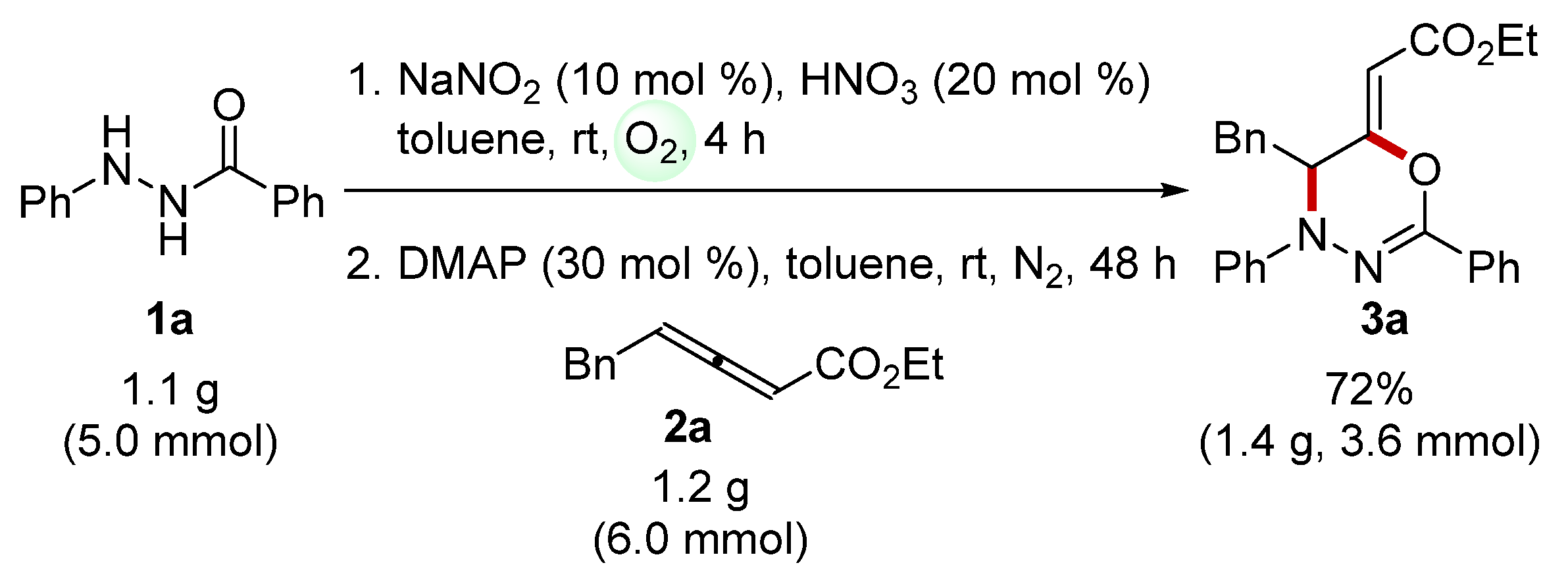{kind=link}
Table 1.
Direct aerobic oxidative cyclization of N′-phenylbenzohydrazide (1a) and benzyl allenoate (2a) a.
Table 1.
Direct aerobic oxidative cyclization of N′-phenylbenzohydrazide (1a) and benzyl allenoate (2a) a.
 | ||
|---|---|---|
| Entry | Catalyst (mol %) | Yield (%) b |
| 1 | CuCl (10)/DMAP (60) | 14 |
| 2 | Fe(Pc) (10)/DMAP (40) | 15 |
| 3 | Mn(Pc) (10)/DMAP (40) | 19 |
| 4 | NaNO2 (10)/HNO3 (20)/DMAP (40) | 30 |
a Reaction conditions: 1a (0.3 mmol), 2a (0.45 mmol), and catalyst in toluene (3.0 mL) under O2 for 48 h. b Isolated yield.
Table 2.
Optimization of the one-pot synthesis of 1,3,4-oxadiazine (3a) from N′-phenylbenzohydrazide (1a) and benzyl allenoate (2a) a.
Table 2.
Optimization of the one-pot synthesis of 1,3,4-oxadiazine (3a) from N′-phenylbenzohydrazide (1a) and benzyl allenoate (2a) a.
 | ||||
|---|---|---|---|---|
| Entry | Catalyst (mol %) | Base | Solvent | Yield (%) b |
| 1 | CuCl (10)/DMAP (20) | DMAP | toluene | 24 |
| 2 | Fe(Pc) (10) | DMAP | toluene | 30 |
| 3 | Mn(Pc) (10) | DMAP | toluene | 13 |
| 4 | NaNO2 (10)/HNO3 (20) | DMAP | toluene | 75 |
| 5 | NaNO2 (10)/HNO3 (20) | pyridine | toluene | 13 |
| 6 | NaNO2 (10)/HNO3 (20) | DBU | toluene | 0 |
| 7 | NaNO2 (10)/HNO3 (20) | DMAP | CH3CN | 20 |
| 8 | NaNO2 (10)/HNO3 (20) | DMAP | CH2Cl2 | 46 |
| 9 | NaNO2 (10)/HNO3 (20) | DMAP | 1,4-dioxane | 45 |
| 10 | NaNO2 (10)/HNO3 (20) | DMAP | EtOH | 25 |
| 11 c | NaNO2 (10)/HNO3 (20) | DMAP | toluene | 76 |
a Reaction conditions: 1a (0.3 mmol) and catalyst in solvent (1.5 mL) under O2 at room temperature for 2 h were used for the first step. The reaction mixture of the first step was added to the base (40 mol %) and 2a (0.45 mmol) in solvent (1.5 mL) under N2 at room temperature; then, the reaction mixture of the second step was stirred for 48 h. b Isolated yield. c The use of 1.2 equiv of 2a and 30 mol % of DMAP.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, S.B.; Maiti, S.; Park, E.S.; Kim, G.Y.; Choun, Y.; Ahn, S.K.; Kim, J.K.; Kim, J. One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates. Molecules 2023, 28, 3815. https://doi.org/10.3390/molecules28093815
AMA Style
Kim SB, Maiti S, Park ES, Kim GY, Choun Y, Ahn SK, Kim JK, Kim J. One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates. Molecules. 2023; 28(9):3815. https://doi.org/10.3390/molecules28093815
Chicago/Turabian StyleKim, Su Been, Santanu Maiti, Eun Sun Park, Ga Young Kim, Yunji Choun, Soon Kil Ahn, Jae Kwang Kim, and Jinho Kim. 2023. "One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates" Molecules 28, no. 9: 3815. https://doi.org/10.3390/molecules28093815